
Abstract
The concept that aneuploidy is a characteristic of malignant cells has long been known; however, the idea that aneuploidy is an active contributor to tumorigenesis, as opposed to being an associated phenotype, is more recent in its evolution. At the same time, we are seeing the emergence of novel roles for tumor suppressor genes and oncogenes in genome stability. These include the adenomatous polyposis coli gene (APC), p53, the retinoblastoma susceptibility gene (RB1), and Ras. Originally, many of these genes were thought to be tumor suppressive or oncogenic solely because of their role in proliferative control. Because of the frequency with which they are disrupted in cancer, chromosome instability caused by their dysfunction may be more central to tumorigenesis than previously thought. Therefore, this review will highlight how the proper function of cell cycle regulatory genes contributes to the maintenance of genome stability, and how their mutation in cancer obligatorily connects proliferation and chromosome instability.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Genome instability is a broad term used to describe the failure of a cell to accurately pass on a copy of its genome to its daughter cells. There are several mechanisms by which this can occur, and these have been grouped into three broad categories. Microsatellite instability is caused by defective mismatch repair that leaves DNA replication errors uncorrected [1–3]. Nucleotide excision repair-related instability arises from defects that prevent removal and replacement of UV-damaged nucleotides [4–6]. The third type of instability, which will be the focus of this review, is chromosome instability (CIN), which can be further dissected into two types, whole chromosome instability (W-CIN) and segmental chromosome instability (S-CIN) (delineated by Geigl et al. [7]). W-CIN arises through the gain and/or loss of whole chromosomes, which, if sustained through successive cell divisions, results in aneuploidy. Additionally, smaller regions of gain or loss, or changes in chromosome structure that do not result in copy number alterations, such as translocations or inversions, are broadly termed S-CIN [7]. A more in-depth discussion of the mechanisms by which CIN arises can be found in recent reviews by Aguilera et al., Holland et al., Tanaka et al., and Schvartzman et al. [8–11].
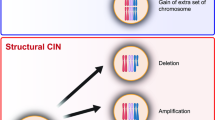
The concept that aneuploidy is a characteristic of malignant cells was first suggested by the work of von Hansemann and Boveri [12–14]. However, this theory was not verified until the early 1950s, when Sajiro Makino, and Levan and Hauschka among others [15–19] demonstrated that malignant cells have a unique chromosome compliment compared to their normal counterparts. Since then, chromosome instability has been observed to be tolerated, and even selected for, in many malignant cell types [20, 21]. Originally, such chromosome instability was thought to be a by-product, or a passenger that accompanied tumorigenesis. In other words, it was a cancer-associated phenotype, not a cancer-causing mechanism (Fig. 1a). However, in recent years, it has become evident that chromosome instability may exhibit a more causative role in the transformation of a normal cell into one that becomes cancerous (Fig. 1b). This shift in thought has been supported by several mouse models in which alterations of the spindle assembly checkpoint lead to higher than normal chromosome segregation errors, and offer proof of principle that chromosome instability alone can be the root cause of spontaneous tumors in mammals (reviewed in [11, 22]). In addition, the combination of these spindle assembly defects with other genetic lesions can enhance tumorigenesis, further demonstrating that CIN can stimulate progression of the disease [22]. Moreover, chromosome instability phenotypes are caused by mutations in tumor suppressor genes whose primary function resides in maintaining genome stability through repair and damage checkpoints, and/or the spindle assembly checkpoint, such as BRCA1, BubR1, and others [23–28]. These tumor suppressors, along with recently reported massive chromosome rearrangements (chromothripsis) that are evident in initial disease, and even in relapse [29], further argue that defects in chromosome stability can be central to cancer pathogenesis.

Schematic diagram of cell cycle deregulation, genomic instability, and tumorigenesis. a Originally, genomic instability was thought of as a cancer associated phenotype—over time, a cell would randomly acquire cell cycle deregulation, leading to cancer formation. Then, as a result of uncontrolled proliferation, genomic instability would inherently arise in these cancer cells. b As discussed in this review, it is becoming apparent that deregulated cell cycle control can compromise genome stability. If the instability is tolerated, in combination with uncontrolled proliferation, a cell may acquire mutations more readily that enhance its tumorigenic potential (e.g., acquire mutations that help the cell evade apoptosis, initiate angiogenesis, and acquire other hallmarks of cancer cells). This in turn facilitates tumorigenesis in a shorter time frame, and, in theory, more quickly than if genome instability was acquired after a cell has become cancerous as shown in (a). c This may be even more true of those individuals born with a predisposing mutation that causes cell cycle deregulation, and, as a consequence, genomic instability. Because these individuals acquire cell cycle deregulation early on, they also have genomic instability, and are predisposed to tumorigenesis at a much earlier age in life. Examples of predisposing mutations are those in APC that lead to familial adenomatous polyposis (FAP), those in p53 that cause Li-Fraumeni syndrome, and mutations in RB1 that cause retinoblastoma
In a manner similar to our shift towards viewing chromosome instability as an active contributor to cancer as opposed to being an associated phenotype, our understanding of many well-known oncogenes and tumor suppressors have followed a similar path. The adenomatous polyposis coli gene (APC), p53, and the retinoblastoma susceptibility gene (RB1) were all initially discovered to function in growth control [30–36]. While this remains true, our expanding knowledge of these genes has revealed roles for them in the maintenance of genome stability and, in many cases, specifically in chromosome stability. As a result, chromosome instability caused by common genetic lesions in cancer may be more central to the process of tumorigenesis than is currently estimated (Fig. 1c).
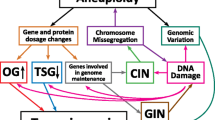
In this review, we will present several examples whereby common tumor suppressor genes or oncogenes, with well-characterized roles in cell cycle control and cancer pathogenesis, also function to maintain a stable genome (Fig. 2). We will highlight how these genes are best known for regulating proliferation, and how they facilitate chromosome stability. In addition, we will explore how chromosome instability enhances the tumorigenic potential beyond deregulated proliferation that is caused by loss of these tumor suppressors, or gain of these oncogenes (Fig. 1c). Lastly, we will conclude by suggesting circumstances during which chromosome instability caused by these mutations may be exploited in the ongoing search for new cancer therapeutic strategies.

Genomic instability goes hand-in-hand with cell cycle deregulation. a In a normal cell, tumor suppressors function to ensure proper regulation of the cell division cycle, and this is the canonical way in which we understand them to prevent cancer. However, many of these tumor suppressors (i APC, ii p53, iii pRB) have been found to also be involved in the maintenance of genome stability— a role that is also important to mediate their anti-tumorigenic effects. Similarly, proper regulation of proto-oncogenes ensures cell proliferation and survival (iv Ras). Upon loss of a tumor suppressor, or activation of a proto-oncogene, cell cycle regulation becomes compromised, and this is accompanied by a loss in genome stability. For tumor suppressors, this is due to a loss in their role in the maintenance of genome stability, and for proto-oncogenes, this results in a potential to promote genome instability as a by-product of uncontrolled proliferation. b Whether there is the loss of a tumor suppressor, or activation of an oncogene, deregulated cell cycle is subsequently followed by an acquisition of genome instability and ultimately cancer. This sequence of events may be true for other tumor suppressors and proto-oncogenes that are, as yet, unidentified, roles in the regulation of genome stability. Green tumor suppressor, yellow oncogene, SAC spindle assembly checkpoint
Adenomatous polyposis coli
Cytoplasmic growth regulator and mitotic spindle component
APC was identified through linkage analysis as the gene responsible for familial adenomatous polyposis, a type of colon cancer [37, 38]. In addition, APC is mutated in most cases of sporadic colon cancer [39]. Investigation into the functions of the APC protein product revealed that it is a cytoplasmic, β-catenin interacting protein [30, 31]. In the absence of stimulation by Wingless family ligands, APC binds to β-catenin leading to its phosphorylation by the GSK3 kinase, and subsequent targeting for degradation [40]. In simple terms, the presence of Wingless signaling inhibits β-catenin phosphorylation, releasing it from APC regulation, thereby allowing it to translocate to the nucleus and activate the transcription of genes that stimulate cell proliferation [40]. Wingless target genes that are stimulated by β-catenin include c-myc [41], n-myc [42], cyclin D [43, 44], survivin [45], and Id2 [46, 47]. Together, these target genes promote cell cycle progression and inhibit apoptosis. In familial colon cancer, germline mutations in APC lead to constitutive activation of these target genes, providing a distinct survival and proliferative advantage to polyps in these patients.
In addition to its role in proliferative control, APC has also been shown to function in the attachment of spindle microtubules to mitotic chromosomes [48]. During mitosis, APC localizes to the kinetochore through its interaction with the microtubule end binding protein, EB1, thereby facilitating the interaction of spindle microtubules with mitotic chromosomes [48–50]. APC mutant cells exhibit numerous unattached microtubules in metaphase and, not surprisingly, display chromosomal instability [51–54]. More recently, it has been proposed that APC also plays a role in regulating the amount of time microtubules persist at the kinetochore, which, when compromised by loss of APC, leads to lagging chromosomes [55].
Separating APC functions and their contribution to the maintenance of genome stability
A key question surrounding APC function is how the regulation of both β-catenin and microtubule attachment relates to chromosome instability, and how each contributes to APC’s tumor suppressor function. A number of mutant forms of APC that can separate function in mitosis from proliferative control have helped to investigate this question. One frequently studied allele of APC, derived from a mutagenesis screen in mice, is designated Min, and encodes a protein containing the first 850 amino acids [56]. This mutation deletes the interaction domains for β-catenin, tubulin, and EB-1 [57]. In addition, a targeted allele that truncates Apc at amino acid 1638 (1638T), and by comparison only eliminates the tubulin and EB-1 interactions, acts as a valuable comparison [58]. Studies using embryonic stem cells homozygous for either of these mutations demonstrate a clear defect in mitotic spindle formation, characterized by improper connections of the mitotic spindle to the kinetochore [59]. This leads to gross aneuploidy (W-CIN) and polyploidy [59]. Moreover, because embryonic stem cells homozygous for the 1638T mutation can still regulate β-catenin, but exhibit chromosomal instability, this illustrates that the role for APC in maintaining chromosome stability is separate from deregulated proliferation caused by the Wingless signaling pathway [59]. In support of these findings, it has been reported that upregulation of β-catenin alone is not sufficient to cause mitotic defects and chromosome instability in 293 cells [60]. Further correlative data demonstrates that cells from the human colon cancer cell line SW480, containing mutations in APC, exhibit multinucleation (as can be caused by cytokinetic failure due to APC-related microtubule defects) [60]. In stark contrast, cells from the HCT116 human colon cancer cell line that has a stabilizing mutation in β-catenin and wild-type APC, do not [60]. Together, these studies strongly suggest that chromosome instability is a distinct effect of APC mutations.
Instability in mouse models of cancer
Given that the proliferative control and mitotic functions of APC are separable, mutant alleles in mice have also provided insight into their relative roles in cancer progression. From these analyses, mouse models generated to investigate the role for APC in colon cancer support a role for W-CIN in the progression of this cancer type. This is best exemplified by the phenotype of the heterozygous mouse strain (Apc Δ716/+), which shows loss of heterozygosity eliminating wild-type Apc in adenomas of the small intestine in a manner similar to patients [61]. Additionally, the Apc Min/+ mouse exhibits similar mitotic defects, including misoriented spindles and misaligned chromosomes in normal crypts with wild-type levels of β-catenin; these cells also exhibited a tetraploid genotype [59, 60]. Moreover, loss of heterozygosity of the wild-type Apc allele has been demonstrated in dysplastic intestinal crypts, revealing that genome instability caused by mutant Apc is evident early in colon cancer development [60]. Because these phenotypes are observed in normal cells, before adenomas form, and before there is upregulation of β-catenin, it argues that CIN contributes to colorectal cancer from the earliest stages.
Interestingly, Apc 1638T/+ mice that exhibit only CIN phenotypes due to defective interactions with microtubules and EB-1, but maintain normal regulation of β-catenin, are not cancer prone [58]. This suggests that, at least in this context, CIN caused by APC mutations is not sufficient to cause cancer and is most likely a contributor to progression. A key experiment, that is needed to fully understand the interplay between proliferative control by APC through β-catenin and maintenance of chromosome stability, is the generation of a mutant mouse model in which regulation of β-catenin is lost, but interactions with microtubules and EB-1 are preserved. Analysis of these mice would offer definitive insight into the degree with which CIN contributes to cancer progression stimulated by β-catenin in Apc mutant mice.
A role for APC in the spindle assembly checkpoint (SAC)
Finally, oncogenic APC mutants have been shown to deregulate the spindle assembly checkpoint; truncated mutants of APC (N-APC) associated with colon cancer have been shown to sequester soluble Mad2, and thereby reduce the potency of Mad2 activation of the SAC. Furthermore, this also reduces the interaction of Cdc20, Mad2, and BubR1, the “wait anaphase” complex [62]. As a result, there is increased CIN in cells expressing the oncogenic N-APC fragment, due to reduced activation of the SAC [63]. Interestingly, this does not occur in cells with complete loss of APC, as full length APC does not appear to bind Mad2, indicating that this is a gain of function of the N-APC truncated mutant [63]. However, it has also been suggested that full length APC plays a role in the regulation of the SAC by binding to Bub1 and Bub3 (SAC proteins), and being a substrate of the Bub1/BubR1 kinases in in vitro experiments [48]. In accordance with this data is the fact that BubR1 +/−;APC min/+ mice develop ten times more colonic tumors than APC min/+ mice alone [64]. While MEFs from the compound mutant mice exhibited higher levels of β-catenin and increased proliferation compared to wild-type and BubR1 +/− MEFs, they were also able to proceed through mitosis when challenged with nocodazole. Compound mutant MEFs and exhibited increased genomic instability (as demonstrated by aneuploidy) in comparison with wild-type, BubR1 +/−, and APC min/+ MEFs [59, 64–66].
APC summary
Regardless of the precise contributions of CIN to APC mutant cancers, the APC gene provides an ideal example for how oncogenesis due to deregulation of wingless signaling is enhanced by a chromosome instability phenotype. Because the vast majority of APC mutations in cancer truncate the protein eliminating both β-catenin and mitotic regulatory domains, the consequences of these deletions obligatorily affect both functions. For this reason, deregulation of proliferative control and CIN are intimately linked in this common type of cancer (Fig. 2ai, b).
Tp53
Tumor suppressor gene, global cell fate regulator
The p53 protein was first discovered because it is bound by a large T antigen from simian virus 40 [67–69]. In addition to its role in the viral transformation of cells, p53 was later discovered to be a potent tumor suppressor that is mutated in most families with Li-Fraumeni syndrome [70–73]. Furthermore, sporadic mutation of the Tp53 gene also takes place in the majority of human cancers [74, 75]. Together, this background has established p53 as a critical target for mutation during tumorigenesis, whose function is compromised in virtually all forms of cancer.
Mechanisms of p53 regulation and function
Under conditions of homeostasis, the p53 protein is continuously translated and targeted for degradation via the 26S proteasome by the E3 ligase MDM2 (HDM2 in humans) [76–78]. Upon genotoxic stresses such as DNA damage or oncogene activation, the p53 protein becomes stabilized through phosphorylation by DNA damage responsive kinases such as Chk1, Chk2, and ATM, that block its interaction with MDM2 [79]. Furthermore, acetylation and methylation facilitate p53-dependent transcriptional activation of target genes to induce a cell cycle arrest or activate apoptotic signaling, among other responses [79]. Additionally, p53 can be stabilized and activated in response to aberrant proliferative signals. For example, E2F1 activates expression of p14ARF (also referred to by its murine nomenclature, p19ARF) [80], which stabilizes p53 by sequestering MDM2 and preventing MDM2-mediated ubiquitination of p53 [81–83].
To mediate either a transient cell cycle arrest or a more permanent senescent arrest, p53 activates the transcription of p21Cip1 [84], which in turn inhibits cyclin-dependent kinases, leading to an arrest of proliferation in any phase of the cell cycle [84–88]. To mediate apoptosis, p53 activates the transcription of genes encoding Bax, Noxa, and Puma, that stimulate apoptosis through the mitochondrial pathway [89–91]. Based on this summary of p53 function, it is clear that there is a direct connection between the maintenance of genome integrity through response to DNA damage and proliferative control, and through regulation of the cell division cycle. The many functions of p53 were conceptualized into a unified purpose when it was proposed to be the ‘guardian of the genome’ [35]. The ensuing paragraphs on p53 function will explain how its loss leads to an intimate association between chromosome instability and deregulated proliferation.
Guardian of the genome
Compromising the role of p53 in cell cycle and apoptotic regulation allows genetic change to accumulate over many cell generations. For example, fibroblasts from patients with Li-Fraumeni syndrome that exhibit LOH eliminating the wild-type Tp53 allele, consequently display a significant increase in aneuploidy and structural chromosomal aberrations [92–94]. Furthermore, the ability to amplify drug resistance gene loci is dramatically higher in cells harboring mutations in Tp53 [95], further indicating that S-CIN is a consequence of defective p53 function. Finally, several studies of human cancers and human cancer-derived cell lines have demonstrated that there is a strong correlation between aneuploidy in tumors and Tp53 mutations [96–98]. Together, these studies argue that defective p53 function leads to chromosome instability.
The ability of p53 to maintain a stable genome was originally thought to be a result of its inhibition of cell cycle progression, or induction of apoptosis in response to cellular stresses such as DNA damage. Failure to repair DNA damage before division, or survival of a cell following high levels of damage, both provide opportunities for chromosomal changes to arise [35]. Moreover, several studies have demonstrated that loss of p53 makes cells tolerant of both tetraploidy and aneuploidy [98–100]. However, other studies have suggested that p53 may play a more active role in the maintenance of genome stability (reviewed in [101]). For example, p53 has been reported to recognize DNA damage-related structures, and bind to ssDNA and dsDNA to promote strand exchange and mediate repair [102–105]; subsequently, a role for p53 in regulating homologous recombination and non-homologous end joining was uncovered [106–116]. p53 is also capable of binding proteins involved in homologous recombination repair and non-homologous end joining [117–124]. Presumably p53’s activities in these different repair processes work to repair damaged chromosomes and prevent their accumulation. Whether p53 functions through active repair versus activating checkpoints and apoptosis is more important for suppressing CIN is not within the purview of this review. Simply, in all cases, cellular studies indicate that a lack of p53 function deregulates proliferation and leads to alterations at the chromosomal level and deficiency in Tp53 is correlated with aneuploidy in tumors.
The genetics of p53 inactivation
In order to understand loss of p53 function in cancer and its impact on chromosome stability, it is also important to describe the mechanisms by which its function is eliminated. p53 is unique among tumor suppressors because it is organized into a homotetrameric transcription factor [125, 126]. This creates the opportunity for its function to be disrupted by dominantly acting mutations. For this reason, one allele of p53 can be eliminated by random mutation, which can then be followed by loss of heterozygosity to remove the remaining wild-type allele, much like other tumor suppressor genes are thought to be lost [70–73]. A number of means by which mutant p53 can gain function have been suggested. For example, mutant p53 can oligomerize with wild-type p53 as well as p53 family members p63 and p73, thereby inhibiting their ability to transactivate target genes [127–135]. Furthermore, p53 mutants have also been shown to activate the transcription of new target genes encoding growth stimulators including c-myc [136], cyclins and cdks [137], and hTert [138], all of which provide survival and proliferative advantages (reviewed by [139, 140]). Alternatively, regulators such as MDM2 can become oncogenically activated leading to loss of p53 function [141, 142]. Cancer patients with missense mutations in Tp53 often have a poorer prognosis than those lacking Tp53 entirely, as the lack of wild-type Tp53 confers only loss of tumor suppressor function [143], while the presence of dominantly mutated p53 not only confers loss of tumor suppressor activity but also provides a gain of function that is selected for in malignancy [143]. This gain of function concept is supported by the fact that Li-Fraumeni patients with mutated Tp53, as opposed to no p53 expression, have a significantly higher incidence of cancer that also occurs at an earlier age of onset [144]. For these reasons, loss of wild-type p53 function is variable in its effects on malignant progression, and this can impact the degree of chromosome instability exhibited from one mutation type to the next.
Instability in Trp53 mouse models of cancer
Numerous transgenic and gene-targeted mouse models that manipulate the murine Tp53 gene (called Trp53) have been generated. In many regards, the analysis of these animals has revealed that mutations in Trp53 predispose to cancer and cause chromosome instability in a manner similar to what is suggested from cell culture and clinical data. However, a small number of reports using gene-targeted Trp53 mice offer a clear demonstration of the connection between defective cell cycle control and CIN, and they are outlined below.
Trp53 knock out mice develop primarily lymphomas in a relatively short period of time [145–147]. Flow cytometric and cytogenetic analyses of these tumors suggests the emergence of aneuploidy, but maintenance of a near diploid karyotype [148–150]. Spectral karyotype analysis of chromosome spreads from these tumors indicates that chromosomal translocations are rare [149]. Depending on perspective, this can be considered either validation that loss of Trp53 results in CIN, or a suggestion that these phenotypes are remarkably mild considering p53’s role in maintaining genome stability. However, as noted above, null alleles are less severe than dominantly acting point mutations. Data on aneuploidy in primary cell culture and tumors from mice harboring the R172H and R270H mutants are scarce. These mutations represent two of the most common dominant negative point mutations found in human p53, and likely offer a better model of how p53 function is most often altered in human cancer [151, 152]. These mouse strains exhibit highly aggressive forms of cancer reminiscent of Li-Fraumeni syndrome, that are distinctly more metastatic than lymphomas found in Trp53 −/− animals [151, 152]. Taken together, this implies that these point mutations in murine p53 recapitulate the human syndrome well, and suggests that they likely accommodate genome instability. Another mouse line that harbors a point mutation in Trp53 and mimics a human cancer-derived allele with dominant properties very elegantly demonstrates the connection between CIN and defective proliferative control by p53. The substitution of R172P (R175P in humans) creates an allele of Trp53 that eliminates p53 dependent apoptosis but retains some transcriptional activation function for the cyclin-dependent kinase inhibitor p21 [153]. Mice homozygous for this mutation are considerably more resistant to spontaneous tumor formation than Trp53 −/− alone [153]. These mice also develop lymphomas, but, surprisingly, their ploidy remains strictly diploid. This suggests that defective apoptotic regulation is not linked to CIN and implicates a role for p53 in cell cycle control as its means to inhibit CIN. Further experimentation with these mice revealed that crossing them to p21-deficient strains advanced the onset of tumor formation and led to widespread aneuploidy in the resulting tumors [154]. Thus, analysis of the R172P mouse offers a very compelling case that defective cell cycle control by p53 is tightly linked to chromosomal aberrations.
Tp53 summary
In sum, discussion of the function of p53 in cell cycle control, the genetics of mutations that inactivate its function, and gene-targeted mouse models, demonstrates linkage between defective cell cycle control and CIN. In Li-Fraumeni patients, Tp53 mutations are the initiating event and this ensures that, in these cancers and likely in many others that suffer sporadic loss of p53, CIN is a reliable companion that can facilitate genetic change on the pathway to tumorigenesis. Considering that point mutations which dominantly inactivate p53 are found in most human cancers, and that these mutations accompany aneuploidy, it suggests that CIN is a frequent consequence of mutations that drive cancer cell proliferation (Fig. 2aii, b).
Retinoblastoma (RB)
The original tumor suppressor
The retinoblastoma susceptibility gene is the prototypical tumor suppressor gene. It was first cloned through positional mapping and identification of regions on chromosome 13q that were deleted in retinoblastomas and osteosarcomas [155]. Initially, it was expected that RB1 would function in a relatively specialized role in the few tissues where its loss of function contributed to hereditary cancer development. However, studies of the transforming activity of oncogenic viral proteins such as adenovirus E1A, simian virus 40 TAg, and human papilloma virus E7 indicated that inactivation of the RB1 protein (pRB) was a requirement for transformation [156]; this work suggested that pRB may function more broadly in an anti-oncogenic manner. Subsequent studies later revealed that pRB regulates the transition from the G1 to S-phase of the cell division cycle and that this universal role in proliferation transcends all cell types [157]. Since deregulated proliferation is key to cancer initiation and progression, genetic alterations that eliminate pRB function are a hallmark of nearly all cancers [158, 159].
The mechanism of pRB cell cycle control
The means by which pRB regulates G1 to S-phase progression in the cell cycle is through the control of E2F transcription factors [157]. In G1, pRB binds to transcriptional activation domains of E2Fs and inhibits the expression of genes that are required for S-phase progression [160, 161]. Concomitantly, pRB interacts with a number of cellular proteins that exhibit enzymatic activity capable of remodeling chromatin including histone deacetylases [162–164], histone methyltransferases [165, 166], DNA methyltransferases [167], and helicases such as BRG1/Brm [168]. This leads to heterochromatinization of E2F-target gene promoters and further inhibition of their expression. Under growth arrest conditions such as quiescence, pRB is hypophosphorylated and binds stably to E2Fs and chromatin regulators, preventing E2F-target gene transcription [157]. Upon mitogenic signaling, cyclinD/cdk4, followed by cyclinE/cdk2 complexes, hyperphosphorylate pRB, releasing both chromatin remodeling proteins and E2F transcription factors; E2Fs are thus free to activate the transcription of genes required for S-phase progression, and this irreversibly drives the cell cycle forward [159].
Inactivation of pRB function in cancer
The regulatory pathway that controls cell cycle advancement through G1 is often referred to as the RB-pathway. It includes cyclin D/cdk4, cyclin-dependent kinase inhibitors (CKIs) such as p16Ink4a, and finally pRB; in the vast majority of cancers, the retinoblastoma signaling pathway is compromised [158, 169]. The means by which it can be disrupted include (1) direct mutation of the retinoblastoma susceptibility gene, rendering it non-functional [170], (2) inactivation and degradation of pRB as caused by the human papilloma virus E7 oncoprotein, with effects similar to those resulting from direct mutation [171], (3) constitutive hyperphosphorylation of pRB in cancer by overexpression of cyclin D/cdk4 complexes, which is most commonly observed [158], or (4) inactivation of CKIs [158]. Functionally, all of these result in the loss of regulation of E2F transcription and inappropriate entry into the cell division cycle.
Inactivation of the RB1 gene in retinoblastoma has become a paradigm for the inactivation of tumor suppressor genes. The original ‘two-hit’ hypothesis predicted the need for both alleles of RB1 to be eliminated in order for its function to be compromised. It posited that elimination of each allele was an independent event [172]. Thus, for RB1 to be inactivated by direct mutation, and deregulate the RB pathway as described above, both copies of the gene need to be affected [173]. Elegant work by the White laboratory has revealed a number of chromosomal aberrations that can facilitate the loss of the remaining wild-type RB1 allele in a heterozygous, premalignant cell that already has one mutated copy of RB1 [174]. Thus, while rare, chromosomal aberrations are intimately linked to the elimination of pRB function in cancer initiation. For these reasons, it is important to distinguish the sporadic chromosomal segregation errors and abnormalities that can lead to loss of wild-type RB1 in RB1 +/− cells, from the CIN phenotype that arises as a consequence of complete deficiency for pRB.
Multiple mechanisms allow the retinoblastoma protein to prevent CIN
As described above, much attention has been focused on pRB’s ability to regulate E2F transcription factors at the G1 to S-phase transition, as this regulates a cell’s commitment to replicate its DNA and divide. There are two general divisions in which to categorize pRB’s functions that maintain genome stability. The first is as a consequence of deregulated E2F transcription. As detailed above, the vast majority of cancers possess mutations that disrupt regulation of the RB pathway, leading to uncontrolled E2F transcription. For this reason, missexpression of genes early in the cell cycle can result in chromosome re-replication or missegregation later, as is the case with deregulation of the E2F-target genes, cyclin E and MAD2, respectively [175, 176]. In addition, E2F-independent regulation of the chromatin structure of mitotic chromosomes has also emerged as a means by which the retinoblastoma protein contributes to the maintenance of genome stability [177–179]; both mechanisms will be discussed below.
Among E2F transcriptional targets, a number stand out as known causes of chromosome instability when overexpressed. First, both cyclin E1 and E2 isoforms are E2F target genes; their stable overexpression leads to abnormally elevated cyclin-dependent kinase activity, ultimately leading to aneuploidy or polyploidy [175]. Furthermore, a number of components of the spindle assembly checkpoint are E2F target genes, including Mad2 and BubR1, whose overexpression leads to enhanced checkpoint activity [11]. This in turn delays progression through mitosis and manifests as chromosome segregation errors [176]. These examples of deregulated E2F target gene-induced expression reveal how elevated levels of these gene products drive CIN, and offer a simple connection between loss of proliferation control and aneuploidy.
The first reports to suggest a role for pRB in maintaining a stable genome independently of E2Fs, and thereby G1 to S-phase regulation, demonstrated defects in chromosome structure or maintenance. In a study by Zheng et al. [180], it was demonstrated that Rb1 +/− and Rb1 −/− mouse embryonic stem cells exhibit a high frequency of loss of a selectable chromosomal marker compared to wild-type. Furthermore, loss of drug resistance was due to complete absence of the selectable marker, implicating chromosomal loss or rearrangement as the explanation for genetic change [180]. Similarly, it was also observed that cells deficient for all pRB family proteins display lengthened telomeres and centromere fusions [165, 181]. Metaphase spreads from these cells are characterized by chromosome fusions and tetraploidy [165].
Interestingly, similar centromere, aneuploidy, polyploidy, W-CIN, and S-CIN phenotypes have been observed in cells with defective condensin I/II complex function [182–186]. The condensin II complex facilitates chromosome condensation during prometaphase, and is important for maintaining chromosome structure and architecture during mitosis, particularly at the centromere [185, 187, 188] (reviewed by [189]). The association of the condensin II complex with pRB is lacking in both RB1 null cells, as well as those containing a targeted mutation in pRB that eliminates just LXCXE type interactions with the pRB pocket domain [177, 178, 190]. Defective condensin II function offers an explanation for the observed hypocondensation at centromeres, centromere fusions, and increase in whole chromosome gains and losses [177, 178, 190]. Presumably, defects in condensation lead to misshapen centromeres and merotelic attachments by spindle microtubules, which leads to missegregation of chromosomes without activating the spindle assembly checkpoint [191, 192]. Defects in S-CIN have also been reported in pRB-family-deficient fibroblasts, suggesting that chromosome structure defects may be more prevalent than at centromeres alone [179].
Defects in E2F regulation lead to elevated levels of aneuploidy because of improper regulation of DNA replication and activation of the spindle assembly checkpoint [175, 176]. The connection between chromosome instability caused by defective condensation in pRB mutants, and deregulated cell cycle control is less clear. For example, wild-type embryonic stem cells lack a pRB-dependent G1 arrest mechanism [193], but display CIN phenotypes caused by pRB deficiency [180]. For this reason, it remains to be determined whether elevated cyclin-dependent kinase activity (the most common way of eliminating pRB function in cancer) compromises pRB’s role in chromosome condensation, beyond causing aneuploidy through elevated E2F-mediated transcription.
Chromosome instability in pRB mouse models of cancer
Heterozygous Rb1 +/− mice are cancer prone and develop pituitary tumors by 1 year of age, the majority of which have lost the remaining wild-type Rb1 allele [194]; in this regard, they recapitulate the steps of the two-hit hypothesis quite faithfully. We are unaware of attempts to evaluate CIN in these tumors, or any others created by conditional deletion of Rb1 in a specific tissue. However, it would be difficult to discern the contribution of CIN to an Rb1-deficient cancer model in isolation from the effects of deregulated proliferation when using null alleles of Rb1.
Two mouse models offer a glimpse at the effects of deregulated E2F target gene expression. One is from transgenic overexpression of a non-degradable cyclin E in the mammary gland, and the other is overexpression of Mad2 in the same tissue; both cause mammary cancer and result in chromosomal abnormalities [195, 196]. While cyclin E clearly causes proliferation, analysis of MMTV-cyclin E;Trp53 +/− mice reveals that elevated cyclin E hastens the loss of the remaining wild-type Trp53 allele.
A recently generated mouse strain called Rb1 ΔL has demonstrated a connection between pRB-mediated chromosome condensation and tumor suppression [177]. This mutation in Rb1 abrogates the ability of cellular and viral proteins with the LXCXE peptide motif to bind to the pocket domain [197]. Importantly, this mutant form of pRB retains the ability to bind to, and regulate, E2F transcription factors such that Mad2 and other E2F target genes are expressed normally in cells from these mice [197]. Rb1 ΔL/ΔL mice do not develop spontaneous tumors; however, cells from these mice exhibit a significant increase in mitotic abnormalities [177, 197]. Rb1 ΔL/ΔL;Trp53 +/− mice succumb to cancer significantly sooner than Trp53 +/− mice, and because both genotypes lose their remaining wild-type Trp53 allele in the process, it implies that CIN caused by the Rb1 ΔL mutation accelerates cancer pathogenesis [177].
RB1 summary
Based on the available data from these mouse models of cancer, loss of pRB function contributes to both deregulated proliferation and chromosome instability. As shown with p53, this section of the review offers another example of a frequent target for inactivation in cancer that facilitates both chromosome instability, and deregulated proliferation in a largely inseparable manner (Fig. 2aiii, b).
Are there roles for oncogenes in genome instability?
So far, the proteins discussed here have been tumor suppressors whose loss leads to increased proliferation. Furthermore, in addition to traditional tumor suppressive mechanisms, their involvement in the maintenance of genome stability is a contributor to their tumor suppressive mechanisms.
What about oncogenes? Cells are unlikely to have a protein whose role is to cause genome instability. As such, it follows that there would be no proto-oncogene whose mutation into an oncogene would be directly involved in generating genome instability. However, it is possible that, by somewhat indirect mechanisms, activation of oncogenes can cause chromosomal instability as a by-product of increased proliferation.
The Ras pathway in proliferation and instability
The prototypical oncogene for this example is Ras. Under normal conditions, mitogenic signaling promotes GTP binding by Ras, and this leads to elevated growth signals through a number of pathways (reviewed in [198]). In cancer, mutations preferentially affect H-Ras or K-Ras and prevent GTP hydrolysis, causing constitutive signaling [198]. As a result, Ras activation by mutation leads to transcriptional activation of many genes, and these downstream effectors drive cell proliferation and survival [199, 200]. The contribution of Ras to chromosome instability comes from two perspectives. First, it has been reported that both human and rodent tumor cells with an activating mutation in one of their Ras isoforms exhibit heteroploidy, as well as chromosome breaks and rearrangements [201–203]. A more mechanistic explanation comes from studies of Ras in oncogene-induced senescence (OIS) [204]. In this case, senescence is induced by a robust DNA damage response (DDR) that becomes activated from partially replicated DNA caused by the repeated firing of DNA replication origins as a result of Ras induction of abnormally high proliferative signals [205]. This partially replicated DNA is a source of S-CIN; accordingly, loss of heterozygosity (LOH) at common fragile sites in OIS has been demonstrated with Ras over-expression [205]. Furthermore, genomic instability resulting from oncogenic Ras can be detected following just one round of DNA replication [201].
The acquisition of mutations that accommodate activated Ras, such as loss of DDR mediators including ATM, p53, and ATR, permit these cells to proliferate [205]. In this way, Ras-induced hyperproliferation can cause chromosome instability as a by-product of its proliferative signals (Fig. 2aiv, b). By extension, cells with oncogenic activation of receptor tyrosine kinases upstream of Ras, or activating mutations in downstream effectors such as Raf, can readily be envisioned to cause similar effects. In this regard, the phenotype of MMTV-cyclin E overexpressing mice, described above as causing over-replication and genome instability, may also exemplify over-active growth stimulating signals, as well as loss of growth inhibiting mechanisms [196].
Summation of proliferative control defects and chromosome instability in cancer
Each of the genes described above represent common mutational events in cancer. Tp53 is the most frequently mutated gene in cancer with mutations present in more than 50% of cases, irrespective of disease site [74, 75]. Direct mutation of pRB is relatively rare, but other alterations to its regulators ensure that it is functionless for E2F regulation in the vast majority of cancers [158]. APC mutations are uncommon outside of colorectal cancer, but occur very frequently in this cancer type [37–39]. Since colorectal cancer is one of the most common cancer types in western nations such as Canada and the USA, this further emphasizes the abundance of tumors with a defect in APC-mediated proliferative control and chromosome stability [133, 206]. Lastly, Ras mutations are present in approximately 10–90% of cancers, depending on disease site [198]. All told, the summation of these common mutation types, and their effect on chromosome instability, indicates that CIN is likely to be as intimately associated with cancer as its most well-known characteristics, such as deregulated proliferative control.
Prospects for harnessing the linkage between chromosome instability and deregulated proliferation
While it is obvious that chromosome instability facilitates the acquisition of potentially oncogenic mutations, it has also been proposed to act as a tumor suppressive mechanism in cell-specific contexts [22, 207]. In such circumstances, cancer-promoting alterations to chromosomes gained in one cell division cycle may rapidly be lost in successive rounds of cell division, resulting in a lack of persistence of a malignant clone. Additionally, as discussed previously, there is presumably a threshold level of DNA damage above which cell cycle arrest and subsequent senescence or apoptosis occur. Therefore, it follows that cells with high levels of chromosome instability may selectively undergo senescence or apoptosis. In this way, high levels of chromosome instability can act as a tumor suppressive mechanism. Taking advantage of chromosome instability for cancer treatment likely involves converting low levels of instability, that are tolerated and permissive to cancer pathogenesis, into higher levels that arrest proliferation or lead to cell death.
An example of how to capitalize on chromosome instability defects and exacerbate them to cause lethality in chromosomally unstable cancer cells has been demonstrated in tumors deficient for BRCA1 or BRCA2. These tumor suppressors function in homologous recombination repair of stalled replication forks (Fig. 3a) [188]. Cancers deficient for BRCA function can be selectively killed by agents that increase fork stalling, such as inhibition of the poly ADP ribose polymerase (PARP) [208]. This predicts that disruption of both repair pathways will be synthetically lethal, and has given rise to the therapeutic approach of treating BRCA1 or BRCA2 mutant cancers with PARP inhibitors (Fig. 3b). The addition of PARP inhibitors to such mutant cancers leads to single strand break accumulation that cannot be fixed during replication by homologous recombination [142, 209]. As a result, these cells die from unrepaired double-stranded breaks; cells with functional homologous recombination are orders of magnitude less sensitive (Fig. 3c). BRCA mutant cancers are relatively rare, and moreover not all BRCA mutant cancers exhibit this synthetic lethality; therefore, this limits the applicability of this therapy. In comparison, the tumor suppressor and oncogene mutations described in this review likely cause CIN on a much larger scale. We suggest that there are likely synthetic lethal combinations between common tumor suppressor pathway mutations that cause CIN, and other as yet to be determined genome maintenance mechanisms (Fig. 3d). We propose that, in addition to further investigation of the tumor suppressor and oncogenes described here and how they cause CIN, future research should also embrace the pursuit of synthetic lethal combinations with these gene mutations. This approach promises to translate our basic knowledge of relatively difficult drug targets such as pRB, Ras, or p53 into future treatment options.

Model for the exploitation of genome instability to induce apoptosis in cancer. a In wild-type cells, DNA damage causing double-strand breaks can be repaired by homologous recombination (HRR) mediated by BRCA1/2. b Inactivating mutations in either BRCA1 or BRCA2 compromise HRR, and DNA damage is more difficult to repair. In the case of BRCA-deficient cancers, repair of single-stranded lesions by PARP-mediated DNA damage repair is critical because subsequent replication fork stalling cannot be repaired by homologous recombination. c New chemotherapeutics for cancers with BRCA1 or BRCA2 mutations include the treatment of these tumors with PARP inhibitors. PARP inhibitors disable one of the remaining mechanisms by which these cancer cells repair DNA. This creates synthetic lethality in which BRCA mutant cells display greatly elevated sensitivity to PARP inhibitors compared to BRCA wild-type controls. d We suggest that this principle of synthetic lethality can be extended to more frequently affected tumor suppressors or oncogenes. There may be signaling pathways that cancer cells with common APC, Tp53, RB1, or Ras mutations rely on to compensate for the chromosome instability that these mutations create. Targeting these pathways with novel chemotherapeutics may lead to more anti-cancer agents akin to PARP inhibitors, except with a broader range of susceptible tumors because of the common nature of linked defects in proliferative control and genome instability described in this review
Abbreviations
- APC:
-
Adenomatous polyposis coli
- CDK:
-
Cyclin dependent kinase
- CIN:
-
Chromosome instability
- DDR:
-
DNA damage response
- LXCXE:
-
Leucine-any amino acid-cysteine-any amino acid-glutamate
- MMTV:
-
Mouse mammary tumor virus promoter
- OIS:
-
Oncogene induced senescence
- PARP:
-
Poly ADP ribose polymerase
- RB:
-
Retinoblastoma
- S-CIN:
-
Segmental chromosome instability
- W-CIN:
-
Whole chromosome instability
References
Grilley M, Holmes J, Yashar B, Modrich P (1990) Mechanisms of DNA-mismatch correction. Mutat Res 236:253–267
Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M (1993) Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 363:558–561
Thibodeau SN, Bren G, Schaid D (1993) Microsatellite instability in cancer of the proximal colon. Science 260:816–819
Sancar A (1996) DNA excision repair. Annu Rev Biochem 65:43–81
Wood RD (1997) Nucleotide excision repair in mammalian cells. J Biol Chem 272:23465–23468
Batty DP, Wood RD (2000) Damage recognition in nucleotide excision repair of DNA. Gene 241:193–204
Geigl JB, Obenauf AC, Schwarzbraun T, Speicher MR (2008) Defining ‘chromosomal instability’. Trends Genet 24:64–69
Aguilera A, Gomez-Gonzalez B (2008) Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet 9:204–217
Holland AJ, Cleveland DW (2009) Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Natl Rev Mol Cell Biol 10:478–487
Tanaka K, Hirota T (2009) Chromosome segregation machinery and cancer. Cancer Sci 100:1158–1165
Schvartzman JM, Sotillo R, Benezra R (2010) Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer 10:102–115
Boveri T (1912) Anton Dohbn. Science 36:453–468
Boveri T (2008) Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci 121(Suppl 1):1–84
SA A, BC H, SS J (2009) Application of UV-spectrophotometric methods for estimation of tenofovir disoproxil fumarate in tablets. Pak J Pharm Sci 22:27–29
Makino S (1952) Cytological studies on cancer. III. The characteristics and individuality of chromosomes in tumor cells of the Yoshida sarcoma which contribute to the growth of the tumor. Gan No Rinsho 43:17–34
Makino S, Nakahara H (1952) Observations on cell division in living tumor cells of ascites-sarcomas of rats. Gan No Rinsho 43:302
Hauschka TS, Levan A (1958) Cytologic and functional characterization of single cell clones isolated from the Krebs-2 and Ehrlich ascites tumors. J Natl Cancer Inst 21:77–135
Hauschka TS (1958) Correlation of chromosomal and physiologic changes in tumors. J Cell Physiol Suppl 52:197–233
Hauschka TS (1953) Cell population studies on mouse ascites tumors. Trans N Y Acad Sci 16:64–73
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P et al (2004) A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 119:591–602
Huen MS, Sy SM, Chen J (2010) BRCA1 and its toolbox for the maintenance of genome integrity. Natl Rev Mol Cell Biol 11:138–148
Joukov V, Groen AC, Prokhorova T, Gerson R, White E, Rodriguez A, Walter JC, Livingston DM (2006) The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell 127:539–552
Stolz A, Ertych N, Kienitz A, Vogel C, Schneider V, Fritz B, Jacob R, Dittmar G, Weichert W, Petersen I et al (2010) The CHK2-BRCA1 tumour suppressor pathway ensures chromosomal stability in human somatic cells. Nat Cell Biol 12:492–499
Choi E, Lee H (2008) Chromosome damage in mitosis induces BubR1 activation and prometaphase arrest. FEBS Lett 582:1700–1706
Elowe S, Dulla K, Uldschmid A, Li X, Dou Z, Nigg EA (2010) Uncoupling of the spindle-checkpoint and chromosome-congression functions of BubR1. J Cell Sci 123:84–94
Wei L, Liang XW, Zhang QH, Li M, Yuan J, Li S, Sun SC, Ouyang YC, Schatten H, Sun QY (2010) BubR1 is a spindle assembly checkpoint protein regulating meiotic cell cycle progression of mouse oocyte. Cell Cycle 9:1112–1121
Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA et al (2011) Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144:27–40
Su LK, Vogelstein B, Kinzler KW (1993) Association of the APC tumor suppressor protein with catenins. Science 262:1734–1737
Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P (1993) Association of the APC gene product with beta-catenin. Science 262:1731–1734
Hinck L, Nathke IS, Papkoff J, Nelson WJ (1994) Dynamics of cadherin/catenin complex formation: novel protein interactions and pathways of complex assembly. J Cell Biol 125:1327–1340
Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW (1991) Participation of p53 protein in the cellular response to DNA damage. Cancer Res 51:6304–6311
Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M (1991) Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 352:345–347
Lane DP (1992) Cancer. p53, guardian of the genome. Nature 358:15–16
Goodrich DW, Wang NP, Qian YW, Lee EY, Lee WH (1991) The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle. Cell 67:293–302
Bodmer WF, Bailey CJ, Bodmer J, Bussey HJ, Ellis A, Gorman P, Lucibello FC, Murday VA, Rider SH, Scambler P et al (1987) Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature 328:614–616
Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D et al (1991) Identification of FAP locus genes from chromosome 5q21. Science 253:661–665
Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW (1992) APC mutations occur early during colorectal tumorigenesis. Nature 359:235–237
Clevers H (2006) Wnt/beta-catenin signaling in development and disease. Cell 127:469–480
He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c-MYC as a target of the APC pathway. Science 281:1509–1512
ten Berge D, Brugmann SA, Helms JA, Nusse R (2008) Wnt and FGF signals interact to coordinate growth with cell fate specification during limb development. Development 135:3247–3257
Tetsu O, McCormick F (1999) Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398:422–426
Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A (1999) The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA 96:5522–5527
Zhang T, Otevrel T, Gao Z, Ehrlich SM, Fields JZ, Boman BM (2001) Evidence that APC regulates survivin expression: a possible mechanism contributing to the stem cell origin of colon cancer. Cancer Res 61:8664–8667
Rockman SP, Currie SA, Ciavarella M, Vincan E, Dow C, Thomas RJ, Phillips WA (2001) Id2 is a target of the beta-catenin/T cell factor pathway in colon carcinoma. J Biol Chem 276:45113–45119
Willert J, Epping M, Pollack JR, Brown PO, Nusse R (2002) A transcriptional response to Wnt protein in human embryonic carcinoma cells. BMC Dev Biol 2:8
Kaplan KB, Burds AA, Swedlow JR, Bekir SS, Sorger PK, Nathke IS (2001) A role for the adenomatous polyposis coli protein in chromosome segregation. Nat Cell Biol 3:429–432
Su LK, Burrell M, Hill DE, Gyuris J, Brent R, Wiltshire R, Trent J, Vogelstein B, Kinzler KW (1995) APC binds to the novel protein EB1. Cancer Res 55:2972–2977
Tirnauer JS, Bierer BE (2000) EB1 proteins regulate microtubule dynamics, cell polarity, and chromosome stability. J Cell Biol 149:761–766
Draviam VM, Shapiro I, Aldridge B, Sorger PK (2006) Misorientation and reduced stretching of aligned sister kinetochores promote chromosome missegregation in EB1- or APC-depleted cells. EMBO J 25:2814–2827
Green RA, Kaplan KB (2003) Chromosome instability in colorectal tumor cells is associated with defects in microtubule plus-end attachments caused by a dominant mutation in APC. J Cell Biol 163:949–961
Green RA, Wollman R, Kaplan KB (2005) APC and EB1 function together in mitosis to regulate spindle dynamics and chromosome alignment. Mol Biol Cell 16:4609–4622
Zhang J, Ahmad S, Mao Y (2007) BubR1 and APC/EB1 cooperate to maintain metaphase chromosome alignment. J Cell Biol 178:773–784
Bakhoum SF, Genovese G, Compton DA (2009) Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr Biol 19:1937–1942
Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF (1992) Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 256:668–670
Harvey M, Vogel H, Lee EY, Bradley A, Donehower LA (1995) Mice deficient in both p53 and Rb develop tumors primarily of endocrine origin. Cancer Res 55:1146–1151
Smits R, Kielman MF, Breukel C, Zurcher C, Neufeld K, Jagmohan-Changur S, Hofland N, van Dijk J, White R, Edelmann W et al (1999) Apc1638T: a mouse model delineating critical domains of the adenomatous polyposis coli protein involved in tumorigenesis and development. Genes Dev 13:1309–1321
Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, van Es JH, Breukel C, Wiegant J, Giles RH et al (2001) Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol 3:433–438
Caldwell CM, Green RA, Kaplan KB (2007) APC mutations lead to cytokinetic failures in vitro and tetraploid genotypes in Min mice. J Cell Biol 178:1109–1120
Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M (1995) Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci USA 92:4482–4486
Sudakin V, Chan GK, Yen TJ (2001) Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol 154:925–936
Zhang J, Neisa R, Mao Y (2009) Oncogenic adenomatous polyposis coli mutants impair the mitotic checkpoint through direct interaction with Mad2. Mol Biol Cell 20:2381–2388
Rao CV, Yang YM, Swamy MV, Liu T, Fang Y, Mahmood R, Jhanwar-Uniyal M, Dai W (2005) Colonic tumorigenesis in BubR1±ApcMin/+ compound mutant mice is linked to premature separation of sister chromatids and enhanced genomic instability. Proc Natl Acad Sci USA 102:4365–4370
Dai W, Wang Q, Liu T, Swamy M, Fang Y, Xie S, Mahmood R, Yang YM, Xu M, Rao CV (2004) Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res 64:440–445
Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P et al (2004) BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet 36:744–749
Kress M, May E, Cassingena R, May P (1979) Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol 31:472–483
Lane DP, Crawford LV (1979) T antigen is bound to a host protein in SV40-transformed cells. Nature 278:261–263
Linzer DI, Levine AJ (1979) Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17:43–52
Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA et al (1990) Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250:1233–1238
Srivastava S, Zou ZQ, Pirollo K, Blattner W, Chang EH (1990) Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 348:747–749
Varley JM, McGown G, Thorncroft M, Santibanez-Koref MF, Kelsey AM, Tricker KJ, Evans DG, Birch JM (1997) Germ-line mutations of TP53 in Li-Fraumeni families: an extended study of 39 families. Cancer Res 57:3245–3252
Santibanez-Koref MF, Birch JM, Hartley AL, Jones PH, Craft AW, Eden T, Crowther D, Kelsey AM, Harris M (1991) p53 germline mutations in Li-Fraumeni syndrome. Lancet 338:1490–1491
Carson DA, Lois A (1995) Cancer progression and p53. Lancet 346:1009–1011
Hollstein M, Sidransky D, Vogelstein B, Harris CC (1991) p53 mutations in human cancers. Science 253:49–53
Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387:296–299
Honda R, Tanaka H, Yasuda H (1997) Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 420:25–27
Wu X, Bayle JH, Olson D, Levine AJ (1993) The p53-mdm-2 autoregulatory feedback loop. Genes Dev 7:1126–1132
Toledo F, Wahl GM (2006) Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer 6:909–923
Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, Vousden KH (1998) p14ARF links the tumour suppressors RB and p53. Nature 395:124–125
Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D (1999) Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol 1:20–26
Midgley CA, Desterro JM, Saville MK, Howard S, Sparks A, Hay RT, Lane DP (2000) An N-terminal p14ARF peptide blocks Mdm2-dependent ubiquitination in vitro and can activate p53 in vivo. Oncogene 19:2312–2323
Hauschka T, Saxe LH Jr, Blair M (1947) Trypanosoma cruzi in the treatment of mouse tumors. J Natl Cancer Inst 7:189–197
el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75:817–825
Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ (1993) The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75:805–816
Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D (1993) p21 is a universal inhibitor of cyclin kinases. Nature 366:701–704
Dulic V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, Elledge SJ, Reed SI (1994) p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 76:1013–1023
Waga S, Hannon GJ, Beach D, Stillman B (1994) The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 369:574–578
Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80:293–299
Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288:1053–1058
Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN et al (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4:321–328
Bischoff FZ, Yim SO, Pathak S, Grant G, Siciliano MJ, Giovanella BC, Strong LC, Tainsky MA (1990) Spontaneous abnormalities in normal fibroblasts from patients with Li-Fraumeni cancer syndrome: aneuploidy and immortalization. Cancer Res 50:7979–7984
Tainsky MA, Bischoff FZ, Strong LC (1995) Genomic instability due to germline p53 mutations drives preneoplastic progression toward cancer in human cells. Cancer Metastasis Rev 14:43–48
Boyle JM, Mitchell EL, Greaves MJ, Roberts SA, Tricker K, Burt E, Varley JM, Birch JM, Scott D (1998) Chromosome instability is a predominant trait of fibroblasts from Li-Fraumeni families. Br J Cancer 77:2181–2192
Livingstone LR, White A, Sprouse J, Livanos E, Jacks T, Tlsty TD (1992) Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell 70:923–935
Campomenosi P, Assereto P, Bogliolo M, Fronza G, Abbondandolo A, Capasso A, Bellomo PF, Monaco R, Rapallo A, Sciutto A et al (1998) p53 mutations and DNA ploidy in colorectal adenocarcinomas. Anal Cell Pathol 17:1–12
Blount PL, Galipeau PC, Sanchez CA, Neshat K, Levine DS, Yin J, Suzuki H, Abraham JM, Meltzer SJ, Reid BJ (1994) 17p allelic losses in diploid cells of patients with Barrett’s esophagus who develop aneuploidy. Cancer Res 54:2292–2295
Thompson SL, Compton DA (2010) Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol 188:369–381
Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D (2005) Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 437:1043–1047
Galipeau PC, Cowan DS, Sanchez CA, Barrett MT, Emond MJ, Levine DS, Rabinovitch PS, Reid BJ (1996) 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett’s esophagus. Proc Natl Acad Sci USA 93:7081–7084
Gatz SA, Wiesmuller L (2006) p53 in recombination and repair. Cell Death Differ 13:1003–1016
Bakalkin G, Yakovleva T, Selivanova G, Magnusson KP, Szekely L, Kiseleva E, Klein G, Terenius L, Wiman KG (1994) p53 binds single-stranded DNA ends and catalyzes DNA renaturation and strand transfer. Proc Natl Acad Sci USA 91:413–417
Lee S, Elenbaas B, Levine A, Griffith J (1995) p53 and its 14 kDa C-terminal domain recognize primary DNA damage in the form of insertion/deletion mismatches. Cell 81:1013–1020
Jean D, Gendron D, Delbecchi L, Bourgaux P (1997) p53-mediated DNA renaturation can mimic strand exchange. Nucleic Acids Res 25:4004–4012
Jayaraman J, Prives C (1995) Activation of p53 sequence-specific DNA binding by short single strands of DNA requires the p53 C-terminus. Cell 81:1021–1029
Saintigny Y, Lopez BS (2002) Homologous recombination induced by replication inhibition, is stimulated by expression of mutant p53. Oncogene 21:488–492
Janz C, Wiesmuller L (2002) Wild-type p53 inhibits replication-associated homologous recombination. Oncogene 21:5929–5933
Elliott B, Jasin M (2001) Repair of double-strand breaks by homologous recombination in mismatch repair-defective mammalian cells. Mol Cell Biol 21:2671–2682
Lee S, Cavallo L, Griffith J (1997) Human p53 binds Holliday junctions strongly and facilitates their cleavage. J Biol Chem 272:7532–7539
Dudenhoffer C, Kurth M, Janus F, Deppert W, Wiesmuller L (1999) Dissociation of the recombination control and the sequence-specific transactivation function of P53. Oncogene 18:5773–5784
Janz C, Susse S, Wiesmuller L (2002) p53 and recombination intermediates: role of tetramerization at DNA junctions in complex formation and exonucleolytic degradation. Oncogene 21:2130–2140
Kumari A, Schultz N, Helleday T (2004) p53 protects from replication-associated DNA double-strand breaks in mammalian cells. Oncogene 23:2324–2329
Squires S, Coates JA, Goldberg M, Toji LH, Jackson SP, Clarke DJ, Johnson RT (2004) p53 prevents the accumulation of double-strand DNA breaks at stalled-replication forks induced by UV in human cells. Cell Cycle 3:1543–1557
Dahm-Daphi J, Hubbe P, Horvath F, El-Awady RA, Bouffard KE, Powell SN, Willers H (2005) Nonhomologous end-joining of site-specific but not of radiation-induced DNA double-strand breaks is reduced in the presence of wild-type p53. Oncogene 24:1663–1672
Lin Y, Waldman BC, Waldman AS (2003) Suppression of high-fidelity double-strand break repair in mammalian chromosomes by pifithrin-alpha, a chemical inhibitor of p53. DNA Repair (Amst) 2:1–11
Morris SM (2002) A role for p53 in the frequency and mechanism of mutation. Mutat Res 511:45–62
Sengupta S, Linke SP, Pedeux R, Yang Q, Farnsworth J, Garfield SH, Valerie K, Shay JW, Ellis NA, Wasylyk B et al (2003) BLM helicase-dependent transport of p53 to sites of stalled DNA replication forks modulates homologous recombination. EMBO J 22:1210–1222
Linke SP, Sengupta S, Khabie N, Jeffries BA, Buchhop S, Miska S, Henning W, Pedeux R, Wang XW, Hofseth LJ et al (2003) p53 interacts with hRAD51 and hRAD54, and directly modulates homologous recombination. Cancer Res 63:2596–2605
Restle A, Janz C, Wiesmuller L (2005) Differences in the association of p53 phosphorylated on serine 15 and key enzymes of homologous recombination. Oncogene 24:4380–4387
Zink D, Mayr C, Janz C, Wiesmuller L (2002) Association of p53 and MSH2 with recombinative repair complexes during S phase. Oncogene 21:4788–4800
Gaymes TJ, North PS, Brady N, Hickson ID, Mufti GJ, Rassool FV (2002) Increased error-prone non homologous DNA end-joining—a proposed mechanism of chromosomal instability in Bloom’s syndrome. Oncogene 21:2525–2533
Albrechtsen N, Dornreiter I, Grosse F, Kim E, Wiesmuller L, Deppert W (1999) Maintenance of genomic integrity by p53: complementary roles for activated and non-activated p53. Oncogene 18:7706–7717
Bertrand P, Saintigny Y, Lopez BS (2004) p53’s double life: transactivation-independent repression of homologous recombination. Trends Genet 20:235–243
Sengupta S, Harris CC (2005) p53: traffic cop at the crossroads of DNA repair and recombination. Natl Rev Mol Cell Biol 6:44–55
Friedman PN, Chen X, Bargonetti J, Prives C (1993) The p53 protein is an unusually shaped tetramer that binds directly to DNA. Proc Natl Acad Sci USA 90:3319–3323
Sakamoto H, Lewis MS, Kodama H, Appella E, Sakaguchi K (1994) Specific sequences from the carboxyl terminus of human p53 gene product form anti-parallel tetramers in solution. Proc Natl Acad Sci USA 91:8974–8978
Shaulian E, Zauberman A, Ginsberg D, Oren M (1992) Identification of a minimal transforming domain of p53: negative dominance through abrogation of sequence-specific DNA binding. Mol Cell Biol 12:5581–5592
Unger T, Mietz JA, Scheffner M, Yee CL, Howley PM (1993) Functional domains of wild-type and mutant p53 proteins involved in transcriptional regulation, transdominant inhibition, and transformation suppression. Mol Cell Biol 13:5186–5194
Chene P (1998) In vitro analysis of the dominant negative effect of p53 mutants. J Mol Biol 281:205–209
Srivastava S, Wang S, Tong YA, Hao ZM, Chang EH (1993) Dominant negative effect of a germ-line mutant p53: a step fostering tumorigenesis. Cancer Res 53:4452–4455
Kern SE, Pietenpol JA, Thiagalingam S, Seymour A, Kinzler KW, Vogelstein B (1992) Oncogenic forms of p53 inhibit p53-regulated gene expression. Science 256:827–830
Strano S, Munarriz E, Rossi M, Cristofanelli B, Shaul Y, Castagnoli L, Levine AJ, Sacchi A, Cesareni G, Oren M et al (2000) Physical and functional interaction between p53 mutants and different isoforms of p73. J Biol Chem 275:29503–29512
Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C (2001) A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol 21:1874–1887
Strano S, Fontemaggi G, Costanzo A, Rizzo MG, Monti O, Baccarini A, Del Sal G, Levrero M, Sacchi A, Oren M et al (2002) Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J Biol Chem 277:18817–18826
Marin MC, Jost CA, Brooks LA, Irwin MS, O’Nions J, Tidy JA, James N, McGregor JM, Harwood CA, Yulug IG et al (2000) A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat Genet 25:47–54
Frazier MW, He X, Wang J, Gu Z, Cleveland JL, Zambetti GP (1998) Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Mol Cell Biol 18:3735–3743
Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, Blandino G, Piaggio G (2006) Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 10:191–202
Scian MJ, Stagliano KE, Deb D, Ellis MA, Carchman EH, Das A, Valerie K, Deb SP, Deb S (2004) Tumor-derived p53 mutants induce oncogenesis by transactivating growth-promoting genes. Oncogene 23:4430–4443
Strano S, Dell’Orso S, Di Agostino S, Fontemaggi G, Sacchi A, Blandino G (2007) Mutant p53: an oncogenic transcription factor. Oncogene 26:2212–2219
Brosh R, Rotter V (2009) When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 9:701–713
Cahilly-Snyder L, Yang-Feng T, Francke U, George DL (1987) Molecular analysis and chromosomal mapping of amplified genes isolated from a transformed mouse 3T3 cell line. Somat Cell Mol Genet 13:235–244
Haines DS (1997) The mdm2 proto-oncogene. Leuk Lymphoma 26:227–238
Sigal A, Rotter V (2000) Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res 60:6788–6793
Birch JM, Blair V, Kelsey AM, Evans DG, Harris M, Tricker KJ, Varley JM (1998) Cancer phenotype correlates with constitutional TP53 genotype in families with the Li-Fraumeni syndrome. Oncogene 17:1061–1068
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, Bradley A (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356:215–221
Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA (1994) Tumor spectrum analysis in p53-mutant mice. Curr Biol 4:1–7
Purdie CA, Harrison DJ, Peter A, Dobbie L, White S, Howie SE, Salter DM, Bird CC, Wyllie AH, Hooper ML et al (1994) Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene 9:603–609
Liao MJ, Zhang XX, Hill R, Gao J, Qumsiyeh MB, Nichols W, Van Dyke T (1998) No requirement for V(D)J recombination in p53-deficient thymic lymphoma. Mol Cell Biol 18:3495–3501
Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA (2000) Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 406:641–645
Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA (2005) Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436:660–665
Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T (2004) Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119:847–860
Chen CY, Hall I, Lansing TJ, Gilmer TM, Tlsty TD, Kastan MB (1996) Separate pathways for p53 induction by ionizing radiation and N-(phosphonoacetyl)-L-aspartate. Cancer Res 56:3659–3662
Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, Multani A, Chang S, Lozano G (2004) Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet 36:63–68
Barboza JA, Liu G, Ju Z, El-Naggar AK, Lozano G (2006) p21 delays tumor onset by preservation of chromosomal stability. Proc Natl Acad Sci USA 103:19842–19847
Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, Dryja TP (1986) A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 323:643–646
Nevins JR (1992) E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science 258:424–429
Dyson N (1998) The regulation of E2F by pRB-family proteins. Genes Dev 12:2245–2262
Sherr CJ (1996) Cancer cell cycles. Science 274:1672–1677
DeCaprio JA, Ludlow JW, Lynch D, Furukawa Y, Griffin J, Piwnica-Worms H, Huang CM, Livingston DM (1989) The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell 58:1085–1095
Diaz-Rodriguez E, Sotillo R, Schvartzman JM, Benezra R (2008) Hec1 overexpression hyperactivates the mitotic checkpoint and induces tumor formation in vivo. Proc Natl Acad Sci USA 105:16719–16724
Hiebert SW, Chellappan SP, Horowitz JM, Nevins JR (1992) The interaction of RB with E2F coincides with an inhibition of the transcriptional activity of E2F. Genes Dev 6:177–185
Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T (1998) Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391:597–601
Luo RX, Postigo AA, Dean DC (1998) Rb interacts with histone deacetylase to repress transcription. Cell 92:463–473
Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, Troalen F, Trouche D, Harel-Bellan A (1998) Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 391:601–605
Gonzalo S, Garcia-Cao M, Fraga MF, Schotta G, Peters AH, Cotter SE, Eguia R, Dean DC, Esteller M, Jenuwein T et al (2005) Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat Cell Biol 7:420–428
Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O’Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE et al (2001) Rb targets histone H3 methylation and HP1 to promoters. Nature 412:561–565
Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP (2000) DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet 25:338–342
Dunaief JL, Strober BE, Guha S, Khavari PA, Alin K, Luban J, Begemann M, Crabtree GR, Goff SP (1994) The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell 79:119–130
Malumbres M, Barbacid M (2001) To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 1:222–231
Lohmann DR (1999) RB1 gene mutations in retinoblastoma. Hum Mutat 14:283–288
Downward J (2003) Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 3:11–22
Knudson AG Jr (1971) Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 68:820–823
Knudson AG (2001) Two genetic hits (more or less) to cancer. Nat Rev Cancer 1:157–162
Cavenee WK, Dryja TP, Phillips RA, Benedict WF, Godbout R, Gallie BL, Murphree AL, Strong LC, White RL (1983) Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature 305:779–784
Spruck CH, Won KA, Reed SI (1999) Deregulated cyclin E induces chromosome instability. Nature 401:297–300
Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, Michel L, Mittal V, Gerald W, Benezra R et al (2004) Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature 430:797–802
Coschi CH, Martens AL, Ritchie K, Francis SM, Chakrabarti S, Berube NG, Dick FA (2010) Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes Dev 24:1351–1363
Manning AL, Longworth MS, Dyson NJ (2010) Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev 24:1364–1376
van Harn T, Foijer F, van Vugt M, Banerjee R, Yang F, Oostra A, Joenje H, te Riele H (2010) Loss of Rb proteins causes genomic instability in the absence of mitogenic signaling. Genes Dev 24:1377–1388
Zheng L, Flesken-Nikitin A, Chen PL, Lee WH (2002) Deficiency of retinoblastoma gene in mouse embryonic stem cells leads to genetic instability. Cancer Res 62:2498–2502
Felsher DW, Zetterberg A, Zhu J, Tlsty T, Bishop JM (2000) Overexpression of MYC causes p53-dependent G2 arrest of normal fibroblasts. Proc Natl Acad Sci USA 97:10544–10548
Ono T, Losada A, Hirano M, Myers MP, Neuwald AF, Hirano T (2003) Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell 115:109–121
Ono T, Fang Y, Spector DL, Hirano T (2004) Spatial and temporal regulation of condensins I and II in mitotic chromosome assembly in human cells. Mol Biol Cell 15:3296–3308
Samoshkin A, Arnaoutov A, Jansen LE, Ouspenski I, Dye L, Karpova T, McNally J, Dasso M, Cleveland DW, Strunnikov A (2009) Human condensin function is essential for centromeric chromatin assembly and proper sister kinetochore orientation. PLoS One 4:e6831
Ribeiro SA, Gatlin JC, Dong Y, Joglekar A, Cameron L, Hudson DF, Farr CJ, McEwen BF, Salmon ED, Earnshaw WC et al (2009) Condensin regulates the stiffness of vertebrate centromeres. Mol Biol Cell 20:2371–2380
Vagnarelli P, Hudson DF, Ribeiro SA, Trinkle-Mulcahy L, Spence JM, Lai F, Farr CJ, Lamond AI, Earnshaw WC (2006) Condensin and Repo-Man-PP1 co-operate in the regulation of chromosome architecture during mitosis. Nat Cell Biol 8:1133–1142
Yong-Gonzalez V, Wang BD, Butylin P, Ouspenski I, Strunnikov A (2007) Condensin function at centromere chromatin facilitates proper kinetochore tension and ensures correct mitotic segregation of sister chromatids. Genes Cells 12:1075–1090
Hagstrom KA, Holmes VF, Cozzarelli NR, Meyer BJ (2002) C. elegans condensin promotes mitotic chromosome architecture, centromere organization, and sister chromatid segregation during mitosis and meiosis. Genes Dev 16:729–742
Hirano T (2005) Condensins: organizing and segregating the genome. Curr Biol 15:R265–R275
Longworth MS, Herr A, Ji JY, Dyson NJ (2008) RBF1 promotes chromatin condensation through a conserved interaction with the condensin II protein dCAP-D3. Genes Dev 22:1011–1024
Thompson SL, Compton DA (2008) Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol 180:665–672
Cimini D, Howell B, Maddox P, Khodjakov A, Degrassi F, Salmon ED (2001) Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol 153:517–527
Frame S, Balmain A (1999) Target genes and target cells in carcinogenesis. Br J Cancer 80(Suppl 1):28–33
Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA (1992) Effects of an Rb mutation in the mouse. Nature 359:295–300
Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-Cardo C, Lowe SW, Benezra R (2007) Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 11:9–23
Francis SM, Bergsied J, Isaac CE, Coschi CH, Martens AL, Hojilla CV, Chakrabarti S, Dimattia GE, Khoka R, Wang JY et al (2009) A functional connection between pRB and transforming growth factor beta in growth inhibition and mammary gland development. Mol Cell Biol 29:4455–4466
Isaac CE, Francis SM, Martens AL, Julian LM, Seifried LA, Erdmann N, Binne UK, Harrington L, Sicinski P, Berube NG et al (2006) The retinoblastoma protein regulates pericentric heterochromatin. Mol Cell Biol 26:3659–3671
Friend SH, Horowitz JM, Gerber MR, Wang XF, Bogenmann E, Li FP, Weinberg RA (1987) Deletions of a DNA sequence in retinoblastomas and mesenchymal tumors: organization of the sequence and its encoded protein. Proc Natl Acad Sci USA 84:9059–9063
Malumbres M, Barbacid M (2003) RAS oncogenes: the first 30 years. Nat Rev Cancer 3:459–465
McCormick F (1999) Signalling networks that cause cancer. Trends Cell Biol 9:M53–M56
Denko NC, Giaccia AJ, Stringer JR, Stambrook PJ (1994) The human Ha-ras oncogene induces genomic instability in murine fibroblasts within one cell cycle. Proc Natl Acad Sci USA 91:5124–5128
Ichikawa T, Kyprianou N, Isaacs JT (1990) Genetic instability and the acquisition of metastatic ability by rat mammary cancer cells following v-H-ras oncogene transfection. Cancer Res 50:6349–6357
Ichikawa T, Schalken JA, Ichikawa Y, Steinberg GD, Isaacs JT (1991) H-ras expression, genetic instability, and acquisition of metastatic ability by rat prostatic cancer cells following v-H-ras oncogene transfection. Prostate 18:163–172
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88:593–602
Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A et al (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444:638–642
Statistics CCSsSCoC (2011) Canadian cancer statistics 2011. Canadian Cancer Society, Toronto
Garbe JC, Holst CR, Bassett E, Tlsty T, Stampfer MR (2007) Inactivation of p53 function in cultured human mammary epithelial cells turns the telomere-length dependent senescence barrier from agonescence into crisis. Cell Cycle 6:1927–1936
Han X, Garcia-Manero G, McDonnell TJ, Lozano G, Medeiros LJ, Xiao L, Rosner G, Nguyen M, Fernandez M, Valentin-Vega YA et al (2007) HDM4 (HDMX) is widely expressed in adult pre-B acute lymphoblastic leukemia and is a potential therapeutic target. Mod Pathol 20:54–62
Hall PA, Meek D, Lane DP (1996) p53—integrating the complexity. J Pathol 180:1–5
Acknowledgments
C.H.C. is a recipient of a CIHR PhD studentship and is a member of the CaRTT training program. Research in F.A.D.’s laboratory is supported by grants from the Canadian Institutes for Health Research and the Canadian Cancer Society.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Coschi, C.H., Dick, F.A. Chromosome instability and deregulated proliferation: an unavoidable duo. Cell. Mol. Life Sci. 69, 2009–2024 (2012). https://doi.org/10.1007/s00018-011-0910-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-011-0910-4