
Abstract
Microglia are key sentinels of central nervous system health, and their dysfunction has been widely implicated in the progressive nature of neurodegenerative diseases. While microglia can produce a host of factors that are toxic to neighboring neurons, NOX2 has been implicated as a common and essential mechanism of microglia-mediated neurotoxicity. Accumulating evidence indicates that activation of the NOX2 enzyme complex in microglia is neurotoxic, both through the production of extracellular reactive oxygen species that damage neighboring neurons as well as the initiation of redox signaling in microglia that amplifies the pro-inflammatory response. More specifically, evidence supports that NOX2 redox signaling enhances microglial sensitivity to pro-inflammatory stimuli, and amplifies the production of neurotoxic cytokines, to promote chronic and neurotoxic microglial activation. Here, we describe the evidence denoting the role of NOX2 in microglia-mediated neurotoxicity with an emphasis on Alzheimer’s and Parkinson’s disease, describe available inhibitors that have been tested, and detail evidence of the neuroprotective and therapeutic potential of targeting this enzyme complex to regulate microglia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inflammation, oxidative stress, microglial activation, and progressive neuron damage are common denominators present in most neurodegenerative diseases and some cases of central nervous system (CNS) damage. Increasing evidence supports that microglia, the resident innate immune cells in the brain, are a chronic source of inflammation and reactive oxygen species (ROS) culpable in progressive neuron damage. The NOX2 enzyme complex is present in microglia, has been implicated as a prominent source of microglial ROS in pathology, and is a key mechanism regulating microglia-mediated neurotoxicity. Here, we will detail the role of NOX2 in microglia-mediated neurotoxicity and explore the evidence implicating NOX2 inhibitors as a plausible neuroprotective approach regulating the neurotoxic microglial response.
Microglia
Microglia are myeloid-derived cells that are essential for normal, healthy central nervous system (CNS) physiology [1]. Comprising approximately 0.5–16.6 % of the adult human brain [2], microglia perform dynamic cellular functions that include synaptic plasticity [3], cleaning of cellular debris [4, 5], neuronal support through the production of growth factors [6–8], wound healing through alternative activation [4, 5], and innate immune defense [9]. In fact, the loss of normal microglial function is deleterious [10]. For example, the inability of microglia to clear toxic disease proteins, such as beta amyloid (Aβ), has also been linked to neurodegenerative diseases [10]. In addition, the absence of microglial cells can also be neurotoxic, as deletion of infiltrating macrophages that supply the expansion of microglial numbers in early stages of spinal injury actually enhances neuron damage [11]. Microglia are even essential for normal behavior, as genetic deletion of a particular microglial subtype has been linked to self-mutilating behavior in mice through compulsive grooming [12]. Thus, therapeutic approaches targeting microglia must focus on regulation of the toxic phenotype with preservation of their beneficial and maintenance functions.
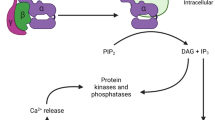
While normal microglial function is mandatory for CNS health, there is increasing evidence that microglia also have an active role in neuropathology in disease, as microglia are activated in several neurodegenerative diseases and are shown to initiate neuron damage, amplify ongoing neurotoxicity, and drive chronic neuron loss over time [13]. As the resident innate immune cell in the brain, microglia actively survey the brain environment and are capable of responding to diverse stimuli, such as neuron damage, disease proteins, and environmental toxins [13]. When activated in response to these assorted triggers, microglia can produce several factors that are toxic to neurons, such as pro-inflammatory factors (TNFα, PGE2, and INFγ) and reactive oxygen species (NO, H2O2, O ·−2 , NOO−) contributing to neurodegeneration [14–16]. While limited production of these factors are expected in normal physiology, robust and chronic activation microglia lead to toxic levels that are believed to drive disease [17]. In particular, microglial NOX2 has been implicated as a key mechanism of microglia-mediated neurotoxicity (Fig. 1).

NOX2 is key for neurotoxic microglial activation. Microglial NOX2 is activated by cytokines, endogenous disease proteins, inflammogens, and soluble neuron injury signals from neuron death/damage (reactive microglisosis), which is implicated in self-propagating neurotoxicity. Thus, specific inhibition of NOX2 is a promising target with the potential to break the cycle of chronic and neurotoxic microglial activation that is implicated in the progressive nature of neurodegenerative diseases
The NOX2 complex
The NOX family of NADPH oxidases is comprised of seven transmembrane proteins (NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1, and DUOX2) that oxidize intracellular NADPH/NADH, causing electron transport across the membrane and the reduction of molecular oxygen to superoxide [18]. The seven NOX proteins are unique in that they are expressed in different cell populations and require unique combinations of accessory proteins to form the complex necessary for activation [19].
NOX2, also known as gp91PHOX, is the catalytic subunit of the phagocyte NADPH oxidase (PHOX) enzyme complex [20]. As the name PHOX suggests, while NOX2 is known to be expressed in multiple cell types, it is highly expressed in phagocytes including neutrophils and microglia [21, 22], where it is involved in host defense [23], proliferation [24, 25], activated morphology [26], and cell signaling [27–29]. NOX2 is heavily glycosylated with six transmembrane domains and a long cytosolic C-terminus that contains both a NADPH-binding domain as well as a FAD-binding domain, which are required for enzymatic activity [30–32]. While NOX2 is necessary for activation of the enzyme complex, it is not sufficient for generation of superoxide from NADPH and O2 in cells [33]. In fact, activation of the NOX2 complex requires a host of organizing and regulating subunits [18, 34].
P22 PHOX is a membrane protein that colocalizes with NOX2 in the phagosome [35] to form the 1:1 heterodimer flavocytochrome b558 [36, 37]. With no catalytic activity of its own, P22PHOX serves to stabilize NOX2 and organize other PHOX subunits for binding and is essential for catalytic activity of NOX2 [38, 39].
Several cytosolic proteins are also necessary for activation of the NOX2 complex and the successful generation of superoxide. P47 PHOX is located in the cytoplasm in quiescent microglia [40], but upon activating stimulus, P47PHOX is phosphorylated [41], then undergoes a conformational change that allows it to translocate to the membrane, binding membrane phospholipids (particularly PI3 K [42, 43]) and the proline-rich C-terminus of P22PHOX [44]. P47 PHOX is considered to be an organizer subunit that recruits the activator subunit P67PHOX [45]. P67 PHOX is located in the cytoplasm prior to activation [46]. Upon activation, P67PHOX interacts with proline-rich repeats of P47PHOX via a C-terminal SH3 domain [47]. Once at the membrane, P67PHOX binds to Rac1/2 via an N-terminal tetratricopeptide repeat [48], and presumably interacts with gp91phox directly in order to activate the catalytic subunit [49]. P47PHOX and P67PHOX are mandatory for microglial activation of NOX2 [50]. P40 PHOX, another cytosolic subunit, also coprecipitates with P47PHOX and P67PHOX [51], but is dispensable for enzyme activity [52]. However, it is not completely innocuous, and has been implicated in both positive and negative modulation of ROS generation, but neither the required modifications nor a mechanism have been elucidated [53]. Meanwhile, Rac1, a small GTPase both tethers P67PHOX to the membrane and is involved in its activation of NOX2 [54]. Thus, the NOX2 complex is comprised of multiple proteins located both in the membrane and cytosol in microglia that contribute to enzymatic activity (Fig. 2).

Inhibitors target microglial NOX2 activation through two mechanisms. Activation of the NOX2 complex occurs in microglia in response to several stimuli and multiple signaling pathways, where activation is due to the assembly of the enzyme complex, as depicted here. Several putative NOX2 inhibitors have been identified that attenuate microglial activation to confer neuroprotection. Here, we classify compounds that have been shown to inhibit microglial NOX2 activity into one of two categories based on whether the molecules were: (1) proposed to directly inhibit the catalytic subunit or (2) indirectly modulate NOX2 through inhibition of phosporylation and translocation of cytosolic subunits or other pathways. Notably, most of these molecules are not specific for NOX2 and the mechanism of NOX2 inhibition are either poorly understood or unknown at this time
Activation of the NOX2 complex occurs in microglia in response to several stimuli (Table 1) and multiple signaling pathways, where activation is due to the assembly of the enzyme complex (Fig. 2). Assembly and activation of the NOX2 complex occurs through sequential events beginning with the phosphorylation of P47PHOX at multiple critical serine residues (serines 208 and 283 in the Src homology 3 domain; serine 384 in the C-terminus) [55]. Several kinases have been implicated in P47PHOX phosphorylation: protein kinase C (PKC) isoform −δ, −θ, or −η [34, 56, 57], protein kinase A (PKA) [58], casein kinase 2 (CK2) [55], and mitogen-activated protein kinases (MAPK) P38 and ERK [58–61]. Following phosphorylation, P47PHOX translocates to cytochrome b558 in the cell membrane and acts as recruitment scaffolding for P67PHOX, the activating subunit. P67PHOX also requires phosphorylation by PKC [62] or the MAPK P38 and ERK2 [63] to initiate activation. Following assembly of the complex, NADPH binds to the c-terminal NADPH binding domain spanning four folds of the c-terminal region of NOX2, and a single FAD molecule binds across two folds [31, 64]. FAD is thought to be transiently reduced by a single electron from NADPH, after which the two heme groups sequentially pass an electron, moving it to the outside of the membrane, where molecular O2 is the final electron acceptor and is converted to superoxide (O ·−2 ) [18].
NOX2 in central nervous system cells
The NOX family of proteins is expressed in diverse cell types throughout the brain in a cell-specific and localized manner [65]. Microglia have been shown to express all the necessary subunits of NOX2 both in vivo and in vitro [66, 67]. Microglial NOX2 is involved in host defense [23], proliferation [24], and regulation of cell signaling via redox signaling mechanisms [27–29]. Neurons, the primary communicators in the CNS which are damaged in both AD and PD, express NOX2 [68, 69] in a variety of brain regions [65], where the enzyme complex has been implicated in neuronal apoptosis [69], learning and memory [70], long-term potentiation [71], and in neuronal myelination signals [72, 73]. Astrocytes, which are involved in maintaining CNS structure, trophic and metabolic support of neurons, neurotransmission [74], and inflammation, also express NOX2 [75], where it is involved in cell signaling [27] and cell survival [76], and may also contribute toward inducible neuroinflammation [75]. At present, whether NOX2 is expressed in oligodendrocytes is unknown. Recent work shows not only that NOX2 is expressed in adult hippocampal stem/progenitor cells but also that NOX2-generated ROS regulate proliferation signals through redox signaling in response to NOX2-derived ROS [77]. For a more detailed review of NOX homologues in the CNS, see [65]. Given that multiple NOX homologues are present in the brain and employ many common subunits for function, and the fact that NOX2 is involved in cellular functions independent of microglia, the specificity of NOX2 inhibitors and the timing of drug administration will be important to confer accuracy when targeting the deleterious microglial response.
Microglial NOX2 is activated by a surprisingly long list of compounds and events (Table 1). As expected, classical triggers of the innate immune response, such as LPS [26, 78–81], Phorbol 12-myristate 13-acetate (PMA) [50, 82, 83], zymosan [50, 84], encephalomyocardus virus [85], and N-formylmethionine leucyl-phenylalanine (fMLP) [83, 86] activate the enzyme complex. Consistent with the expected phenotype of phagocytic cells, cytokines are also reported to initiate microglial NOX2 activation, including TNFα [25], Interleukin-1β [25], Interleukin-4 [87], and Interleukin-13 [88]. However, disease proteins found in the CNS, such as Aβ [43, 89], α synuclein [90, 91], myelin [92], HIV tat [93, 94], and fibrillogenic prion peptide PrP106-126 [95], are also known to initiate microglial superoxide production through NOX2 activation. In fact, NOX2 is implicated in reactive microgliosis (the microglial response to neuronal death/damage), a mechanism contributing to the progressive nature of many neurodegenerative diseases [96]. More specifically, several neuron injury factors have been identified that activate microglial NOX2 to further propagate additional neurotoxicity, such as matrix metalloproteinase-3 (MMP3) [97], μ calpain [98], neuromelanin [99], and α synuclein [90, 91]. Even endogenous neuropeptides are capable of activating microglial NOX2, including angiotensin II [100] and substance P [101], or inhibiting it, such as dynorphin [101], suggesting this enzyme may be tightly regulated in the CNS under normal physiological conditions. Environmental toxins have also been reported to reach the brain and activate microglial NOX2 to produce ROS, including paraquat [102], rotenone [103], dieldrin [104], diesel exhaust particles [105], lindane [106], mancozeb [107], and maneb [107]. Thus, triggers of microglial NOX2 activation extend well past traditional immunological stimuli and include environmental toxins, neuromodulators, neuronal death, and CNS disease pathways (Table 1).
Microglial NOX2: dual modes of neurotoxicity
There is increasing evidence that activation of microglial NOX2 is culpable in neuronal damage. Microglial NOX2-induced neurotoxicity is believed to occur through two mechanisms: the production of extracellular ROS that directly damages neurons and intracellular signaling that primes microglia to enhance the pro-inflammatory response and propagate neurotoxicity (Fig. 1).
Microglial NOX2 has been implicated as chronic source of ROS in pathological CNS conditions. NOX2 generates extracellular superoxide (O ·−2 ), which is highly reactive and is rapidly dismuted, either spontaneously or by the enzyme superoxide dismutase (SOD) [108], to yield the cell-soluble molecule hydrogen peroxide (H2O2). However, H2O2 is reduced to water and the hydroxyl radical (-OH) through the Fenton reaction [109, 110]. The hydroxyl radical is one of the strongest known oxidizing species and a powerful host-defense mechanism because of its ability to damage pathogens [109, 110]. NOX2 ROS is generated both in the phagosome and at the membrane in phagocytes, where concentrations of superoxide in the phagosome can surpass 1 M in addition to other reactive species [111]. Because NOX2 is located at the cellular membrane, NOX2 activation has been implicated in damage in surrounding tissues, particularly neurons [112, 113]. Peroxynitrite (ONOO−), a product of NO and superoxide, is also noted to be toxic to neurons [113, 114]. Excessive levels of ROS that exceed endogenous, compensatory anti-oxidant levels can lead to oxidative modification in neurons responsible for dysfunction of proteins, nucleic acids, and lipids [113, 115]. While many neurons can adjust to and regulate spikes in ROS, there are select populations of neurons in the brain that are vulnerable, which is implicated in neurodegenerative disease [115].
Redox signaling occurs when a biological signaling pathway is modified by free radical species such as a ROS or nitric oxide in a manner characteristic of a messenger, modulating or impacting the signaling in a way that is not explicable simply by oxidative tissue damage [116]. As ROS and other free radical species are highly reactive in comparison to uncharged molecules, redox signaling occurs frequently within a cell, less frequently between cells that are in contact, and rarely between cells separated by a space of even a few microns. One mechanism by which ROS can influence signaling is through redox-sensitive target proteins such as protein tyrosine phosphatases [117], which can be reversibly oxidized at vulnerable cysteine residues, temporarily inactivating proteins. In addition, many transcription factors have been shown to be redox-sensitive, including members of the AP-1 [118] and NF-κB [119] families, Nrf2 [120], p53 [121], and glucocorticoid receptor [122]. There is evidence for ROS involvement in cell–cell contact [123, 124], in regulation of NOX2 subunits Rac1 and P47PHOX [125], and in many other cellular processes.
Importantly, NOX2 redox signaling has been shown to play an essential role in the microglial pro-inflammatory response and associated neurotoxicity. More specifically, NOX2 has been shown regulate intracellular ROS levels in microglia to result in both amplification of the production of pro-inflammatory cytokines, such as TNF α [26] or PGE2 [126] and priming of microglia to be sensitive to additional stimuli [13]. For example, NOX2 inhibitors suppress LPS-induced expression of cytokines (IL-1β, IL-6, and TNFα), iNOS expression, MAP kinases, and NFκB phosphorylation [27]. Further supporting this premise, NOX2−/− mice have shown drastically reduced microglial activation, cytokine levels, and neurotoxicity with intranigral LPS injection [26]. In addition, evidence supports activation of NOX2 shifts the microglia to a primed phenotype that can result in a neurotoxic response to otherwise benign stimuli. Ongoing neuron damage and rotenone [127, 128], both of which activate NOX2 (Table 1), have been shown to synergystically amplify the microglial pro-inflammatory response to LPS and neurotoxicity in vitro. Interestingly, paraquat, another initiator of microglial NOX2 (Table 1), appears to exert toxicity in vivo only when combined with more than one stimulus of microglial activation. Rather, priming appears to be necessary for neuronal damage, where paraquat must be administered in at least two doses or in combination with another pro-inflammatory stimulus, such as LPS [129]. Notably, paraquat priming only occurs in mice with functional NOX2 [129]. Thus, current evidence supports that NOX2 regulates microglial priming, enhancing the microglial response to diverse stimuli, which may contribute to both the magnitude of the microglial response and the chronic nature of microglial activation in the brain.
Microglial NOX2 in neurodegenerative diseases
Microglial NOX2 is implicated in the ongoing pathology of several neurodegenerative diseases and CNS disorders/conditions, including amyotrophic lateral sclerosis [130, 131], multiple sclerosis [132], pathologically altered behavior [133], ischemic stroke [134, 135], neuropathic pain [136, 137], HIV-associated dementia [93, 138], neurotrauma [67, 139], schizophrenic behavior [140, 141], Alzheimer’s disease (AD) [142], and Parkinson’s disease (PD) [143]. In the current review, we focus on the relevance of NOX2 inhibition for the two most prevalent neurodegenerative diseases, AD and PD. Notably, as a common cause of dementia in the elderly and the most prevalent neurodegenerative disease [144], AD affects more than 4 million people in the United States and an estimated 27 million are affected worldwide [145]. PD is a devastating movement disorder and is the second most prevalent neurodegenerative disease [144], affecting 1–2 % of the population over the age of 50 [146]. These numbers for both of these diseases are expected to swell in the future with the aging population. Analysis of post-mortem brains from both AD and PD patients reveal activated microglia [14], where this microglial activation has been implicated in the progressive nature of each disease [16, 96, 147, 148]. Interestingly, observations in AD brains found marked increases in P47PHOX and P67PHOX translocation to the membrane in microglia when compared to control brains [142]. Further, mild cognitive impairment has been associated with increased expression of P40PHOX, P47PHOX, and P67PHOX in AD brains [149]. In PD brains, the NOX2 protein itself is upregulated in the brain region that is selectively damaged, the substantia nigra [143]. Given the number of individuals affected by AD and PD, the strong link between microglial NOX2 and ongoing pathology driving these diseases, and the fact that current treatment is only palliative, targeting of NOX2 may be especially beneficial for AD and PD. However, the majority of mechanistic information regarding the role of NOX2 in microglia-mediated neurotoxicity has been derived from animal and cell culture studies.
NOX2 in Parkinson’s disease models
Increasing evidence points to NOX2 as a critical mechanism of neuron damage in several models of PD (Table 2). 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is a classic in vivo and in vitro model used to explore selective dopaminergic (DA) neurotoxicity, a predominating characteristic in PD [150]. MPTP-induced parkinsonian symptoms were first observed in human patients by Langston et al. in 1984, and experiments in C57 mice followed [151, 152]. MPTP is converted by monoamine oxidase B, to 1-methyl-4-phenylpyridine (MPP+) by astrocytes, which is a metabolite that is then selectively toxic to DA neurons. Selectivity is conferred when the dopamine active transporter moves MPP+ into the DA neuron, where it interferes with complex I and III of the mitochondrial electron transport chain to cause DA neuron death [153, 154]. Microglia are clearly activated in vivo in response to MPTP-induced lesions in the SN [97, 155], and NOX2 is activated in microglia that are localized with the neuron damage [143]. Reconstituted cell cultures have shown that while microglia are not activated by MPP+ alone, microglia are activated in the presence of MPP+ damaged neurons [98, 156]. Thus, it is not surprising that MPP+ in cultures is more toxic to DA neurons in the presence of microglia [98, 157]. However, NOX2 inhibitors or genetic deletion of the catalytic subunit NOX2 show that this additional microglia-mediated neurotoxicity is dependent on NOX2 [98, 143, 157–159]. Together, these studies indicate that the microglial response to neuronal damage (reactive microgliosis) is toxic and point to an active role for microglial NOX2 in chronic neurotoxicity (Fig. 2).
2,4,5-trihydroxyphenylethylamine (6-OHDA) is another established model used in the study of PD. 6-OHDA does not cross the blood–brain barrier, but upon direct injection into the brain, it efficiently produces lesions of DA neurons in the SNPC and has been used for decades to model PD [160]. Interestingly, 6-OHDA is selectivley toxic to DA neurons by more than one route. More specificially, 6-OHDA is internalized by the dopamine active transporter [161], where once inside the neuron it can both directly induce oxidative stress [162] and dysregulate complexes I and IV in the mitochondrial transport chain [163]. Recent studies show that neuron damage due to 6-OHDA activates microglia, induces expression of NOX2 subunits, induces apocynin-inhibitable NOX2 activity, and causes superoxide production, where these phenomenon cause additional DA neurotoxicity [164, 165]. Thus, evidence supports that neuronal damage in response to 6-OHDA also initates neurotoxic reactive microgliosis, which is regulated by NOX2.
In addition, several environmental toxins have been linked to Parkinson’s disease and as such, are used as experimental models for this disease [166]. N,N′-dimethyl-4,4′-bipyridinium dichloride (paraquat) is a widely used herbicide structurally similar to MPTP, but in vitro studies reveal that unlike MPTP, lower concentrations of paraquat are not toxic to DA neurons in the absence of microglia [167]. In fact, paraquat is shown to be a potent inducer of microglial NOX2 activity and consequent superoxide production, where both in vivo [129] and in vitro [167] studies confirm this is the primary mechanism of DA neurotoxicity. Rotenone is another environmental compound associated with PD and is a plant-derived insecticide that is known to inhibit the transfer of electrons from iron–sulfur centers in complex I of the MTC to ubiquinone (Coenzyme Q10). However, in vitro evidence suggests that, at low concentrations, rotenone is selectively toxic to DA neurons, but only in the presence of microglia [168]. Further analysis showed that microglia are activated by rotenone to produce superoxide, which is selectively toxic to DA neurons, and completely dependent on NOX2, as NOX2−/− cultures are protected from rotenone [128, 169]. With different molecular stuctures and predicted sites of action, it may at first seem surprising that rotenone and paraquat converge on the common NOX2 pathway of microglia-mediated neurotoxicity. However, several other stucturally unrelated environmental toxins have also been identified that are selectively toxic to DA neurons through NOX2, as noted in Table 1, supporting that NOX2 activation may be a common mechanism of neurotoxic microglial activation.
Other PD models that use pro-inflammitory triggers of microglial NOX2 activation are also available. Bacterial lipopolysaccharide (LPS) is a structural component of the cell wall of Gram-negative bacteria and a potent inflammogen. Importantly, LPS is not directly toxic to DA neurons and LPS activates microglia to produce ROS through iNOS [170] and NADPH oxidase [128], where the role of NOX2 in LPS-induced neurotoxicity is confirmed in vivo [26]. α Synuclein is a disease protein linked to PD, where A30P and A53T α synuclein mutant mice are a common genetic model for PD. The mutations result in elevated and advanced aggregation of α-synuclein with age. Aggregated wild-type, mutant A30P, and mutant A53T α synuclein have been shown in vitro to activate microglia via the Mac1 receptor [91]. These α synuclein aggregates were not toxic to DA neurons in the absence of microglia, nor in microglia deficient in NOX2 catalytic subunit of NAPDH oxidase [90]. Together, these findings further support the common mechanism of microglial NOX-mediated neurotoxicity.
NOX2 in Alzheimer’s disease models
Microglia are localized in and around senile plaques and neurofibrillary tangles [171, 172] in AD, where they become activated [173], and are noted to produce neurotoxic factors, including nitric oxide (NO) [174], superoxide [89, 175], and TNFα [176]. The amyloid hypothesis holds that Aβ, a key component of senile plaques, has a causative role in AD pathology, where microglia-mediated neurotoxicity has been implicated as a key mechanism [89, 177]. Indeed, Aβ recruits and activates microglia [172, 173], further supporting a role for both Aβ and microglia in AD progression [178]. Unfortunately, the same receptor complex necessary for microglia to recognize and phagocytize Aβ fibrils is also culpable in activation of microglial NOX2 and the consequent production of superoxide [179, 180], supporting that microglia NOX2 is a source of oxidative stress in AD [180]. Although not all available animal AD models are based on the amyloid hypothesis, the ones we will discuss here employ some derivation of this principle. Of note, choosing the correct AD model to test anti-inflammatory compounds, such as NOX2 inhibitors, is complicated and fraught with problems, as many mouse AD models fail to exhibit neuroinflammation at levels comparable to human patients [148], and the timeline and degree of inflammatory parameters may vary by model. Further, while some studies have explored the effects of NOX2 inhibitors on plaque load, neuroprotection, and memory, few AD animal models can directly confirm NOX2 activation in the brain, let alone specific NOX2 activation in microglia that is similar to AD patients, despite clear elevation of Aβ and plaque deposition. However, regardless of these limitations, with judicious use and cautious interpretation, appropriate and interesting models are available to test anti-inflammatory compounds.
Several AD animal models show that inhibition of NOX2 is neuroprotective (Tables 2, 3). For example, neocortical superfusion of Aβ1–40 causes free radical production and cerebrovascular dysregulation in the wild-type mouse cortex, where studies have shown ROS were not generated in response to Aβ in NOX2−/− mice [181]. The Tg(APPSWE)2576Kha (TG2576) mouse model is a commonly modified transgenic mouse that over-expresses the human APP695.SWE mutation (K670N and M671L) amyloid precursor protein (APP) gene under the control of the hamster prion protein promoter [182]. These mice are normal at 3 months, but acquire learning and memory in spatial reference deficit after 9 months, concomitant with augmented Aβ accumulation and plaque formation [183]. In a recent in vivo study using the Tg2576 mice crossed with Cybb−/− mice (mice missing functional NOX2), analyses showed that cerebrovascular alterations, amyloid deposition, and behavioral deficits observed in 12-15 month old TG2576 mice were absent in TG2576/Cybb−/− mice. Interestingly, the plaque load was not affected in TG2576/Cybb−/− mice, indicating that behavior deficits in this mouse model were somehow independent of plaque deposition [184]. Another genetic AD mouse model, the Tg(PRNP-APPSweInd)19959Dwst (TG19959) mouse, expresses mutant human amyloid protein precursor with both the Swedish (K670N/M671L) and the Indiana (V717F) mutations, under the control of the Syrian hamster prion protein promoter. This particular model demonstrates cognitive deficits and Aβ pathology quite early, at 3 months of age [185]. Microglial activation of NOX2 has not been established in these mice, but apocynin did show protection from AD pathology (both plaque load and cognitive deficit) [185]. Further, the N141I-PS2 presenilin mutant animal model of AD demonstrates that the NOX2 inhibitors, DPI and apocynin, abrogated neurotoxicity was in these mice as well [186]. Thus, while NOX2 inhibition has an effect in most animal AD models tested, each model seems to have a unique phenotype of AD pathology, and NOX2 activation has not been confirmed in most models tested.
The importance of assessing NOX2 activation prior to initiation of testing NOX2 inhibition was evidenced by a recent study using transgenic mice overexpressing human amyloid precursor protein with the Swedish and London mutations (hAPP(751)SL). Amyloid depositions begin to occur as plaques 3–4 months in hAPP(751)SL mice, with accumulation in the hippocampus commencing at 7 months [187–189]. During this study, mice were given apocynin from 4 to 8 months of age, before significant damage had occurred.
Apocynin was shown to significantly reduce microglial number and Aβ plaque size, with no effects found on measures of synaptic damage or learning and memory, supporting a disconnect between plaque deposition and neuronal damage/behavior in this model. Upon closer analysis, despite the high level of Aβ, accumulation of Aβ protein deposits, and behavioral deficits associated with this model [187–189], hAPP(751)SL mice showed little evidence of neuroinflammation and oxidative stress in saline control animals at 8 months, making reduction by any inhibitors improbable. Thus, the effects of apocynin were likely independent of any anti-inflammatory effects, emphasizing the importance of choice in the AD animal model when testing NOX2 inhibitors.
In vitro studies modeling components of AD and microglial NOX2 activation tend to be more consistent and clear cut when compared to animal models. There is extensive direct and indirect support that aggregated/fibrillar Aβ activates NOX2 in microglia in vitro and that NOX2 inhibition reduce superoxide production [24, 89, 190, 191]. In addition to basic primary cell culture studies, more creative in vitro models are also available. For example, NOX2 in a monocyte-lineage cell was firmly implicated in neurotoxicity in response to Aβ in a co-culture study with the neuroblastoma cell line SH-SY5Y [190]. More specifically, SH-SY5Y cells expressing APP with three aggregation-inducing mutations (Swedish double mutation at positions 670/671 combined with the London V717I mutation) were co-cultured with microglia from wildtype or NOX2−/− microglia cell lines (PLB-985-wt or PLB-985 X-CGD). Wild-type microglia were activated by unidentified factors, presumably due to chronic Aβ production, and the neuroblastoma cells were killed. However, microglia deficient in functional NOX2 were incapable of killing the neuroblastoma cells [192], supporting the role of phagocytic NOX2 in neurotoxicity. Thus, multiple tools are available to test the ability of NOX2 inhibitors to modulate microglia-mediated neurotoxicity in AD models.
Classification of microglial NOX2 inhibition
At present, current treatment for both AD and PD is largely unable to halt disease progression, emphasizing the need for new approaches to both disease prevention and intervention. Inhibition of NOX2 has been implicated as an ideal therapeutic target in diverse diseases, as it is involved in a variety of pathological processes throughout the body in addition to CNS effects [193]. However, given the strong support for the role of NOX2 in pathological microglial activation driving CNS disease, here we report on potential NOX2 inhibitors tested in AD and PD models. Most of the compounds discussed are considered potential inhibitors because the mechanism of actions have yet to be confirmed as directly interacting with members of the NOX2 complex [193]. The potential NOX2 inhibitors discussed here have demonstrated the ability to modify general measures of NOX2 activity (i.e., ROS production and assembly of the NOX2 enzyme complex), attenuate neurotoxic microglial activation, promote beneficial microglial activities (i.e., Aβ clearance and phagocytosis), and/or are neuroprotective through inhibition of microglial activation (Table 3). We have classified these potential NOX2 inhibitors by two general molecular approaches to the inhibition (Fig. 2): (1) direct NOX2 inhibition involving documented/hypothesized interaction with the NOX2 catalytic subunit to impede enzymatic activation; and (2) indirect NOX2 inhibition, which refers to everything else, including prevention of P47PHOX/P67PHOX phosphorylation and translocation, where the lack of assembly of the enzyme complex fails to activate NOX2, but the precise mechanisms are unknown. While these categories likely oversimplify NOX2 inhibition, the majority of studies attempting to identify the mechanism of inhibition are limited by current understanding of the enzyme, in addition to the lack of specificity of available inhibitors, and this classification serves to help organize this rapidly expanding class of compounds.
Direct inhibition of microglial NOX2
Gp91ds-tat & DPI
At present, no compounds have been tested to conclusively show that they affect microglial NOX2 enzyme function through direct interactions with the catalytic subunit. However, several potential molecules have been proposed to work through this mechanism. For example, DPI is an established inhibitor of flavoproteins that was once implicated as a specific, direct inhibitor of NOX2. While DPI clearly modulates microglial function and attenuates NOX2-derived ROS [194], it is now well known that the inhibitor is not specific for NOX2 and likely impacts NOX2 through multiple other mechanisms. Interestingly, gp91ds-tat is a peptide inhibitor of NOX2 (gp91ds-tat) that is believed to directly bind to NOX2 to impair enzymatic activity [184]. Unfortunately, at present, no studies have assessed the efficacy of gp91ds-tat on microglial function. We mention it here, because gp91ds-tat has been tested in an in vivo AD disease model with short-term administration, where gp91ds-tat treatment was sufficient to rescue to the diseased phenotype of aged TG2576 mice, suggesting that further inquiry may be warranted [184].
Opioids and related molecules
The majority of compounds tested in microglia that are proposed to work through direct interaction with NOX2 can best be classified as opiods and opiod-related molecules. However, as increasing numbers of related molecules are identified, the structural similarities remain, but the related compounds have extended past strict morphinan and opiod classifications. All the referenced compounds are structurally similar molecules with similar anti-inflammatory properties, neuroprotective effects, and a unique bi-modal dose response curve. Yet some of the molecules are opioid receptor agonists, while others are antagonists. Further, the concentrations at which they are effective are very tightly maintained across the range of molecules, suggesting a common mode of action. The compounds in this category include: both l- and d-morphine [195]; the morphinan analog sinomenine [196]; the mu-opioid receptor competitive antagonist naloxone [197]; the l-isomer of levorphanol dextromorphan [198]; the dynorphin opioid peptide and its minimal 3-residue peptide gly-gly-phe (GGF) [101, 197]; enkephalin (a natural opioid peptide similar to dynorphin) [199]; pituitary adenylate cyclase-activating polypeptide (PACAP) 38, PACAP 27, and its minimal internal peptide, gly-ile-phe (GIF) [200]; the natural opioid peptide leu-enkephalin [199]; DT-leu-enkephalin, which is unable to bind the kappa opiod receptor [199]; and verapamil [201]. Many of the molecules were analyzed for chemical similarities in various studies using pharmacophore representations of essential features, yielding the common model features of a hydrogen bond acceptor, hydrogen bond donor, ionizable, and sometimes hydrophobic moieties [101]. This suggests a common binding site(s), perhaps on NOX2 itself, rather than an opioid receptor and subsequent signaling, especially considering that both opiod receptor agonists and antagonists have the same effect, and that both naloxone [198, 199] and verapamil [201] have been found bound to NOX2 [198, 199]. Memantine, an NMDA receptor antagonist used in AD treatment, is protective of DA neurons in LPS-mediated toxicity and has been suggested to belong to this class of compounds [202]. However, because memantine alone significantly increased dopamine uptake in neuron/glia cultures (a measure of DA neuron health and function) and an increase in glia-derived neurotrophic factor (GDNF) expression, it is also proposed that much of memantine’s protective effect is due to induction of neurotrophic factors from astroglia [202]. Regardless, it is clear these compounds are not specific for NOX2, as they are known ligands for several other receptors with documented effects, and some of the inhibitors reported in this category also impair the NOX2 complex assembly.
Indirect inhibition of microglial NOX2
This is the largest and most diverse category of potential microglial NOX2 inhibitors (Fig. 2), as targeting this aspect of NOX2 activation could occur through a multitude of pathways that include inhibiting the phosphorylation and translocation of the cytosolic subunits directly, ablating receptor signaling, or impairing kinase signaling that leads to initiation of NOX2 complex assembly. Of course, the disadvantage of this NOX2 inhibition strategy is the risk of losing specificity and gaining the potential for unwanted side-effects. Indeed, most inhibitors in this category are not specific for NOX2 effects. None the less, several promising molecules in this category have been identified.
Natural compounds and herbal supplements
Apocynin (4′-Hydroxy-3′-methoxyacetophenone) was originally isolated from the medicinal plant Picrorhiza kurroa and is widely used as a non-specific NOX2 inhibitor that prevents translocation of P47PHOX and P67PHOX to the membrane. However, apocynin has also been implicated as an antioxidant because it inhibits ROS in nonphagocytic cells [203], although there is some support that the molecule may exert predominantly NOX2 effects in vivo [193]. Apocynin has been tested in several AD and PD models, as noted above. In addition, the squamosamide derivative FLZ is another molecule in this category, as it has been shown to impede P47PHOX translocation in microglia [28]. FLZ was protective in vitro against LPS-induced DA neurotoxicity and in vivo in the MPTP model of DA neuron loss in the substantia nigra, pars compacta (SNpc) [28]. Melatonin (N-acetyl-5-methoxytryptamine) is another commercially available supplement found to be effective in preventing phosphorylation of P47PHOX, translocation of P47PHOX and P67PHOX to the membrane, and generation of ROS in primary microglia by aggregated Aβ. In addition, EUK134 [204], the green tea polyphenol (−)-epigallocatechin-3-gallate [205], has also been shown to inhibit microglial ROS production. Further, docosahexaenoic acid (DHA) is an omega III fatty acid component of fish oil and a peroxisome proliferation-activating receptor (PPAR) agonist [206]. DHA inhibits NOX2 activation induced by angiotensin II in models of hypertension, although, interestingly, it may activate NOX2 in polymorphonuclear monocytes [207]. In microglia, DHA suppresses neuroinflammation [208], possibly by integrating into the phospholipids bilayer and modifying presentation of receptors like TLR4 and CD14, which form a receptor complex that can activate NOX2 in response to LPS [209]. The capacity of DHA to act as a ligand to PPAR and to integrate into and modify dynamics of lipid bilayers leaves a wide field of possible routes to modify NOX2 activity.
Statins
Statins are cholesterol-lowering drugs that have been proposed as potential therapeutics in PD and AD. While there are conflicting reports, some epidemiological studies show that individuals taking statins for cholesterol are at lower risk for PD [210] and AD [211]. Mechanistically, statins compete with hydroxymethylglutaryl-coenzyme A (HMG CoA), which generates mevalonic acid. HMG CoA activity causes prenylation of Rac1, impairing GTPase activity, providing a route for statin modulation of NOX2 activity [212–214]. Simvastatin has been shown to reduce glial activation, oxidative stress, and DA neurotoxicity [215]. Futher, the statins lovastatin and simvastatin were found to be protective of Aβ-induced microglial activation and superoxide generation through inhibition of Rac1 activation and subsequent NADPH oxidase activity [216]. However, direct interaction with the specific members of the NOX2 enzyme complex has yet to be identified, and the effects on Rac1 support an indirect mechanism of statin NOX2 inhibition.
Non-steroidal anti-inflammatory drugs (NSAIDs)
There has been increasing interest in the role of NSAIDs in PD and AD. Epidemiological studies conflict, with some concluding no correlation [217] and others reporting marginal protection from PD by ibuprofen but not other NSAID, aspirin, or acetaminophen [218–220]. Mechanistically, experiments performed by Landreth et al. show that ibuprofen inhibits activation of iNOS in microglia, as well as phosphorylation of P38 MAPK and Rac1 activation, both of which have been shown to be upstream of NOX2 [221, 222]. Importantly, the beneficial effects of NSAIDs in AD mouse models was confirmed with ibuprofen in R1.40 human swiss mutation APP-over-expressing mice. Administration of ibuprofen for 9 months ameliorated measures of oxidative stress and plaque load [222]. While the effect on cognitive decline was not reported, they did demonstrate in microglial cultures that ibuprofen inhibited assembly of the NOX2 complex, independent of COX2 inhibition [222]. While the mechanisms of action are clearly not specific for NOX2, these findings warrant further inquiry into the potential therapeutic efficacy of NSAIDs.
Targeting upstream kinases
The major inhibitable activating pathways upstream of NOX2 consists of the MAPK family of kinases [58–61], PKC [34, 56, 57], PAK [223], PKA [58], and CK2 [55]. Of these, the greatest amount of work has been done with the MAPK and PKC families. The MAPK P38, ERK1/2, and JNK are phosphorylated (and presumably activated) in rat primary microglia-enriched neuron glia cultures by LPS. This phosphorylation is coincident with microglia- and NOX2- mediated DA neuron-specific toxicity. The DA-specific toxicity is ameliorated with comparable efficiency by NOX2 inhibitors, inhibitors of the MAPK, and the thus far targetless anti-inflammatory molecules compound A and resveratrol [224, 225]. For example both the above-mentioned inhibitors have been shown to inhibit cyclooxygenase-1 and -2 expression at 20 μM [226], and SB203580 has been shown to inhibit phosphoinositide-dependent protein kinase 1 (PDK1) at 3–5 μM [227]. In fact, many molecules may impact these signaling pathways to modify microglial NOX2. For example, transforming growth factor β1 (TGFβ1) is a neuroprotective cytokine tested in vitro in LPS and MPP+ models of DA neurotoxicity [228]. More specifically, TGFβ1 has been shown to attenuate LPS-induced phosphorylation of ERK 1 and 2 and alleviate membrane translocation of P47PHOX in microglia [228]. Further analysis revealed that the ERK phosphorylation inhibitor U0126I was also protective of LPS-induced DA neurotoxicity [228]. While targeting signaling upstream of NOX2 may be therapeutically relevant, this approach to NOX2 inhibition will likely lead to many non-specific effects.
Iron chelation
Iron accumulation in the substantia nigra is implicated in the pathology of PD and iron chelators have been proposed as therapeutics [229]. With reference to NOX2, the catalytic subunits require the occupation of two heme groups with iron atoms which are used to convey the electron through the membrane to extracellular molecular oxygen. Thus, iron sequestration with chelating molecules is a strategy for inhibition of NOX2 activity [230]. The iron chelator desferrioxamine (DFO) not only ameliorates mircrobe- and LPS-induced NADPH oxidase activity in vivo, but it suppressed expression of P22PHOX in subcutaneous tissue, though this may have been a secondary effect of decreased ROS and altered redox signaling [231, 232]. Inhibition of NADPH oxidase by iron chelation in microglia has not been investigated, but may prove to be an important part of the observed benefit of iron chelation in PD.
Summary and conclusions
NOX2 may be an ideal therapeutic target for promoting healthy microglial function and attenuating the chronic production of neurotoxic microglial ROS underlying progressive neuron damage, which is particularly relevant to Parkinson’s disease and Alzheimer’s disease. More specifically, current data support that inhibition of microglial NOX2 is neuroprotective and can halt the progression of chronic neuron damage. However, while in vitro and animal studies using potential NOX2 inhibitors hold promise, most available compounds indirectly target NOX2, increasing the probability of side effects, and potentially decreasing consistency across experimental models and reducing therapeutic utility. Thus, future efforts must focus on the development of more specific and perhaps potent NOX2 inhibitors that are able to reach the brain and are safe for human use.
References
Graeber MB (2010) Changing face of microglia. Science 330(6005):783–788
Mittelbronn M et al (2001) Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol (Berl) 101(3):249–255
Tremblay ME, Majewska AK (2011) A role for microglia in synaptic plasticity? Commun Integr Biol 4(2):220–222
Rivest S (2011) The promise of anti-inflammatory therapies for CNS injuries and diseases. Expert Rev Neurother 11(6):783–786
Neumann H, Kotter MR, Franklin RJ (2009) Debris clearance by microglia: an essential link between degeneration and regeneration. Brain 132(Pt 2):288–295
Muller FJ, Snyder EY, Loring JF (2006) Gene therapy: can neural stem cells deliver? Nat Rev Neurosci 7(1):75–84
Morgan SC, Taylor DL, Pocock JM (2004) Microglia release activators of neuronal proliferation mediated by activation of mitogen-activated protein kinase, phosphatidylinositol-3-kinase/Akt and delta-Notch signalling cascades. J Neurochem 90(1):89–101
Liao H et al (2004) Tenascin-R plays a role in neuroprotection via its distinct domains coordinately modulating the microglia function. J Biol Chem 280:8316–8323
Loane DJ, Byrnes KR (2010) Role of microglia in neurotrauma. Neurotherapeutics 7(4):366–377
Streit WJ, Xue QS (2009) Life and death of microglia. J Neuroimmune Pharmacol 4(4):371–379
Shechter R et al (2009) Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med 6(7):e1000113
Chen SK et al (2010) Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell 141(5):775–785
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8(1):57–69
McGeer PL et al (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38(8):1285–1291
Gao HM et al (2002) Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson’s disease. J Neurochem 81(6):1285–1297
Block ML, Hong JS (2005) Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76(2):77–98
Perry VH, Nicoll JA, Holmes C (2010) Microglia in neurodegenerative disease. Nat Rev Neurol 6(4):193–201
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87(1):245–313
Sareila O et al (2011) NOX2 complex-derived ROS as immune regulators. Antioxid Redox Signal 15:2197–2208
Zhen L et al (1993) Gene targeting of X chromosome-linked chronic granulomatous disease locus in a human myeloid leukemia cell line and rescue by expression of recombinant gp91phox. Proc Natl Acad Sci USA 90(21):9832–9836
Dinauer MC et al (1987) The glycoprotein encoded by the X-linked chronic granulomatous disease locus is a component of the neutrophil cytochrome B complex. Nature 327(6124):717–720
Sankarapandi S et al (1998) Measurement and characterization of superoxide generation in microglial cells: evidence for an NADPH oxidase-dependent pathway. Arch Biochem Biophys 353(2):312–321
Babior BM (1978) Oxygen-dependent microbial killing by phagocytes (first of two parts). N Engl J Med 298(12):659–668
Jekabsone A et al (2006) Fibrillar beta-amyloid peptide Abeta1-40 activates microglial proliferation via stimulating TNF-alpha release and H2O2 derived from NADPH oxidase: a cell culture study. J Neuroinflammation 3:24
Mander PK, Jekabsone A, Brown GC (2006) Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J Immunol 176(2):1046–1052
Qin L et al (2004) NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem 279(2):1415–1421
Pawate S et al (2004) Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J Neurosci Res 77(4):540–551
Anilkumar N et al (2008) Nox4 and nox2 NADPH oxidases mediate distinct cellular redox signaling responses to agonist stimulation. Arterioscler Thromb Vasc Biol 28(7):1347–1354
Li Q et al (2009) Endosomal Nox2 facilitates redox-dependent induction of NF-kappaB by TNF-alpha. Antioxid Redox Signal 11(6):1249–1263
Bjorgvinsdottir H, Zhen L, Dinauer MC (1996) Cloning of murine gp91phox cDNA and functional expression in a human X-linked chronic granulomatous disease cell line. Blood 87(5):2005–2010
Segal AW et al (1992) Cytochrome b-245 is a flavocytochrome containing FAD and the NADPH-binding site of the microbicidal oxidase of phagocytes. Biochem J 284(Pt 3):781–788
Henderson LM, Banting G, Chappell JB (1995) The arachidonate-activable, NADPH oxidase-associated H + channel. Evidence that gp91-phox functions as an essential part of the channel. J Biol Chem 270(11):5909–5916
Maly FE et al (1993) Restitution of superoxide generation in autosomal cytochrome-negative chronic granulomatous disease (A22(0) CGD)-derived B lymphocyte cell lines by transfection with p22phax cDNA. J Exp Med 178(6):2047–2053
Nauseef WM et al (1991) Assembly of the neutrophil respiratory burst oxidase. Protein kinase C promotes cytoskeletal and membrane association of cytosolic oxidase components. J Biol Chem 266(9):5911–5917
Jesaitis AJ et al (1990) Ultrastructural localization of cytochrome b in the membranes of resting and phagocytosing human granulocytes. J Clin Invest 85(3):821–835
Chanock SJ et al (1992) O2- production by B lymphocytes lacking the respiratory burst oxidase subunit p47phox after transfection with an expression vector containing a p47phox cDNA. Proc Natl Acad Sci USA 89(21):10174–10177
Huang J, Hitt ND, Kleinberg ME (1995) Stoichiometry of p22-phox and gp91-phox in phagocyte cytochrome b558. Biochemistry 34(51):16753–16757
Parkos CA et al (1989) Absence of both the 91kD and 22kD subunits of human neutrophil cytochrome b in two genetic forms of chronic granulomatous disease. Blood 73(6):1416–1420
DeLeo FR et al (2000) Processing and maturation of flavocytochrome b558 include incorporation of heme as a prerequisite for heterodimer assembly. J Biol Chem 275(18):13986–13993
Clark RA et al (1990) Two cytosolic components of the human neutrophil respiratory burst oxidase translocate to the plasma membrane during cell activation. J Clin Invest 85(3):714–721
Inanami O et al (1998) Activation of the leukocyte NADPH oxidase by phorbol ester requires the phosphorylation of p47PHOX on serine 303 or 304. J Biol Chem 273(16):9539–9543
Baumer AT et al (2008) Phosphatidylinositol 3-kinase-dependent membrane recruitment of Rac-1 and p47phox is critical for alpha-platelet-derived growth factor receptor-induced production of reactive oxygen species. J Biol Chem 283(12):7864–7876
Zhang D et al (2011) Microglial MAC1 receptor and PI3K are essential in mediating beta-amyloid peptide-induced microglial activation and subsequent neurotoxicity. J Neuroinflammation 8(1):3
Cheng G, Lambeth JD (2004) NOXO1, regulation of lipid binding, localization, and activation of Nox1 by the Phox homology (PX) domain. J Biol Chem 279(6):4737–4742
DeLeo FR et al (1999) NADPH oxidase activation and assembly during phagocytosis. J Immunol 163(12):6732–6740
Dusi S, Donini M, Rossi F (1996) Mechanisms of NADPH oxidase activation: translocation of p40phox, Rac1 and Rac2 from the cytosol to the membranes in human neutrophils lacking p47phox or p67phox. Biochem J 314(Pt 2):409–412
de Mendez I, Homayounpour N, Leto TL (1997) Specificity of p47phox SH3 domain interactions in NADPH oxidase assembly and activation. Mol Cell Biol 17(4):2177–2185
Koga H et al (1999) Tetratricopeptide repeat (TPR) motifs of p67(phox) participate in interaction with the small GTPase Rac and activation of the phagocyte NADPH oxidase. J Biol Chem 274(35):25051–25060
Nisimoto Y et al (1999) The p67(phox) activation domain regulates electron flow from NADPH to flavin in flavocytochrome b(558). J Biol Chem 274(33):22999–23005
Lavigne MC et al (2001) Genetic requirement of p47phox for superoxide production by murine microglia. FASEB J 15(2):285–287
Lapouge K et al (2002) Architecture of the p40-p47-p67phox complex in the resting state of the NADPH oxidase. A central role for p67phox. J Biol Chem 277(12):10121–10128
Massenet C et al (2005) Effects of p47phox C terminus phosphorylations on binding interactions with p40phox and p67phox. Structural and functional comparison of p40phox and p67phox SH3 domains. J Biol Chem 280(14):13752–13761
Cross AR (2000) p40(phox) Participates in the activation of NADPH oxidase by increasing the affinity of p47(phox) for flavocytochrome b(558). Biochem J 349(Pt 1):113–117
Sarfstein R et al (2004) Dual role of Rac in the assembly of NADPH oxidase, tethering to the membrane and activation of p67phox: a study based on mutagenesis of p67phox-Rac1 chimeras. J Biol Chem 279(16):16007–16016
Park HS et al (2001) Phosphorylation of the leucocyte NADPH oxidase subunit p47(phox) by casein kinase 2: conformation-dependent phosphorylation and modulation of oxidase activity. Biochem J 358(Pt 3):783–790
Bey EA et al (2004) Protein kinase C delta is required for p47phox phosphorylation and translocation in activated human monocytes. J Immunol 173(9):5730–5738
Dang PM et al (2001) Protein kinase C zeta phosphorylates a subset of selective sites of the NADPH oxidase component p47phox and participates in formyl peptide-mediated neutrophil respiratory burst. J Immunol 166(2):1206–1213
El Benna J et al (1996) Phosphorylation of the respiratory burst oxidase subunit p47phox as determined by two-dimensional phosphopeptide mapping. Phosphorylation by protein kinase C, protein kinase A, and a mitogen-activated protein kinase. J Biol Chem 271(11):6374–6378
Ward RA, Nakamura M, McLeish KR (2000) Priming of the neutrophil respiratory burst involves p38 mitogen-activated protein kinase-dependent exocytosis of flavocytochrome b558-containing granules. J Biol Chem 275(47):36713–36719
Dewas C et al (2000) The mitogen-activated protein kinase extracellular signal-regulated kinase 1/2 pathway is involved in formyl-methionyl-leucyl-phenylalanine-induced p47phox phosphorylation in human neutrophils. J Immunol 165(9):5238–5244
El Benna J et al (1996) Activation of p38 in stimulated human neutrophils: phosphorylation of the oxidase component p47phox by p38 and ERK but not by JNK. Arch Biochem Biophys 334(2):395–400
Benna JE et al (1997) Phosphorylation of the respiratory burst oxidase subunit p67(phox) during human neutrophil activation. Regulation by protein kinase C-dependent and independent pathways. J Biol Chem 272(27):17204–17208
Dang PM et al (2003) Phosphorylation of the NADPH oxidase component p67(PHOX) by ERK2 and P38MAPK: selectivity of phosphorylated sites and existence of an intramolecular regulatory domain in the tetratricopeptide-rich region. Biochemistry 42(15):4520–4526
Rotrosen D et al (1992) Cytochrome b558: the flavin-binding component of the phagocyte NADPH oxidase. Science 256(5062):1459–1462
Sorce S, Krause KH (2009) NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal 11(10):2481–2504
Green SP et al (2001) Induction of gp91-phox, a component of the phagocyte NADPH oxidase, in microglial cells during central nervous system inflammation. J Cereb Blood Flow Metab 21(4):374–384
Dohi K et al (2010) Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. J Neuroinflammation 7:41
Tejada-Simon MV et al (2005) Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol Cell Neurosci 29(1):97–106
Tammariello SP, Quinn MT, Estus S (2000) NADPH oxidase contributes directly to oxidative stress and apoptosis in nerve growth factor-deprived sympathetic neurons. J Neurosci 20(1):RC53
Thiels E, Klann E (2002) Hippocampal memory and plasticity in superoxide dismutase mutant mice. Physiol Behav 77(4–5):601–605
Knapp LT, Klann E (2002) Role of reactive oxygen species in hippocampal long-term potentiation: contributory or inhibitory? J Neurosci Res 70(1):1–7
Atkins CM, Sweatt JD (1999) Reactive oxygen species mediate activity-dependent neuron-glia signaling in output fibers of the hippocampus. J Neurosci 19(17):7241–7248
Atkins CM et al (1997) Increased phosphorylation of myelin basic protein during hippocampal long-term potentiation. J Neurochem 68(5):1960–1967
Pascual O et al (2011) Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci USA 109(4):E197–E205
Abramov AY et al (2005) Expression and modulation of an NADPH oxidase in mammalian astrocytes. J Neurosci 25(40):9176–9184
Abramov AY, Canevari L, Duchen MR (2004) Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim Biophys Acta 1742(1–3):81–87
Dickinson BC et al (2011) Nox2 redox signaling maintains essential cell populations in the brain. Nat Chem Biol 7(2):106–112
Clement HW et al (2010) Lipopolysaccharide-induced radical formation in the striatum is abolished in Nox2 gp91phox-deficient mice. J Neural Transm 117(1):13–22
Qin L et al (2005) Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia 52(1):78–84
Bal-Price A, Matthias A, Brown GC (2002) Stimulation of the NADPH oxidase in activated rat microglia removes nitric oxide but induces peroxynitrite production. J Neurochem 80(1):73–80
Oh YT et al (2008) Lipopolysaccharide induces hypoxia-inducible factor-1 alpha mRNA expression and activation via NADPH oxidase and Sp1-dependent pathway in BV2 murine microglial cells. Neurosci Lett 431(2):155–160
Ueyama T et al (2004) Superoxide production at phagosomal cup/phagosome through beta I protein kinase C during Fc gamma R-mediated phagocytosis in microglia. J Immunol 173(7):4582–4589
Rasmussen I et al (2010) Effects of F/G-actin ratio and actin turn-over rate on NADPH oxidase activity in microglia. BMC Immunol 11:44
Harrigan TJ et al (2008) Activation of microglia with zymosan promotes excitatory amino acid release via volume-regulated anion channels: the role of NADPH oxidases. J Neurochem 106(6):2449–2462
Ano Y et al (2010) Oxidative damage to neurons caused by the induction of microglial NADPH oxidase in encephalomyocarditis virus infection. Neurosci Lett 469(1):39–43
Gao X et al (2008) Formyl-methionyl-leucyl-phenylalanine-induced dopaminergic neurotoxicity via microglial activation: a mediator between peripheral infection and neurodegeneration? Environ Health Perspect 116(5):593–598
Park KW, Baik HH, Jin BK (2008) Interleukin-4-induced oxidative stress via microglial NADPH oxidase contributes to the death of hippocampal neurons in vivo. Curr Aging Sci 1(3):192–201
Park KW, Baik HH, Jin BK (2009) IL-13-induced oxidative stress via microglial NADPH oxidase contributes to death of hippocampal neurons in vivo. J Immunol 183(7):4666–4674
Qin L et al (2002) Microglia enhance beta-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J Neurochem 83(4):973–983
Zhang W et al (2005) Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J 19(6):533–542
Zhang W et al (2007) Microglial PHOX and Mac-1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant alpha-synuclein. Glia 55(11):1178–1188
Liu Y et al (2006) Suppression of microglial inflammatory activity by myelin phagocytosis: role of p47-PHOX-mediated generation of reactive oxygen species. J Neurosci 26(50):12904–12913
Turchan-Cholewo J et al (2009) NADPH oxidase drives cytokine and neurotoxin release from microglia and macrophages in response to HIV-Tat. Antioxid Redox Signal 11(2):193–204
Gupta S et al (2010) HIV-Tat elicits microglial glutamate release: role of NAPDH oxidase and the cystine-glutamate antiporter. Neurosci Lett 485(3):233–236
Mander P, Brown GC (2005) Activation of microglial NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: a dual-key mechanism of inflammatory neurodegeneration. J Neuroinflammation 2:20
Block ML, Hong JS (2007) Chronic microglial activation and progressive dopaminergic neurotoxicity. Biochem Soc Trans 35(Pt 5):1127–1132
Kim YS et al (2007) A pivotal role of matrix metalloproteinase-3 activity in dopaminergic neuronal degeneration via microglial activation. FASEB J 21(1):179–187
Levesque S et al (2010) Reactive microgliosis: extracellular micro-calpain and microglia-mediated dopaminergic neurotoxicity. Brain 133(Pt 3):808–821
Zhang W et al (2011) Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: implications for progression of Parkinson’s disease. Neurotox Res 19(1):63–72
Griendling KK et al (1994) Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 74(6):1141–1148
Block ML et al (2006) Potent regulation of microglia-derived oxidative stress and dopaminergic neuron survival: substance P vs. dynorphin. FASEB J 20(2):251–258
Wu XF et al (2005) The role of microglia in paraquat-induced dopaminergic neurotoxicity. Antioxid Redox Signal 7(5–6):654–661
Gao HM, Liu B, Hong JS (2003) Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. J Neurosci 23(15):6181–6187
Mao H et al (2007) Induction of microglial reactive oxygen species production by the organochlorinated pesticide dieldrin. Brain Res 1186:267–274
Block ML et al (2004) Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: the role of microglia, phagocytosis, and NADPH oxidase. FASEB J 18(13):1618–1620
Mao H, Liu B (2008) Synergistic microglial reactive oxygen species generation induced by pesticides lindane and dieldrin. Neuro Rep 19(13):1317–1320
Domico LM et al (2007) Reactive oxygen species generation by the ethylene-bis-dithiocarbamate (EBDC) fungicide mancozeb and its contribution to neuronal toxicity in mesencephalic cells. Neurotoxicology 28(6):1079–1091
McCord JM, Fridovich I (1969) Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 244(22):6049–6055
Cederbaum AI, Dicker E, Cohen G (1980) Role of hydroxyl radicals in the iron-ethylenediaminetetraacetic acid mediated stimulation of microsomal oxidation of ethanol. Biochemistry 19(16):3698–3704
Morehouse KM, Mason RP (1988) The transition metal-mediated formation of the hydroxyl free radical during the reduction of molecular oxygen by ferredoxin–ferredoxin: NADP + oxidoreductase. J Biol Chem 263(3):1204–1211
Hampton MB, Kettle AJ, Winterbourn CC (1998) Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood 92(9):3007–3017
Sundar IK et al (2010) Oxidative stress, thiol redox signaling methods in epigenetics. Methods Enzymol 474:213–244
Brown GC, Neher JJ (2010) Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol 41(2–3):242–247
Kuhn DM et al (2004) Nitrotyrosine as a marker for peroxynitrite-induced neurotoxicity: the beginning or the end of the end of dopamine neurons? J Neurochem 89(3):529–536
Wang X, Michaelis EK (2010) Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci 2:12
Ushio-Fukai M (2009) Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal 11(6):1289–1299
Chiarugi P, Cirri P (2003) Redox regulation of protein tyrosine phosphatases during receptor tyrosine kinase signal transduction. Trends Biochem Sci 28(9):509–514
Abate C et al (1990) Redox regulation of fos and jun DNA-binding activity in vitro. Science 249(4973):1157–1161
Galter D, Mihm S, Droge W (1994) Distinct effects of glutathione disulphide on the nuclear transcription factor kappa B and the activator protein-1. Eur J Biochem 221(2):639–648
Bloom D, Dhakshinamoorthy S, Jaiswal AK (2002) Site-directed mutagenesis of cysteine to serine in the DNA binding region of Nrf2 decreases its capacity to upregulate antioxidant response element-mediated expression and antioxidant induction of NAD(P)H: quinone oxidoreductase1 gene. Oncogene 21(14):2191–2200
Hainaut P, Milner J (1993) Redox modulation of p53 conformation and sequence-specific DNA binding in vitro. Cancer Res 53(19):4469–4473
Bodwell JE, Holbrook NJ, Munck A (1984) Sulfhydryl-modifying reagents reversibly inhibit binding of glucocorticoid-receptor complexes to DNA-cellulose. Biochemistry 23(7):1392–1398
Lin MT et al (2003) Inhibition of vascular endothelial growth factor-induced angiogenesis by resveratrol through interruption of Src-dependent vascular endothelial cadherin tyrosine phosphorylation. Mol Pharmacol 64(5):1029–1036
Nawroth R et al (2002) VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J 21(18):4885–4895
Wu RF et al (2005) Subcellular targeting of oxidants during endothelial cell migration. J Cell Biol 171(5):893–904
Wang T et al (2004) Role of reactive oxygen species in LPS-induced production of prostaglandin E2 in microglia. J Neurochem 88(4):939–947
Terry MJ, Maines MD, Lagarias JC (1993) Inactivation of phytochrome- and phycobiliprotein-chromophore precursors by rat liver biliverdin reductase. J Biol Chem 268(35):26099–26106
Gao HM et al (2003) Synergistic dopaminergic neurotoxicity of the pesticide rotenone and inflammogen lipopolysaccharide: relevance to the etiology of Parkinson’s disease. J Neurosci 23(4):1228–1236
Purisai MG et al (2007) Microglial activation as a priming event leading to paraquat-induced dopaminergic cell degeneration. Neurobiol Dis 25(2):392–400
Wu DC et al (2006) The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc Natl Acad Sci USA 103(32):12132–12137
Li Q et al (2011) Alsin and SOD1(G93A) proteins regulate endosomal reactive oxygen species production by glial cells and proinflammatory pathways responsible for neurotoxicity. J Biol Chem 286(46):40151–40162
Chechneva OV et al (2011) Low dose dextromethorphan attenuates moderate experimental autoimmune encephalomyelitis by inhibiting NOX2 and reducing peripheral immune cells infiltration in the spinal cord. Neurobiol Dis 44(1):63–72
Schiavone S et al (2009) Involvement of NOX2 in the development of behavioral and pathologic alterations in isolated rats. Biol Psychiatry 66(4):384–392
Chen H et al (2011) NADPH oxidase is involved in post-ischemic brain inflammation. Neurobiol Dis 42(3):341–348
De Silva TM et al (2011) Nox2 Oxidase activity accounts for the oxidative stress and vasomotor dysfunction in mouse cerebral arteries following ischemic stroke. PLoS ONE 6(12):e28393
Kim D et al (2010) NADPH oxidase 2-derived reactive oxygen species in spinal cord microglia contribute to peripheral nerve injury-induced neuropathic pain. Proc Natl Acad Sci USA 107(33):14851–14856
Berger JV et al (2011) Enhanced neuroinflammation and pain hypersensitivity after peripheral nerve injury in rats expressing mutated superoxide dismutase 1. J Neuroinflammation 8:33
Chakrabarti L et al (1991) Early viral replication in the brain of SIV-infected rhesus monkeys. Am J Pathol 139(6):1273–1280
Lo W et al (2007) NADPH oxidase inhibition improves neurological outcomes in surgically-induced brain injury. Neurosci Lett 414(3):228–232
Behrens MM et al (2007) Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 318(5856):1645–1647
Sorce S et al (2010) The NADPH oxidase NOX2 controls glutamate release: a novel mechanism involved in psychosis-like ketamine responses. J Neurosci 30(34):11317–11325
Shimohama S et al (2000) Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem Biophys Res Commun 273(1):5–9
Wu DC et al (2003) NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc Natl Acad Sci USA 100(10):6145–6150
Hirtz D et al (2007) How common are the “common” neurologic disorders? Neurology 68(5):326–337
Wimo A, Jonsson L, Winblad B (2006) An estimate of the worldwide prevalence and direct costs of dementia in 2003. Dement Geriatr Cogn Disord 21(3):175–181
Thomas B, Beal MF (2007) Parkinson’s disease. Hum Mol Genet 2:R183–R194
Lull ME, Block ML (2010) Microglial activation and chronic neurodegeneration. Neurotherapeutics 7(4):354–365
McGeer EG, McGeer PL (2010) Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. J Alzheimers Dis 19(1):355–361
Ansari MA, Scheff SW (2011) NADPH-oxidase activation and cognition in Alzheimer disease progression. Free Radic Biol Med 51(1):171–178
Jackson-Lewis V, Przedborski S (2007) Protocol for the MPTP mouse model of Parkinson’s disease. Nat Protoc 2(1):141–151
Langston JW, Ballard P (1984) Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): implications for treatment and the pathogenesis of Parkinson’s disease. Can J Neurol Sci 11(1 Suppl):160–165
Langston JW, Irwin I, DeLanney LE (1987) The biotransformation of MPTP and disposition of MPP+: the effects of aging. Life Sci 40(8):749–754
Zhao Y et al (2009) Effects of CYP3A5, MDR1 and CACNA1C polymorphisms on the oral disposition and response of nimodipine in a Chinese cohort. Eur J Clin Pharmacol 65(6):579–584
Gandhi S et al (2009) PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell 33(5):627–638
Hu X et al (2008) Macrophage antigen complex-1 mediates reactive microgliosis and progressive dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. J Immunol 181(10):7194–7204
Gao HM et al (2003) Critical role of microglial NADPH oxidase-derived free radicals in the in vitro MPTP model of Parkinson’s disease. FASEB J 17(13):1954–1956
McCarty MF, Barroso-Aranda J, Contreras F (2010) Oral phycocyanobilin may diminish the pathogenicity of activated brain microglia in neurodegenerative disorders. Med Hypotheses 74(3):601–605
Chung YC et al (2011) Cannabinoid receptor type 1 protects nigrostriatal dopaminergic neurons against MPTP neurotoxicity by inhibiting microglial activation. J Immunol 187(12):6508–6517
Chung YC et al (2011) Fluoxetine prevents MPTP-induced loss of dopaminergic neurons by inhibiting microglial activation. Neuropharmacology 60(6):963–974
Ungerstedt U (1968) 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur J Pharmacol 5(1):107–110
Luthman J et al (1989) Effects of d-amphetamine and methylphenidate on hyperactivity produced by neonatal 6-hydroxydopamine treatment. Psychopharmacology 99(4):550–557
Mazzio EA, Reams RR, Soliman KF (2004) The role of oxidative stress, impaired glycolysis and mitochondrial respiratory redox failure in the cytotoxic effects of 6-hydroxydopamine in vitro. Brain Res 1004(1–2):29–44
Glinka Y, Gassen M, Youdim MB (1997) Mechanism of 6-hydroxydopamine neurotoxicity. J Neural Transm Suppl 50:55–66
Rodriguez-Pallares J et al (2007) Mechanism of 6-hydroxydopamine neurotoxicity: the role of NADPH oxidase and microglial activation in 6-hydroxydopamine-induced degeneration of dopaminergic neurons. J Neurochem 103(1):145–156
Rodriguez-Pallares J et al (2008) Brain angiotensin enhances dopaminergic cell death via microglial activation and NADPH-derived ROS. Neurobiol Dis 31(1):58–73
Schapira AH, Jenner P (2011) Etiology and pathogenesis of Parkinson’s disease. Mov Disord 26(6):1049–1055
Mangano EN et al (2011) Interferon-gamma plays a role in paraquat-induced neurodegeneration involving oxidative and proinflammatory pathways. Neurobiol Aging. doi:10.1016/j/neurobiolaging.2011.02.016
Danilov CA et al (2009) Sulforaphane protects astrocytes against oxidative stress and delayed death caused by oxygen and glucose deprivation. Glia 57(6):645–656
Hu LF et al (2010) Neuroprotective effects of hydrogen sulfide on Parkinson’s disease rat models. Aging Cell 9(2):135–146
Possel H et al (2000) Selective upregulation of inducible nitric oxide synthase (iNOS) by lipopolysaccharide (LPS) and cytokines in microglia: in vitro and in vivo studies. Glia 32(1):51–59
McGeer PL et al (1987) Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett 79(1–2):195–200
Sasaki A et al (1997) Microglial activation in early stages of amyloid beta protein deposition. Acta Neuropathol (Berl) 94(4):316–322
Meda L et al (1995) Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature 374(6523):647–650
Li M, Sunamoto M, Ohnishi K, Ichimori Y (1996) Beta-Amyloid protein-dependent nitric oxide production from microglial cells and neurotoxicity. Brain Res 720(1–2):93–100
Wilkinson BL, Landreth GE (2006) The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer’s disease. J Neuroinflammation 3:30
Dheen ST et al (2004) Retinoic acid inhibits expression of TNF-alpha and iNOS in activated rat microglia. Glia 50:21–31
Combs CK et al (2000) Inflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci 20(2):558–567
Griffin WS et al (1998) Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol 8(1):65–72
Reed-Geaghan EG et al (2009) CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci 29(38):11982–11992
Wilkinson BL, Landreth GE (2006) The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer’s disease. J Neuroinflammation 3(1):30
Park L et al (2005) NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J Neurosci 25(7):1769–1777
Hsiao KK et al (1995) Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron 15(5):1203–1218
Hsiao K et al (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274(5284):99–102
Park L et al (2008) Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci USA 105(4):1347–1352
Chishti MA et al (2001) Early onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 276(24):21562–21570
Hashimoto Y et al (2002) Neurotoxic mechanisms by Alzheimer’s disease-linked N141I mutant presenilin 2. J Pharmacol Exp Ther 300(3):736–745
Rockenstein E et al (2001) Early formation of mature amyloid-beta protein deposits in a mutant APP transgenic model depends on levels of Abeta(1-42). J Neurosci Res 66(4):573–582
Hutter-Paier B et al (2004) The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease. Neuron 44(2):227–238
Willis M et al (2007) Localization and expression of substance P in transgenic mice overexpressing human APP751 with the London (V717I) and Swedish (K670M/N671L) mutations. Brain Res 1143:199–207
Qin B et al (2006) A key role for the microglial NADPH oxidase in APP-dependent killing of neurons. Neurobiol Aging 27(11):1577–1587
Wilkinson B et al (2006) Fibrillar beta-amyloid-stimulated intracellular signaling cascades require Vav for induction of respiratory burst and phagocytosis in monocytes and microglia. J Biol Chem 281(30):20842–20850
de la Monte SM, Wands JR (2006) Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J Alzheimers Dis 9(2):167–181
Jaquet V et al (2009) Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid Redox Signal 11(10):2535–2552
Qian L et al (2007) NADPH oxidase inhibitor DPI is neuroprotective at femtomolar concentrations through inhibition of microglia over-activation. Parkinsonism Relat Disord 13(Suppl 3):S316–S320
Qian L et al (2007) Microglia-mediated neurotoxicity is inhibited by morphine through an opioid receptor-independent reduction of NADPH oxidase activity. J Immunol 179(2):1198–1209
Qian L et al (2007) Sinomenine, a natural dextrorotatory morphinan analog, is anti-inflammatory and neuroprotective through inhibition of microglial NADPH oxidase. J Neuroinflammation 4:23
Wang XJ et al (2011) Impaired CD200-CD200R-mediated microglia silencing enhances midbrain dopaminergic neurodegeneration: Roles of aging, superoxide, NADPH oxidase, and p38 MAPK. Free Radic Biol Med 50(9):1094–1106
Li G et al (2005) Femtomolar concentrations of dextromethorphan protect mesencephalic dopaminergic neurons from inflammatory damage. FASEB J 19(6):489–496
Qin L et al (2005) Microglial NADPH oxidase mediates leucine enkephalin dopaminergic neuroprotection. Ann NY Acad Sci 1053:107–120
Yang S et al (2006) Pituitary adenylate cyclase-activating polypeptide (PACAP) 38 and PACAP4-6 are neuroprotective through inhibition of NADPH oxidase: potent regulators of microglia-mediated oxidative stress. J Pharmacol Exp Ther 319(2):595–603
Liu Y et al (2011) Verapamil protects dopaminergic neuron damage through a novel anti-inflammatory mechanism by inhibition of microglial activation. Neuropharmacology 60(2–3):373–380
Wu HM et al (2009) Novel neuroprotective mechanisms of memantine: increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology 34(10):2344–2357
Heumuller S et al (2008) Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 51(2):211–217
Ma T et al (2011) Amyloid {beta}-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J Neurosci 31(15):5589–5595
He Y et al (2011) Prolonged exposure of cortical neurons to oligomeric amyloid-beta impairs NMDA receptor function via NADPH oxidase-mediated ROS production: protective effect of green tea (−)-epigallocatechin-3-gallate. ASN Neuro 3:e00050
Diep QN et al (2002) PPARalpha activator effects on Ang II-induced vascular oxidative stress and inflammation. Hypertension 40(6):866–871
Wu GS, Rao NA (1999) Activation of NADPH oxidase by docosahexaenoic acid hydroperoxide and its inhibition by a novel retinal pigment epithelial protein. Invest Ophthalmol Vis Sci 40(5):831–839
Lu DY et al (2010) Docosahexaenoic acid suppresses neuroinflammatory responses and induces heme oxygenase-1 expression in BV-2 microglia: implications of antidepressant effects for omega-3 fatty acids. Neuropsychopharmacology 35(11):2238–2248
De Smedt-Peyrusse V et al (2008) Docosahexaenoic acid prevents lipopolysaccharide-induced cytokine production in microglial cells by inhibiting lipopolysaccharide receptor presentation but not its membrane subdomain localization. J Neurochem 105(2):296–307
Wahner AD et al (2008) Statin use and the risk of Parkinson disease. Neurology 70(16 Pt 2):1418–1422
Reiss AB, Wirkowski E (2009) Statins in neurological disorders: mechanisms and therapeutic value. Sci World J 9:1242–1259
Wassmann S et al (2001) Inhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: involvement of angiotensin AT1 receptor expression and Rac1 GTPase. Mol Pharmacol 59(3):646–654
Roy A, Pahan K (2011) Prospects of statins in Parkinson disease. Neuroscientist 17(3):244–255
Molnar G et al (2001) Role of prenylation in the interaction of Rho-family small GTPases with GTPase activating proteins. Biochemistry 40(35):10542–10549
Ghosh A et al (2009) Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurosci 29(43):13543–13556
Cordle A, Landreth G (2005) 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors attenuate beta-amyloid-induced microglial inflammatory responses. J Neurosci 25(2):299–307
Becker C, Jick S, Meier CR (2011) NSAID use and risk of Parkinson disease: a population-based case-control study. Eur J Neurol 18(11):1336–1342
Becker C, Jick SS, Meier CR (2011) NSAID use and risk of Parkinson disease: a population-based case-control study. Eur J Neurol 18(11):1336–1342
Schiess M (2003) Nonsteroidal anti-inflammatory drugs protect against Parkinson neurodegeneration: can an NSAID a day keep Parkinson disease away? Arch Neurol 60(8):1043–1044
Samii A et al (2009) NSAID use and the risk of Parkinson’s disease: systematic review and meta-analysis of observational studies. Drugs Aging 26(9):769–779
Raz L et al (2010) Role of Rac1 GTPase in NADPH oxidase activation and cognitive impairment following cerebral ischemia in the rat. PLoS ONE 5(9):e12606
Wilkinson BL et al. (2012) Ibuprofen attenuates oxidative damage through NOX2 inhibition in Alzheimer’s disease. Neurobiol Aging 33(1):197.e21–197.e32
Martyn KD et al (2005) p21-activated kinase (Pak) regulates NADPH oxidase activation in human neutrophils. Blood 106(12):3962–3969
Zhang F et al (2010) Resveratrol protects dopamine neurons against lipopolysaccharide-induced neurotoxicity through its anti-inflammatory actions. Mol Pharmacol 78(3):466–477
Zhang F et al (2010) Inhibition of IkappaB kinase-beta protects dopamine neurons against lipopolysaccharide-induced neurotoxicity. J Pharmacol Exp Ther 333(3):822–833
Tominaga K et al (2004) Correlation of MAP kinases with COX-2 induction differs between MKN45 and HT29 cells. Aliment Pharmacol Ther 20(Suppl 1):143–150
Lali FV et al (2000) The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in interleukin-2-stimulated T cells independently of p38 mitogen-activated protein kinase. J Biol Chem 275(10):7395–7402
Qian L et al (2008) Potent anti-inflammatory and neuroprotective effects of TGF-beta1 are mediated through the inhibition of ERK and p47phox-Ser345 phosphorylation and translocation in microglia. J Immunol 181(1):660–668
Riederer P et al (1989) Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J Neurochem 52(2):515–520
Biberstine-Kinkade KJ et al (2001) Heme-ligating histidines in flavocytochrome b(558): identification of specific histidines in gp91(phox). J Biol Chem 276(33):31105–31112
Li L, Frei B (2006) Iron chelation inhibits NF-kappaB-mediated adhesion molecule expression by inhibiting p22(phox) protein expression and NADPH oxidase activity. Arterioscler Thromb Vasc Biol 26(12):2638–2643
Collins HL, Kaufmann SH, Schaible UE (2002) Iron chelation via deferoxamine exacerbates experimental salmonellosis via inhibition of the nicotinamide adenine dinucleotide phosphate oxidase-dependent respiratory burst. J Immunol 168(7):3458–3463
Jin J et al (2007) Prostaglandin E2 receptor subtype 2 (EP2) regulates microglial activation and associated neurotoxicity induced by aggregated alpha-synuclein. J Neuroinflammation 4:2
Choi SH et al (2005) Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: role of microglial NADPH oxidase. J Neurosci 25(16):4082–4090
Won SY, Choi SH, Jin BK (2009) Prothrombin kringle-2-induced oxidative stress contributes to the death of cortical neurons in vivo and in vitro: role of microglial NADPH oxidase. J Neuroimmunol 214(1–2):83–92
Min KJ et al (2004) Gangliosides activate microglia via protein kinase C and NADPH oxidase. Glia 48(3):197–206
Gao HM et al (2011) HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J Neurosci 31(3):1081–1092
Kauppinen TM et al (2008) Zinc triggers microglial activation. J Neurosci 28(22):5827–5835
Yang CS et al (2007) Reactive oxygen species and p47phox activation are essential for the Mycobacterium tuberculosis-induced pro-inflammatory response in murine microglia. J Neuroinflammation 4:27
Lull ME et al (2011) Chronic apocynin treatment attenuates beta amyloid plaque size and microglial number in hAPP(751)(SL) mice. PLoS ONE 6(5):e20153
Moon JH et al (2008) Activation of nicotinic acetylcholine receptor prevents the production of reactive oxygen species in fibrillar beta amyloid peptide (1-42)-stimulated microglia. Exp Mol Med 40(1):11–18
Zhou J et al (2008) Melatonin impairs NADPH oxidase assembly and decreases superoxide anion production in microglia exposed to amyloid-beta1-42. J Pineal Res 45(2):157–165
Qian L et al (2006) Interleukin-10 protects lipopolysaccharide-induced neurotoxicity in primary midbrain cultures by inhibiting the function of NADPH oxidase. J Pharmacol Exp Ther 319(1):44–52
Qin L et al (2005) Microglial NADPH oxidase is a novel target for femtomolar neuroprotection against oxidative stress. FASEB J 19(6):550–557
Zhang W et al (2004) Neuroprotective effect of dextromethorphan in the MPTP Parkinson’s disease model: role of NADPH oxidase. FASEB J 18(3):589–591
Lin YC et al (2007) Neuroprotective effects of glyceryl nonivamide against microglia-like cells and 6-hydroxydopamine-induced neurotoxicity in SH-SY5Y human dopaminergic neuroblastoma cells. J Pharmacol Exp Ther 323(3):877–887
Choi SH et al (2005) Inhibition of thrombin-induced microglial activation and NADPH oxidase by minocycline protects dopaminergic neurons in the substantia nigra in vivo. J Neurochem 95(6):1755–1765
Chung YC, Kim SR, Jin BK (2010) Paroxetine prevents loss of nigrostriatal dopaminergic neurons by inhibiting brain inflammation and oxidative stress in an experimental model of Parkinson’s disease. J Immunol 185(2):1230–1237
Li Y et al (2009) Nimodipine protects dopaminergic neurons against inflammation-mediated degeneration through inhibition of microglial activation. Neuropharmacology 56(3):580–589
Murotomi K et al (2010) mGluR1 antagonist decreased NADPH oxidase activity and superoxide production after transient focal cerebral ischemia. J Neurochem 114(6):1711–1719
Chern, C.M., et al., (2011) Andrographolide inhibits PI3 K/AKT-dependent NOX2 and iNOS expression protecting mice against hypoxia/ischemia-induced oxidative brain injury. Planta Med 77:1–11
Woodfin A et al (2011) Acute NADPH oxidase activation potentiates cerebrovascular permeability response to bradykinin in ischemia-reperfusion. Free Radic Biol Med 50(4):518–524
Chen H, Song YS, Chan PH (2009) Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab 29(7):1262–1272
Hong H et al (2006) Atorvastatin protects against cerebral infarction via inhibition of NADPH oxidase-derived superoxide in ischemic stroke. Am J Physiol Heart Circ Physiol 291(5):H2210–H2215
Kato TA et al (2011) Aripiprazole inhibits superoxide generation from phorbol-myristate-acetate (PMA)-stimulated microglia in vitro: Implication for antioxidative psychotropic actions via microglia. Schizophr Res 129(2–3):172–182
Loane DJ et al (2009) Activation of metabotropic glutamate receptor 5 modulates microglial reactivity and neurotoxicity by inhibiting NADPH oxidase. J Biol Chem 284(23):15629–15639
El-Remessy AB et al (2008) Neuroprotective effects of cannabidiol in endotoxin-induced uveitis: critical role of p38 MAPK activation. Mol Vis 14:2190–2203
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Surace, M.J., Block, M.L. Targeting microglia-mediated neurotoxicity: the potential of NOX2 inhibitors. Cell. Mol. Life Sci. 69, 2409–2427 (2012). https://doi.org/10.1007/s00018-012-1015-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-012-1015-4