
Abstract
Human natural killer-1 (HNK-1) carbohydrate, comprising a unique trisaccharide HSO3-3GlcAβ1-3Galβ1-4GlcNAc, shows well-regulated expression and unique functions in the nervous system. Recent studies have revealed sophisticated and complicated expression mechanisms for HNK-1 glycan. Activities of biosynthetic enzymes are controlled through the formation of enzyme-complexes and regulation of subcellular localization. Functional aspects of HNK-1 carbohydrate were examined by overexpression, knockdown, and knockout studies of these enzymes. HNK-1 is involved in several neural functions such as synaptic plasticity, learning and memory, and the underlying molecular mechanisms have been illustrated upon identification of the target carrier glycoproteins of HNK-1 such as the glutamate receptor subunit GluA2 or tenascin-R. In this review, we describe recent findings about HNK-1 carbohydrate that provide further insights into the mechanism of its expression and function in the nervous system.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The expression and cellular functions of proteins are regulated by post-translational modifications such as phosphorylation, proteolytic cleavage, ubiquitination, and acetylation. Glycosylation is the most frequent protein modification in mammals, with over half of proteins estimated to be glycosylated [1]. Glycans expressed on proteins have enormous structural diversity and play pivotal roles in many physiological processes at molecular, cellular and individual levels [2]. Also, a specific glycan and the enzyme producing it are expressed in a cell-type-dependent manner, which is closely related to a specific function such as sialyl 6-sulfo LewisX in lymph node and a specific N-glycan branch in pancreatic beta-cells [3, 4]. Cell-specific glycans are also used as cell markers, such as SSEAs for stem cells [5]. In the nervous system, unique glycans such as polysialic acid are involved in many neural functions, and the impaired functioning of neural specific glycans causes neural disorders like schizophrenia, hydrocephalous and neuropathy [6–9].
Human natural killer-1 (HNK-1) is one such neural glycan epitope that is predominantly expressed in brain and peripheral nervous tissues. HNK-1 is composed of a sulfated trisaccharide, HSO3-3GlcAβ1-3Galβ1-4GlcNAc- [10], found at the non-reducing terminus of glycans (Fig. 1a). This glycan epitope is biosynthesized in the Golgi apparatus the same as many other glycosylation reactions. A terminal sulfated glucuronic acid is quite rare among glycans. The glycan terminus is usually capped with another acidic monosaccharide, sialic acid, in mammals [11, 12]. HNK-1 epitope is mainly expressed in N-glycan and in O-mannose glycan [13–15], and is not found in other types of O-glycan under physiological conditions. In addition, HNK-1 glycan has some structural varieties such as non-sulfated form or 6-sulfation of the inner GlcNAc residue [16, 17]. During the last few decades, we and others have reported that this structurally unique glycan plays physiologically important functions in the nervous system revealed by using specific antibody, gene-knockout, and knockdown studies. Also, the identification and cDNA cloning of key biosynthetic enzymes for HNK-1 glycan have been elucidating well-controlled expression mechanisms for this unique glycan. Here, we focus on how HNK-1 glycan is expressed and exerts its functions in the nervous system, and also discuss newly emerging issues.

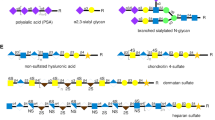
Structure of HNK-1 epitope and biosynthetic enzymes. a Structure of HNK-1 and the enzymes that biosynthesize it. b Structure of the proteoglycan linkage tetrasaccharide. Sulfation of this glycan by HNK-1ST may be an off-switch for further biosynthesis of glycosaminoglycan
Characteristic expression of HNK-1 glycan in the nervous system
HNK-1 was first identified as an antigen at the surface of human natural killer cells in 1981 [18], and a HNK-1 monoclonal antibody was widely used in subsequent studies. The epitope of the HNK-1 antibody was also designated as CD57 and is still used as a marker for some T cell populations in immunology [19]. However, later studies revealed that HNK-1 antigen is highly expressed in the nervous system including in chick embryonic neural crest cells [20] and mammalian brain [21], while not detected in other tissues. Now, with the identification of neural-specific biosynthetic enzymes, HNK-1 epitope is considered to be predominantly expressed in the nervous system.
In the central nervous system, HNK-1 is widely distributed over most of mouse brain [22], with some characteristic expression patterns such as parasagittal stripes in cerebellum [23]. In the peripheral nervous system, HNK-1 was found in myelinating Schwann cells that are associated with motor axon but not in cells that are associated with sensory axon [24], suggesting that HNK-1 carbohydrate may be specifically involved in axon outgrowth/regrowth of motor neurons. Of note, expression of HNK-1 in Schwann cells seems to vary between human and rodents [25]. These previous studies as well as recent studies often used specific antibodies, and some kinds of monoclonal antibodies commonly recognize the HNK-1 epitope with slight different binding specificities. For example, 412 mAb and M6749 mAb can detect both sulfated and non-sulfated form of HNK-1 carbohydrate [16, 26].
Interestingly, HNK-1 glycan is only carried by certain kinds of molecules, indicating that this glycan is biosynthesized in the Golgi in a tightly regulated manner. HNK-1-carrying molecules identified in the nervous system to date include Ig-superfamily adhesion molecules (NCAM, L1, P0, MAG, etc. [21, 27]), a specific glutamate receptor subunit (GluA2) [28], a GPI-anchored protein (CD24) [29], soluble extracellular proteins (tenascin-R, tenascin-C and phosphacan [30–32]), and glycolipids [10]. The fact that even a highly homologous glycoprotein like GluA1 forming a complex with GluA2 is not modified with HNK-1 in vivo suggests that the expression mechanism of HNK-1 is elaborate. Previously, we compared specific activities of the biosynthetic glucuronyltransferase toward certain glycoprotein acceptors. Surprisingly, however, the enzyme similarly transferred glucuronic acid to both a natural acceptor protein (NCAM or L1) and a non-natural acceptor (asialo-orosomucoid), suggesting that the HNK-1-synthesizing enzyme does not recognize their polypeptide sequence at least in vitro [33]. Thus we now speculate that HNK-1-carrying molecules are somehow directed to a specific route for HNK-1 modification in the Golgi apparatus in living cells, but the molecular mechanism by which HNK-1 modification occurs on specific molecules in vivo has yet to be elucidated.
Biosynthetic enzymes of HNK-1 glycan
Identification of biosynthetic enzymes
The most unique structural feature of the HNK-1 epitope is a β3-linked glucuronic acid attached to the non-reducing end of a galactose residue (Fig. 1a). Our group had set out to biochemically identify the glucuronyltransferase responsible. Sequential column chromatography procedures successfully isolated one glucuronyltransferase from rat brain that we named GlcAT-P (B3GAT1 gene) [34, 35]. This enzyme completely satisfied the biochemical requirements for a HNK-1-synthesizing enzyme such as predominant mRNA expression in the brain, a type-II membrane topology typical of a glycosyltransferase, and a mildly acidic optimal pH suitable for reactions in the Golgi. The transfection of GlcAT-P actually elicited ectopic expression in HNK-1 antigen in non neural cells like COS-1 cells [34]. Also, a GlcAT-P gene-knockout study clearly showed that it is the major enzyme responsible for HNK-1 synthesis in the brain as described below. After that, we and another group independently cloned a homologous glucuronyltransferase designated as GlcAT-S (or GlcAT-D) (B3GAT2) from a rat brain cDNA library that also showed HNK-1-synthesizing activity [36, 37]. Although GlcAT-P and -S showed similar activity when overexpressed in cultured cells [38], they differed in their expression patterns, substrate glycan specificity and physiological functions.
Another homologous β1, 3-glucuronyltransferase was also identified, GlcAT-I (B3GAT3) [39]. GlcAT-I is involved in the synthesis of a linkage tetrasaccharide of glycosaminoglycan (Fig. 1b), GlcAβ1-3Galβ1-3Galβ1-4Xyl-Ser, which is commonly found in chondroitin or heparan sulfate [40]. GlcAT-I also displayed HNK-1-synthesizing activity only when overexpressed in cultured cells [40, 41], but its ubiquitous expression, biochemical analysis and knockout experiments all showed that the main physiological function of this enzyme is to biosynthesize the linkage structure for glycosaminoglycan [42].
The other structural determinant of HNK-1 is the terminal sulfate group. We and Dr. Fukuda’s group independently identified the same sulfotransferase involved in this sulfation reaction, which is now called HNK-1ST (CHST10) [43, 44]. Although other sulfotransferases such as chondroitin sulfotransferases show amino acid sequence similarity to this enzyme, biochemical properties and knockout experiments suggested that HNK-1ST is the sole sulfotransferase for HNK-1 synthesis in vivo [45]. Interestingly, however, HNK-1ST is expressed in non-neural tissues where GlcAT-P and -S are not expressed [44]. In addition, HNK-1ST can form enzyme complex with GlcAT-I despite no apparent usefulness for HNK-1 synthesis [38]. These results suggest that this sulfotransferase has additional functions other than in HNK-1 biosynthesis. Actually, we and Dr. Sugahara’s group demonstrated that HNK-1ST can use a glycosaminoglycan linkage tetrasaccharide on thrombomodulin as an acceptor substrate to form an unusual sulfated structure (Fig. 1b) [46, 47]. The sulfated linkage region on thrombomodulin was indeed identified in human urine, SO4-3GlcAβ1-3Galβ1-3Galβ1-4Xyl [48], and a non-elongated form of proteoglycan is known to exist as a part-time proteoglycan [49, 50]. The sulfated linkage region of thrombomodulin can no longer be used for the further elongation of chondroitin sulfate [46], indicating that the action of HNK-1ST may be an off-switch for glycosaminoglycan biosynthesis. It is an intriguing question whether this unusual sulfated linkage region is also expressed in other proteoglycan molecules or in brain under physiological conditions.
Substrate specificity of GlcAT-P and GlcAT-S
The enzymatic properties of GlcAT-P and -S have been characterized using recombinant enzymes [33, 51]. Both enzymes efficiently transfer glucuronic acid from UDP-GlcA to the non-reducing terminus of the acceptor glycan of glycoprotein, glycolipid and oligosaccharide substrates. However, they differ in features like acceptor specificity. Our detailed enzymatic characterization revealed that while GlcAT-P strictly recognizes the N-acetyllactosamine structure (Galβ1-4GlcNAc) as an acceptor which is the known inner structure of HNK-1 glycan, GlcAT-S can efficiently transfer GlcA to a lacto-N-biose structure (Galβ1-3GlcNAc) as well as N-acetyllactosamine. In addition, GlcAT-P exhibited almost equally efficient activity toward bi-, tri- and tetra-antennary N-glycans [52], while GlcAT-S preferred tri-antennary N-glycan [51]. The presence of a bisecting GlcNAc or core α1,6-fucose in N-glycan had little effect on the activities of GlcAT-P(S), indicating that the glycan structure of the non-reducing terminus is the factor most critical to the specificity of GlcAT-P and -S. Notably, the activities of these glucuronyltransferases are regulated differently by phospholipids. For example, sphingomyelin increased GlcAT-P activity toward the glycoprotein acceptor by five-fold but had no effect on GlcAT-S activity [51]. These results suggest the activities of these enzymes to be regulated by the surrounding microenvironment in the Golgi apparatus.
Crystal structure and catalytic reaction of GlcAT-P and -S
To elucidate the reaction mechanisms of GlcAT-P and -S in more detail, the crystal structures of these two enzymes were solved [53, 54]. Recombinant catalytic domains of human GlcAT-P and -S were expressed in and purified from E. coli, and these soluble enzymes showed levels of activity in vitro comparable to those of the native enzymes [53]. Crystallographic studies revealed that the two enzymes had a similar overall structure including GT-A type folding with a Rossman-like fold, a tight homodimeric form, and a conserved DXD motif commonly found in UDP-sugar-utilizing glycosyltransferases [55]. Binding sites for the donor substrate (UDP-GlcA) were almost completely conserved between GlcAT-P and -S, but the two differed in how they recognized acceptor substrates. Key residues of GlcAT-P for binding to Galβ1-4GlcNAc are Phe245 and Val320, which form parallel stacking interaction and hydrophobic interaction with the GlcNAc moiety, respectively. Computer-aided modeling of the docking of Galβ1-3GlcNAc (poor substrate) with GlcAT-P showed that Phe245 may form poorer stacking interaction, and Val320 no longer interacts with GlcNAc, which is consistent with the strict acceptor specificity of GlcAT-P toward Galβ1-4GlcNAc. In the case of GlcAT-S, Phe245 and Val320 of GlcAT-P correspond to Trp234 and Ala309, which probably allows GlcAT-S to recognize Galβ1-3GlcNAc as well as Galβ1-4GlcNAc. Consistent with this, a mutant GlcAT-S in which Trp234 and Ala309 were replaced by Phe and Val, respectively, showed a drastic reduction in activity toward Galβ1-3GlcNAc, resembling GlcAT-P, which supports our observation that these residues are critical for acceptor recognition. From a structural point of view, a catalytic mechanism was also proposed in which an acidic residue (Glu284 for GlcAT-P and Glu273 for GlcAT-S) served as a catalytic base to deprotonate the 3-position of Gal and then the nucleophilic deprotonated Gal attacks the C1 carbon of GlcA in UDP-GlcA to create GlcAβ1-3Gal.
Interestingly, both GlcAT-P and -S have a disordered mobile loop region that shows no electron density. Most glycosyltransferases have such a region, called a flexible loop, considered to be involved in substrate binding [56]. In the case of β4GalT1, where the flexible loop can be traced even without substrates, conformational change from an opened to closed form occurs upon binding with UDP-Gal. After the binding to UDP-Gal, the flexible loop moves and acts as a lid to cover the binding pocket. In the case of GnT-I, the loop was disordered in the apo-form, but structured in the donor-bound form [57]. In GlcAT-P, the loop was still invisible even in a co-crystal with a donor-substrate. Deletion of the entire loop (seven amino acids) or a point mutation of two acidic amino acids in the GlcAT-P loop almost completely abrogated the activity in cells [58]. Although the loop obviously plays a pivotal role in GlcAT-P activity, a more detailed analysis is needed to uncover its function.
Regulation of HNK-1 expression in cells
Gene regulation of biosynthetic enzymes
As described above, HNK-1 glycan is predominantly expressed in the nervous system. Consistent with this, GlcAT-P and -S showed tightly regulated expression patterns with high levels in brain tissue [34, 36]. Studies have partially revealed the transcriptional control mechanisms for these glucuronyltransferase genes. Our analysis suggested the cis-element in the GlcAT-P promoter including the binding site for Sp1 and Krox20 to be required for neural expression of the mouse GlcAT-P gene [59]. An in vivo study showed that in Pax6 mutant rats, HNK-1 and GlcAT-P gene expression is ectopically upregulated, suggesting that Pax6 is a negative regulator for the GlcAT-P gene in rat embryonic brain [60]. Another report demonstrated that TNFα signaling up-regulates GlcAT-P(S) gene expression in brain endothelial cells, which supports a role for HNK-1 in inflammatory responses through interaction between HNK-1 and selectin [61]. More recently, we found the brain-specific expression of the mouse GlcAT-P gene to be highly correlated with epigenetic histone activation marks, indicating that the GlcAT-P gene is epigenetically regulated [62].
One interesting finding is that the GlcAT-S gene is highly expressed in mouse kidney as well as brain [16], while GlcAT-P and HNK-1ST are not expressed in mouse kidney, suggesting a specific role for GlcAT-S in kidney even without the sulfotransferase action. Actually, expression of a non-sulfated form of HNK-1 (GlcAβ1-3Galβ1-4GlcNAc) in mouse kidney was confirmed by a specific antibody and tandem mass-spectrometry, and we identified three major glycoproteins carrying this glycan (CD13, meprin-α and laminin-111) [16, 63]. CD13 and meprin are metalloproteases highly expressed in kidney brush border membrane [64, 65], and laminin-111 is a well-characterized component of basement membranes that serves as a ligand for integrin or dystroglycan [66]. Another group reported that GlcA in the N-glycan of a calcium channel, TRPV5, in kidney is required for its retention at the cell surface and for calcium homeostasis [67], but we have not detected the expression of a non-sulfated HNK-1 on TRPV5 in mouse kidney (unpublished results). In addition, HNK-1 expression in kidney seems to differ among species. In mouse, we found non-sulfated HNK-1 was expressed on apical membranes of proximal tubules and thin ascending limb in medulla, but in other species sulfated HNK-1 is expressed in thin ascending loop of Henle and may be carried by proteoglycan molecules [68]. So far, it is still unclear how the non-sulfated HNK-1 regulates the functions of these glycoproteins. We have generated GlcAT-S deficient mice and confirmed the disappearance of the non-sulfated HNK-1 in kidney (unpublished results), and are currently investigating the role of GlcAT-S in kidney as well as in brain.
Regarding HNK-1ST, there has been almost no promoter analysis. However, one intriguing report showed that the HNK-1ST gene is a target for a transcription factor, retinoic acid receptor-gamma, and upregulation of this gene through this pathway in melanoma cells is functionally involved in suppression of invasiveness (as described below) [69]. HNK-1ST gene transcription must be regulated in a different manner from GlcAT-P(S), and an analysis of its gene regulation mechanism would be an interesting future study.
Functional enzyme complex for HNK-1 synthesis
Another regulatory mechanism for HNK-1 expression is that intracellular activities of biosynthetic enzymes are controlled by the formation of a protein complex or fine subcellular localization in living cells. The idea that GlcAT-P(S) might interact with HNK-1ST first came from the fact that almost all the HNK-1 structures identified so far are sulfated. Thus we hypothesized that HNK-1ST associated with GlcAT-P(S) to efficiently transfer a sulfate group to GlcA. This idea also suggests the functional importance of the sulfate moiety in HNK-1. Actually, using co-immunoprecipitation experiments we found that GlcAT-P(S) and HNK-1ST physically interact in living cells when they are transiently expressed (Fig. 2a) [38]. This interaction is highly specific because we did not detect the binding using either a sialyltransferase instead of GlcA-P(S) or other sulfotransferases instead of HNK-1ST. Functionally, we revealed that the specific activity of HNK-1ST is upregulated by interaction with GlcAT-P(S), whereas the activity of GlcAT-P(S) is not influenced by HNK-1ST [38]. This result is consistent with our assumption that the enzyme complex enables efficient sulfation as soon as glucuronylation occurs during HNK-1 synthesis.

Regulation of HNK-1 expression in cells. a Complex of HNK-1-related enzymes comprising β4GalT2, GlcAT-P(S) and HNK-1ST. This specialized complex may work for efficient biosynthesis of HNK-1 carbohydrate. b Two GlcAT-P isoforms. The longer isoform has an additional 13 amino acids in its cytosolic tail. A dibasic motif exists in the cytosolic tail in both isoforms. c Distinct ER exiting processes for two GlcAT-P isoforms. The short isoform can be easily bound by Sar1 to be conveyed from the ER to Golgi, which results in higher intracellular activity in the Golgi. In the case of the long form, the additional 13 amino acids weaken the interaction between Sar1 and the dibasic motif, which retains GlcAT-P in the ER leading to lower intracellular activity
We also focused on the biosynthesis of the inner N-acetyllactosamine structure (Galβ1-4GlcNAc). This carbohydrate structure is used for a wide variety of glycans and is synthesized by a β1,4-galactosyltransferase (β4GalT). The β4GalT family is composed of seven isozymes (β4GalT1 to β4GalT7), some of which are involved in the synthesis of Galβ1-4GlcNAc in N-glycan [70, 71]. Although it had been unknown which β4GalT is responsible for synthesis of the inner Galβ1-4GlcNAc of HNK-1 glycan, we found that HNK-1 expression almost completely disappeared in β4GalT2 deficient mouse brain [72]. In contrast, knockout of β4GalT1, a major and ubiquitous β4GalT, did not affect HNK-1 expression at all [13, 73]. These results strongly indicated that β4GalT2 is a key enzyme for HNK-1 synthesis and led us to hypothesize that β4GalT2 forms a specific complex with GlcAT-P(S) (and HNK-1ST). When co-expressed with GlcAT-P, β4GalT2 but not β4GalT1 can be co-immunoprecipitated with GlcAT-P [13]. In addition, expression of an ER-retained mutant of GlcAT-P redistributed β4GalT2 but not β4GalT1 from the Golgi to the ER, again indicating that β4GalT2 specifically associates with GlcAT-P in cells. Functionally, addition of β4GalT2 upregulated the specific activity of GlcAT-P in vitro, and co-expression of β4GalT2 and GlcAT-P enhanced HNK-1 expression in cells compared to that of only GlcAT-P. All these results support that β4GalT2 plays a key role in HNK-1 synthesis by forming a complex with GlcAT-P.
These studies imply that a specific enzyme complex composed of β4GalT2, GlcAT-P(S) and HNK-1ST is present and that this specialized machinery may be placed in a defined compartment in the Golgi (Fig. 2a). Only specific acceptor molecules can pass by this compartment to become modified with HNK-1 glycan. Dr. Goto’s group proposed that in drosophila, functionally related molecules form a distinct functional unit in Golgi, called the Golgi unit [74]. The mammalian glycosylation machinery might be similarly organized with different functional units. Unfortunately, however, endogenous levels of HNK-1-synthesizing enzymes are quite low under physiological conditions. This is also the case for many glycosyltransferases. Investigation of the endogenous molecular features of glycosyltransferases and their complexes in the Golgi would be a difficult but fascinating challenge.
Regulated subcellular localization of GlcAT-P
The intracellular activity of GlcAT-P is also regulated by controlling its subcellular localization. We found that isoforms of GlcAT-P generated by alternative splicing showed distinct localization patterns which are well correlated with their intracellular activities [75]. These isoforms had previously been identified in mice and humans [59, 76], differing in only the presence or absence of 13N-terminal amino acids (Fig 2b). The expression of these two isoforms (short GlcAT-P, sGlcAT-P and long GlcAT-P, lGlcAT-P) resulted in a clearly different expression of the HNK-1 epitope in cells, and sGlcAT-P exhibited quite high intracellular activity despite having the same catalytic domain and similar specific activity in vitro [75]. More detailed analysis clarified that sGlcAT-P is localized to the Golgi but lGlcAT-P resides in both the Golgi and the ER. ER-distributed lGlcAT-P is assumed not to act as an enzyme because of a lack of the acceptor Galβ1-4GlcNAc in the ER, which accounts for the weak intracellular activity of lGlcAT-P. ER-exit of some glycosyltransferases is dependent on binding with COP-II vesicle component Sar1 that directly recognizes a dibasic motif [K/R](X)[K/R] of the cytosolic tail of glycosyltransferases [77]. GlcAT-P also has this motif in its N-terminal tail and is actually bound by Sar1, but lGlcAT-P showed weaker binding with Sar1 than sGlcAT-P presumably because of the close existence of the 13 additional amino acids (Fig. 2c) [75].
Although we still do not know how these strong and weak GlcAT-Ps are used in nature, both isoforms are indeed expressed in brain. β4GalT1 has two similar isoforms with or without an additional 13 amino acids in the cytoplasmic tail, which are distributed differently in cells and regulated differentially at the transcriptional level. In somatic cells the longer mRNA variant is dominant, while in mammary gland the shorter variant is upregulated [78]. It would be interesting to examine how transcription of the two GlcAT-P mRNAs is regulated in specific brain regions, neural cell types, and developmental periods.
In vivo HNK-1 function
Role of HNK-1 during development
HNK-1 had received attention from developmental biologists because of its characteristic expression in chick neural crest cells [20], which show a highly dynamic migration during development [79]. Application of an HNK-1 antibody perturbed the migration of cranial neural crest cells in the chick embryo, indicating that HNK-1 glycan is functionally involved in neural cell migration [80]. Although the expression of HNK-1 in rodent neural crest cells is still enigmatic [81], overexpression of GlcAT-P in rat neural crest cells also affected the migratory distance and pattern [82], suggesting the importance of HNK-1 in cell migration during neural development.
Another HNK-1 function was reported in neural stem cells [32]. HNK-1 glycan was expressed in undifferentiated mouse neural stem cells and down-regulated upon differentiation. The carrier glycoprotein for HNK-1 was identified as tenascin-C, and only the largest isoform seemed to be modified with HNK-1. By depleting HNK-1 expression in neural stem cells using siRNA for HNK-1ST, it was demonstrated that HNK-1 on tenascin-C is functionally involved in the proliferation of neural stem cells via modulation of EGF receptor expression.
To examine the role of HNK-1 during development at the individual level in vertebrates, we performed knockdown and overexpression of GlcAT-P(S) in medaka fish, a well-established model for developmental studies [83]. In medaka, HNK-1 expression is weak at the gastrulation stage and gradually increases in neural tissues during development. We first cloned cDNAs of GlcAT-P and -S that are conserved also in medaka. Overexpression of each glucuronyltransferase affected embryogenesis along with ectopic overexpression of HNK-1 glycan. In particular, abnormal curvature in the head region was observed with a severer phenotype on the overexpression of GlcAT-S than of GlcAT-P. In knockdown experiments using morpholino-oligonucleotides, depletion of GlcAT-P caused severe abnormalities in head formation, while knockdown of GlcAT-S resulted in no obvious phenotype, suggesting that these two glucuronyltransferases have different functions in vivo. These results indicate that GlcAT-P is essential for medaka development especially for the formation of the head region and that well-controlled expression of HNK-1 is required for proper embryogenesis in fish development.
Phenotypes of enzyme gene-deficient mice
We generated GlcAT-P gene-deficient mice to examine the physiological roles of HNK-1 in mammals [84]. HNK-1 expression in the knockout mouse brain almost disappeared except in specific regions such as the perineuronal net (described below), indicating that GlcAT-P is the major glucuronyltransferase in mouse brain. Despite the marked loss of HNK-1 expression in the mutant mice, normal birth and overall brain morphology were observed. However, electrophysiological analysis and behavioral tests revealed that these knockout mice showed abnormal brain functions such as impairment of synaptic plasticity and spatial learning. Frequent stimulation of afferent fibers in the hippocampus evoked long lasting changes in synaptic strength (synaptic plasticity) called LTP (long term potentiation) that is considered to underlie memory function [85]. The magnitude of LTP in the hippocampal CA1 region was significantly reduced in GlcAT-P-deficient mice, suggesting that HNK-1 glycan is required for synaptic plasticity and memory formation [84]. In addition, in the Morris water maze test and water-filled multiple T-maze test, GlcAT-P-deficient mice showed reduced spatial learning and memory, which is consistent with impaired synaptic plasticity [84]. Knockout of the HNK-1ST gene also caused a reduction of LTP in the hippocampus and poor performance in the water maze tests [45]. In addition, β4GalT2-deficient mice, which showed a marked reduction of HNK-1 expression as described above, also exhibited impaired performance in the water maze test [72]. All these results support that HNK-1 glycan is required for learning and memory formation. β4GalT2 knockout mice also showed retarded motor learning as assessed by the rota-rod test [72], while GlcAT-P knockout mice performed normally suggesting that this phenotype is caused by a loss of intrinsic β4GalT2 function other than the loss of HNK-1 expression.
Mechanism underlying reduced synaptic plasticity caused by the loss of HNK-1 expression
HNK-1 stabilizes GluA2 on the neuronal cell surface
To examine the molecular mechanism underlying the reduced synaptic plasticity and memory formation in GlcAT-P-deficient mice, we set out to identify the molecule responsible. We found a specific glycoprotein carrying HNK-1 to be highly concentrated in the hippocampal postsynaptic density fraction (PSD). This glycoprotein was immunoaffinity-purified and identified as a subunit of AMPA-type glutamate receptors, GluA2 [28]. AMPA-type glutamate receptors mediate most of the fast excitatory synaptic transmission in the mammalian brain and control synaptic strength. The regulated trafficking of AMPA receptors to the postsynaptic membrane is thought to be a major mechanism contributing to long-lasting changes in synaptic strength including LTP [86]. In mature pyramidal neurons, GluA1 and GluA2 are predominantly expressed, and the great majority of AMPA receptors contain GluA2 subunits. GluA2 subunits have important roles in the assembly and trafficking of AMPA-type glutamate receptors, and recent studies also indicated that the redistribution of GluA2 from the intracellular compartment to synaptic membrane underlies the mechanism of synaptic plasticity [86]. Thus, we hypothesized that impaired trafficking of GluA2 may be a molecular mechanism of reduced LTP in GlcAT-P-deficient mice. Actually, GluA2 was more prone to be internalized in GlcAT-P knockout hippocampal neurons than wild-type neurons under basal and stimulated conditions, though expression of the GluA2 protein itself was normal in the knockout neurons [28]. Moreover, we found that the interaction of GluA2 with N-cadherin was impaired in GlcAT-P knockout neurons, conversely, the interaction was enhanced by the overexpression of HNK-1 in cultured cells [28]. Since it was reported that GluA2-N-cadherin interaction is required for neuronal surface expression of GluA2 and its spine-promoting activity [87], our results suggest that HNK-1 glycan stabilizes GluA2 on the synaptic membrane through enhancement of the interaction with N-cadherin (Fig. 3a). The unstable surface expression of GluA2 in GlcAT-P-deficient neurons may be a molecular mechanism of the impaired induction of LTP in GlcAT-P knockout brain.

HNK-1 on GluA2 regulates its synaptic membrane residency and spine maturation process. a A glutamate receptor subunit, GluA2, interacts with N-cadherin at the postsynaptic membrane. In GlcAT-P knockout neurons, loss of HNK-1 on GluA2 results in weaker interaction with N-cadherin and enhanced internalization of GluA2. This phenomenon could be one mechanism behind the reduced long-term potentiation in GlcAT-P knockout neurons. b In GlcAT-P knockout neurons, immature thin spines are observed. Also, loss of HNK-1 on GluA2 changes its localization from spines to dendritic shafts
Requirement of HNK-1 for dendritic spine maturation
Meanwhile, we examined the morphological features of dendritic spines and synapses in GlcAT-P-deficient neuron, because morphological maturation of dendritic spines is highly related to synaptic plasticity [88]. Dendritic spines are specialized actin-rich mushroom-like protrusions where an input from another neuron (synapse) is received. One explanation of their origin is the maturation of filopodium-like immature protrusions [89]. The abundance of AMPA receptors at the postsynapse is related to spine size, and it was reported that overexpression of GluA2 resulted in promotion of spine morphogenesis due to interaction with N-cadherin. [87]. Given that loss of HNK-1 impairs GluA2’s association with N-cadherin and stabilization at the cell surface in GlcAT-P knockout neurons, we hypothesized that spine morphogenesis may be impaired in these mutant mice. Expectedly, spines in both GlcAT-P knockout mouse hippocampus and cultured neurons showed an immature long and thin morphology compared to the mature mushroom-like spines in wild-type mouse hippocampus and neurons (Fig. 3b) [90]. In contrast, spine density in the knockout neurons was almost the same as in the wild-type, suggesting that the later maturation step in spinogenesis but not early budding step is impaired in GlcAT-P knockout neurons. Consistent with the increased number of immature spines in knockout neurons, the localization of GluA2 also changed from spines to dendritic shafts (Fig. 3b). Also, the promoting effect on spine maturation by GluA2 overexpression was smaller in GlcAT-P knockout neurons. All these results strongly suggest that the HNK-1 glycan on GluA2 is required for normal spine maturation in developing hippocampal neurons. Although direct evidence that impaired spine maturation is the main cause of reduced LTP in GlcAT-P-deficient adult brain is still missing, loss of HNK-1 may also contribute to the impaired GluA2 localization under LTP-inducing conditions.
Modulation of perisomatic inhibition through HNK-1 on tenascin-R (TN-R)
Dr. Schachner’s group extensively studied the role of HNK-1 in perisomatic inhibition. The first evidence came from the application of a HNK-1 mAb to hippocampal slices, which reduced perisomatic inhibitory postsynaptic currents (pIPSCs), suggesting that HNK-1 glycan regulates perisomatic inhibition in hippocampal pyramidal cells [30]. This effect of the HNK-1 mAb was not observed in TN-R-deficient mice but was observed in NCAM-deficient mice, suggesting that HNK-1 on TN-R but not on other HNK-1-carrying molecules is responsible for the decreased inhibitory transmission by the mAb. Also, similar reduced perisomatic inhibitory currents were actually observed in TN-R-deficient mice. Subsequent study using a synthetic HNK-1 carbohydrate or a peptide mimetic for HNK-1 proposed a mechanism underlying the reduced inhibitory transmission whereby the HNK-1 carbohydrate directly inhibited GABAB receptors by binding to them [91]. In the presence of the HNK-1 mAb, the constitutive inhibition of the GABAB receptor by HNK-1 glycan is probably canceled out, leading to elevated extracellular K+ concentrations and a reduction in the amount of GABA released. Although reduced perisomatic inhibition may be expected to promote LTP in vivo, a reduction of LTP in CA1 was observed in TN-R-deficient and HNK-1ST-deficient mice [45, 92]. This can be explained by a complicated metaplastic mechanism in which reduced inhibitory synaptic activity and elevated excitatory transmission in the CA1 region in TN-R knockout brain may lead to an increase in the threshold for LTP induction and impaired LTP [93]. More detailed analyses are needed to clarify how GluA2 and TN-R studies are combined and how HNK-1 glycan regulates the function of each carrier glycoprotein.
HNK-1 in perineuronal nets
Although HNK-1 expression is markedly reduced in the GlcAT-P knockout brain, it still occurs in some neurons in characteristic regions. HNK-1 immunoreactivity in GlcAT-P knockout mice is mainly detected in the perineuronal net (PN), a specialized lattice-like extracellular matrix around a subset of neurons (Fig. 4) [22, 84]. PNs, comprising TN-R, chondroitin sulfate proteoglycans such as aggrecan and phosphacan, and hyaluronan, are formed 2–5 weeks after birth in mice. Although the precise functions of PNs have not yet been elucidated, numerous studies support that PNs are involved in synaptic plasticity. For example, degradation of PNs by chondroitinase ABC (ChABC) restores the ocular dominance plasticity in visual cortex, an experience-dependent plasticity only observed in the juvenile period (called critical period), suggesting that the maturation of PNs reduces the potential plasticity of neurons [94]. Another example was recently reported about fear memory [95]. Fear memories cannot be erased as easily in adulthood as during the postnatal period. ChABC treatment renders acquired fear memories susceptible to erasure even in adults, suggesting that PNs in the adult brain protect fear memories.

HNK-1 expression at perineuronal nets (PNs) in GlcAT-P knockout brain. a Mouse brain cortex was immunostained with a HNK-1 mAb. HNK-1 expression was markedly reduced in the knockout mice but still detected in PNs. b Magnified images of HNK-1 expression at PNs in cultured GlcAT-P knockout neurons. PNs were stained with Wisteria Floribunda lectin
Among the PN components, TN-R, phosphacan and aggrecan are modified with HNK-1. A deficiency of TN-R caused morphological alterations of PNs [96], while the knockout of GlcAT-P did not alter PN morphology itself [22], suggesting that the altered PN structure in TN-R knockout mice is caused by loss of the core protein but not HNK-1 glycan. Phosphacan and aggrecan are suggested to have HNK-1 glycans on their O-glycans [97], and the major HNK-1-carrying glycoprotein between these two probably switches from phosphacan in juveniles to aggrecan in adults along with the maturation of PNs [97]. This study was dependent on a monoclonal antibody, Cat-315, whose epitope partially overlapped with HNK-1 mAb, though its precise structure has not been clarified. Based on these results, a major HNK-1-carrying molecule in GlcAT-P knockout PNs is likely aggrecan. The HNK-1-containing glycan structures on these glycoproteins remain to be determined, and the function of HNK-1 on aggrecan in mature brain is also unclear. Further study using our knockout mouse brains would be useful to address these issues.
Involvement in disease
Although HNK-1 expression is tightly regulated in a nervous system-specific manner, it was reported that this glycan is unusually expressed in some cancer cells especially in melanoma cells [98]. An immunohistochemical study suggested that the expression of HNK-1 correlated with metastatic potential and poor survival [99]. In contrast, another study showed that transcriptional upregulation of the HNK-1ST gene via the direct binding of retinoic acid receptor gamma is involved in reduced metastasis of murine melanoma cells, suggesting that HNK-1 glycan may suppress metastasis [69]. Recently, Dr. Fukuda’s group reported that HNK-1 may act as a tumor suppressor in the case of gliomas through attenuation of the ERK pathway [100]. Roles of HNK-1 glycan in cancer cells are still controversial. It would be interesting to identify the HNK-1-carrying glycoproteins in melanoma cells to examine in detail how this glycan affects their metastatic potential.
HNK-1 is also suggested to be involved in other diseases. HNK-1 glycan is highly antigenic and one of the epitopes for auto-antibodies in peripheral demyelinating neuropathy. The epitope for IgM M-protein in some paraproteinemia patients was characterized as HNK-1 expressed on MAG and glycolipid [101, 102]. The injection of an anti-HNK-1/MAG IgM M-protein into cat sciatic nerve or sensitization of rabbits and rats with a HNK-1-carrying glycolipid demonstrated the involvement of this glycan in demyelination [103, 104].
It was also proposed that HNK-1 or the enzymes that biosynthesize it may be involved in schizophrenia or Alzheimer’s disease (AD). GlcAT-P and -S (B3GAT1 and 2, respectively) emerged as a candidate for a risk factor for schizophrenia [105, 106]. Another study found GlcAT-P to be a target of oxidative stress induced by the stimulation of neurons with amyloid-beta, a peptide causative of AD [107]. It suggested that the development of AD may lead to oxidation of GlcAT-P, which in turn causes downregulation of GlcAT-P activity and HNK-1 expression, leading to impaired memory and learning function mediated by the loss of HNK-1 glycan. Although these studies have revealed clinically interesting features of HNK-1 glycan, more detailed molecular and cellular experiments should be done to clarify how HNK-1 is involved in these diseases.
Dr. Schachner’s group developed peptide mimetics of HNK-1 carbohydrate [108, 109], and they showed their potential for clinical use. The peptides were applied to reconstruction of femoral nerves in mice and primates after nerve injury [110, 111]. Muscle function recovery was greatly improved after glycomimetic application compared to control, supporting their idea that HNK-1 glycan is involved in axon outgrowth of motor neurons. These mimic peptides would be clinically useful for treatment of some nerve injury.
Future perspectives
In this review, we focused on HNK-1 glycan in terms of its well-regulated expression, functions in vivo, and underlying molecular mechanisms. Although the gene knockout of HNK-1-synthesizing enzymes has provided much information about the functions of HNK-1 in the nervous system, the molecular mechanisms by which this glycan regulates each carrier and which carrier is really responsible for each phenomenon observed in knockout experiments have yet to be elucidated. This is because of a common difficulty in glycobiological studies. That is, one glycosyltransferase is in charge of glycosylation for many proteins, and knockout (knockdown) of the enzyme causes loss (downregulation) of glycans on all carrier molecules, which makes our interpretation more difficult. Furthermore, even if the glycan structures are the same, the glycans on different glycoproteins probably have different functions. Thus, we have to investigate and identify a target carrier glycoprotein, which mainly contributes to the phenomenon observed. In our case, we found a great loss of HNK-1 expression on many glycoproteins in the GlcAT-P knockout brain. Thus, we have to seek for a target in the brain (GluA2, TN-R, etc.), which is really involved in synaptic plasticity in vivo.
One way of overcoming such a hurdle is to completely clarify the expression mechanism of a given glycan. Glycosylation is not a simple process but a multi-regulated event that is controlled by the expression level of the enzyme, substrate specificity of the enzyme, sugar-nucleotide level, enzyme-complex, subcellular-localization of the enzyme, etc. as described above. Also, we propose here that specific multi-enzyme complexes may exist in defined Golgi compartments to form specific glycosylation machineries. And this may enable a specific glycoprotein to get a specific glycan by selectively passing by one of the multi-enzyme complexes. If we know the identity of such glycosylation machinery and elucidate the complete mechanism of glycan biosynthesis, we might create a situation in cells where only one glycoprotein is not modified with a specific glycan without knocking out the biosynthetic enzyme. We believe that examination of the regulatory mechanisms of HNK-1 expression in cells will uncover the whole picture of HNK-1 biosynthesis, providing us with further insight into the functions of HNK-1 glycan on each carrier glycoprotein.
References
Apweiler R, Hermjakob H, Sharon N (1999) On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta 1473:4–8
Ohtsubo K, Marth JD (2006) Glycosylation in cellular mechanisms of health and disease. Cell 126:855–867
Mitoma J, Bao X, Petryanik B, Schaerli P, Gauguet JM, Yu SY, Kawashima H, Saito H, Ohtsubo K, Marth JD, Khoo KH, von Andrian UH, Lowe JB, Fukuda M (2007) Critical functions of N-glycans in l-selectin-mediated lymphocyte homing and recruitment. Nat Immunol 8:409–418
Ohtsubo K, Chen MZ, Olefsky JM, Marth JD (2011) Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nat Med 17:1067–1075
Tang C, Lee AS, Volkmer JP, Sahoo D, Nag D, Mosley AR, Inlay MA, Ardehali R, Chavez SL, Pera RR, Behr B, Wu JC, Weissman IL, Drukker M (2011) An antibody against SSEA-5 glycan on human pluripotent stem cells enables removal of teratoma-forming cells. Nat Biotechnol 29:829–834
Kleene R, Schachner M (2004) Glycans and neural cell interactions. Nat Rev Neurosci 5:195–208
Isomura R, Kitajima K, Sato C (2011) Structural and functional impairments of polysialic acid by a mutated polysialyltransferase found in schizophrenia. J Biol Chem 286:21535–21545
Weinhold B, Seidenfaden R, Rockle I, Muhlenhoff M, Schertzinger F, Conzelmann S, Marth JD, Gerardy-Schahn R, Hildebrandt H (2005) Genetic ablation of polysialic acid causes severe neurodevelopmental defects rescued by deletion of the neural cell adhesion molecule. J Biol Chem 280:42971–42977
Kaida K, Ariga T, Yu RK (2009) Antiganglioside antibodies and their pathophysiological effects on Guillain–Barre syndrome and related disorders—a review. Glycobiology 19:676–692
Chou DK, Ilyas AA, Evans JE, Costello C, Quarles RH, Jungalwala FB (1986) Structure of sulfated glucuronyl glycolipids in the nervous system reacting with HNK-1 antibody and some IgM paraproteins in neuropathy. J Biol Chem 261:11717–11725
Brockhausen I, Schachter H, Stanley P (2009) Essentials of Glycobiology, 2nd edn. O-GalNAc Glycans
Stanley P, Schachter H, Taniguchi N (2009) Essentials of Glycobiology, 2nd edn. N-Glycans
Kouno T, Kizuka Y, Nakagawa N, Yoshihara T, Asano M, Oka S (2011) Specific enzyme complex of β-1,4-galactosyltransferase-II and glucuronyltransferase-P facilitates biosynthesis of N-linked human natural killer-1 (HNK-1) carbohydrate. J Biol Chem 286:31337–31346
Yuen CT, Chai W, Loveless RW, Lawson AM, Margolis RU, Feizi T (1997) Brain contains HNK-1 immunoreactive O-glycans of the sulfoglucuronyl lactosamine series that terminate in 2-linked or 2,6-linked hexose (mannose). J Biol Chem 272:8924–8931
Chai W, Yuen CT, Kogelberg H, Carruthers RA, Margolis RU, Feizi T, Lawson AM (1999) High prevalence of 2-mono- and 2,6-di-substituted manol-terminating sequences among O-glycans released from brain glycopeptides by reductive alkaline hydrolysis. Eur J Biochem 263:879–888
Tagawa H, Kizuka Y, Ikeda T, Itoh S, Kawasaki N, Kurihara H, Onozato ML, Tojo A, Sakai T, Kawasaki T, Oka S (2005) A non-sulfated form of the HNK-1 carbohydrate is expressed in mouse kidney. J Biol Chem 280:23876–23883
Gallego RG, Blanco JL, Thijssen-van Zuylen CW, Gotfredsen CH, Voshol H, Duus JO, Schachner M, Vliegenthart JF (2001) Epitope diversity of N-glycans from bovine peripheral myelin glycoprotein P0 revealed by mass spectrometry and nano probe magic angle spinning 1H NMR spectroscopy. J Biol Chem 276:30834–30844
Abo T, Balch CM (1981) A differentiation antigen of human NK and K cells identified by a monoclonal antibody (HNK-1). J Immunol 127:1024–1029
Focosi D, Bestagno M, Burrone O, Petrini M (2010) CD57+ T lymphocytes and functional immune deficiency. J Leukoc Biol 87:107–116
Bronner-Fraser M (1986) Analysis of the early stages of trunk neural crest migration in avian embryos using monoclonal antibody HNK-1. Dev Biol 115:44–55
Kruse J, Mailhammer R, Wernecke H, Faissner A, Sommer I, Goridis C, Schachner M (1984) Neural cell adhesion molecules and myelin-associated glycoprotein share a common carbohydrate moiety recognized by monoclonal antibodies L2 and HNK-1. Nature 311:153–155
Inoue M, Kato K, Matsuhashi H, Kizuka Y, Kawasaki T, Oka S (2007) Distributions of glucuronyltransferases, GlcAT-P and GlcAT-S, and their target substrate, the HNK-1 carbohydrate epitope in the adult mouse brain with or without a targeted deletion of the GlcAT-P gene. Brain Res 1179:1–15
Eisenman LM, Hawkes R (1993) Antigenic compartmentation in the mouse cerebellar cortex: zebrin and HNK-1 reveal a complex, overlapping molecular topography. J Comp Neurol 335:586–605
Martini R, Schachner M, Brushart TM (1994) The L2/HNK-1 carbohydrate is preferentially expressed by previously motor axon-associated Schwann cells in reinnervated peripheral nerves. J Neurosci 14:7180–7191
Levi AD, Guenard V, Aebischer P, Bunge RP (1994) The functional characteristics of Schwann cells cultured from human peripheral nerve after transplantation into a gap within the rat sciatic nerve. J Neurosci 14:1309–1319
Tsvetkov YE, Burg-Roderfeld M, Loers G, Arda A, Sukhova EV, Khatuntseva EA, Grachev AA, Chizhov AO, Siebert HC, Schachner M, Jimenez-Barbero J, Nifantiev NE (2012) Synthesis and molecular recognition studies of the HNK-1 trisaccharide and related oligosaccharides. The specificity of monoclonal anti-HNK-1 antibodies as assessed by surface plasmon resonance and STD NMR. J Am Chem Soc 134:426–435
Burger D, Simon M, Perruisseau G, Steck AJ (1990) The epitope(s) recognized by HNK-1 antibody and IgM paraprotein in neuropathy is present on several N-linked oligosaccharide structures on human P0 and myelin-associated glycoprotein. J Neurochem 54:1569–1575
Morita I, Kakuda S, Takeuchi Y, Itoh S, Kawasaki N, Kizuka Y, Kawasaki T, Oka S (2009) HNK-1 glyco-epitope regulates the stability of the glutamate receptor subunit GluR2 on the neuronal cell surface. J Biol Chem 284:30209–30217
Bleckmann C, Geyer H, Lieberoth A, Splittstoesser F, Liu Y, Feizi T, Schachner M, Kleene R, Reinhold V, Geyer R (2009) O-glycosylation pattern of CD24 from mouse brain. Biol Chem 390:627–645
Saghatelyan AK, Gorissen S, Albert M, Hertlein B, Schachner M, Dityatev A (2000) The extracellular matrix molecule tenascin-R and its HNK-1 carbohydrate modulate perisomatic inhibition and long-term potentiation in the CA1 region of the hippocampus. Eur J Neurosci 12:3331–3342
Maeda N, Hamanaka H, Oohira A, Noda M (1995) Purification, characterization and developmental expression of a brain-specific chondroitin sulfate proteoglycan, 6B4 proteoglycan/phosphacan. Neuroscience 67:23–35
Yagi H, Yanagisawa M, Suzuki Y, Nakatani Y, Ariga T, Kato K, Yu RK (2010) HNK-1 epitope-carrying tenascin-C spliced variant regulates the proliferation of mouse embryonic neural stem cells. J Biol Chem 285:37293–37301
Kakuda S, Oka S, Kawasaki T (2004) Purification and characterization of two recombinant human glucuronyltransferases involved in the biosynthesis of HNK-1 carbohydrate in Escherichia coli. Protein Expr Purif 35:111–119
Terayama K, Oka S, Seiki T, Miki Y, Nakamura A, Kozutsumi Y, Takio K, Kawasaki T (1997) Cloning and functional expression of a novel glucuronyltransferase involved in the biosynthesis of the carbohydrate epitope HNK-1. Proc Natl Acad Sci USA 94:6093–6098
Terayama K, Seiki T, Nakamura A, Matsumori K, Ohta S, Oka S, Sugita M, Kawasaki T (1998) Purification and characterization of a glucuronyltransferase involved in the biosynthesis of the HNK-1 epitope on glycoproteins from rat brain. J Biol Chem 273:30295–30300
Seiki T, Oka S, Terayama K, Imiya K, Kawasaki T (1999) Molecular cloning and expression of a second glucuronyltransferase involved in the biosynthesis of the HNK-1 carbohydrate epitope. Biochem Biophys Res Commun 255:182–187
Shimoda Y, Tajima Y, Nagase T, Harii K, Osumi N, Sanai Y (1999) Cloning and expression of a novel galactoside β1, 3-glucuronyltransferase involved in the biosynthesis of HNK-1 epitope. J Biol Chem 274:17115–17122
Kizuka Y, Matsui T, Takematsu H, Kozutsumi Y, Kawasaki T, Oka S (2006) Physical and functional association of glucuronyltransferases and sulfotransferase involved in HNK-1 biosynthesis. J Biol Chem 281:13644–13651
Kitagawa H, Tone Y, Tamura J, Neumann KW, Ogawa T, Oka S, Kawasaki T, Sugahara K (1998) Molecular cloning and expression of glucuronyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. J Biol Chem 273:6615–6618
Tone Y, Kitagawa H, Imiya K, Oka S, Kawasaki T, Sugahara K (1999) Characterization of recombinant human glucuronyltransferase I involved in the biosynthesis of the glycosaminoglycan-protein linkage region of proteoglycans. FEBS Lett 459:415–420
Wei G, Bai X, Sarkar AK, Esko JD (1999) Formation of HNK-1 determinants and the glycosaminoglycan tetrasaccharide linkage region by UDP-GlcUA:Galactose β1, 3-glucuronosyltransferases. J Biol Chem 274:7857–7864
Izumikawa T, Kanagawa N, Watamoto Y, Okada M, Saeki M, Sakano M, Sugahara K, Sugihara K, Asano M, Kitagawa H (2010) Impairment of embryonic cell division and glycosaminoglycan biosynthesis in glucuronyltransferase-I-deficient mice. J Biol Chem 285:12190–12196
Bakker H, Friedmann I, Oka S, Kawasaki T, Nifant’ev N, Schachner M, Mantei N (1997) Expression cloning of a cDNA encoding a sulfotransferase involved in the biosynthesis of the HNK-1 carbohydrate epitope. J Biol Chem 272:29942–29946
Ong E, Yeh JC, Ding Y, Hindsgaul O, Fukuda M (1998) Expression cloning of a human sulfotransferase that directs the synthesis of the HNK-1 glycan on the neural cell adhesion molecule and glycolipids. J Biol Chem 273:5190–5195
Senn C, Kutsche M, Saghatelyan A, Bosl MR, Lohler J, Bartsch U, Morellini F, Schachner M (2002) Mice deficient for the HNK-1 sulfotransferase show alterations in synaptic efficacy and spatial learning and memory. Mol Cell Neurosci 20:712–729
Nakagawa N, Izumikawa T, Kitagawa H, Oka S (2011) Sulfation of glucuronic acid in the linkage tetrasaccharide by HNK-1 sulfotransferase is an inhibitory signal for the expression of a chondroitin sulfate chain on thrombomodulin. Biochem Biophys Res Commun 415:109–113
Hashiguchi T, Mizumoto S, Nishimura Y, Tamura J, Yamada S, Sugahara K (2011) Involvement of human natural killer-1 (HNK-1) sulfotransferase in the biosynthesis of the GlcUA(3-O-sulfate)-Gal-Gal-Xyl tetrasaccharide found in α-thrombomodulin from human urine. J Biol Chem 286:33003–33011
Wakabayashi H, Natsuka S, Mega T, Otsuki N, Isaji M, Naotsuka M, Koyama S, Kanamori T, Sakai K, Hase S (1999) Novel proteoglycan linkage tetrasaccharides of human urinary soluble thrombomodulin, SO4-3GlcAβ1-3Galβ1-3(±Siaα2-6)Galβ1-4Xyl. J Biol Chem 274:5436–5442
Aono S, Tokita Y, Shuo T, Yamauchi S, Matsui F, Nakanishi K, Hirano K, Sano M, Oohira A (2004) Glycosylation site for chondroitin sulfate on the neural part-time proteoglycan, neuroglycan C. J Biol Chem 279:46536–46541
Nadanaka S, Kitagawa H, Sugahara K (1998) Demonstration of the immature glycosaminoglycan tetrasaccharide sequence GlcAβ1-3Galβ1-3Galβ1-4Xyl on recombinant soluble human α-thrombomodulin. An oligosaccharide structure on a “part-time” proteoglycan. J Biol Chem 273:33728–33734
Kakuda S, Sato Y, Tonoyama Y, Oka S, Kawasaki T (2005) Different acceptor specificities of two glucuronyltransferases involved in the biosynthesis of HNK-1 carbohydrate. Glycobiology 15:203–210
Oka S, Terayama K, Imiya K, Yamamoto S, Kondo A, Kato I, Kawasaki T (2000) The N-glycan acceptor specificity of a glucuronyltransferase, GlcAT-P, associated with biosynthesis of the HNK-1 epitope. Glycoconj J 17:877–885
Kakuda S, Shiba T, Ishiguro M, Tagawa H, Oka S, Kajihara Y, Kawasaki T, Wakatsuki S, Kato R (2004) Structural basis for acceptor substrate recognition of a human glucuronyltransferase, GlcAT-P, an enzyme critical in the biosynthesis of the carbohydrate epitope HNK-1. J Biol Chem 279:22693–22703
Shiba T, Kakuda S, Ishiguro M, Morita I, Oka S, Kawasaki T, Wakatsuki S, Kato R (2006) Crystal structure of GlcAT-S, a human glucuronyltransferase, involved in the biosynthesis of the HNK-1 carbohydrate epitope. Proteins 65:499–508
Breton C, Bettler E, Joziasse DH, Geremia RA, Imberty A (1998) Sequence–function relationships of prokaryotic and eukaryotic galactosyltransferases. J Biochem 123:1000–1009
Qasba PK, Ramakrishnan B, Boeggeman E (2005) Substrate-induced conformational changes in glycosyltransferases. Trends Biochem Sci 30:53–62
Unligil UM, Zhou S, Yuwaraj S, Sarkar M, Schachter H, Rini JM (2000) X-ray crystal structure of rabbit N-acetylglucosaminyltransferase I: catalytic mechanism and a new protein superfamily. EMBO J 19:5269–5280
Kizuka Y, Nakagawa N, Morita I, Oka S (2012) Requirement of acidic amino acids in the Glucuronyltransferase-P (GlcAT-P) flexible loop for its enzymatic activity. In: Mora-Montes HM (ed) Glycans: Biochemistry, Characterization and Applications. Nova Publishers, New York
Yamamoto S, Oka S, Saito-Ohara F, Inazawa J, Kawasaki T (2002) Molecular cloning and genomic analysis of mouse glucuronyltransferase involved in biosynthesis of the HNK-1 epitope. J Biochem 131:337–347
Nagase T, Nakamura S, Harii K, Osumi N (2001) Ectopically localized HNK-1 epitope perturbs migration of the midbrain neural crest cells in Pax6 mutant rat. Dev Growth Differ 43:683–692
Dasgupta S, Yanagisawa M, Krishnamurthy K, Liour SS, Yu RK (2007) Tumor necrosis factor-α up-regulates glucuronosyltransferase gene expression in human brain endothelial cells and promotes T-cell adhesion. J Neurosci Res 85:1086–1094
Kizuka Y, Kitazume S, Yoshida M, Taniguchi N (2011) Brain-specific expression of N-acetylglucosaminyltransferase IX (GnT-IX) is regulated by epigenetic histone modifications. J Biol Chem 286:31875–31884
Kizuka Y, Kobayashi K, Kakuda S, Nakajima Y, Itoh S, Kawasaki N, Oka S (2008) Laminin-1 is a novel carrier glycoprotein for the nonsulfated HNK-1 epitope in mouse kidney. Glycobiology 18:331–338
Bond JS, Matters GL, Banerjee S, Dusheck RE (2005) Meprin metalloprotease expression and regulation in kidney, intestine, urinary tract infections and cancer. FEBS Lett 579:3317–3322
Tauc M, Chatelet F, Verroust P, Vandewalle A, Poujeol P, Ronco P (1988) Characterization of monoclonal antibodies specific for rabbit renal brush-border hydrolases: application to immunohistological localization. J Histochem Cytochem 36:523–532
Sasaki T, Fassler R, Hohenester E (2004) Laminin: the crux of basement membrane assembly. J Cell Biol 164:959–963
Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG (2005) The β-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science 310:490–493
Allory Y, Commo F, Boccon-Gibod L, Sibony M, Callard P, Ronco P, Debiec H (2006) Sulfated HNK-1 epitope in developing and mature kidney: a new marker for thin ascending loop of Henle and tubular injury in acute tubular necrosis. J Histochem Cytochem 54:575–584
Zhao X, Graves C, Ames SJ, Fisher DE, Spanjaard RA (2009) Mechanism of regulation and suppression of melanoma invasiveness by novel retinoic acid receptor-gamma target gene carbohydrate sulfotransferase 10. Cancer Res 69:5218–5225
Hennet T (2002) The galactosyltransferase family. Cell Mol Life Sci 59:1081–1095
Lo NW, Shaper JH, Pevsner J, Shaper NL (1998) The expanding β4-galactosyltransferase gene family: messages from the databanks. Glycobiology 8:517–526
Yoshihara T, Sugihara K, Kizuka Y, Oka S, Asano M (2009) Learning/memory impairment and reduced expression of the HNK-1 carbohydrate in β4-galactosyltransferase-II-deficient mice. J Biol Chem 284:12550–12561
Kido M, Asano M, Iwakura Y, Ichinose M, Miki K, Furukawa K (1998) Presence of polysialic acid and HNK-1 carbohydrate on brain glycoproteins from β-1,4-galactosyltransferase-knockout mice. Biochem Biophys Res Commun 245:860–864
Yano H, Yamamoto-Hino M, Abe M, Kuwahara R, Haraguchi S, Kusaka I, Awano W, Kinoshita-Toyoda A, Toyoda H, Goto S (2005) Distinct functional units of the Golgi complex in Drosophila cells. Proc Natl Acad Sci USA 102:13467–13472
Kizuka Y, Tonoyama Y, Oka S (2009) Distinct transport and intracellular activities of two GlcAT-P isoforms. J Biol Chem 284:9247–9256
Mitsumoto Y, Oka S, Sakuma H, Inazawa J, Kawasaki T (2000) Cloning and chromosomal mapping of human glucuronyltransferase involved in biosynthesis of the HNK-1 carbohydrate epitope. Genomics 65:166–173
Giraudo CG, Maccioni HJ (2003) Endoplasmic reticulum export of glycosyltransferases depends on interaction of a cytoplasmic dibasic motif with Sar1. Mol Biol Cell 14:3753–3766
Shaper NL, Charron M, Lo NW, Shaper JH (1998) β1,4-galactosyltransferase and lactose biosynthesis: recruitment of a housekeeping gene from the nonmammalian vertebrate gene pool for a mammary gland specific function. J Mammary Gland Biol Neoplasia 3:315–324
Le Douarin NM (2004) The avian embryo as a model to study the development of the neural crest: a long and still ongoing story. Mech Dev 121:1089–1102
Bronner-Fraser M (1987) Perturbation of cranial neural crest migration by the HNK-1 antibody. Dev Biol 123:321–331
Tucker GC, Delarue M, Zada S, Boucaut JC, Thiery JP (1988) Expression of the HNK-1/NC-1 epitope in early vertebrate neurogenesis. Cell Tissue Res 251:457–465
Nagase T, Sanai Y, Nakamura S, Asato H, Harii K, Osumi N (2003) Roles of HNK-1 carbohydrate epitope and its synthetic glucuronyltransferase genes on migration of rat neural crest cells. J Anat 203:77–88
Anzai D, Tonoyama Y, Ikeda A, Kawasaki T, Oka S (2009) Regulated expression of the HNK-1 carbohydrate is essential for medaka (Oryzias latipes) embryogenesis. Glycobiology 19:868–878
Yamamoto S, Oka S, Inoue M, Shimuta M, Manabe T, Takahashi H, Miyamoto M, Asano M, Sakagami J, Sudo K, Iwakura Y, Ono K, Kawasaki T (2002) Mice deficient in nervous system-specific carbohydrate epitope HNK-1 exhibit impaired synaptic plasticity and spatial learning. J Biol Chem 277:27227–27231
Debanne D (1996) Associative synaptic plasticity in hippocampus and visual cortex: cellular mechanisms and functional implications. Rev Neurosci 7:29–46
Isaac JT, Ashby MC, McBain CJ (2007) The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54:859–871
Saglietti L, Dequidt C, Kamieniarz K, Rousset MC, Valnegri P, Thoumine O, Beretta F, Fagni L, Choquet D, Sala C, Sheng M, Passafaro M (2007) Extracellular interactions between GluR2 and N-cadherin in spine regulation. Neuron 54:461–477
Segal M (2005) Dendritic spines and long-term plasticity. Nat Rev Neurosci 6:277–284
Hering H, Sheng M (2001) Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci 2:880–888
Morita I, Kakuda S, Takeuchi Y, Kawasaki T, Oka S (2009) HNK-1 (human natural killer-1) glyco-epitope is essential for normal spine morphogenesis in developing hippocampal neurons. Neuroscience 164:1685–1694
Saghatelyan AK, Snapyan M, Gorissen S, Meigel I, Mosbacher J, Kaupmann K, Bettler B, Kornilov AV, Nifantiev NE, Sakanyan V, Schachner M, Dityatev A (2003) Recognition molecule associated carbohydrate inhibits postsynaptic GABA(B) receptors: a mechanism for homeostatic regulation of GABA release in perisomatic synapses. Mol Cell Neurosci 24:271–282
Saghatelyan AK, Dityatev A, Schmidt S, Schuster T, Bartsch U, Schachner M (2001) Reduced perisomatic inhibition, increased excitatory transmission, and impaired long-term potentiation in mice deficient for the extracellular matrix glycoprotein tenascin-R. Mol Cell Neurosci 17:226–240
Bukalo O, Schachner M, Dityatev A (2007) Hippocampal metaplasticity induced by deficiency in the extracellular matrix glycoprotein tenascin-R. J Neurosci 27:6019–6028
Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L (2002) Reactivation of ocular dominance plasticity in the adult visual cortex. Science 298:1248–1251
Gogolla N, Caroni P, Luthi A, Herry C (2009) Perineuronal nets protect fear memories from erasure. Science 325:1258–1261
Weber P, Bartsch U, Rasband MN, Czaniera R, Lang Y, Bluethmann H, Margolis RU, Levinson SR, Shrager P, Montag D, Schachner M (1999) Mice deficient for tenascin-R display alterations of the extracellular matrix and decreased axonal conduction velocities in the CNS. J Neurosci 19:4245–4262
Dino MR, Harroch S, Hockfield S, Matthews RT (2006) Monoclonal antibody Cat-315 detects a glycoform of receptor protein tyrosine phosphatase beta/phosphacan early in CNS development that localizes to extrasynaptic sites prior to synapse formation. Neuroscience 142:1055–1069
Tang NE, Luyten GP, Mooy CM, Naus NC, de Jong PT, Luider TM (1996) HNK-1 antigens on uveal and cutaneous melanoma cell lines. Melanoma Res 6:411–418
Thies A, Schachner M, Berger J, Moll I, Schulze HJ, Brunner G, Schumacher U (2004) The developmentally regulated neural crest-associated glycotope HNK-1 predicts metastasis in cutaneous malignant melanoma. J Pathol 203:933–939
Suzuki-Anekoji M, Suzuki M, Kobayashi T, Sato Y, Nakayama J, Suzuki A, Bao X, Angata K, Fukuda M (2011) HNK-1 glycan functions as a tumor suppressor for astrocytic tumor. J Biol Chem 286:32824–32833
Ilyas AA, Quarles RH, MacIntosh TD, Dobersen MJ, Trapp BD, Dalakas MC, Brady RO (1984) IgM in a human neuropathy related to paraproteinemia binds to a carbohydrate determinant in the myelin-associated glycoprotein and to a ganglioside. Proc Natl Acad Sci USA 81:1225–1229
Ariga T, Kohriyama T, Freddo L, Latov N, Saito M, Kon K, Ando S, Suzuki M, Hemling ME, Rinehart KL Jr et al (1987) Characterization of sulfated glucuronic acid containing glycolipids reacting with IgM M-proteins in patients with neuropathy. J Biol Chem 262:848–853
Hays AP, Latov N, Takatsu M, Sherman WH (1987) Experimental demyelination of nerve induced by serum of patients with neuropathy and an anti-MAG IgM M-protein. Neurology 37:242–256
Maeda Y, Brosnan CF, Miyatani N, Yu RK (1991) Preliminary studies on sensitization of Lewis rats with sulfated glucuronyl paragloboside. Brain Res 541:257–264
Jeffries AR, Mungall AJ, Dawson E, Halls K, Langford CF, Murray RM, Dunham I, Powell JF (2003) β-1,3-Glucuronyltransferase-1 gene implicated as a candidate for a schizophrenia-like psychosis through molecular analysis of a balanced translocation. Mol Psychiatry 8:654–663
Kahler AK, Djurovic S, Rimol LM, Brown AA, Athanasiu L, Jonsson EG, Hansen T, Gustafsson O, Hall H, Giegling I, Muglia P, Cichon S, Rietschel M, Pietilainen OP, Peltonen L, Bramon E, Collier D, St Clair D, Sigurdsson E, Petursson H, Rujescu D, Melle I, Werge T, Steen VM, Dale AM, Matthews RT, Agartz I, Andreassen OA (2011) Candidate gene analysis of the human natural killer-1 carbohydrate pathway and perineuronal nets in schizophrenia: B3GAT2 is associated with disease risk and cortical surface area. Biol Psychiatry 69:90–96
Thomas SN, Soreghan BA, Nistor M, Sarsoza F, Head E, Yang AJ (2005) Reduced neuronal expression of synaptic transmission modulator HNK-1/neural cell adhesion molecule as a potential consequence of amyloid β-mediated oxidative stress: a proteomic approach. J Neurochem 92:705–717
Simon-Haldi M, Mantei N, Franke J, Voshol H, Schachner M (2002) Identification of a peptide mimic of the L2/HNK-1 carbohydrate epitope. J Neurochem 83:1380–1388
Bhunia A, Vivekanandan S, Eckert T, Burg-Roderfeld M, Wechselberger R, Romanuka J, Bachle D, Kornilov AV, von der Lieth CW, Jimenez-Barbero J, Nifantiev NE, Schachner M, Sewald N, Lutteke T, Hans-Joachim G, Siebert HC (2010) Why structurally different cyclic peptides can be glycomimetics of the HNK-1 carbohydrate antigen. J Am Chem Soc 132:96–105
Irintchev A, Wu MM, Lee HJ, Zhu H, Feng YP, Liu YS, Bernreuther C, Loers G, You SW, Schachner M (2011) Glycomimetic improves recovery after femoral injury in a non-human primate. J Neurotrauma 28:1295–1306
Simova O, Irintchev A, Mehanna A, Liu J, Dihne M, Bachle D, Sewald N, Loers G, Schachner M (2006) Carbohydrate mimics promote functional recovery after peripheral nerve repair. Ann Neurol 60:430–437
Acknowledgments
This work was supported in part by Grant-in-Aid for Scientific Research (B) 21370053 (to S. O.) from JSPS and for Scientific Research on Innovative Areas 23110006 (to S.O.) from MEXT, and by the Mizutani Foundation for Glycoscience (to S. O.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kizuka, Y., Oka, S. Regulated expression and neural functions of human natural killer-1 (HNK-1) carbohydrate. Cell. Mol. Life Sci. 69, 4135–4147 (2012). https://doi.org/10.1007/s00018-012-1036-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-012-1036-z