
Abstract
The identification of chemokines in blood platelets has strengthened our view of these cells as participants in immune host defense. Platelet chemokines representing prestored and rapidly releasable proteins may play a major role as first-line inflammatory mediators. This is evident from their capability to recruit early inflammatory cells such as neutrophil granulocytes and monocytes and even to exhibit direct antimicrobial activity. However, insight is growing that platelet chemokines may be also long-term regulators, e.g., by activating T lymphocytes, by modulating the formation of endothelium and even thrombocytopoiesis itself. This review deals with the individual and cooperative functionality of platelet chemokines, as well as their potential as a basis for therapeutic intervention in the pathology of inflammation, infection, allergy and tumors. Within this context, therapeutic strategies based on the use of antibodies, modified chemokines, chemokine-binding proteins and chemokine receptor antagonists as well as first clinical studies will be addressed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Their peculiar morphologic and functional features, together with their ubiquitous abundance and immediate availability within the circulation, define blood platelets as perfect adaptations to their primary tasks as surveillants of hemostasis and organizing elements in vascular tissue repair. However, several recent discoveries have given reason to broadening this classical view to several surprising aspects. These demonstrate that platelet activation may entail immediate as well as far-reaching consequences in immunity, comprising the establishment of innate antimicrobial host defense to the point of modulating the adaptive immune response. In fact, platelets now are viewed as innate inflammatory cells with acute host defense functions as well as long-term regulatory roles (for a review see [1]). Additionally it has become clear that platelets also constitute powerful autoregulators of their formation from precursor cells.
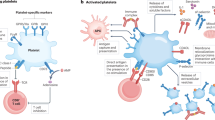
In recent years, two novel families of mediators have been recognized to play major roles for the communication of platelets with vascular cells and blood leukocytes. One of these is the tumor necrosis factor (TNF) family, where several members, such as CD40L, FasL and LIGHT, were shown to act as platelet-associated ligands of their counter-receptors on a variety of immune and vascular wall cells, resulting in a broad spectrum of inflammatory responses [2, 3]. Interestingly, members of the TNF family are also inducers of chemokines, which represent a second, even larger family of multifunctional mediators, some of which appear to be specifically platelet-associated and functionally adapted to a role in the vascular tissue. According to recent evidence, these platelet-derived chemokines not only constitute first-line mediators in inflammation, but apart from initiating host defense responses, are also involved in the prolongation and limitation of defence processes. Remarkably, some of these chemokines appear to be involved in the regulation of platelet formation from megakaryocytes and their progenitors. Although the spectrum of chemokines [both C-X-C motif ligand (CXCL) and C–C motif ligand (CCL)] that has been found to be produced and secreted by platelets has been extended by several molecular species, such as macrophage inflammatory protein alpha (MIP-1α/CCL3), growth-regulated protein alpha (GROα/CXCL1) and epithelial neutrophil-activating peptide 78 (ENA-78/CXCL5) [4, 5], the major platelet chemokines are still represented by beta thromboglobulin (β-TG/CXCL7) and platelet factor 4 (PF4/CXCL4), and regulated upon activation and normal T cell expressed and secreted (RANTES/CCL5) [6, 7]. Therefore, these latter members of the chemokine family will represent the main focus of this review. Following a brief update of their classic roles in host defense and hemostasis, several of their still less prominent and on some occasions unexpected features will be addressed and discussed. These will include recent discoveries about their cell and receptor interaction and novel cellular sources, as well as their emerging role as antimicrobial agents (Fig. 1).

Roles of major platelet-derived chemokines in the recruitment of blood leukocytes and the regulation of megakaryocytopoiesis and angiogenesis. Following the formation of a thrombus at an injured vessel wall, activated platelets rapidly secrete CXCL4, CCL5 and PBP/CTAP-III, two precursors of the active CXCL7 chemokine NAP-2. Following processing of these precursors by thrombus-associated neutrophils (PMN), i.e., in an environment depleted of protease inhibitors, the arising NAP-2 cooperates with CXCL4 in negative regulation of megakaryocytopoiesis, thus counter-acting CXCL12, an inducer of TPO. Both CXCL12 and NAP-2 participate in stimulating angiogenesis, while CXCL4 and especially CXCL4L1 represent strong inhibitors of angiogenesis. Although not chemotactic itself, CXCL4 cooperates with NAP-2 and CCL5 in recruiting PMN and monocytes (Mo), respectively, to the vessel wall. Both CCL5 and CXCL4 also induce adherence of eosinophils (Eo), while CCL5 recruits T cells (T). Transmigration of the cells is stimulated by extravascular NAP-2 (PMN) and CCL5 (Mo, Eo, T)
Platelet chemokines: a set of complementary chemoattractants with specific roles in host defense and hemostasis
Platelet chemokines, like many other secretable platelet proteins including growth factors and various cytokines, are located within α-granules. Notably, a common three-dimensional granule targeting motif has been identified within CXCL7, CXCL4 and CCL5 that appears to be responsible for the localization of these major platelet chemokines within their storage compartments [8]. Upon proper platelet activation (e.g., by thrombin) their release from α-granules into the extracellular environment constitutes a rapid process that is initiated within seconds. Together with the ensuing clot formation and platelet aggregation, this process brings forth not only the mechanical sealing of vascular wounding, but also marks the onset of tissue repair and establishes a proinflammatory environment culminating in the recruitment and activation of various types of immune cells. The roles of platelet chemokines in these events are complex and often interdependent, suggesting the existence of a system of thrombocytic mediators with structural and functional adaptations to the conditions found within and around the vascular tissue.
Platelet chemokines and immune cell recruitment
Cellular targets of platelet chemokines
Regarding their differing functional properties, it appears not surprising that the three major platelet chemokines each belong to a different branch of the chemokine family and altogether form a repertoire of functionally diverse although complementary chemoattractants. Thus, CXCL7 as a member of the CXC chemokine subfamily constitutes a potent chemoattractant and activator for neutrophil granulocytes [9–11], consequently, recruiting those blood leukocytes to sites of injury that are responsible for the immediate establishment of an antimicrobial line of defense. As CXCL7 belongs to the subgroup of ELR+ CXC chemokines, it functions through interaction with the two Gi protein-coupled seven-transmembrane domain (7-TMD) receptors, CXCR1 and CXCR2 [12, 13], with CXCR1 representing a low affinity and CXCR2 a high affinity receptor for this chemokine [14–16]. Conversely, the lack of the ELR+ tripeptide motif in CXCL4, allocating this protein to the ELR− CXC chemokine subgroup, prevents the binding of CXCL4 to the above receptors [17]. This correlates with the absence of chemotactic activity not only for neutrophils [9, 18] but also for any other blood cell population so far tested [19, 20] and renders CXCL4 a somewhat unusual chemokine (chemotactic cytokine), despite its clear structural relationship to other members of this cytokine family. Notably CXCL4 participates in the recruitment and activation of neutrophils and even more so of monocytes by acting through a variety of other mechanisms, which include its specific binding to and signaling through integral chondroitin sulfate proteoglycans [21–23] as well as the formation of heterooligomers with other chemokines. So far, there is only one report suggesting the existence of Gi protein-coupled 7-TMD receptor(s) (CXCR3) for CXCL4 [24]. However, this finding with human T cells has neither been confirmed by others since then nor could we reproduce the data describing CXCL4 chemotactic activity for human T cells (F. Petersen, Research Center Borstel; unpublished results). By contrast, CCL5, a member of the CC-subfamily of chemokines, has been found to interact with at least three species of Gi protein-coupled receptors (CCR1, CCR3, CCR5) that are differentially expressed on a variety of leukocytes and lymphocytes [25]. Thus, CCL5 considerably adds to the repertoire of immune cells that become attracted and recruited by platelet-secreted chemokines, by acting on monocytes [25], resting and activated T cells [26, 27], eosinophils [28, 29], basophils [30], natural killer (NK) cells [31] and dendritic cells (DC) [32]. Altogether the three major platelet chemokines form a set of rapidly releasable mediators having important roles in host defence by addressing cells involved in innate as well as adaptive immunity. In the following some examples are given of how platelet-derived chemokines act in concert on distinct leukocyte populations.
Interaction of CXCL7 and CXCL4 in neutrophil recruitment
As a chemoattractant and activator for neutrophils [9–11], CXCL7 is crucial in establishing the first line of cellular defense following vascular injury. It is comprehensible that such a signal marking the onset of an inflammatory reaction must be under tight control. Thus, different from other chemokines, CXCL7 is secreted as a chemotactically inactive precursor molecule that requires proteolytic processing to become active [33, 34]. Human platelets secrete two CXCL7 precursors, termed platelet basic protein (PBP, 94aa) and connective tissue-activating peptide III (CTAP-III, 85 aa), which represent N-terminally truncated derivatives of pre-PBP (128 aa), the primary CXCL7 translation product bearing a 34 aa leader sequence [6, 35–38]. Interestingly, platelets themselves do not have the capacity to process PBP or CTAP-III, but conversion of these precursors into the chemotactically active neutrophil-activating peptide 2 (NAP-2, 70 aa) is dependent on the presence of neutrophils, the NAP-2 target cells themselves, which expose cell-associated cathepsin G on their surface [33, 39, 40]. Cleavage of PBP/CTAP-III by this serine protease behind the only tyrosine residue in these molecules removes their inhibitory N terminus that was found to fold back over the ELR+ motif and to prevent interaction of the molecules with receptors CXCR1/2 [41]. The peculiarity of this way of NAP-2 generation consists in its potential to release a self-amplifying process, consisting in consecutive steps of neutrophil attraction and enhanced CXCL7 activation. Among the mechanisms involved in controlling these events, inhibition of the PBP/CTAP-III-processing enzyme by plasma proteinase inhibitors is of prime importance. Thus, we found plasma down to 0.01% to completely inhibit CXCL7 processing by suspended neutrophils [39]. However, the same cells captured within aggregates of activated platelets exhibited hardly impaired processing capacity even in the presence of up to 40% plasma (own unpublished results). This demonstrates that within thrombi plasma proteinase inhibitors may become excluded and/or inactivated [42, 43], allowing the establishment of a proinflammatory microenvironment. Thus, as far as the formation of NAP-2 is concerned, plasma inhibitors appear to control NAP-2 formation at its onset in order to prevent inadequate neutrophil activation, while limitation of the reaction once initiated appears to be brought about by other factors. One of these is likely CXCL4, the second major platelet chemokine simultaneously released from the same storage granules. Notably we found CXCL4 to act as a potent inhibitor of neutrophil as well as cathepsin G-catalyzed CXCL7 processing [44]. All evidence suggests that CXCL4 acts as a direct inhibitor of the processing enzyme and does not function through forming heterooligomers with CXCL7 or through activating neutrophils to release inhibitory factors. Interestingly, CXCL4 is inhibitory at concentrations far below those required to induce neutrophil activation on its own. As a consequence of blocked CXCL7 processing, CXCL4 acts also as a strong inhibitor of NAP-2-induced neutrophil adhesion to endothelial cells, although it does not inhibit NAP-2-dependent transendothelial migration [45]. Although, as mentioned above, CXCL4 is not a chemotaxin, it may assist in the recruitment of neutrophils by inducing a very strong adhesion to resting endothelial cells, and may even activate exocytosis of secondary granule contents in adherent or TNF-primed neutrophils in the absence of primary granule activation [19, 46]. Notably, we even found CXCL4 at suboptimal concentrations to positively cooperate with NAP-2 in inducing neutrophil adhesion to endothelial cells [45]. These observations and the fact that CXCL4 exhibits its activating functions at much higher concentrations than its inhibitory ones suggest a regulatory role for this chemokine consisting in the sharpening of proinflammatory signals by suppressing weak ones and enhancing those that are beyond threshold.
Interaction of CXCL4 and CCL5 in monocyte recruitment
While CCL5 is known as a potent monocyte chemoattractant, CXCL4 and CXCL7 do not have chemotactic activity for these cells. Nevertheless, all three chemokines have been shown to support monocyte recruitment by their capacity to bind to activated human microvascular endothelial cells, which increases surface immobilization and subsequent monocyte arrest [47]. Inhibition studies using corresponding antibodies revealed that the chemokines cooperated in an additive manner. Deposition of platelet-derived chemokines on endothelium may occur through differing ways, including release of these mediators by activated platelets or through delivery by platelet microparticles, which is favored by flow and may also serve as a transcellular delivery system [48]. The fact that especially platelet-derived chemokines but not, e.g., CXCL5 have a prominent capacity to bind to endothelium highlights a special importance of these mediators in intravascular cell recruitment and suggests the expression of proteoglycans in the vasculature that may selectively capture such chemokines. Interestingly, activated platelets have been found to secrete proteoglycans such as soluble chondroitin sulfate A by themselves. The platelet-derived, fully sulfated form of this molecular species, but not the less-sulfated molecule from plasma, was observed to prevent sequestration of CCL5 on the vascular surface as well as CCL5-mediated firm adhesion of monocytes to endothelial cells under physiological flow conditions [49]. A further indication that this proteoglycan may represent an efficient counter-regulatory element to confine leukocyte recruitment was supported by the fact that it was capable of inhibiting CCL5-elicited influx of calcium in CCR5-transfected CHO cells. On the other hand, binding of platelet-derived chemokines to chondroitin sulfate proteoglycans expressed on the monocyte surface was revealed to provide a basis for cooperative action between these mediators. Such cooperation exists between CXCL4 and CCL5, where CXCL4 alone was not able to stimulate arrest of monocytes to activated endothelium under flow conditions, but greatly enhanced cell arrest induced by CCL5 [50]. Vice versa, CCL5 enhanced binding of CXCL4 to monocytes. Interestingly, these effects were dependent on the ability of CCL5 to form oligomers of higher order, which relies on certain structural motifs and on its binding to proteoglycans and may facilitate the formation of CCL5/CXCL4 heterooligomers. As CCL5 was shown to signal monocyte arrest through CCR1 instead of CCR5 (the chemotaxis receptor) [51], it was proposed that heterophilic interaction of these chemokines promotes signaling through the former receptor [50]. The physiological relevance of platelet-derived chemokines for monocyte recruitment was demonstrated by experiments with activated full blood where Met-RANTES (an antagonist to CCL5) and antibodies to CXCL4 and CXCL7 inhibited the arrest of monocytes to activated human endothelium [48].
Platelets and their progenitors as targets of chemokines
Chemokine receptors on mature platelets and their involvement in platelet function
There are various reports describing message as well as surface expression for a limited set of chemokine receptors in mature human platelets. Among these are receptors for CXC and CC chemokines, however, functional effects of chemokines on platelets appear to be dependent on several parameters. In one of the earliest studies, Power et al. [5] found mRNA expression for IL-8R A and CKR K5.5, now termed CXCR1 and CCR4, respectively. In a more comprehensive study, this was confirmed by Clemetson et al. [52]; however, surface expression was only found for CCR4, and moreover for several additional receptors, comprising CCR1, CCR3, CCR4 and CXCR4, while CXCR1, CXCR2, CXCR3 and CCR5 were all negative. Evidence for functionality of surface-expressed chemokine receptors, as seen by intracellular Ca2+ signals and platelet aggregation, was implicated by the responsiveness of platelets to corresponding ligands, i.e., to chemokines MIP-1α (CCR1), RANTES (CCR1, CCR3), eotaxin (CCR3), the thymus and activation-regulated chemokine TARC/CCL17 and the macrophage-derived chemokine MDC/CCL22 (CCR4), as well as to the stromal cell-derived factor SDF-1/CXCL12 (CXCR4). Notably, aggregation responses to chemokines were relatively weak as compared to thrombin and were partly dependent on the liberation of ADP as a secondary signal, as well as on the interaction with platelet surface proteoglycans [52]. More direct evidence for the involvement of CCR4 and CXCR4 was provided by other authors, showing that specific antibodies to these receptors could prevent functional signaling by its respective ligands [53, 54]. Also, CCL17 and CCL22 as well as CXCL12 were clearly shown to require the presence of low amounts of primary platelet agonists thrombin or ADP to induce maximal platelet aggregation under arterial flow conditions, and ADP potentiates shape change, aggregation and adhesion to collagen [55]. Inhibition of chemokine-induced aggregation by apyrase [55] or by ADP-receptor antagonists [56] again highlighted the importance of ADP as a cooperative signal. Responsiveness of platelets to CCL17 and CCL22, which are Th2-type cytokine-induced mediators, has been taken to suggest a role for these chemokines in platelet activation seen in Th2 diseases, such as asthma and atopic dermatitis. Correspondingly, the identification of highly expressed CXCL12 in endothelium and atherosclerotic plaques has suggested a role for CXCL12 in the pathogenesis of atherosclerosis and thrombo-occlusive diseases. CCL22 may as well participate in these events, as it has been shown that the chemokine may cooperate in thrombus formation with oxidized low-density lipoprotein, exposed during atherosclerotic plaque rupture [57]. Interestingly, it has also been found that platelet-derived chemokines such as CCL5 may modulate the platelet response to external chemokines. Thus, it was shown that CCL5 abrogated CXCL12-induced fibrinogen binding as well as CXCL12-promoted whole blood platelet adhesion to endothelial cells under venous flow conditions [58]. Also, CXCL12-induced platelet aggregation was inhibited to a certain extent, but not that induced by thrombin receptor-activating peptide or ADP, altogether suggesting that CCL5 represents a non-competitive antagonist to CXCL12 and thus may play a regulatory role in the platelet response to inflammation.
Chemokines involved in platelet development and maturation
First evidence that chemokines could play a role in platelet development came from the observation that the major platelet chemokines CXCL4 and CXCL7 effected negative autocrine regulation of human megakaryocytopoiesis [59, 60]. Both CXCL4 and the CTAP-III species of CXCL7 in normal marrow cultures caused a decrease in mixed-meg cultures (BFU-meg and large CFU-meg). This suggested that both proliferation and maturation of megakaryocyte (MK) progenitor cells were inhibited, with a predominant effect on earlier progenitor cells [60]. Subsequently others described that the NAP-2 species of CXCL7 was two to three orders of magnitude more potent than CXCL4 in inhibiting formation of MK colonies and that this depended on the presence of the ELR-motif in NAP-2 [61]. NAP-2/CXCL4 chimera containing the ELR-motif were equally potent to NAP-2, and also CXCL8, another ELR+ CXC chemokine, was effective. While this indicated the involvement of chemokine receptors CXCR1 and/or CXCR2, also several CC chemokines produced by ancillary marrow cells (CCL3, MIP-1β/CCL4) had comparable activity, suggesting both autocrine and paracrine control of megakaryocytopoiesis by chemokines. Further evidence for the involvement of CXCR1 and CXCR2 came from the observation that antibodies to these receptors increased proliferation and differentiation and restored polyploidization in MK precursors from patients with myeloid metaplasis with myelofibrosis [62].
Interestingly, it was found that MK maturation is associated with the expression of CTAP-III, the latter being especially high in mature human bone MK, where it localizes to the platelet-producing zones in the cytoplasm [63]. Thrombopoietin (TPO) is a strong inducer of CXCL7 as well as CXCL4, and as is also seen in mice, both chemokines act as negative feed-back regulators by suppressing polyploidization in the presence of TPO and pro-platelet formation during MK maturation [64].
As another major chemokine player in platelet development, although with quite opposite functions than the above mediators, the ELR- CXC chemokine CXCL12 has been recognized. According to observations by several authors, CXCR4, the major and monospecific receptor for CXCL12, is expressed on the MK lineage from early progenitors to fully differentiated platelets. These reports comprise CXCR4 mRNA expression in CD34(+) bone marrow cells, CD16(+) cells, platelets, MK leukemia cells and CFU-Meg [65], as well as surface expression of the receptor on mature polyploidal MK [66], MK precursors, and platelets [67]. Despite clear-cut expression of CXCR4 throughout all developmental stages, effects of its ligand CXCL12 differ with respect to the differentiation state of the respective cell population. As a general outline, immature MKs were found to readily respond to CXCL12 by cytosolic Ca2+ elevation and migration, while mature MK and platelets did not [65, 67, 68]. There is evidence that the upregulation in mature MK of RGS16, a negative regulator of G protein signaling interfering with CXCR4 signal transduction, is responsible for these discrepancies [68]. The role of CXCL12 as an enhancer of MK development and platelet formation is based on observations by several authors, describing its stimulation of immature megakaryoblast formation as well as its enhancing effect on thrombopoietin (TPO)-induced development of MK precursors [69, 70]. The impact of CXCL12 appears to be selective, since in the murine system stem cell factor (SCF)- and IL-3-dependent effects are not supported and CXCR4 inhibitors reduce TPO-dependent growth [70]. Moreover, CXCL12 and TPO obviously regulate distinct aspects of human megakaryopoiesis since CXCL12, unlike TPO, does not stimulate alpha(IIb)beta(3)(+) cell proliferation or differentiation or have an antiapoptotic effect, whereas both increase adhesion of these cells to fibrinogen and vitronectin [71]. Thus, CXCL12, platelet chemokines CXCL4/CXCL7 and TPO altogether appear to form part of a balanced system in the regulation of platelet development and maturation. Interestingly, recent evidence suggests that CXCL12, so far believed to stem from external cellular sources, may also be secreted by platelets themselves [72], which circumstance would allow for an additional autoregulatory circuit in platelet development.
Platelet chemokines in vascular repair and angiogenesis
Vascular damage constitutes an inevitable consequence of wounding and inflammation. Apart from initiating the inflammatory process through the recruitment and activation of blood leukocytes, platelet chemokines were also shown to participate in physiological as well as pathological aspects of vascular repair and tissue remodeling. According to observations in mice, injured arteries may become recolonized not only by lateral outgrowth of resident vascular cells, but also by bone marrow-derived progenitors of smooth muscle and endothelial cells that are recruited to thrombi by platelet-secreted CXCL12 [72]. In this respect it may also be interesting that some platelet-derived growth factors like vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) induce massive expression of CXCR4 on human EC, leading to enhanced chemotactic migration of these cells in response to CXCL12 and to in vivo neovascularization as observed in rats [73]. Concomitantly, growth factors and hematopoietic cytokines induce CXCL12 secretion in various cells leading to enforcement of neoangiogenic processes, which demonstrates that CXCL12 is essential in mediating the angiogenic effects of growth factors. In fact, as seen in CXCL12 and CXCR4-deficient mice, the chemokine has a crucial role in trafficking, engraftment and differentiation of hematopoietic stem cells, and moreover as a product of tumor cells is responsible for neoangiogenesis in cancer (reviewed in [74]). Furthermore, Hristov et al. [75] have recently described that circulating endothelial progenitor cells may contribute to reendothelialization of wounded arterial areas by their expression of chemokine receptor CXCR2, allowing for chemotactic migration and recruitment of these cells to fibronectin or platetet-covered surfaces through chemokines CXCL1 and CXCL7. Interestingly, expression of these two chemokines was not only found in platelets but also in denuded vascular smooth muscle cells soon after injury. It is suggested that colonization by endothelial precursor cells may be instrumental in preventing neointimal growth induced by, e.g., CCL5 through monocyte attraction following endothelial injury [76]. Meanwhile it has been recognized that CXCR2 is not only important for cell recruitment but also for transmitting growth signals in endothelial cells. This was first suggested upon the observation that all ELR+ CXC chemokines are able to induce proliferation in uninflamed microvascular endothelium [77] and that blocking of CXCR2 in different animal models inhibited chemotaxis as well as angiogenesis in response to ELR+ CXC chemokines [78]. Similarly, indicating the importance of the CXCR2/CXCR2-ligand axis in pathology, human lung transplantation ischemia-reperfusion injury was found to be associated with elevated levels of CXCL3, CXCL7 and CXCL8, and in a rat orthotopic single lung transplantation model of cold ischemia-reperfusion injury the same authors demonstrated that CXCR2 receptor blockade reduced lung neutrophil sequestration and attenuated post-lung transplantation cold ischemia-reperfusion injury [79]. While it is known that apart from several ELR+ CXC chemokines growth factors such as VEGF, bFGF, platelet-derived growth factor (PDGF) and heparanase contribute to the proangiogenic potential of platelets, the non-ELR chemokine CXCL4 has been identified as the major anti-angiogenic component contained in platelet releasates [80]. In fact anti-angiogenic activity of CXCL4 has been reported by several authors previously [81–83], and this observation is in good accordance with findings describing that other non-ELR CXC chemokines such as Mig/CXCL9, IP-10/CXCL10 and I-TAC/CXCL11 similarly have a potential to inhibit endothelial cell proliferation [77, 84, 85]. Due to the angiostatic properties of CXCL4 and the latter, interferon (IFN)-inducible chemokines have come into focus regarding their potential benefit for cancer treatment and other diseases promoted by neovascularization [86–88]. As a clue to exploiting the operating principle, receptor-ligand interactions have been intensely investigated. These studies, especially those addressing CXCL4, resulted in the discovery of variable and complex mechanisms being involved in the chemokines’ mode of action. Competable activity and binding of CXCL4 and other non-ELR chemokines to endothelial cell surface heparan sulfate proteoglycans (HSPG) [81, 82, 84] suggested that these might function either directly as signaling receptors or indirectly as bystander molecules. Although this may be true for CXCL4-mediated activation of monocytes and neutrophils that do not express G protein-coupled receptors (GPCR) for this chemokine [20, 21], microvascular endothelial cells meanwhile were shown to express an alternatively spliced variant of CXCR3, referred to as CXCR3-B, that is coupled to Gs proteins instead of Gi proteins and mediates the anti-angiogenic effect of CXCL4 as well as that of CXCL9, CXCL10 and CXCL11 [85, 89, 90]. Nevertheless, it is not yet clear whether the angiostatic effect of the three IFN gamma-inducible chemokines and CXCL4 is exclusively mediated by CXCR3-B, as these mediators are angiostatic also in mice [91], despite there being no evidence for the expression of CXCR3-B in mice. Especially for CXCL4 the utilization of several concurrent mechanisms in addition to CXCR3-B activation has become evident. These are based on competition with growth factors such as bFGF or VEGF for binding to endothelial cell surface HSPG [82, 92] or inhibition of growth factor dimerization by complex formation with ligands, as seen in the murine system with FGF-2 [93]. Further studies revealed, that CXCL4 may inhibit angiogenic growth signals in a dual manner by interfering with growth factor-mediated receptor activation and signaling as well as by eliciting HSPG and growth factor receptor-independent inhibition by disrupting the MAP-kinase cascade in endothelial cells [94]. Because the latter study also demonstrated that CXCR3-B activation was not involved in these processes, the existence of so far unidentified CXCL4 receptor(s) was postulated. Interestingly very recently an additional potential mechanism for anti-angiogenesis operating via binding to integrins has come into focus. According to Aidoudi et al. [95], soluble CXCL4 binds to integrins (preferentially αvβ3 and some related) on endothelial cells, thereby inhibiting their adhesion to RGD matrix proteins as well as cell migration, i.e., two prerequisits required for neovasularization. Novel features concerning CXCL4-mediated anti-angiogenesis have not only been considered at the receptor level, but were recently also recognized to exist at the level of ligands. Thus, although known for about 20 years already [96], a non-allelic variant of the CXCL4 molecule termed CXCL4L1 (PF4var1, PF4alt) has come into focus only recently. Differing from CXCL4 by the exchange of only three amino acids within the C terminus, CXCL4L1 was found to be 30-fold more active in inhibiting endothelial cell chemotaxis and comparably more active as an inhibitor of angiogenesis, as seen in a rat cornea micropocket assay [97]. Later on the variant’s enhanced ability to prevent angiogenesis was also recognized to be responsible for its higher inhibitory efficiency for melanoma and lung carcinoma growth and metastasis [91]. Thus, taken together in vascular damage, platelet chemokines play important roles not only in the trafficking and activation of inflammatory cells but also in the recruitment and growth regulation of endothelial cells during vascular repair.
Thrombicidins, platelet chemokines with direct antimicrobial activity.
Not until a few years ago was a quite unexpected feature of chemokines discovered, extending their role from classic cell-directed cytokines to that of mediators being directly involved in antimicrobial host defense. First evidence for a role of chemokines as antimicrobial agents came from studies investigating rabbit platelets for their content of so-called “platelet microbicidal proteins” (PMPs), resulting in the discovery of two thrombin-releasable cationic proteins with potent in vitro activity against gram-positive and gram-negative bacteria as well as Candida albicans blastopores [98]. Intriguingly these proteins later turned out to be orthologs of human CXCL4 [99], while in the meantime corresponding studies on human platelets identified CXCL7, CCL5 as well as CXCL4 as novel antimicrobial agents [100, 101]. Whereas antimicrobial chemokines in general are now termed “kinocidins,” those stemming from platelets are also known as “thrombocidins” (TC). Altogether thrombocidins were found to display a rather broad spectrum of antimicrobial activity, including Escherichia coli, Staphylococcus aureus, and fungi Cryptococcus neoformans and Candida albicans (with the latter being however resistant to CXCL7). Notably, among 22 different chemokines tested, CCL5 (besides CCL3 and CCL4) was found to exhibit significant antiviral activity against HSV-1 [99]. On the other hand, thrombocidins exhibit characteristic differences regarding the conditions required for activity. While CCL5, CXCL4 and the CTAP-III species of CXCL7 exert significant microbicidity at slightly acidic pH only, the PBP species of CXCL7 is also active under neutral pH conditions [101]. Lacking antimicrobial activity at neutral pH in CXCL4 as well as CXCL7 species CTAP-III and NAP-2 has been described by Krijgsveld et al. [100] before. However, concerning CXCL7-based thrombocidins, platelets were found to contain derivatives of NAP-2 and CTAP-III (TC-1 and TC-2, respectively) with potent microbicidal activity under neutral conditions. TC-1 and TC-2 differed from their parent molecules by the lack of two amino acids at the C terminus, suggesting that limited C-terminal truncation is a prerequisite for their antimicrobial activity. Despite their structural similarity and functional overlap with defensins, TC-1 and TC-2 appear to utilize a killing mechanism different from pore formation, most likely by interacting with intracellular target(s) [102]. A membrane-disrupting mechanism by rabbit PMPs different from that exerted by defensins was also suggested by a previous study by Yeaman et al. [103]. Taken together, the major platelet chemokines represent a distinct category of kinocidins that may be active under variable environmental conditions to support host defense via the recruitment of inflammatory cells and by the direct killing of microorganisms at the same time. As secreted products their direct antimicrobial activity may become effective extracellularly in inflammatory exudates as well as intracellularly following opsonization and subsequent ingestion by phagocytes.
Expression of platelet chemokines by other cell types
Whereas until recently it was believed that CXCL7 and CXCL4 constitute specific marker chemokines of the megakaryocytic lineage, there is increasing evidence for their expression in and secretion by a variety of other cell types. Although being secreted to quantities far below those stored and released by platelets, several investigations have uncovered their involvement in rather tissue-specific functions within the environment of the respective producer cells. In early work Iida et al. reported the induction by LPS of leukocyte-derived growth factor (LDGF), a cytokine identical in structure to pre-PBP, the 128 aa precursor of CXCL7 in human monocytes. Upon recombinant expression this protein was found to act as a fibroblast mitogenic factor in a PDGF-like manner [104]. This could however not be reproduced by others, who described the induction of CXCL7 in monocytes upon coculture with stromal bone marrow cells, leading to the secretion of PBP and NAP-2 protein [105]. Moreover human monocytes were shown to constitutively transcribe and express CXCL7 at the protein level, the intracellular molecule being localized to granular structures and existing as derivatives of variable size [106]. While stimulation of monocytes with IL-4, IL-10 or glucocorticoids led to down-regulation [106], stimulation with LPS, zymosan or synthetic activating peptides for proteinase-activated receptor 1 (PAR-1) effected increased synthesis of CXCL7 and derivatives. Localization of these molecules to phagolysomes raised speculations that CXCL7 proteins could constitute part of the antimicrobial arsenal of human monocytes [107]. This view may be enforced by the finding that correspondingly stimulated monocytes also underwent induction and increased synthesis of the microbicidal platelet chemokines CXCL4 and CCL5 [108]. At least an indirect role for monocyte-derived CXCL7 in antimycobacterial defense appears likely according to the finding that enhanced synthesis of CXCL7 by BCG-stimulated M-CSF-induced human macrophages correlated with increased resistance of these cells to M. tuberculosis and that CXCL7 was able to increase superoxide production as well as resistance to M. tuberculosis in the more susceptible GM-CSF-induced macrophages [109]. Apart from monocytes/macrophages also several types of tissue cells have been reported to express CXCL7. Increased synthesis was found in intestinal epithelial cells from patients with ulcerative colitis, and matrix metalloproteinases from lamina propria myofibroblasts were found to act as proteolytic activators of the chemokine [110]. A novel role for CXCL7 in metabolism of the bone was recently established by showing that hypoxic stress (i.e., IGF2-mediated giant osteoclast formation) is promoted by stromal bone marrow cell-secreted CXCL7 and becomes enhanced by CXCL12 from osteoblastic cells [111]. Concerning CXCL4 both the original chemokine and its variant CXCL4L1 have not only been found in platelets, monocytes and T cells, but are also expressed in human microvascular ECs, in human aortic as well as coronary smooth muscle cells, and a human colonic epithelial cell line [112]. Interestingly, mRNA expression of the two genes differed in all cell populations, being throughout more prevalent for CXCL4 than for CXCL4L1, except in smooth muscle cells, which generally also exhibited significantly enhanced secretion of the proteins. The large (38%) difference of CXCL4 and CXCL4L1 in the signal peptide region suggested individual mechanisms of secretion in the variant proteins. This was confirmed upon transfection into different cells, where CXCL4 was observed to be stored in secretory granules and to become released upon activation of protein kinase C, whereas CXCL4L1 was continuously synthesized and secreted through a constitutive pathway [113]. Similar findings by others with human osteosarcoma cells also argued for de novo synthesis instead of release from intracellular stores of CXCL4L1 and moreover exemplified coinduction of the (strongly angiostatic) variant but not (the less angiostatic) CXCL4 with (the strongly angiogenic) granulocyte chemotactic protein 2 (GCP-2/CXCL6) [112]. These and additional findings argue for an intricate regulation of angiogenesis and may facilitate the potential use of CXCL4L1 in cancer therapy, as outlined later on in this review.
Principles of chemokine-based therapeutic intervention in the pathology of infection, allergy and tumors
At the onset of these considerations it should be recalled that under physiological conditions the chemokine-chemokine receptor system has self-regulatory properties. In the early event of inflammation, thrombus formation occurs, and as mentioned before, the thrombus provides a proinflammatory environment in which proteinase inhibitors preventing the formation of CXCL7 are inactivated or neutralized [39, 42, 43]. Furthermore, platelet-derived CXCL4 inhibits cathepsin G-catalyzed processing of CXCL7 by directly blocking/acting on the processing enzyme, and thus inhibits CXCL7-induced adhesion of neutrophils to vascular endothelial cells [44, 45]. This inhibition of neutrophil adhesion occurs at low concentration of CXCL4, whereas at high concentrations CXCL4 cooperates with CXCL7 in inducing adherence of neutrophils to endothelial cells. This dual role of CXCL4 points to its regulatory role in determining the proinflammatory microenvironment. Furthermore, platelet agonists affecting the release of the above chemokines, such as thrombin or ADP, also induce maximal platelet aggregation under arterial flow conditions. In addition, ADP potentiates shape change, aggregation and adhesion to collagen induced by the chemokines CCL17, CCL22 and CXCL12 [54–56]. Chemokines function as key regulators of cell migration and therefore represent proteins that might be exploited to therapy. Their therapeutic potential includes autoimmune diseases such as rheumatoid arthritis, multiple sclerosis, systemic lupus erythematodes, psoriasis as well as allergic disorders such as allergic dermatitis, asthma, inflammatory bowel disease, transplant rejection, cancer and infectious diseases including AIDS. Results of the last 10 years of research using knockout mice, specific monoclonal antibodies and receptor antagonists have shown that despite the redundancy of chemokines and promiscuity of ligands binding to receptors in inflammatory processes, therapeutic compounds blocking individual chemokines and/or receptors could be developed in animal models of disease.
Small molecule chemokine receptor antagonists
The approach to block the interaction of a chemokine with its receptor by small molecule chemokine receptor antagonists was reported to be successful in vivo in an animal model in which neutrophil accumulation in the lungs of hyperoxia-exposed newborn rats was prevented by a non-peptide CXCR2 antagonist [114]. A synthetic non-peptide CXCR2 antagonist blocked macrophage inflammatory protein 2 (MIP-2/CXCL2)-induced neutrophil migration in vivo in mice [115]. In the meantime, based on the structure of reparixin, a small molecular weight allosteric inhibitor of CXCR1, a dual inhibitor of CXCRl and CXCR2 with a longer half-life, DF2156A has been developed, which in a cerebral ischemia-reperfusion rat model inhibited the infiltration of neutrophils, reduced infarct size and improved neurological functions [116]. In an athymic nude-mouse model of human melanoma cell tumor growth, small molecule CXCR2 and CXCR1 inhibitors increased melanoma cell apoptosis in treated compared to control mice [117]. In models of multiple sclerosis, a neurodegenerative disease characterized by demyelination/remyelination episodes, the CXCL1-CXCR2 system is involved in the regulation of migration, proliferation and differentiation of immune and neural cells. The authors showed that treatment of myelin oligodendrocyte glycoprotein peptide 35–55-induced EAE mice with small molecule antagonists against CXCR2 results in decreased lesions and enhanced remyelination, suggesting that inhibition of CXCR2 signaling may provide a therapeutic target for demyelinating disease [118]. In chronic obstructive pulmonary disorders, interference with CXCR2 receptor function has demonstrated inhibition of pulmonary damage induced by neutrophils, antigen or irritant induced goblet cell hyperplasia, and angiogenesis/collagen deposition caused by lung injury [119, 120]. These studies hold considerable promise for identifying novel and efficacious treatments of pulmonary disorders. Clinical trials evaluating small molecule CXCR2 antagonists in COPD, asthma and cystic pulmonary fibrosis are underway. Therapeutic targets include cognate receptors for CCL5 as well as for ELR+ CXC chemokines (Table 1).
Anti-CCR5/CXCR4 strategies in HIV
The human chemokine receptors CCR5 and CXCR4 are attractive targets for small molecule antagonists that inhibit HIV-1 infection. In the overall process of viral entry, the point of attack of such antagonists occurs after the HIV envelope gp120 has interacted with its primary receptor CD4, but before the HIV envelope gp41 inserts the fusion peptide into the host cell membrane. HIV-1 strains can be categorized by co-receptor tropism, that is, their ability to use CXCR4 or CCR5 or both for entry into cells. CCR5 has received more attention as a potential target, because a natural deletion (DELTA 32) in the CCR5 gene confers natural resistance to infection by CCR5-tropic strains, which are most frequently sexually transmitted [122]. CXCR4 has received less attention, although a switch from CCR5 to CXCR4 tropism may occur spontaneously in approximately 50% of HIV-infected patients and has been associated with HIV progression [123]. The most advanced CXCR4 antagonist AMD3100 (Mozobil) has not been further pursued as a therapeutic modality for HIV infections, but as a highly selective CXCR4 antagonist it has proved to be particularly promising for the mobilization of hematopoietic stem cells in patients with hematologic malignancies such as non-Hodgkin’s lymphoma (NHL) [124]. Three CCR5 antagonists have been evaluated in clinical trials—maraviroc, vicriviroc and aplaviroc. The further development of aplaviroc was interrupted in 2005 following the observation of elevated liver enzymes and total bilirubin in one of the HIV-infected patients receiving the compound [123]. Maraviroc is highly selective for CCR5 indicating potential for a clinical safety profile [125]. It was FDA-approved in August 2007 (Selcentry), and a 10-day monotherapy with maraviroc at twice daily doses of about 100 mg achieved a mean reduction of approximately 1.6 log10 viral RNA copies per ml at a median of 10–15 days, thus providing proof-of-concept that CCR5 antagonism is a valuable antiretroviral therapeutic approach [126]. There is no clear evidence for a co-receptor switch from CCR5 to CXCR4 under selection pressure from maraviroc, as virus strains that have become resistant to maraviroc still use inhibitor-bound receptor for entry [127]. Like maraviroc, vicriviroc was given to patients with HIV during a 14-day monotherapy at doses of 25 or 50 mg twice daily. Reductions of 1.0–1.5 log10 HIV RNA copies per ml were achieved [128]. In another study involving HIV-infected treatment-experienced patients, the reduction in viral load was sustained throughout a 24-week treatment period with doses as low as 10 or 15 mg [129]. Malignancies occurred in a number of patients; however, the relationship of vicriviroc and malignancy has remained uncertain [130]. Infections with a dual (CCR5 and CXCR4)-tropic HIV-1 population or several HIV-1 populations are common among highly treatment-experienced patients, but how these dual-tropic or mixed HIV-1 populations respond to these compounds in vivo remains to be investigated. Ultimately, combinations of CCR5 and CXCR4 inhibitors will be needed to cope with the dual-tropic or mixed-tropic HIV-1 infections. It is of interest to note that small molecule CCR5-antagonists and monoclonal antibodies that recognize different epitopes on CCR5 have shown potent synergy [131, 132]. These findings could be the basis for a new strategy for HIV-1 therapy. The co-receptors of HIV-1, CCR5 and CXCR4 have been targeted in T cells using ribozyme, RNAi, single-chain antibodies (intrabodies) and zinc-finger DNA-binding proteins (ZFNs) [132, 133, 134]. As a variation of this, trans-dominant mutant variants of CCR5 also interfere with HIV infection of CD4+ T cells [135]. Lentiviral vectors expressing a single-chain antibody against CCR5 in primary CD4+ T cells disrupt CCR5 cell surface expression and provide protection from CCR5-tropic viral isolates [136]. Single-chain antibodies targeted to CXCR4 and cyclin T1 inhibit the replication of various HIV strains [122, 137]. Further testing is required, however, to show that targeting such cellular factors to lymphoid cells will not result in further immunodeficiency.
A different approach in HIV-1 therapy was taken by Sangamo BioSciences (Richmond, CA). This company is developing a biologic that targets the gene encoding CCR5, preventing cells from making this co-receptor protein. A zinc-finger DNA-binding endonuclease (ZFN) specifically cleaves the CCR5-DNA [138]. The adenovirus vector-borne product would be administered ex vivo to T cells extracted from HIV-infected patients, part of these treated cells would be re-infused into the patient, while the remainder would be stored as a reservoir of uninfectable T cells.
Addressing CXCR4/CXCL12 in angiogenesis/cancer
As mentioned before, the CXCL12-CXCR4 axis is involved in the mobilization and recruitment of vascular endothelial precursor cells at the site of neoangiogenesis. CXCL12 induces, controls and regulates vascularization of damaged tissues and of tumors. Its angiogenic property consists of inducing proliferation, differentiation and of preventing apoptosis of endothelial precursor cells [139–141]. The central role of the CXCL12-CXCR4 axis has prompted investigators to examine the effects of manipulating CXCL12-CXCR4 signaling as a possible therapeutic target. Recent studies indicate that the CXCL12-CXCR4 interaction is involved in several inflammatory conditions such as in the pathophysiology of inflammatory bowel disease (IBD) [142]. The expression of CXCR4 on peripheral T cells was found to be significantly higher in patients with active ulcerative colitis compared with normal controls and the levels of expression correlated with disease activity. In the murine model of dextran sulfate sodium (DSS)-induced colitis, colonic tissue expressed CXCR4 and CXCL12. Administration of CXCR4 antagonists ameliorated colonic inflammation in DSS-induced colitis. These results suggest that blockade of the CXCL12-CXCR4 axis might have potential as a therapeutic target for treatment of IBD. Several CXCR4 antagonists are currently being validated for clinical use [124, 141, 143]. Chronic treatment with the CXCR4 antagonist AMD 3100 blocks CXCL12-mediated angiogenesis [144, 145]. In tumor models inhibition of the CXCL12-CXCR4 pathway by chronic treatment with AMD 3100 leads to efficient reduction of tumor growth and angiogenesis by inhibiting proliferation of CXCR4-expressing tumor cells, diminishing recruitment of CXCR4+ angiogenic cells and inhibiting CXCR4-dependent metastasis. Since the inhibition of VEGF-A signaling as an anti-angiogenic strategy in clinical protocols was only moderately successful, the central role of CXCL12 in revascularization may be a more successful target in cancer therapy. Alternatively, combining CXCR4 blockade and VEGF-A signaling blockade with chemotherapy could represent an alternative therapeutic approach in tumor therapy [146].
Within this context, it appears interesting that the widely clinical use of stem cell mobilization by G-CSF not only mobilizes CD34+ CD133+ VEGFR2+ endothelial precursor cells, but also induces upregulation of CXCL12 [147–149]. Therefore, the promising efficacy of G-CSF in supporting organ regeneration in animal studies is currently being tested in clinical trials [150]. Alternatively, promotion of CXCL12 signaling is being induced by fucoidan-mediated CXCL12 elevation [151], or small peptide analogs to CXCL12 activating the CXCR4 signaling pathway [152], or with FTY 720, a sphingosine-1-phosphate analog that activates CXCR4 [153] or soluble molecules priming CXCR4+ cells such as C3a [154]. Careful preclinical studies are necessary considering also side effects of excessive angiogenesis and atherosclerosis [155].
Modified chemokines
Natural modified chemokine CXCL4L1
As mentioned before, platelets release a non-allelic variant of the chemokine CXCL4 named CXCL4L1 differing from CXCL4 by the exchange of three amino acids within the C terminus [97]. Recombinant CXCL4L1 exhibited higher angiostatic activity than CXCL4 in various test systems including wound-healing and migration assays for microvascular endothelial cells and the rat cornea micropocket assay for angiogenesis [91]. Furthermore, CXCL4L1 was more efficient in animal models of melanoma and lung carcinoma than CXCL4 in inhibiting tumor growth, development of microvasculature and metastasis to various organs. Preclinical trials have not been reported.
Analogs of CCL5
A further approach to block the chemokine-receptor interaction is using chemically modified chemokines that retain high affinity binding to their receptor but have lost signaling properties, i.e., they represent receptor antagonists. The extension of the N terminus by retaining the initial methionine residue in CCL5 during recombinant protein production in bacterial cells led to the antagonist Met-CCL5 (Met-RANTES), which prevents diseases in animal models [156]. Under physiological conditions Met-CCL5 inhibits arrest of monocytes in whole blood to human vascular endothelial cells [48]. A further CCL5 variant was generated by extension of the N terminus with aminooxypentane (AOP). AOP-RANTES prevents HIV infectivity by abolishing receptor recycling [157]. This prompted the development of an even more potent CCL5 N-terminally modified analog, PSC-RANTES, which was shown to prevent Simian HIV transmission in Rhesus macaques, indicating its potential of a topically protective compound [158]. On the other hand, N-terminal truncation may also generate receptor antagonists. One of these N-terminally truncated chemokines with antagonistic properties is 9–76-monocyte chemotactic protein-1 (MCP-1/CCL2), which is capable of preventing spontaneous arthritis in MRL-lpr mice [159]. In the context of mechanisms relying on the prevention of receptor cycling, it should be mentioned that surface expression and reexpression of a functional CXCR2, the receptor for NAP-2 and CXCL8, was found to be dependent on N-glycosylation of the receptor. Even partial deglycosylation of the receptor resulted in its serine protease-mediated disappearance from the cell surface. It thus appears that glycosylation protects CXCR2 from proteolytic attack and is required to maintain the responsiveness of neutrophils to ELR-CXC chemokines during inflammatory reactions [160].
Recently, a new anti-inflammatory therapeutic strategy has been developed. Based on the mutation of the heparin-binding site of the proinflammatory chemokine CCL5, the ability of the protein to form higher order of oligomers was abrogated, yet the receptor activation was retained [161, 162]. This non-glycosaminoglycan (GAG)-binding variant of CCL5 exhibits potent inhibitory activity on endogenous CCL5-mediated cellular recruitment. This does not occur through desensitization of target cells but rather by prevention of higher-order oligomer formation necessary for the biological activity of CCL5 in vivo. The mutant forms non-functional heterodimers with the native chemokine CCL5, which, by lack of their deposition on vascular cells, are not functional in recruiting immune cells under flow conditions. Thus, although the mutant chemokine retains its receptor binding activity, the mutation of the GAG-associated interactive site renders it a “dominant negative inhibitor” of recruitment and localization of inflammatory cells at the inflammatory site. This CCL5 mutant deficient in GAG binding also abolishes recruitment of inflammatory cells induced by non-specific agents such as thioglycolate. In the murine model of multiple sclerosis, murine experimental autoimmune encephalomyelitis (EAE), it reduces the clinical score of hind limb paralysis in these animals when given prior to disease onset. In the context of GAGs it should be mentioned that association of some chemokines with cell surface GAGs compromising their accessibility for antibodies may also be a reason for the inefficacy of some antibody-based therapies of chemokine inhibition, as indicated by lacking recognition of GAG-bound CXCL12 by antibodies directed to this region [163].
PGP: a collagen-derived inflammatory analog
A molecule that may perhaps also be addressed as a modified chemokine is a novel CXCR ligand derived from extracellular matrix degradation [164]. This tripeptide N-acetyl Pro-Gly-Pro (acPGP) shares sequence and structural homology with the GP motif, a domain of all ELR-positive alpha chemokines. It causes chemotaxis and production of superoxide by neutrophils through binding to CXCR1 and CXCR2. acPGP could be considered as part of a regulatory circuit in which CXC chemokines induce a direct first wave of neutrophil influx followed by neutrophil protease-mediated generation of acPGP, which in turn induces a second influx of neutrophils followed by monocytes and macrophages into the inflamed tissue. The known decrease of neutrophils in concert with increased numbers of monocytes and macrophages may be explained by the inactivation of chemotactic factors for neutrophils. The authors also present preliminary evidence that acPGP has a role in chronic obstructive pulmonary disease (COPD).
Neutralizing monoclonal antibodies
Effects in inflammation
A third approach is the development of neutralizing monoclonal antibodies. Monoclonal antibodies to CXCL4 and CXCL7 revealed that these chemokines, although not chemotactic for human monocytes, in early events of inflammation nevertheless promote monocyte recruitment by binding to proteoglycans of endothelial cells and induce monocyte arrest [48]. As mentioned before, activated platelets themselves were shown to secrete fully sulfated proteoglycans such as soluble chondroitinsulfate A, which under physiological flow conditions prevents sequestration of CCL5 on vascular cell surfaces and CCL5-mediated adhesion of monocytes [49]. The approach of neutralizing monoclonal antibodies, although blocking inflammation in a variety of animal models, had at first, most likely due to species differences, not reached the same efficacy in human diseases. Indeed, neutralization of chemokines CCL5 and CXCL9 with antisera has been reported to have negative effects in Theiler’s virus infection [165]. A more advanced anti-chemokine antibody is an anti-CXCL8 monoclonal antibody that was tested in a phase II trial of COPD and showed improvement in the transition dyspnoea index (TDI) but no improvement in the lung function, health status, 6-min walking distance and adverse events between groups of patients [166].
Effects in hematopoiesis
CXCL8 and other ELR chemokines (like CXCL7/NAP-2) are also involved in the proliferation and mobilization of hematopoietic progenitor cells [62 ]. The level of CXCL8 is significantly increased in the serum of patients with myeloid metaplasia with myelofibrosis (idiopathic myelofibrosis, MMM). Hematopoietic cells including platelets participate in the production of CXCL8. Inhibition of autocrine CXCL8 expressed by CD34-positive cells with either a neutralizing antibody or antisense anti-CXCL8 treatment increased the proliferation of MMM-derived CD34-positive cells and stimulated their differentiation into megakaryocytes (MK). Anti-CXCR1- or anti-CXCR2 antibodies to MMM-derived CD34-positive cells cultured under MK liquid culture conditions increased proliferation and differentiation of MMM CD41-positive MK and restored their defective polyploidization. Thus, the CXCL8-CXCL8 receptor system is involved in the inhibition of MK growth characteristic of MMM. This finding opens new therapeutic prospects for this still incurable disease. In contrast to the negative effects of the CXCL8-CXCL8 receptor system, positive effects on hematopoiesis have been reported for CXCR4 and its ligand CXCL12. As an adaptive process to tissue injury and inflammation in the periphery, the chemokine receptor CXCR4 expressed on bone marrow endothelial and stromal cells has been shown to internalize and re-secrete functional circulating CXCL12 [167]. Thus, chemokine translocation across the blood-bone marrow barrier allows effective transfer of functional CXCL12 from the periphery to the stem cell niche in the bone marrow during homeostasis and “alarm” situations. This translocation of CXCL12 is similar to that shown for CXCL8 across dermal blood vessels [168]. Certain physiological and pathophysiological conditions are characterized by an increase in CXCL12 in the bone marrow and other organs; these include ischemia due to injury [169], inflammation induced by infection [170], or total body irradiation and chemotherapy [170]. Successful recovery in these pathological conditions relies on the mobilization and local recruitment of committed stem cells [171]. To our knowledge, no therapeutic strategies targeting this chemokine-chemokine receptor system have been developed, although animal experimental evidence has been obtained in the mouse [142].
Effects in HIV
Monoclonal antibodies (mAb) targeting CCR5 have been developed to prevent infection of T cells with HIV [172]. Several of these products have entered clinical trials. A humanized monoclonal antibody (PRO 140) developed by Progenics Pharmaceuticals (Tarrytown, NY) entered a phase 1b clinical trial. It was claimed to bind to the same amino acids of CCR5 as HIV, which means that it does not interrupt the functions of the receptor [138]. Human Genome Sciences (Rockville, MD) developed several anti-CCR5 monoclonal antibodies. mAb 004 went into phase 1 clinical trial in January 2005 and was reported in 2007 to be safe and well tolerated in 63 HIV patients [138].
Effects in angiogenesis/fibroproliferation
Monoclonal antibodies targeting chemokines or chemokine receptors involved in angiogenesis have been used to modulate a dysbalance of angiogenesis and angiostasis under pathological conditions [173]. Idiopathic pulmonary fibrosis, an often fatal pulmonary fibroproliferative lung disease, is characterized by a dysregulation of tissue repair and neovascularisation as reflected by overexpression in the lung of the angiogenic ELR+ CXC chemokine CXCL8 as compared to the angiostatic non-ELR CXC chemokine CXCL10 [174]. In the mouse model of bleomycin-induced pulmonary fibrosis, a depletion of the endogenous angiogenic ELR+ CXC chemokine CXCL2/3 or administration of exogenous non-ELR CXCL10 resulted in marked attenuation of pulmonary fibrosis, which was attributable to a reduction in angiogenesis in the lung [175, 176]. The human lung disorder bronchiolitis obliterans syndrome is characterized by increased fibroproliferation around small airways and represents the most important cause of long-term organ dysfunction in lung transplant patients. The elevated ELR+ CXC chemokine ligand levels as demonstrated in the corneal micro-pocket model can be decreased by monoclonal antibody-mediated neutralization of CXCR2 [78]. An important feature of the human disorder, acute respiratory distress syndrome, is the dysbalance of angiogenic ELR+ CXC chemokines and angiostatic non-ELR chemokines [177]. On this basis, ELR+ CXC chemokines and their receptors represent potential therapeutic targets for the treatment of chronic inflammatory/fibroproliferative disorders with aberrant angiogenesis. In disorders induced by malignant tumors, CXCR3 and its ligands both inhibit angiogenesis and mediate TH1-type cell-mediated immunity via recruitment of CXCR3-expressing T cells [178]. In this context it should be mentioned that injection of 6Ckine (CCL21) into tumors induced tumor regression, reduction of angiogenesis and influx of CXCR3-expressing CD4+ and CD4+ T cells and NK cells and DC into both the tumor and the draining lymph nodes, an effect that requires the presence of CCL21-induced IFNgamma, CXCL9 and CXCL10 [179, 180]. Taken together, these findings support the notion that CXCR3 and its ligands contribute to anti-tumor defense by two mechanisms, namely inhibition of angiogenesis and mediation of direct anti-tumor immunity.
Effects on arteriosclerosis
In the pathogenesis of arteriosclerosis the recruitment of monocytes to the intima and their differentiation into macrophages are the primary cellular events during the initiation of a vascular lesion, and this recruitment continues during the expansion and progression of the arteriosclerotic lesion. Numerous mediators that attract monocytes to developing lesions have been identified. One of the early steps of this process is the binding of the chemoattractant CCL2. Recent studies have defined distinct subsets of monocytes that are preferentially recruited to lesions [181, 182]. Using an antibody directed against cell surface molecules called Ly6C antigens, a subset of monocytes was found to accumulate preferentially in arteriosclerotic lesions: these cells display large amounts of Ly6C at the cells surface as well as the chemokine receptors CCR2, CCR5 and CX3CR1. Thus, it might be possible to inhibit selectively the recruitment of discrete monocyte subpopulations into the arterial cell wall, avoiding the potentially adverse consequences of broadly inhibiting monocyte recruitment. This finding points to the notion that CCR2, CCR5 and CX3CR1 might represent targets for therapy of arteriosclerosis. Recent studies have begun to investigate how mast cells [183] and platelets [184] might also contribute to atherosclerosis. Concerning platelets, these may contribute by secreting chemokines (e.g., CCL2 and CCL5) that affect recruitment and differentiation of monocytes as well as E-selectin expression by endothelium. Of particular interest is the finding that DC can exit from lesions; this egress depends on CCR7 signaling and might promote lesion regression through the removal of lipid from the lesion [185, 186]. These findings provide a new model of how lesions can be modulated and indicate that activation of CCR7 could promote regression of arteriosclerosis.
Chemokine-binding proteins
The human organism, probably as reflection of environmental stress, contains in its genome binding proteins that cope with inflammation by neutralizing proinflammatory cytokines. Among these are: IL-1 receptor alpha, TNF-binding protein 1 and -2, interferon receptor alpha and IL-18 binding protein [187–190]. There is, as yet, no evidence for the existence of secreted chemokine-binding proteins in humans. There are, however, two naturally occurring, cell membrane-integrated chemokine-binding proteins in humans with a possible role in the pathophysiology of human diseases, the Duffy antigen (DARC) and D6.
The Duffy antigen (DARC)
The Duffy antigen, a minor blood group antigen, is a chemokine-binding protein that binds selectively CXC and CC chemokines with high affinity, in particular CXC chemokines that contain the tripeptide motif glutamyl-leucyl-arginine (ELR) [191], among those the platelet-derived chemokines NAP-2/CXCL7 and CXCL8, but also the CC chemokine CCL5. Duffy antigen is expressed on the surface of erythrocytes and postcapillary venular endothelial cells. Two in vivo animal studies have shown that Duffy knock-out mice can exhibit differential responses to an inflammatory stimulus such as lipopolysaccharide (LPS) and indicate a complex role that Duffy antigen may play during inflammation. Dawson et al. [192] observed an enhanced influx of neutrophils into the livers and lungs of Duffy knock-out mice 2 h after intraperitoneal injection of LPS. This suggests that absence of the chemokine “sink” effect associated with circulating erythrocytes may promote distant organ inflammation after LPS administration. However, Luo et al. [193] observed impaired neutrophil recruitment into the peritoneum, intestine and lungs 24 h after intraperitoneal injection of LPS into Duffy knock-out mice. They obtained similar results when thioglycolate was used as inflammatory stimulus. Furthermore, transgenic mice expressing enhanced Duffy antigen in vascular endothelial cells exhibited diminished angiogenetic response to mouse CXCL2, and enhanced hepatocellular toxicity and necrosis in response to a hepatotoxic drug [194]. In a mouse model of spontaneous prostate cancer, mice of a Duffy antigen receptor for chemokines (DARC)-deficient background developed larger and more aggressive tumors [195]. This associated with increased intratumor levels of angiogenic ELR+ CXC chemokines and blood vessel density, supporting the hypothesis that red blood cell expression of DARC sequesters angiogenic chemokines thereby inhibiting tumor growth. In a clinical study the expression of Duffy antigen was found to be enhanced in the vascular beds and alveolar septa of lung parenchyma from patients with suppurative pneumonia [196]. All these findings suggest that Duffy antigen may participate in the regulation of neutrophil migration. This is in keeping with the concept that on endothelial cells Duffy antigen may serve as a mediator of chemokine transcytosis, thus facilitating neutrophil recruitment [168]. The biological role of Duffy antigen in human suppurative pneumonia is not defined. It could bind excessive inflammatory chemokines and thus reduce neutrophil recruitment. Alternatively, enhanced expression of Duffy antigen could serve a proinflammatory role in lungs by providing chemokine transport across vascular endothelium and thereby facilitating neutrophil recruitment into organ specific sites.
D6
The second naturally occuring chemokine-binding protein is D6. This promiscuous receptor binds several inflammatory CC chemokines such as (also platelet-derived) CCL5, and CCL2, CCL4, the monocyte chemotactic protein MCP-3/CCL7 and the macrophage inflammatory protein MIP-1alpha P/CCL3L1, a CCL3 variant with a proline residue in position 2. The latter variant, different from genuine human CCL3, exhibits high affinity binding to D6. D6 is expressed in circulating cells and in some hematopoietic cell lines, but highly expressed in lymphatic, but not in vascular endothelial cells [197]. More recently, the expression of D6 on leukocytes, such as myeloid progenitor cells, circulating DC and B cells in inflamed tissue was stressed [198]. The possibility was considered that D6 expressed on leukocytes patrolling along the luminal face of the vasculature might remove and degrade chemokines presented on glycosaminoglycans of vascular endothelial cells. Furthermore, D6-expressing leukocytes appear to soak up most of the chemokines at the inflammatory site, whereas D6 expressed on lymphatic endothelial cells might restrict CCR7-mediated lymph node drainage of inflammatory leukocytes. In vitro studies have shown that D6 as a constitutively recycling receptor actively internalizes its ligands and targets them probably to endolysosomal compartments where they are degraded [199, 200]. More recently, the resolution of an inflammatory response in the skin was shown to be closely connected with the function of D6. Unlike wild-type mice, D6-deficient mice were unable to mount a transient inflammatory response in the skin after induction by phorbol esters (TPA), but rather exhibited a chronic inflammation with excess of residual chemokines, which caused an inflammatory skin pathology with similarities of human psoriasis, [201] production of MIP-2/CXCL2 and the murine homolog of human CXCL8. The expression of MIP-2/CXCL2 in the epidermis of D6-deficient mice corresponds to human skin pathology of psoriasis, which is also characterized by the expression of CXCL8 mRNA in the epidermis [202]. CXCL8 may be involved in the recruitment of neutrophils and possibly T cells and in the regulation of keratinocyte proliferation. The number of neutrophils in the skin was comparable in wild-type and D6-deficient mice, which does not correspond to the increased amount of MIP-2/CXCL8 in D6−/− mice suggesting that keratinocyte-derived MIP-2/CXCL2 may not reach the luminal face of the vessel to recruit passing neutrophils. MIP-2/CXCL2 is not a ligand of D6, and the increase in the epidermis may be secondary to increased numbers of T cells and mast cells. Since the capacity of chemokines to bind to proteoglycans in the context of inflammatory responses is not fully understood [159], it is conceivable to assume that in a given tissue environment some CC chemokines may be protected from D6-dependent degradation through proteoglycan binding. A further protection from D6-dependent degradation is given by the proline residue at position 2 of mature inflammatory beta-chemokines, which is on the one hand required for binding to D6 [203] and on the other hand provides susceptibility to cleavage by the enzyme dipeptidyl-peptidase IV (CD26) [204]. Thus, CCL5, eotaxin (CCL11), CCL22 and CXCL12 are N-terminally truncated by CD26, which results in impaired inflammatory properties of these chemokines, whereas N-terminally truncated chemokine (C–C motif) ligand 3-like 1 (LD78beta/CCL3L1) exhibits increased chemotactic activity compared to intact CCL3L1 [205]. Taken together, the local concentrations of some chemokines appear to be regulated by a dynamic and delicate system involving both CD26-dependent down-regulation of D6-mediated degradation and of up- or down-regulation of receptor affinity of certain chemokines, including CCL3L1 and CCL5, CCL11, CCL22 and CXCL12, respectively.
Regulation by CXCR1 and fragments
Previous work of our own had suggested that CXCR1 is generally more resistant to desensitization and down-regulation, facilitates neutrophil migration to sites of high NAP-2 and CXCL1 concentration [206] and, according to findings by others [207], is more involved in the generation of antimicrobial responses and respiratory burst, whereas CXCR2 seems to mediate mainly the initial recruitment of neutrophils from the circulation after which the receptor loses signaling capacity and its surface expression is rapidly down-regulated. Hartl et al. [208] recently provided evidence that at the inflammatory site, fragments of CXCR1 are generated by neutrophil proteases elastase and cathepsin G probably from residual viable neutrophils, thus impairing the activation of their bactericidal pathways. The fragments of CXCR1 were found to boost CXCL8 production through their ability to activate TLR2 signaling on bronchial epithelial cells. The binding to TLR2 was claimed to occur via the glycan moiety of the glycosylated receptor fragment. Thus, the soluble receptor moiety of CXCR1 was supposed to act as an alarmin to activate the innate immune system and warn of tissue damage and inflammation [209]. In the chronic inflammatory disease such as cystic fibrosis, the immune system is incapacitated by Pseudomonas aeruginosa. These bacteria impair neutrophil bactericidal functions by producing toxins and secondary metabolites including pyocyanin that accelerates neutrophil death and inhibit removal of apoptotic neutrophils by macrophages [210]. This accelerated neutrophil death, and failed clearance of neutrophils has two consequences: a decreased pool of viable neutrophils able to kill P. aeruginosa and the release of various toxic antimicrobial molecules including proteases, which are also involved in cleavage of CXCR1. Hartl et al. [208] showed that treating patients with cystic fibrosis with the antiprotease alpha 1-antitrypsin resulted in improvement of bacterial killing capacity of airway neutrophils and increased expression of CXCR1.
Pathogen-derived immunomodulatory molecules
Viral chemokine homologues
Viruses have been found to synthesize and secrete a variety of chemokine homologues and binding proteins [211]. The viral chemokine homologues MIP-II (vMIP-2) and MC 148 are encoded by human herpes virus 8 (HHV8) and by Molluscum contagiosum poxvirus, respectively. Both prolong cardiac allograft survival in mice by interfering with chemokine-mediated responses [212]. A further chemokine homologue is C18, an 18-kDa cyclophilin produced by Toxoplasma gondii, which binds to CCR5, thus modulating the activation of DC [213]. As CCR5 is a co-receptor for HIV, C18 could be an immunotherapeutic drug for HIV [214]. Similar candidates are vMIP-1, also expressed by HHV8, and vMIP-2, which likewise block HIV infections [215]. More recent studies elucidated the specificity of binding of C18 and CCR5 to improve anti-HIV therapy. However, the recent demonstration of the important protective role of CCR5 in nerve tissue against West Nile virus infection might counteract the endeavor of an effective therapy based on blockade of CCR5 [216].
Pathogen-derived chemokine-binding proteins
There are also several pathogen-derived chemokine-binding proteins. Viral chemokine-binding proteins have been identified in pox virus and herpes viruses and bind a broad range of chemokines [217, 218], while human cytomegalovirus encodes a highly specific CCL5 decoy receptor [219]. Viral chemokine-binding proteins have been demonstrated to exhibit anti-inflammatory activity in animal models of inflammatory diseases. The myxoma virus-derived M-T7 reduces macrophage and T cell invasion at sites of vascular injury and the 35-kDa chemokine-binding protein of poxvirus decreases inflammatory activity in models of chronic transplant vasculopathy and airway inflammation [220]. Recently, a novel chemokine-binding domain has been identified in the tumor necrosis factor receptor produced by variola (smallpox) virus [221]. This smallpox virus-encoded chemokine receptor (SECRET) domain binds chemokines involved in skin and mucosal inflammation and is part of the virus-encoded TNF receptors called cytokine response modifier B (CrmB) and CrmD. The finding of an additional chemokine-binding domain present in its soluble TNF receptor structure suggests that the anti-inflammatory properties of these soluble TNF receptors are enhanced and, in fact, represent a novel viral strategy. Furthermore Schistosoma mansoni secretes a chemokine-binding protein with anti-inflammatory activity [222]. An anti-CXCL8 activity was also described in the saliva of ticks [223]. Possibly, the parasite prevents local inflammatory reactions by neutrophils to protect its germs. Saliva from ticks (Rhipicephalus sanguineus) was demonstrated to inhibit the chemotaxis induced by CCL3 and to impair chemotaxis of immature DC by down-regulating cell surface CCR5 [224]. A chemokine-binding protein of ticks was cloned, characterized and termed evasin-1, and was shown to inhibit chemotaxis induced by CCL3, CCL4 and the closely related chemokine CCL18 [225]. In addition, evasin-3 structurally unrelated to evasin-1 was found to bind to CXCL8 and CXCL1 [226]. Administration of recombinant evasin-1 and -3 in vivo was demonstrated to exhibit potent anti-inflammatory properties in mouse models of inflammatory diseases such as psoriasis, bleomycin-induced pulmonary fibrosis and antigen-induced arthritis [226]. The high selectivity of some evasins for distinct chemokines renders them promising tools for potential therapeutic intervention.
Concluding remarks
In recent years platelet-derived chemokines such as CXCL1, CXCL4, CXCL5, CXCL7 and CCL3 and CCL5 and their receptors have been structurally and functionally characterized. This structural and functional analysis has revealed their role in inflammatory conditions, bacterial and viral infections, tumor development and autoimmune disorders. To get an insight into the role of platelet-derived chemokines and their receptors in physiological and pathophysiological conditions, it was necessary to define the producer cell, the target cell and target structure, the cells and their cellular products that are involved in the generation and processing of platelet chemokines, and the elements regulating these processes.
A general conclusion from all experimental studies is that the presence, function and biological activity of a chemokine is dependent on three parameters: the causative stimulus, the local environment and the time kinetics of the ensuing recruitment of cells.
A first conclusion from studies on platelets is that these cells actively participate as mediators of a first-line defense by early release of chemokines and chemokine precursors and thus are engaged in the recruitment of myeloid and lymphoid cells. In this process the generation of CXCL7 from its precursor is regulated by the chemokine itself, by other platelet chemokines, by the net activity of proteinases/anti-proteinases and by free and cell-associated chondroitin sulfate proteoglycan, in the local environment at a defined time. Second, it is shown that the chemokines CXCL4, CXCL7 and CXCL12, in addition to thrombopoietin, are regulatory components in platelet development and maturation. Third, vascular repair and neoangiogenesis in wound healing, inflammation and cancer are mediated by CXCL1 and CXCL7, which were shown to be expressed also in vascular smooth muscle cells and to recruit endothelial precursor cells by binding to CXCR2. Fourth, the exciting finding of thrombocidins exhibiting antimicrobial activity may lead to their topical application in infectious conditions. The molecular analysis of cellular sources, cell-associated or soluble target structures and biological activities of chemokines led to studies on their roles in pathophysiological conditions and to the development of concepts of therapeutic intervention in disease models and in a variety of diseases. The principles underlying such strategies have been dealt with in this review. They comprise small molecule chemokine receptor antagonists, chemically modified chemokines, neutralizing monoclonal antibodies, chemokine-binding proteins and pathogen-derived immunomodulatory, i.e., chemokine-chemokine receptor interaction modifying molecules. Although most of these principles still await careful assessment in clinical trials, alone or in combinations, they open new avenues of therapy in view of the observation of increasing resistance of microbial pathogens to antibiotics virostatics and of cancer to anti-cancer drugs.
References
Weyrich AS, Zimmerman GA (2004) Platelets: signaling cells in the immune continuum. Trends Immunol 25:489–495
Elzey BD, Tian J, Jensen RJ, Swanson AK, Lees JR, Lentz SR, Stein CS, Nieswandt B, Wang Y, Davidson BL, Ratliff TL (2003) Platelet-mediated modulation of adaptive immunity. A communication link between innate and adaptive immune compartments. Immunity 19:9–19
Otterdal K, Smith C, Oie E, Pedersen TM, Yndestad A, Stang E, Endresen K, Solum NO, Aukrust P, Damas JK (2006) Platelet-derived LIGHT induces inflammatory responses in endothelial cells and monocytes. Blood 108:928–935
Klinger MH, Wilhelm D, Bubel S, Sticherling M, Schroder JM, Kuhnel W (1995) Immunocytochemical localization of the chemokines RANTES and MIP-1 alpha within human platelets and their release during storage. Int Arch Allergy Immunol 107:541–546
Power CA, Clemetson JM, Clemetson KJ, Wells TN (1995) Chemokine and chemokine receptor mRNA expression in human platelets. Cytokine 7:479–482
Brandt E, Ludwig A, Petersen F, Flad HD (2000) Platelet-derived CXC chemokines: old players in new games. Immunol Rev 177:204–216
Weber C (2005) Platelets and chemokines in atherosclerosis: partners in crime. Circ Res 96:612–616
El Golli N, Issertial O, Rosa JP, Broquet-Laugier V (2005) Evidence for a granule targeting sequence within platelet factor 4. J Biol Chem 280:30329–30335
Walz A, Dewald B, von Tscharner V, Baggiolini M (1989) Effects of the neutrophil-activating peptide NAP-2, platelet basic protein, connective tissue-activating peptide III and platelet factor 4 on human neutrophils. J Exp Med 170:1745–1750
Walz A, Baggiolini M (1989) A novel cleavage product of beta-thromboglobulin formed in cultures of stimulated mononuclear cells activates human neutrophils. Biochem Biophys Res Commun 159:969–975
Walz A, Meloni F, Clark-Lewis I, von Tscharner V, Baggiolini M (1991) [Ca2+]i changes and respiratory burst in human neutrophils and monocytes induced by NAP-1/interleukin-8, NAP-2, and gro/MGSA. J Leukoc Biol 50:279–286
Yan Z, Zhang J, Holt JC, Stewart GJ, Niewiearowski S, Poncz M (1994) Structural requirements of platelet chemokines for neutrophil activation. Blood 84:2329–2339
Clark-Lewis I, Dewald B, Loetscher M, Moser B, Baggiolini M (1994) Structural requirements for interleukin-8 function identified by design of analogs and CXC chemokine hybrids. J Biol Chem 269:16075–16081
Petersen F, Flad HD, Brandt E (1994) Neutrophil-activating peptides NAP-2 and IL-8 bind to the same sites on neutrophils but interact in different ways. Discrepancies in binding affinties, receptor densities, and biologic effects. J Immunol 152:2467–2478
Loetscher P, Seitz M, Clark-Lewis I, Baggiolini M, Moser B (1994) Both interleukin-8 receptors independently mediate chemotaxis. Jurkat cells transfected with IL-8R1 or IL-8R2 migrate in response to IL-8, GRO alpha and NAP-2. FEBS Lett 341:187–192
Ahuja SK, Murphy PM (1996) The CXC chemokines growth-related oncogene (GRO) alpha, GRObeta, GROgamma, neutrophil-activating peptide-2, and epithelial cell-derived neutrophil-activating peptide-78 are potent agonists for the type B, but not the typeA, human interleukin-8 receptor. J Biol Chem 271:20545–20550
Clark-Lewis I, Dewald B, Geiser T, Moser B, Baggiolini M (1993) Platelet factor 4 binds to interleukin 8 receptors and activates neutrophils when its N terminus is modified with Glu-Leu-Arg. Proc Natl Acad Sci USA 90:3574–3577
Petersen F, Ludwig A, Flad HD, Brandt E (1996) TNF-alpha renders human neutrophils responsive to platelet factor 4. Comparison of PF-4 and IL-8 reveals different activity profiles of the two chemokines. J Immunol 156:1954–1962
Fleischer J, Grage-Griebenow E, Kasper B, Heine H, Ernst M, Brandt E, Flad HD, Petersen F (2002) Platelet factor 4 inhibits proliferation and cytokine release of activated human T cells. J Immunol 169:770–777
Pervushina O, Scheuerer B, Reiling N, Behnke L, Schroder JM, Kasper B, Brandt E, Bulfone-Paus S, Petersen F (2004) Platelet factor 4/CXCL4 induces phagocytosis and the generation of reactive oxygen metabolites in mononuclear phagocytes independently of Gi protein activation or intracellular calcium transients. J Immunol 173:2060–2067
Petersen F, Bock L, Flad HD, Brandt E (1998) A chondroitin sulfate proteoglycan on human neutrophils specifically binds platelet factor 4 and is involved in cell activation. J Immunol 161:4347–4355
Kasper B, Brandt E, Bulfone-Paus S, Petersen F (2004) Platelet factor 4 (PF-4)-induced neutrophil adhesion is controlled by src-kinases while PF-4-mediated exocytosis requires the additional activation of p38 MAP kinase and phosphatidylinositol 3-kinase. Blood 103:1602–1610
Kasper B, Brandt E, Ernst M, Petersen F (2006) Neutrophil adhesion to endothelial cells induced by platelet factor 4 requires sequential activation of Ras, Syk, and JNK MAP kinases. Blood 107:1768–1775
Mueller A, Meiser A, McDonagh EM, Fox JM, Petit GX, Williams TJ, Pease JM (2008) CXCL4-induced migration of activated T lymphocytes is mediated by the chemokine receptor CXCR3. J Leukoc Biol 83:875–882
Schall TJ, Greaves DR (2000) RANTES/CCL5. In: Oppenheim JJ, Feldman M (eds.-in-chief), Durum SK, Hirano T, Vilcek J, Nicola NA (eds) Cytokine reference. A Compendium of cytokines and other mediators of host defense, vol 1: ligands. Academic Press, London, pp 1161–1166
Schall TJ, Bacon K, Toy KJ, Goeddel DV (1990) Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature 347:669–671
Taub DD, Lloyd ER, Wang JM, Oppenheim JJ, Kelvin DJ (1993) The effects of human recombinant MIP-1 alpha, MIP-1 beta, and RANTES on the chemotaxis and adhesion of T cell subsets. Adv Exp Med Biol 351:139–146
Rot A, Krieger M, Brunner T, Bischoff SC, Schall TJ, Dahinden CA (1992) RANTES and macrophage inflammatory protein 1 alpha induce the migration and activation of normal human eosinophil granules. J Exp Med 176:1489–1495
Kameyoshi Y, Dörschner A, Mallet AI, Christophers E, Schröder JM (1992) Cytokine RANTES released by thrombin-stimulated platelets is a potent attractant for human eosinophils. J Exp Med 176:587–592
Kuna P, Reddigari SR, Schall TJ, Rucinski D, Viksman MY, Kaplan AP (1992) RANTES, a monocyte and T lymphocyte chemotactic cytokine releases histamine from human basophils. J Immunol 49:636–642
Loetscher P, Seitz M, Clark-Lewis I, Baggiolini M, Moser B (1996) Activation of NK cells by CC chemokines. Chemotaxis, Ca2+ mobilization, and enzyme release. J Immunol 156:322–327
Sozzani S, Luini W, Borsatti A, Polentarutti N, Zhou D, Piemonti L, D`Amico G, Power CA, Wells TN, Gobbi M, Allavena P, Mantovani A (1997) Receptor expression and responsiveness of human dendritic cells to a defined set of CC and CXC chemokines. J Immunol 159:1993–2000
Brandt E, Van Damme J, Flad HD (1991) Neutrophils can generate their activator neutrophil-activating peptide 2 by proteolytic cleavage of platelet-derived connective tissue-activating peptide III. Cytokine 3:311–321
Walz A, Baggiolini M (1990) Generation of the neutrophil-activating peptide NAP-2 from platelet basic protein or connective tissue-activating peptide III through monocyte proteases. J Exp Med 171:449–454
Begg GS, Pepper DS, Chesterman CN, Morgan FJ (1978) Complete covalent structure of human beta-thromboglobulin. Biochemistry 17:1739–1744
Holt JC, Rabellino EM, Gewirtz AM, Gunkel LM, Rucinski B, Niewiarowski S (1988) Occurrence of platelet basic protein, a precursor of low affinity platelet factor 4 and beta-thromboglobulin, in human platelets and megakaryocytes. Exp Hematol 16:302–306
Majumdar S, Gonder D, Koutsis B, Poncz M (1991) Characterization of the human beta-thromboglobulin gene. Comparison with the gene for platelet factor 4. J Biol Chem 266:5785–5789
Zhang C, Gadue P, Scott E, Atchison M, Poncz M (1997) Activation of the megakaryocyte-specific gene platelet basic protein (PBP) by the Ets family factor PU.1. J Biol Chem 272:26236–26246
Harter L, Petersen F, Flad HD, Brandt E (1994) Connective tissue-activating peptide III desensitizes chemokine receptors on neutrophils. Requirement for proteolytic formation of the neutrophil-activating peptide 2. J Immunol 153:5698–5708
Car BD, Baggiolini M, Walz A (1991) Formation of neutrophil-activating peptide 2 from platelet-derived connective-tissue-activating peptide III by different tissue proteinases. Biochem J 275:581–584
Malkowski MG, Lazar JB, Johnson PH, Edwards BF (1997) The amino-terminal residues in the crystal structure of connective tissue activating peptide-III (des10) block the ELR chemotactic sequence. J Mol Biol 266:367–380
Campbell EJ, Senior RM, McDonald JA, Cox DL (1982) Proteolysis by neutrophils. Relative importance of cell–substrate contact and oxidative inactivation of proteinase inhibitors in vitro. J Clin Invest 70:845–852
Travis J, Salvesen GS (1983) Human plasma proteinase inhibitors. Annu Rev Biochem 52:655–709
Schiemann F, Grimm TA, Hoch J, Gross R, Lindner B, Petersen F, Bulfone-Paus S, Brandt E (2006) Mast cells and neutrophils proteolytically activate chemokine precursor CTAP-III and are subject to counterregulation by PF-4 through inhibition of chymase and cathepsin G. Blood 107:2234–2242
Schenk BI, Petersen F, Flad HD, Brandt E (2002) Platelet-derived chemokines CXC chemokine ligand (CXCL)7, CTAP-III, and CXCL4 differentially affect and cross-regulate neutrophil adhesion and transendothelial migration. J Immunol 169:2602–2610
Petersen F, Bock L, Flad HD, Brandt E (1999) Platelet factor 4-induced neutrophil-endothelial cell interaction: involvement of mechanisms and functional consequences different from those elicited by interleukin-8. Blood 94:4020–4028
Baltus T, von Hundelshausen P, Mause SF, Buhre W, Rossaint R, Weber C (2005) Differential and additive effects of platelet-derived chemokines on monocyte arrest under flow conditions. J Leukoc Biol 78:435–441
Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C (2005) Platelet microparticles: a transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol 25:1512–1518
Mack M, Pfirstinger J, Weber C, Weber KS, Nelson PJ, Rupp T, Maletz K, Brühl H, Schlöndorff D (2002) Chondroitin sulfate A released from platelets blocks RANTES presentation on cell surfaces and RANTES-dependent firm adhesion of leukocytes. Eur J Immunol 32:1012–1020
von Hundelshausen P, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE, Hackeng TM, Weber C (2005) Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood 105:924–930
Baltus T, Weber KS, Johnson Z, Proudfoot AE, Weber C (2003) Oligomerization of RANTES is required for CCR1-mediated arrest but not CCR5-mediated transmigration of leukocytes on inflamed endothelium. Blood 102:1985–1988
Clemetson KJ, Clemetson JM, Proudfoot AE, Power CA, Baggiolini M, Wells TN (2000) Functional expression of CCR1, CCR3, CCR4, and CXCR4 chemokine receptors on human platelets. Blood 96:4046–4054
Abi-Younes S, Sauty A, Mach F, Sukhova GK, Libby P, Luster AD (2000) The stromal cell-derived factor-1 chemokine is a potent platelet agonist highly expressed in atherosclerotic plaques. Circ Res 86:131–138
Abi-Younes S, Si-Tahar M, Luster AD (2001) The CC chemokines MDC and TARC induce platelet activation via CCR4. Thromb Res 101:279–289
Gear AR, Suttitanamongkol S, Viisoreanu D, Polanowska-Grabowska RK, Raha S, Camerini D (2001) Adenosine diphosphate strongly potentiates the ability of the chemokines MDC, TARC, and SDF-1 to stimulate platelet function. Blood 97:937–945
Suttitanamongkol S, Gear AR (2001) ADP receptor antagonists inhibit platelet aggregation induced by the chemokines SDF-1, MDC and TARC. FEBS Lett 490:84–87
Coleman LG Jr, Polanowska-Grabowska RK, Marcinkiewicz M, Gear AR (2004) LDL oxidized by hypochlorous acid causes irreversible platelet aggregation when combined with low levels of ADP, thrombin, or macrophage-derived chemokine (CCL22). Blood 104:380–389
Shenkman B, Brill A, Brill G, Lider O, Savion N, Varon D (2004) Differential response of platelets to chemokines: RANTES non-competitively inhibits stimulatory effect of SDF-1 alpha. J Thromb Haemost 2:154–160
Han ZC, Bellucci S, Tenza D, Caen JP (1990) Negative regulation of human megakaryocytopoiesis by human platelet factor 4 and beta thromboglobulin: comparative analysis in bone marrow cultures from normal individuals and patients with essential thrombocythaemia and immune thrombocytopenic purpura. Br J Haematol 74:395–401
Han ZC, Bellucci S, Walz A, Baggiolini M, Caen JP (1990) Negative regulation of human megakaryocytopoiesis by human platelet factor 4 (PF4) and connective tissue-activating peptide (CTAP-III). Int J Cell Cloning 8:253–259
Gewirtz AM, Zhang J, Ratajczak J, Ratajczak M, Park KS, Li C, Yan Z, Poncz M (1995) Chemokine regulation of human megakaryocytopoiesis. Blood 86:2559–2567
Emadi S, Clay D, Desterke C, Guerton B, Maquarre E, Charpentier A, Jasmin C, Le Bousse-Kerdilès MC (2005) IL-8 and its CXCR1 and CXCR2 receptors participate in the control of megakaryocytic proliferation, differentiation, and ploidy in myeloid metaplasia with myelofibrosis. Blood 105:464–473
Deutsch V, Bitan M, Friedmann Y, Eldor A, Vlodavsky I (2000) Megakaryocyte maturation is associated with expression of the CXC chemokine connective tissue-activating peptide CTAP III. Br J Haematol 111:1180–1189
Oda M, Kurasawa Y, Todokoro K, Nagata Y (2003) Thrombopoietin-induced CXC chemokines, NAP-2 and PF4, suppress polyploidization and proplatelet formation during megakaryocyte maturation. Genes Cells 8:9–15
Wang JF, Liu ZY, Groopman JE (1998) The alpha-chemokine receptor CXCR4 is expressed on the megakaryocytic lineage from progenitors to platelets and modulates migration and adhesion. Blood 92:756–764
Hamada T, Möhle R, Hesselgesser J, Hoxie J, Nachman RL, Moore MA, Rafii S (1998) Transendothelial migration of megakaryocytes in response to stromal cell-derived factor 1 (SDF-1) enhances platelet formation. J Exp Med 188:539–548
Kowalska MA, Ratajczak J, Hoxie J, Brass LF, Gewirtz A, Poncz M, Ratajczak MZ (1999) Megakaryocyte precursors, megakaryocytes and platelets express the HIV co-receptor CXCR4 on their surface: determination of response to stromal-derived factor-1 by megakaryocytes and platelets. Br J Haematol 104:220–229
Berthebaud M, Riviere C, Jarrier P, Foudi A, Zhang Y, Compagno D, Galy A, Vainchenker W, Louache F (2005) RGS16 is a negative regulator of SDF-1-CXCR4 signaling in megakaryocytes. Blood 106:2962–2968
Secchiero P, Celeghini C, Cutroneo G, Baldassarre Di, Rana R, Zauli G (2000) Differential effects of stromal derived factor-1 alpha (SDF-1 alpha) on early and late stages of human megakaryocytic development. Anat Rec 260:141–147
Hodohara K, Fujii N, Yamamoto N, Kaushansky K (2000) Stromal cell-derived factor-1 (SDF-1) acts together with thrombopoietin to enhance the development of megakaryocytic progenitor cells (CFU-MK). Blood 95:769–775
Majka M, Janowska-Wieczorek A, Ratajczak J, Kowalska MA, Vilaire G, Pan ZK, Honczarenko M, Marquez LA, Poncz M, Ratajczak MZ (2000) Stromal-derived factor 1 and thrombopoietin regulate distinct aspects of human megakaryopoiesis. Blood 96:4142–4151
Maassberg S, Konrad I, Schürzinger K, Lorenz M, Schneider S, Zohlnhoefer D, Hoppe K, Schiemann M, Kennerknecht E, Sauer S, Schulz C, Kerstan S, Rudelius M, Seidl S, Sorge F, Langer H, Peluso M, Goyal P, Vestweber D, Emambokus NR, Busch DH, Frampton J, Gawaz M (2006) Platelets secrete stromal cell-derived factor1alpha and recruit bone marrow-derived progenitor cells to arterial thrombi in vivo. J Exp Med 203:1221–1233
Salcedo R, Wasserman K, Young HA, Grimm MC, Howard OM, Anver MR, Kleinman HK, Murphy WJ, Oppenheim JJ (1999) Vascular endothelial growth factor and basic fibroblast growth factor induce expression of CXCR4 on human endothelial cells: In vivo neovascularization induced by stromal-derived factor-1alpha. Am J Pathol 154:1125–1135
Petit I, Jin D, Rafii S (2007) The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol 28:299–307
Hristov M, Zernecke A, Bidzhekov K, Liehn EA, Shagdarsuren E, Ludwig A, Weber C (2007) Importance of CXC chemokine receptor 2 in the homing of human peripheral blood endothelial progenitor cells to sites of arterial injury. Circ Res 100:590–597
Schober A, Manka D, von Hundelshausen P, Huo Y, Hanrath P, Sarembock IJ, Ley K, Weber C (2002) Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation 106:1523–1529
Strieter RM, Polverini PJ, Kunkel SL, Arenberg DA, Burdick MD, Kasper J, Dzuiba J, Van Damme J, Walz A, Mariott D, Chan SY, Roczniak S, Shanafelt AB (1995) The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem 270:27348–27357
Addison CL, Daniel TO, Burdick MD, Liu H, Ehlert JE, Xue YY, Buechi L, Walz A, Richmond A, Strieter RM (2000) The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. J Immunol 165:5269–5277
Belperio JA, Keane MP, Burdick MD, Gomperts B, Xue YY, Hong K, Mestas J, Ardehali A, Mehrad B, Saggar R, Lynch JP, Ross DJ, Strieter RM (2005) Role of CXCR2/CXCR2 ligands in vascular remodeling during bronchiolitis obliterans syndrome. J Clin Invest 115:1150–1162
Brill A, Elinav H, Varon D (2004) Differential role of platelet granular mediators in angiogenesis. Cardiovasc Res 63:226–235
Maione TE, Gray GS, Petro J, Donner AL, Bauer SI, Carson HF, Sharpe RJ (1990) Inhibition of angiogenesis by recombinant human platelet factor-4 and related peptides. Science 247:77–79
Watson JB, Getzler SB, Mosher DF (1994) Platelet factor 4 modulates the mitogenic activity of basic fibroblast growth factor. J Clin Invest 94:261–268
Gentilini G, Kirschbaum NE, Augustine JA, Aster RH, Visentin GP (1999) Inhibition of human umbilical vein endothelial cell proliferation by the CXC chemokine, platelet factor 4 (PF4), is associated with impaired downregulation of p21(Cip1/WAF1). Blood 93:25–33
Luster AD, Greenberg SM, Leder P (1995) The IP-10 chemokine binds to a specific cell surface heparan sulfate site shared with platelet factor 4 and inhibits endothelial cell proliferation. J Exp Med 182:219–231
Romagnani P, Annunziato F, Lasagni L, Lazzeri E, Beltrame C, Francalanci M, Uguccioni M, Galli G, Cosmi L, Maurenzi L, Baggiolini M, Maggi E, Romagnani S, Serio M (2001) Cell cycle-dependent expression of CXC chemokine receptor 3 by endothelial cells mediates angiostatic activity. J Clin Invest 107:53–63
Tanaka T, Manome Y, Wen P, Kufe DW, Fine HA (1997) Viral vector–mediated transduction of a modified platelet factor 4 cDNA inhibits angiogenesis and tumor growth. Nat Med 3:437–442
Arenberg DA, Kunkel SL, Polverini PJ, Morris SB, Burdick MD, Glass MC, Taub DT, Iannettoni MD, Whyte RI, Strieter RM (1996) Interferon-γ-inducible protein 10 (IP-10) is an angiostatic factor that inhibits human non-small cell lung cancer (NSCLC) tumorigenesis and spontaneous metastases. J Exp Med 184:981–992
Vandercappellen J, Van Damme J, Struyf S (2008) The role of CXC chemokines and their receptors in cancer. Cancer Lett 28:226–244
Yang J, Richmond A (2004) The angiostatic activity of interferon-inducible protein-10/CXCL10 in human melanoma depends on binding to CXCR3 but not to glycosaminoglycan. Mol Ther 9:846–855
Lasagni L, Francalanci M, Annunziato F, Lazzeri E, Giannini S, Cosmi L, Sagrinati C, Mazzinghi B, Orlando C, Maggi E, Marra F, Romagnani S, Serio M, Romagnani P (2003) An alternatively spliced variant of CXCR3 mediates the the inhibition of endothelial cell growth induced by IP-10, Mig, and I-Tac and acts as functional receptor for platelet factor 4. J Exp Med 197:1537–1549
Struyf S, Burdick MD, Peeters E, Van den Broeck K, Dillen C, Proost P, Van Damme J, Strieter RM (2007) Platelet factor-4 variant chemokine CXCL4L1 inhibits melanoma and lung carcinoma growth and metastasis by preventing angiogenesis. Cancer Res 67:5940–5948
Gengrinovitch S, Greenberg SM, Cohen T, Gitary-Goren H, Rockwell P, Maione TE, Levi BZ, Neufeld G (1995) Platelet factor-4 inhibits the mitogenic activity of VEGF121 and VEGF165 using several concurrent mechanisms. J Biol Chem 270:15059–15065
Perollet C, Han ZC, Savona C, Caen JP, Bikfalvi A (1998) Platelet factor 4 modulates fibroblast growth factor 2 (FGF-2) activity and inhibits FGF-2 dimerization. Blood 91:3289–3299
Sulpice E, Contreres JO, Lacour J, Bryckaert M, Tobelem G (2004) Platelet factor 4 disrupts the intracellular signalling cascade induced by vascular endothelial growth factor by both KDR dependent and independent mechanisms. Eur J Biochem 271:3310–3318
Aidoudi S, Bujakowska K, Kieffer N, Bikfalvi A (2008) The CXC chemokine CXCL4 interacts with integrins implicated in angiogenesis. PloS ONE 3:e2657
Green CJ, Charles RS, Edwards BF, Johnson PH (1989) Identification and characterization of PF4var1, a human gene variant of platelet factor 4. Mol Cell Biol 9:1445–1451
Struyf S, Burdick MD, Proost P, Van Damme J, Strieter RM (2004) Platelets release CXCL4L1, a nonallelic variant of the chemokine platelet factor-4/CXCL4 and potent inhibitor of angiogenesis. Circ Res 95:855–857
Yeaman MR, Tang YQ, Shen AJ, Bayer AS, Selsted ME (1997) Purification and in vitro activities of rabbit platelet microbicidal proteins. Infect Immun 65:1023–1031
Yeaman MR, Yount NY, Waring AJ, Gank KD, Kupferwasser D, Wiese R, Bayer AS, Welch WH (2007) Modular determinants of antimicrobial activity in platelet factor-4 family kinocidins. Biochim Biophys Acta 1768:609–619
Krijgsveld J, Zaat SAJ, Meeldijk J, van Veelen PA, Fang G, Poolman B, Brandt E, Ehlert J, Kuijpers AJ, Engbers GHM, Feijen J, Dankert J (2000) Thrombocidins, microbicidal proteins from human blood platelets, are C-terminal deletion products of CXC chemokines. J Biol Chem 275:20374–20381
Tang YQ, Yeaman MR, Selsted ME (2002) Antimicrobial peptides from human platelets. Infect Immun 70:6524–6533
Nakayama T, Shirane J, Hieshima K, Shibano M, Watanabe M, Jin Z, Nagakubo D, Saito T, Shimomura Y, Yoshie O (2006) Novel antiviral activity of chemokines. Virology 350:484–492
Yeaman MR, Bayer AS, Koo SP, Foss W, Sullam PM (1998) Platelet microbicidal proteins and neutrophil defensin disrupt the Staphylococcus aureus cytoplasmic membrane by distinct mechanisms of action. J Clin Invest 101:178–187
Iida N, Haisa M, Igarashi A, Pencev D, Grotendorst GR (1996) Leukocyte-derived growth factor links the PDGF and CXC chemokine families of peptides. FASEB J 10:1336–1345
Pillai MM, Iwata M, Awaya N, Graf L, Torok-Storb B (2006) Monocyte-derived CXCL7 peptides in the marrow microenvironment. Blood 107:3520–3526
El-Gedaily A, Schoedon G, Schneemann M, Schaffner A (2004) Constitutive and regulated expression of platelet basic protein in human monocytes. J Leukoc Biol 75:495–503
Schaffner A, King CC, Schaer D, Guiney DG (2004) Induction and antimicrobial activity of platelet basic protein derivatives in human monocytes. J Leukoc Biol 76:1010–1018
Schaffner A, Rhyn P, Schoedon G, Schaer DJ (2005) Regulated expression of platelet factor 4 in human monocytes–role of PARs as a quantitatively important monocyte activation pathway. J Leukoc Biol 78:202–209
Khajoee V, Saito M, Takada H, Nomura A, Kusuhara K, Yoshida SI, Yoshikai Y, Hara T (2006) Novel roles of osteopontin and CXC chemokine ligand 7 in the defence against mycobacterial infection. Clin Exp Immunol 143:260–268
Kruidenier L, MacDonald TT, Collins JE, Pender SL, Sanderson IR (2006) Myofibroblast matrix metalloproteinases activate the neutrophil chemoattractant CXCL7 from intestinal epithelial cells. Gastroenterology 130:127–136
Nakao K, Aoyama M, Fukuoka H, Fujita M, Miyazawa K, Asai K, Goto S (2009) IGF2 modulates the microenvironment for osteoclastogenesis. Biochem Biophys Res Commun 378:462–466
Vandercapellen J, Noppen S, Verbeke H, Put W, Conings R, Gouwy M, Schutyser E, Proost P, Sciot R, Geboes K, Opdenakker G, Damme Van, Struyf S (2007) Stimulation of angiostatic platelet factor-4 variant (CXCL4L1/PF-4var) versus inhibition of angiogenic granulocyte chemotactic protein 2 (CXCL6/GCP-2) in normal and tumoral mesenchymal cells. J Leukoc Biol 82:1519–1530
Lasagni L, Grepin R, Mazzinghi B, Lazzeri E, Meini C, Sagrinati C, Liotta F, Frosali F, Ronconi E, Alain-Courtois N, Ballerini L, Netti GS, Maggi E, Annunziato F, Serio M, Romagnani S, Bikfalvi A, Romagnani P (2007) PF-4/CXCL4 and CXCL4L1 exhibit distinct subcellular localization and a differentially regulated mechanism of secretion. Blood 109:4127–4134
Auten RL, Richardson RM, White JR, Mason SN, Vozzelli MA, Whorton MH (2001) Nonpeptide CXCR2 antagonist prevent neutrophil accumulation in hypoxia-exposed newborn rats. J Pharmacol Exp Ther 299:90–95
Matzer SP, Zombou J, Sarau HM, Röllinghoff M, Beuscher HU (2004) A synthetic, non-peptide CXCR2 antagonist blocks MIP-2-induced neutrophil migration in mice. Immunobiology 209:225–233
Garau A, Bertini R, Mosca M, Bizzarri C, Anacardio R, Triulzi S, Allegretti M, Ghezzi P, Villa P (2006) Development of a systemically-active dual CXCR1/CXCR2 allosteric inhibitor and its efficacy in a model of transient cerebral ischemia in the rat. Eur Cytokine Netw 17:35–41
Singh S, Sadanandam A, Nannuru KC, Varney ML, Mayer-Ezell R, Bond R, Singh RK (2009) Small molecule antagonists for CXCR2 and CXCR1 inhibit human melanoma growth by decreasing tumor cell proliferation, survival and angiogenesis. Clin Cancer Res 15:2380–2386
Kerstetter AE, Padovani-Claudio DA, Bai L, Miller RH (2009) Inhibition of CXCR2 signaling promotes recovery in models of multiple sclerosis. Exp Neurol 120:44–56
Chapman RW, Phillips JE, Hipkin RW, Curran AK, Lundell D, Fine JS (2009) CXCR2 antagonists for the treatment of pulmonary disease. Pharmacol Ther 121:55–68
Zarbock A, Allegretti M, Ley K (2008) Therapeutic inhibition of CXCR2 by Reparixin attenuates acute lung injury in mice. Br J Pharmacol 155:357–364
Johnson Z, Schwarz M, Power CA, Wells TN, Proudfoot AE (2005) Multi-faceted strategies to combat disease by interference with the chemokine system. Trends Immunol 26:268–274
Ford SL, Reddy YS, Anderson MT, Murray SC, Fernandez P, Stein DS, Johnson MA (2006) Single-dose safety and pharmacokinetics of brecanavir, a novel human immunodeficiency virus protease inhibitor. Antimicrob Agents Chemother 50:2201–2206
Westby M, van der Ryst E (2005) CCR5 antagonists: host-targeted antivirals for the treatment of HIV infection. Antiviral Chem Chemother 16:339–354
De Clercq E (2003) The bicyclam AMD3100 story. Nat Rev Drug Discov 2:581–587
Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C, Webster R, Armour D, Price D, Stammen B, Wood A, Perros M (2005) Maraviroc (UK427857), a potent bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother 49:4721–4752
Fätkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AI, Saag MS, Goebel FD, Rockstroh JK, Dezube BJ, Jenkins TM, Medhurst C, Sullivan JF, Ridgway C, Abel S, James IT, Youle M, van der Ryst E (2005) Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist. Nat Med 11:1170–1172
Westby M, Smith-Burchnell C, Mori J, Lewis M, Mosley M, Stockdale M, Dorr P, Ciaramella G, Perros M (2007) Reduced maximal inhibition in phenotypic susceptibility assays indicate that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J Virol 81:2359–2571
Schürmann D, Fätkenheuer G, Reynes J, Michelet C, Raffi F, van Lier J, Caceres M, Keung A, Sansone-Parsons A, Dunkle LM, Hoffmann C (2007) Antiviral activity, pharmacokinetics and safety of vicriviroc, an oral CCR5 antagonist, during 14-day monotherapy in HIV-infected adults. AIDS 21:1295–1299
Gulick RM, Su Z, Flexner C, Hughes MD, Skolnik R, Wilkin TJ, Gross R, Krambrink A, Coakley E, Greaves WL, Zolopa A, Reichman R, Godfrey C, Hirsch M, Kuritzkes DR, AIDS Clinical Trials Group 5211 Team (2007) Phase 2 Study of the safety and efficacy of Vicriviroc, a CCR5 inhibitor, in HIV-1-infected, treatment-experienced patients: AIDS Clinical Trial Group 5211. J Infect Dis 196:304–312
Wilkin TJ, Su Z, Kuritzkes DR, Hughes M, Flexner C, Gross R, Coakley E, Greaves W, Godfrey C, Skolnik PR, Timpone J, Rodriguez B, Gulick RM (2007) HIV type 1 chemokine coreceptor use among antiretroviral-experienced patients screened for a clinical trial of a CCR5 inhibitor: AIDS Clinical Trial Group A5211. J Clin Infect Dis 44:591–595
Murga JD, Franti M, Pevear DC, Maddon PJ, Olson W (2006) Potent antiviral synergy between monoclonal antibody and small-molecule CCR5 inhibitors of human immunodeficiency virus type 1. Antimicrob Agents Chemother 50:3289–3296
Ji C, Zhang J, Dioszegi M, Chiu S, Rao E, Derosier A, Cammack N, Brandt M, Sankuratri S (2007) CCR5 small-molecule antagonists and monoclonal antibodies exert potent synergistic antiviral effects by co-binding to the receptor. Mol Pharmacol 72:18–28
Strayer DS, Akkina R, Bunnell BA, Dropulic B, Planelles V, Pomerantz RJ, Rossi JJ, Zaia JA (2005) Current status of gene therapy strategies to treat HIV/AIDS. Mol Ther 11:823–842
Swan CH, Torbett BE (2006) Can gene delivery close the door to HIV-1 entry after escape? J Med Primatol 11:236–247
Luis Abad J, Gonzalez MA, del Real G, Mira E, Manes S, Serrano F, Bernad A (2003) Novel interfering bifunctional molecules against CCR5 coreceptor are efficient inhibitors of HIV infection. Mol Ther 8:478–484
Swan CH, Buhler B, Tschan MP, Barbas CF, Torbett BE (2006) T-cell protection and enrichment through lentiviral CCR5 intrabody gene delivery. Gene Ther 13:1480–1492
Bai J, Sui J, Zhu RY, Tallarico AS, Gennari F, Zhang D, Marasco WA (2003) Inhibition of Tat-mediated transactivation and HIV-1 replication by human anti-hCyclinT1 intrabodies. J Biol Chem 278:1433–1442
Fox JL (2007) Antivirals become a broader enterprise. Nat Biotechnol 25:1395–1402
Salvucci O, Yao L, Villalba S, Sajewicz A, Pittaluga S, Tosato G (2002) Regulation of endothelial cell branching morphogenesis by endogenous chemokine stromal-derived factor-1. Blood 99:2703–2711
Yamaguchi J, Kusano KF, Masuo O, Kawamoto A, Silver M, Murasawa S, Bosch-Marce M, Masuda H, Losordo DW, Isner JM, Asahara T (2003) Stromal cell-derived factor-1 effects on ex vivo expanded endothelial progenitor cell recruitment for ischemic neovascularisation. Circulation 107:1322–1328
Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 254:436–444
Mikami S, Nakase H, Yamamoto S, Takeda Y, Yoshino T, Kasahara K, Ueno S, Uza N, Oishi S, Fujii N, Nagasawa T, Chiba TJ (2008) Blockade of CXCL12/CXCR4 axis ameliorates murine experimental colitis. Pharmacol Exp Ther 327:383–392
Tsutsumi H, Tanaka T, Ohashi N, Masuno H, Tamamura H, Hiramatsu K, Araki T, Ueda S, Oishi S, Fujii N (2006) Therapeutic potential of the chemokine receptor CXCR4 antagonists as multifunctional agents. Biopolymers 88:279–289
Jin DK, Shido K, Kopp HG, Petit I, Shmelkov SV, Young LM, Hooper AT, Amano H, Avecilla ST, Heissig B, Hattori K, Zhang F, Hicklin DJ, Wu Y, Zhu Z, Dunn A, Salari H, Werb Z, Hackett NR, Crystal RG, Lyden D, Rafii S (2006) Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med 12:557–567
Grunewald M, Avraham I, Dor Y, Bachar-Lustig E, Itin A, Chimenti S, Landsman L, Abramovitch R, Keshet E (2006) VEGF-induced adult neovascularisation: recruitment, retention, and role of accessory cells. Cell 124:175–189
Redjal N, Chan JA, Segal RA, Kung AL (2006) CXCR4 inhibition synergizes with cytotoxic chemotherapy in gliomas. Clin Cancer Res 12:6765–6771
Ohki Y, Heissig B, Sato Y, Akiyama H, Zhu Z, Hicklin DJ, Shimada K, Ogawa H, Daida H, Hattori K, Ohsaka A (2005) Granulocyte colony-stimulating factor promotes neovascularisation by releasing vascular endothelial growth factor from neutrophils. FASEB J 19:2005–2007
Körbling M, Reuben JM, Gao H, Lee BN, Harris DM, Cogdell D, Giralt SA, Khouri IF, Saliba RM, Champlin RE, Zhang W, Estrov Z (2006) Recombinant human granulocyte-colony-stimulating factor-mobilized and apheresis-collected endothelial progenitor cells: a novel blood cell component for therapeutic vasculogenesis. Transfusion 46:1795–1802
Misao Y, Takemura G, Arai M, Ohno T, Onogi H, Takahashi T, Minatoguchi S, Fujiwara T, Fujiwara H (2006) Importance of recruitment of bone-marrow-derived CXCR4+ cells in post-infarct cardiac repair mediated by G-CSF. Cardiovasc Res 71:455–465
Kovacic JC, Muller DW, Graham RM (2007) Actions and therapeutic potential of G-CSF and GM-CSF in cardiovascular disease. J Mol Cell Cardiol 42:19–33
Luyt CE, Meddahi-Pellé A, Ho-Tin-Noe B, Colliec-Jouault S, Guezennec J, Louedec L, Prats H, Jacob MP, Osborne-Pellegrin M, Letourneur D, Michel JB (2003) Low-molecular-weight fucoidan promotes therapeutic revascularization in a rat model of critical hindlimb ischemia. J Pharmacol Exp Ther 305:24–30
Zhong R, Law P, Wong D, Merzouk A, Salari H, Ball ED (2004) Small peptide analogs to stromal-derived factor-1 enhance chemotactic migration of human and mouse hematopoietic cells. Exp Hematol 32:470–475
Walter DH, Rochwalsky U, Reinhold J, Seeger F, Aicher A, Urbich C, Spyridopoulos I, Chun J, Brinkmann V, Keul P, Levkau B, Zeiher AM, Dimmeler S, Haendeler J (2007) Sphingosine-1-phosphate stimulates the functional capacity of progenitor cells by activation of the CXCR4-dependent signalling pathway via the S1p3 receptor. Arterioscler Thromb Vasc Biol 27:275–282
Ratajczak MZ, Reca R, Wysoczynski M, Yan J, Ratajczak J (2006) Modulation of the SDF-1–CXCR4+ axis by the third complement component (C3)—implications for trafficking of CXCR4+ stem cells. Exp Hematol 34:986–995
Schober A, Karshovska E, Zernecke A, Weber C (2006) SDF-1 alpha-mediated tissue repair by stem cells: a promising tool in cardiovascular medicine? Trends Cardiovasc Med 16:103–108
Proudfoot AE (2002) Chemokine receptors: multifaceted therapeutic targets. Nat Rev Immunol 2:106–115
Simmons G, Clapham PR, Picard L, Offord RE, Rosenkilde MM, Schwartz TW, Buser R, Wells TN, Proudfoot AE (1997) Potent inhibition of HIV-1 infectivity in macrophages and lymphocytes by a novel CCR5 antagonist. Science 276:276–279
Lederman MM, Veazey RS, Offord R, Mosier DE, Dufour J, Mefford M, Piatak M Jr, Lifson JD, Salkowitz JR, Rodriguez B, Blauvelt A, Hartley O (2004) Prevention of vaginal SHIV transmission in rhesus macaques through inhibition of CCR5. Science 306:485–487
Gong JH, Ratkay LG, Waterfield JD, Clark-Lewis I (1997) An antagonist of monocyte chemoattractant protein 1 (MCP-1) inhibits arthritis in MRL-lpr mouse model. J Exp Med 186:131–137
Ludwig A, Ehlert JE, Flad HD, Brandt E (2000) Identification of distinct surface-expressed and intracellular CXC chemokine receptor 2 glycoforms in neutrophils: N-glycosylation is essential for maintenance of receptor surface expression. J Immunol 165:1044–1052
Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TN, Kosco-Vilbois MH (2003) Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci USA 100:1885–1890
Johnson Z, Kosco-Vilbois MH, Herren S, Cirillo R, Muzio V, Zaratin P, Carbonatto M, Mack M, Smailbegovic A, Rose M, Lever R, Page C, Wells TN, Proudfoot AE (2004) Interference with heparin binding and oligomerization creates a novel anti-inflammatory strategy targeting the chemokine system. J Immunol 173:5776–5785
Netelenbos T, van den Born J, Kessler FL, Zweegman S, Merle PA, van Oostveen JW, Zwaginga JJ, Huijgens PC, Dräger AM (2003) Proteoglycans on bone marrow endothelial cells bind and present SDF-1 towards hematopoietic cells. Leukemia 17:175–184
Weathington NM, van Houvelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G, Nijkamp FP, Blalock JE (2006) A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med 12:317–323
Ure DR, Lane TE, Liu MT, Rodriguez M (2005) Neutralization of chemokines RANTES and MIG increases virus antigen expression and spinal cord pathology during Theiler’s virus infection. Int Immunol 17:569–579
Mahler DA, Huang S, Tabrizi M, Bell GM (2004) Efficacy and safety of a monoclonal antibody recognizing interleukin-8 in COPD: a pilot study. Chest 126:926–934
Dar A, Goichberg P, Shinder V, Kalinkovich A, Kollet O, Netzer N, Margalit R, Zsak M, Nagler A, Hardan I, Resnick I, Rot A, Lapidot T (2005) Chemokine receptor CXCR4-dependent internalisation and resecretion of functional chemokine SDF-1 by bone marrow endothelial and stromal cells. Nat Immunol 6:1038–1146
Middleton J, Neil S, Wintle J, Clark-Lewis I, Moore H, Lam C, Auer M, Hub E, Rot A (1997) Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell 91:385–395
Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, Gurtner GC (2004) Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 10:858–864
Kollet O, Shivtiel S, Chen YQ, Suriawinata J, Thung SN, Dabeva MD, Kahn J, Spiegel A, Dar A, Samira S, Goichberg P, Kalinkovich A, Arenzana-Seisdedos F, Nagler A, Hardan I, Revel M, Shafritz DA, Lapidot T (2003) HGF, SDF-1 and MMP-9 are involved in stress-induced human CD34+ stem cell recruitment to the liver. J Clin Invest 112:160–169
Ponomaryov T, Peled A, Petit I, Taichman RS, Habler L, Sandbank J, Arenzana-Seisdedos F, Magerus A, Caruz A, Fujii N, Nagler A, Lahav M, Szyper-Kravitz M, Zipori D, Lapidot T (2000) Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function J. Clin Invest 106:1331–1339
Reichert JM (2007) Trends in the development and approval of monoclonal antibodies for viral infections. BioDrugs 21:1–7
Mehrad B, Keane MP, Strieter RM (2007) Chemokines as mediators of angiogenesis. Thromb Haemost 97:755–762
Keane MP, Arenberg DA, Lynch JP 3rd, Whyte RI, Iannettoni MD, Burdick MD, Wilke CA, Morris SB, Glass MC, DiGiovine B, Kunkel SL, Strieter RM (1997) The CXC chemokines, IL-8 and IP-10, regulate angiogenic activity in idiopathic pulmonary fibrosis. J Immunol 159:1437–1443
Keane MP, Belperio JA, Arenberg DA, Burdick MD, Xu ZJ, Xue YY, Strieter RM (1999) IFN-gamma-inducible protein-10 atenuates bleomycin-induced pulmonary fibrosis via inhibition of angiogenesis. J Immunol 163:5686–5692
Keane MP, Belperio JA, Moore TA, Moore BB, Arenberg DA, Smith RE, Burdick MD, Kunkel SL, Strieter RM (1999) Neutralization of the CXC chemokine macrophage inflammatory protein-2 attenuates bleomycin-induced pulmonary fibrosis. J Immunol 162:5511–5518
Keane MP, Donelly SC, Belperio JA, Goodman RB, Dy M, Burdick MD, Fishbein MC, Strieter RM (2002) Imbalance in the expression of CXC chemokines correlates with bronchoalveolar lavage fluid angiogenic activity and procollagen levels in acute respiratory distress syndrome. J Immunol 169:6515–6521
Pan J, Burdick MD, Belperio JA, Xue YY, Gerard C, Sharma S, Dubinett SM, Strieter RM (2006) CXCR3 ligand biological axis impairs RENCA tumor growth by a mechanism of immunoangiostasis. J Immunol 176:1456–1464
Sharma S, Stolina M, Luo J, Strieter RM, Burdick M, Zhu LX, Batra RK, Dubinett SM (2000) Secondary lymphoid tissue chemokine mediates T cell-dependent antitumor responses in vivo. J Immunol 164:4558–4563
Sharma S, Yang SC, Hillinger S, Zhu LX, Huang M, Batra RK, Lin JF, Burdick MD, Strieter RM, Dubinett SM (2003) SLC/CCL21–mediated anti-tumor responses require IFN-gamma, MIG/CXCL9 and IP-10/CXCL10. Mol Cancer 2:22
Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ (2007) Ly 6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest 117:195–205
Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ (2007) Monocyte subsets differentially employ CCR2, CCR5, and CX3 CR1 to accumulate within artherosclerotic plaques. J Clin Invest 117:185–194
Kovanen PT (2007) Mast cells: multipotent local effector cells in atherothrombosis. Immunol Rev 217:105–122
Davi G, Patrono C (2007) Platelet activation and atherothrombosis. N Engl J Med 357:2482–2494
Llodrá J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ (2004) Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive plaques. Proc Natl Acad Sci USA 101:11779–11784
Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, Randolph GJ, Fisher EA (2006) Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci USA 103:3781–3786
Eisenberg SP, Evans RJ, Arend WP, Verderber E, Brewer MT, Hannum CH, Thompson RC (1990) Primary structure and functional expression from complementary DNA of a human interleukin-1 receptor antagonist. Nature 343:341–346
Seckinger P, Isaaz S, Dayer JM (1988) A human inhibitor of tumor necrosis factor alpha. J Exp Med 167:1511–1516
Novick D, Cohen B, Tal N, Rubinstein M (1995) Soluble and membrane-anchored forms of the human IFN alpha/beta receptor. J Leukoc Biol 57:712–718
Novick D, Kim H, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M (1999) Interleukin-18 binding protein: a novel modulator of the TH1 cytokine response. Immunity 10:127–136
Murphy PM, Baggiolini M, Charo IF, Hébert CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ, Power CA (2000) International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev 52:145–176
Dawson TC, Lentsch AB, Wang Z, Cowhig JE, Rot A, Maeda N, Peiper SC (2000) Exaggerated response to endotoxin in mice lacking the Duffy antigen receptor for chemokines (DARC). Blood 96:1681–1684
Luo H, Chaudhuri A, Zbrzezna V, He Y, Pogo AO (2000) Deletion of murine Duffy gene (Dfy) reveils that the Duffy receptor is functionally redundant. Mol Cell Biol 20:3091–3101
Du J, Luan J, Liu H, Daniel TO, Peiper S, Chen TS, Yu Y, Horton LW, Nanney LB, Strieter RM, Richmond A (2002) Potential role for Duffy antigen chemokine-binding protein in angiogenesis and maintenance of homeostasis in response to stress. J Leukoc Biol 71:41–53
Shen H, Schuster R, Stringer KF, Waltz SE, Lentsch AB (2006) The Duffy antigen/receptor for chemokines (DARC) regulates prostate tumor growth. FASEB J 20:59–64
Lee JS, Frevert CW, Thorning DR, Segerer S, Alpers CE, Cartron JP, Colin Y, Wong VA, Martin TR, Goodman RB (2003) Enhanced expression of Duffy antigen in the lungs during suppurative pneumonia. J Histochem Cytochem 51:159–166
Nibbs RJ, Kriehuber E, Ponath PD, Parent D, Qin S, Campbell JD, Henderson A, Kerjaschki D, Maurer D, Graham GJ, Rot A (2001) The beta-chemokine receptor D6 is expressed by lymphatic endothelium and a subset of vascular tumors. Am J Path 158:867–877
McKimmie CS, Fraser AR, Hansell C, Gutiérrez L, Philipsen S, Connell L, Rot A, Kurowska-Stolarska M, Carreno P, Pruenster M, Chu CC, Lombardi G, Halsey C, McInnes IB, Liew FY, Nibbs RJ, Graham GJ (2008) Hemopoietic cell expression of the chemokine decoy receptor D6 is dynamic and regulated by GATA1. J Immunol 181:3353–3363 (Erratum in: J Immunol 181, 8170. Corrected and republished in: J Immunol 181, 8171–8181)
Frau AM, Locati M, Otero K, Sironi M, Signorelli P, Massardi ML, Gobbi M, Vecchi A, Sozzani S, Mantovani A (2003) Cutting edge: scavenging of inflammatory CC chemokines by the promiscuous putatively silent chemokine receptor D6. J Immunol 170:2279–2282
Nibbs RJ, Graham G, Rot A (2003) Chemokines on the move: control by the chemokine “interceptors” Duffy blood group antigen and D6. Semin Immunol 15:287–294
Jamieson T, Cook DN, Nibbs RJ, Rot A, Nixon C, McLean P, Alcami A, Lira SA, Wiekowski M, Graham GJ (2005) The chemokine receptor D6 limits the inflammatory response in vivo. Nat Immunol 6:403–411
Bowcock AM, Shannon W, Du F, Duncan J, Cao K, Aftergut K, Catier J, Fernandez-Vina MA, Menter A (2001) Insights into psoriasis and other inflammatory diseases from large scale gene expression studies. Hum Mol Genet 10:1793–1805
Nibbs RJ, Yang J, Landau NR, Mao JH, Graham GJ (1999) LD78beta, a non-allelic variant of human MIP1alpha (LD78alpha), has enhanced receptor interactions and potent HIV suppressive activity. J Biol Chem 274:17478–17483
Proost P, Mahieu F, Schutyser E, Van Damme J (2004) Posttranslational processing of chemokines. Meth Mol Biol 239:27–44
Proost P, Menten P, Struyf S, Schutyser E, De Meester I, Van Damme J (2000) Cleavage by CD26//dipeptidyl peptidase IV converts the chemokine LD78beta into a most efficient monocyte attractant and CCR1 agonist. Blood 96:1674–1680
Ludwig A, Petersen F, Zahn S, Götze O, Schröder JM, Flad HD, Brandt E (1997) The chemokine neutrophil-activating peptide-2 induces two distinct optima of neutrophil chemotaxis by differential interaction with interleukin-8 receptors CXCR1 and CXCR2. Blood 90:4588–4597
Jones SA, Wolf M, Qin S, Mackay CR, Baggiolini M (1996) Different functions for the interleukin 8 receptors (IL-8R) of human leukocytes: NADPH oxidase and phospholipase D are activated through IL-8R1 but not IL-8R2. Proc Natl Acad Sci USA 93:6682–6686
Hartl D, Latzin P, Hordijk P, Marcos V, Rudolph C, Woischnik M, Krauss-Etschmann S, Koller B, Reinhardt D, Roscher AA, Roos D, Griese M (2007) Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat Med 13:1423–1430
Bianchi ME (2007) DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 81:1–5
Bianchi SM, Prince LR, McPhillips K, Allen L, Marriott HM, Taylor GW, Hellewell PG, Sabroe I, Dockrell DH, Henson PW, Whyte MK (2008) Impairment of apoptotic cell engulfment by pyocyanin, a toxic metabolite of Pseudomonas aeruginosa. Am J Respir Crit Care Med 177:35–43
Alcami A (2003) Viral mimicry of cytokines, chemokines and their receptors. Nat Rev Immunol 3:36–50
De Bruyne LA, Li K, Bishop DK, Bromberg JS (2000) Gene transfer of virally encoded chemokine antagonists vMIP-II and MC 148 prolongs cardiac allograft survival and inhibits donor-specific immunity. Gene Ther 7:575–582
Aliberti J, Valenzuela JG, Carruthers VB, Hieny S, Andersen J, Charest H, Reis, e Sousa C, Fairlamb A, Ribeiro JM, Sher A (2003) Molecular mimicry of a CCR5 binding-domain in the microbial activation of dendritic cells. Nat Immunol 4:485–490
Golding H, Aliberti J, King LR, Manischewitz J, Andersen J, Valenzuela J, Landau NR, Sher A (2003) Inhibition of HIV infection by a CCR5 binding cyclophilin from Toxoplasma gondii. Blood 102:3280–3286
Murphy PM (2001) Viral exploitation and subversion of the immune system through chemokine mimicry. Nat Immunol 2:116–122
Lim K, Glass WG, McDermott DH, Murphy PM (2006) CCR5: no longer a “good for nothing” gene–chemokine control of the West Nile virus infection. Trends Immunol 27:308–312
Seet B, McFadden G (2002) Viral chemokine-binding proteins. J Leukoc Biol 72:24–34
Webb LM, Alcami A (2005) Virally encoded chemokine binding proteins. Mini Rev Med Chem 5:833–848
Alexander JM, Nelson CA, van Berkel V, Lau EK, Studts JM, Brett J, Speck SH, Handel TM, Virgin HW, Fremont DH (2002) Structural basis of chemokine sequestration by a herpesvirus decoy receptor. Cell 111:343–356
Liu L, Dai E, Miller L, Seet B, Lalani A, Macauley C, Li X, Virgin HW, Bunce C, Turner P, Moyer R, McFadden G, Lucas A (2004) Viral chemokine binding proteins inhibit inflammatory responses and aortic allograft transplant vasculopathy in rat models. Transplantation 77:1653–1660
Alejo A, Ruiz-Argüello MB, Ho Y, Smith VP, Saraiva M, Alcami A (2006) A chemokine binding domain in the tumor necrosis factor receptor from variola (smallpox) virus. Proc Natl Acad Sci USA 103:5995–6000
Smith P, Fallon RE, Mangan NE, Walsh CM, Saraiva M, Sayers JR, McKenzie AN, Alcami A, Fallon PG (2005) Schistosoma mansoni secretes a chemokine binding protein with antiinflammatory activity. J Exp Med 202:1319–1335
Kocáková P, Sláviková M, Hajnická V, Slovák M, Gasperík J, Vancová I, Fuchsberger N, Nuttall PA (2003) Effect of fast protein liquid chromatography fractionated salivary gland extracts from different ixodic tick species on interleukin-8 binding to its cell receptors. Folia Parasitol (Praha) 50:79–84
Oliveira CJF, Cavassani KA, Moré DD, Garlet GP, Aliberti JC, Silva JS, Ferreira BR (2008) Tick saliva inhibits the chemotactic function of MIP-1α and selectively impairs chemotaxis of immature dendritic cells by down-regulating cell-surface CCR5. Int J Parasitol 38:705–716
Frauenschuh A, Power CA, Déruaz M, Ferreira BR, Silva JS, Teixeira MM, Dias JM, Martin T, Wells TNC, Proudfoot AEI (2007) Molecular cloning and characterization of a hihgly selective chemokine-binding protein from zhe tick Rhipicephalus sanguineus. J Biol Chem 282:27250–27258
Déruaz M, Frauenschuh A, Alessandri AL, Dias JM, Coelho FM, Russo RC, Ferreira BR, Graham GJ, Shaw JP, Wells TNC, Teixeira MM, Power CA, Proudfoor AEI (2008) Ticks produce highly selective chemokine binding proteins with antiinflammatory activity. J Exp Med 205:2019–2031
Acknowledgments
The authors gratefully acknowledge support by Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 367, Projekt C4 (to H.D.F. and E.B.) and Sonderforschungsbereich, Transregio 22, Projekt A11 (to E.B.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Flad, HD., Brandt, E. Platelet-derived chemokines: pathophysiology and therapeutic aspects. Cell. Mol. Life Sci. 67, 2363–2386 (2010). https://doi.org/10.1007/s00018-010-0306-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-010-0306-x