
Abstract
Resistance to tetracycline emerged soon after its discovery six decades ago. Extensive clinical and non-clinical uses of this class of antibiotic over the years have combined to select for a large number of resistant determinants, collectively termed the tetracycline resistome. In order to impart resistance, microbes use different molecular mechanisms including target protection, active efflux, and enzymatic degradation. A deeper understanding of the structure, mechanism, and regulation of the genes and proteins associated with tetracycline resistance will contribute to the development of tetracycline derivatives that overcome resistance. Newer generations of tetracyclines derived from engineering of biosynthetic genetic programs, semi-synthesis, and in particular recent developments in their chemical synthesis, together with a growing understanding of resistance, will serve to retain this class of antibiotic to combat pathogens.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The tetracycline antibiotics offer a primer in the rise, fall, and resurgence of an antibiotic scaffold in the face of selection and emergence of resistance. As such, the tetracycline saga provides important insight into the impact of resistance on an antibiotic class and the options and strategies to researchers and clinicians to retain a valued group of drugs.
Discovered 60 years ago as the first class of broad-spectrum antibiotics (i.e. effective versus both Gram positive and Gram negative bacterial pathogens), tetracyclines found rapid use in the clinic and in agriculture. As a class, they have an excellent therapeutic index and many are orally available. These characteristics made them first choice antibiotics for many outpatient treatments. Unfortunately, following the ‘use it and lose it’ rule of antibiotics, the extensive use of tetracycline antibiotics in medicine and agriculture have maintained a continuous selective pressure for tetracycline resistance in previously susceptible pathogenic organisms. Fortunately, this was balanced for over two decades by the discovery of new analogues from producing soil bacteria and medicinal chemical modification of the chemical scaffolds to generate next generation antibiotics that circumvented resistance. This cycle of resistance coupled to the introduction of new derivatives reached an asymptote about 20 years ago when newer tetracyclines with growth inhibition activity versus the large number of resistance determinants circulating in microbes were increasingly difficult to identify. The contemporaneous emergence of the fluroquinolone class of antibiotics in the 1970s and 1980s with a different mode of action but similar broad-spectrum pathogen profile and pharmacology prevented a health care crisis that may otherwise have followed the decline in utility of tetracyclines. As a result, the use of tetracycline antibiotics in medicine decreased significantly over subsequent years. Recently, the growing problem of fluroquinolone resistance and the introduction of a novel class of third generation semi-synthetic tetracycline antibiotics have returned attention to this class of antibiotic. This review will focus on the tetracycline antibiotic resistance genes, their evolution, and their selection in the face of the introduction of new antibiotics.
Chemical diversity of tetracycline antibiotics
Following the pioneering work of contemporaries in academe including Walksman (aminoglycosides such as streptomycin) and Dubos (peptides such as gramicidin), researchers at various pharmaceutical firms in the US identified the first tetracycline antibiotics over 50 years ago. These antibiotics were the products of actinomycete secondary metabolism and included chlortetracycline (Lederle, 1948), oxytetracycline (Pfizer, 1950), tetracycline (Pfizer, 1953), and 6-demethylchlortetracycline (Lederle, 1957) (Fig. 1). These compounds were discovered by systematic sampling of the fermentation products of spore-forming soil bacteria (and, in the case of tetracycline, also by chemical transformation of chlortetracycline) and were rapidly introduced into clinical practice and, in many cases, into agriculture as feed additives.

Chemical structures of various tetracycline antibiotics. a Structure of tetracycline, b advances in new generation tetracycline development over a period of time
The core structure of an active tetracycline antibiotic includes a four-ring system (labeled A–D, Fig. 1) where ring D is aromatic but rings A, B, and C include saturated carbon centers. As a result, although the tetracyclines appear planar in two-dimensional structure images, they in fact have a significant bend and a unique 3D-structure (Fig. 2). The antibiotic properties are largely retained following chemical modification of the ‘top’ of the molecule (positions 4–7). On the other hand, the ‘bottom’ half of the structure (positions 10–12 and 1) is intolerant of substitution. In particular, the beta-diketone region consisting of carbons 11 and 12 comprises a divalent metal chelation site that is characteristic of the class and essential for biological activity (vide infra).

Three-dimensional architecture of tetracycline and chelocardin. Tetracycline binding to the ribosome is achieved by its unique three-dimensional architecture including the kink between rings A and B. In contrast, chelocardin is a more planar molecule than tetracycline and consequently inhibits bacterial growth by not binding to the ribosome but most likely due to interference with the cell membrane. Tetracycline structure taken from Hinrichs et al. [69] (PDB file *2trt). Chelocardin generated using Chem3D Pro 7.0 and MOPAC minimization [70]
The emergence of resistance to this ‘first generation’ of tetracyclines spurred an effort to chemically synthesize derivatives with improved pharmacological properties and spectrum versus resistant bacteria. These second generation tetracyclines include minocycline (Lederle, 1972) and doxycycline (Pfizer, 1967), which continue to find clinical use across the globe. Meta-substitution of ring D has resulted in a third generation of tetracyclines exemplified by the glycylcyclines and the first in class drug tigecycline (Tigacyl™) from Wyeth approved by the FDA in 2005. These newer antibiotics have a similar mode of action as first and second generation tetracyclines but are insensitive to common mechanisms of resistance such as efflux [1].
Second and third generation tetracyclines are the products of semi-synthesis, i.e. synthetic organic manipulation of natural product antibiotic scaffolds obtained by fermentation. It was not until 2005 that the Myers group at Harvard reported an efficient total synthesis of tetracycline antibiotics [2, 3] thereby enabling the dramatic expansion of tetracycline antibiotic chemical diversity. This advance holds significant promise for the generation of future tetracyclines (Fig. 3).

Newly developed tetracycline analogs. a Promising anticancer drug candidate COL-3, a chemically modified tetracycline. b Recently developed new tetracycline derivatives using the Myers total synthesis approach, which are inaccessible using conventional semi-synthetic approach [71]
Additionally, there have been extensive efforts to explore tetracyclines and their analogs with a goal of expanding bioactivity to cancer targets (Fig. 3). Early studies revealed that certain tetracyclines inhibited cancer cell growth, in particular metastasis. These compounds show a different structure activity relationship profile than the antibacterial compounds and continue to be explored as drug candidates [4]. COL-3 (metastat), a chemically modified tetracycline, has emerged as a promising candidate for treatment of AIDS-related Kaposi’s sarcoma and has successfully cleared Phase II clinical trials [5].
Mode of action
Tetracyclines exert their antibiotic activity by binding to the bacterial ribosome and thereby interfering with protein translation. There are several tetracycline binding sites on the ribosome, but the key interfering binding appears to be in the region of the tRNA acceptor site (A-site) [6–8]. Recent molecular dynamics studies support the high affinity TET1 site as the primary antibiotic binding site [9]. Docking of cognate aminoacyl-tRNAs with mRNA occurs at this site of the ribosome and tetracyclines in complex with Mg2+ bind to the 30S subunit at the apex of the A-site. This binding sterically blocks aminoacyl-tRNA binding and as a result inhibits protein synthesis [8]. The result is generally bacteriostatic (arrest of cell growth) rather than cell death.
Tetracyclines enter the cell in Mg2+ bound form via outer membrane porins in Gram negative bacteria, followed by passive diffusion of the metal-free antibiotic across the cell membrane [10]. Thus, divalent metal binding is vital to both antibiotic delivery into the cell and binding to the target.
The tetracycline resistome
The resistome concept refers to the aggregate of all antibiotic resistance mechanisms [11, 12]. These not only comprise those found in pathogens but also resistance genes in environmental organisms not normally associated with disease. This includes, for example, antibiotic producers that must co-evolve resistance with production in order to avoid auto-toxicity. It has been shown repeatedly that resistance genes that emerge clinically likely have their origins in non-pathogenic bacteria: for example, aminoglycoside resistance via inactivating enzymes [13] and ribosomal methylation [14], glycopeptide resistance via altered cell wall biosynthesis [15], and β-lactam resistance by production of the CTX class of extended spectrum β-lactamases [16] to name but a few. By understanding antibiotic resistance in its totality and not just focusing on mechanisms that have already emerged as clinical problems, we can better understand the evolution of resistance, anticipate the emergence of new mechanisms from the environment into the clinic, and use this information to guide new drug discovery.
Resistance to tetracyclines was first reported in 1953, shortly after their first clinical use [17]. In the following decades, resistance increased rapidly in many bacterial species as a result of horizontal exchange of resistance genes on mobile genetic elements such as plasmids and transposons. Like most antibiotics, resistance to tetracyclines can occur through a number of mechanisms. These include active efflux of tetracycline from the cell, the production of ribosomal protection proteins, decreased drug permeability, target mutation, and enzymatic degradation of the antibiotics (Fig. 4). The first two mechanisms currently predominate in clinical settings.

Diversity and abundance of tetracycline resistance determinants found in nature: the tetracycline resistome. Asterisk indicates activity not confirmed with pure protein
Tetracycline efflux
There are 26 different classes of efflux pumps present in Gram negative and Gram positive bacteria of which 18 classes of tetracycline efflux pumps were classified into 6 groups by Chopra and Roberts [18] and an additional 5 were classified later by Roberts [19]. Similar classification based on protein phylogeny has been suggested by Guillaume et al. [20]. According to this classification, Group-1 consists of drug-H+ antiporters with 12 transmembrane sequences (TMS) namely, Tet(A), Tet(B), Tet(C), Tet(D), Tet(E), Tet(G), Tet(H), Tet(J), Tet(Y), Tet(Z), Tet(30), Tet(31), and Tet(33). Members with 14 TMSs were divided into 2 groups due to the low homology of genes between Streptomyces and non-Streptomyces strains. While Tet(L) and Tet(K) were classified under Group-2, the resistance genes of Streptomyces origin (otrB and tcr3), with high G+C DNA, were kept separately under Group-3. TetA(P) has 12 TMS but a very different hydrophobicity pattern from other Group-1 members, and also lacks conserved major facilitator superfamily (MFS) motifs and so forms sole member of Group-4. Another atypical MFS (see below) protein with at least ten TMS is Tet(V) from Mycobacteria, which is placed in Group-5. Tet(35) is member of Group-6, with only nine TMS and does not belong to the MFS family. The other Group-6 member is OtrC, a predicted ABC transporter from the oxytetracycline producer Streptomyces rimosus. The determinants discovered since these classifications were examined for their similarity to other tetracycline resistance elements using the antibiotic resistance gene database (ardb.cbcb.umd.edu). Tet(39) [21], Tet(41) [22], and Tet(42) [23] showed high similarity among themselves and to Tet(C) and so can be added to Group-1, whereas Tet(40) [24] with 43.4% average similarity to TetA(P) is placed in Group-4. Tet(38) [25] is another 14 TMS protein. It shares only 29% homology and 50% similarity to Tet(K) (Table 1) and should be classified as Group-7.
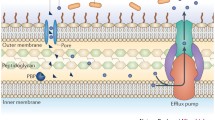
Efflux of tetracyclines predominantly occurs via proteins that are members of the MFS group of integral membrane transporters [26]. These efflux pumps are found across several microbial genera and are integral membrane proteins that span the lipid bilayer of the inner cell membrane 12–14 times. There are well over two dozen MFS tetracycline transporters known (Table 1). Based on homology to other known transporters, the membrane spanning regions of the protein are predicted to be helical. An atomic resolution structure of a tetracycline efflux pump is not yet available; however, mutagenesis and structure–function studies predict a water-filled channel surrounded by six transmembrane helices (Fig. 5). The antibiotic is predicted to pass through this channel and is exchanged for H+ [27]. It is this vectorial flow of protons through the channel, down the pH gradient, which provides the energy required to pump the antibiotic from the cell.

Model of the TetA(B) efflux pump. a Orientation of 12 transmembrane helices to form water-filled channel for export of tetracycline. b Amino acids D66, R101, G247, and D285 implicated to be vital for efflux activity are shown
There is a conserved sequence motif, GXXXXRXGRR, common to tetracycline efflux proteins, glucose uniporters, and sugar/H+ symporters [28]. Much of our understanding of the molecular basis of tetracycline efflux comes from detailed studies of the Tet(B) protein from Tn10 [sometimes referred to as TetA(B)]. Saturation mutagenesis studies of the 62GKMSDRFGRR71 motif of Tet(B) have revealed D66, located in the cytoplasmic loop connecting helices 2 and 3, to be essential for activity [29]. Other key residues identified to be essential for Tet(B) activity include R101 in helix 4, G247 in helix 8, and D285 in helix 9 (Fig. 5) [27]. Residue H257 has been implicated in proton flux in Tet(B), but is not essential for full functionality of the protein [30].
Regulation of expression for these MFS proteins is often tightly coupled to a repressor protein of the TetR family. TetR proteins bind to the upstream operator region of genes encoding efflux determinants and negatively regulate efflux protein expression. Tetracycline also binds to TetR resulting in a conformational change that weakens its interaction with DNA resulting in dissociation from the operator region, which allows efflux protein synthesis [31, 32]. This allows ‘just in time’ expression of resistance only when the antibiotic is present. The interaction of tetracyclines with TetR is not exclusively shared with portions of the molecule that are essential for interaction with the ribosome (Fig. 6). Therefore, modification of the tetracycline scaffold can inhibit TetR interaction, and thus maintain repression of Tet(A) expression while retaining antibiotic activity.

Schematic diagram illustrating interactions of magnesium bound tetracycline with a the TetR homodimer [69] and b primary binding site in 16S rRNA of the small ribosomal subunit [8]. While TetR interactions surround the tetracycline scaffold from all directions, the ribosome binding sites are confined to the bottom portion alone, providing possible sites for modifications within the top portion (dotted ellipse) of the molecule. These modifications may be exploited to create interference in TetR tetracycline docking, and thereby overcome TetR mediated efflux resistance without compromising its ribosome binding antibiotic property
Ribosomal protection proteins
Ribosomal protection proteins (RPP) represent a widely distributed class of tetracycline resistance determinants. There are 11 different types of RPPs spanning Gram positive as well as Gram negative bacterial genera. Tet(O) and Tet(M) are the most prevalent and the best studied class of RPPs. While Tet(M) is detected in 24 genera, Tet(O) is found in 8 different bacterial genera. otrA is believed to be the ancestor of some other RPPs found in pathogens like Mycobacteria [33]. RPPs are divided into three groups based on the amino acid sequence of encoded proteins. Group-1 includes tet(M), tet(O), tet(S), tet(W), tet(32), and tet(36), whereas Group-2 includes tetB(P), otr(A), and tet. Group-3 is represented by tet(Q) and tet(T).
RPPs are approximately 72.5 kDa in size and share high homology to translation elongation factors EF-Tu and EF-G GTPases [34]. RPPs are proposed to be EF paralogs that have evolved through duplication and divergence of an ancient GTPase [35]. RPPs were earlier proposed to work as tetracycline resistant elongation factors to carry out protein synthesis in the presence of tetracycline, but substitution experiments have ruled out this hypothesis [36]. Tetracycline resistance is achieved by weakening the interaction of tetracycline and the ribosome with subsequent antibiotic release [37] (Fig. 7). This then frees the ribosome from the inhibitory effects of the drug, such that aa-tRNA can bind to the A-site and protein synthesis can continue. By this mechanism, they exhibit resistance against first as well as second generation tetracyclines, but not to third generation compounds. In fact, tigecycline has been shown to effectively exhibit antibacterial activity against Tet(M)-protected ribosomes possibly due to stronger binding of the drug to its target [38].

Ribosomal protection protein-mediated tetracycline resistance. a Tetracycline binds the ribosome at the apex of the A-site which in turn sterically blocks the aminoacyl-tRNA binding site and inhibits protein synthesis. b When bound by tetracycline, RPPs along with bound GTP will associate with the ribosome which results in tetracycline release from the A-site. c Upon tetracycline release, GTP is hydrolyzed and the RPP subsequently dissociates from the ribosome which restores protein synthesis
Mosaic elements
A new class of tetracycline resistance determinants has recently been discovered. Mosaic derivatives of known resistance genes, in which at least one part of the gene shares over 80% homology to a known tetracycline resistance gene and the other part with homology to other known or new determinant, have been found. These mosaic genes are products of the RPP group of resistance determinants. So far, the Tet(M), Tet(W), and Tet(O) classes of genes have been predominantly found to form such mosaic patterns. The best example comes from tet(32), a known ribosomal protection gene isolated originally from Clostridium strain K10, which shares 60% identity with tet(O) [39]. It has been reclassified as tet(O/32/O), since the novel tet(32) sequence is limited to the central portion of the gene product, flanked by known tet(O) sequence. Resistance-imparting wild-type tet(32), devoid of any tet(O) sequences flanking it, has been subsequently discovered from human saliva [40, 41]. This indicates the presence of intact genes, which form the source of hybrid genes, also prevailing in similar niches. As more mosaic genes are discovered, their complexity and abundance is becoming evident. Intra-class mosaic genes have been described for tet(M) [42]. Also, there is ample evidence for inter-class mosaic genes. Mosaic tet(O/W/O) genes are prevalent in Megasphaera elsdenii isolates from swine farms in the US. These hybrid genes conferred a higher level of resistance (128 to >256 μg/ml) as compared to 64 μg/ml conferred by tet(O) or tet(W) genes [43]. This suggests that not all mosaic determinants obtained are significantly active, but some can even be superior and exceed the resistance levels of their parental determinants [40]. A recent study on DNA isolated from animal and human fecal samples suggest that mosaic tetracycline resistance genes comprised of tet(O), tet(W), and tet(32) sequences were abundant [40]. More surprisingly, an identical hybrid determinant tet(O/W/32/O/W/O) has been isolated from two independent studies [40, 44]. Initially, the occurrence of such mosaic genes was hypothesized to be restricted to a particular niche [19], but these recent reports suggest that such genes are more prevalent than anticipated and their representation in the environment is significant. Most of the bacteria harboring mosaic genes so far have been sourced from either farm animals or humans, which is probably a reflection of the opportunity for the evolution of resistance in the presence of drug. Interestingly, the mosaic genes found so far have shown different patterns of mosaicism but the final size of the genes has remained unaltered. These determinants definitely impart a strong signal for evolution of resistance determinants at a rapid rate. It will be of particular interest to explore the driving force that determines the specific recombination events and presence of hotspots for crossover, leading to formation of active mosaic determinants. These mosaic genes might have been circulating for a number of years, but escaped detection due to use of well-defined class specific primers for the RPP group of determinants. More rigorous investigation may reveal the abundance and spread of these determinants. The high frequency at which they have been discovered so far strongly suggests that these genes be considered in future surveillance programs.
Tetracycline inactivation
Unlike other antibiotic classes such as the β-lactams and aminoglycosides where drug inactivation and modification predominates, there are relatively few reported tetracycline inactivators. Only three genes in the entire tetracycline resistome, tet(X), tet(34), and tet(37), have so far been associated with enzymatic inactivation of tetracyclines. Tet(X) was the first tetracycline inactivator to be discovered in 1989 from a strain of Bacteroides fragilis [45]. Introduction of the tet(X) gene into E. coli resulted in the presence of an oxygen-dependent color change in the growth media to a black color in the presence of tetracycline. We showed that Tet(X) is a flavin-dependent monooxygenase that inactivates tetracycline by regioselectively adding a hydroxyl group to the C-11a position of the antibiotic [46]. This results in an unstable compound that undergoes non-enzymatic decomposition to a black polymer. Further, not only does Tet(X) act on first and second generation tetracyclines by this mechanism, but it is also active against the recently approved third generation antibiotic tigecycline [47]. Under aerobic conditions, Tet(X) utilizes NADPH in presence of magnesium and converts tigecycline to 11a-hydroxytigecycline (Fig. 8). The modified molecule has weaker binding to magnesium, which is essential for its binding to ribosome.

TetX mediated tigecycline inactivation. The enzymatic inactivation of tigecycline is mediated by TetX, a flavin-dependent monooxygenase. In the presence of oxygen, TetX catalyze the regiospecific hydroxylation of tigecycline at position 11a producing 11a-hydroxytigecycline
Oxidation and reduction are generally not prevalent mechanisms of antibiotic inactivation in bacteria. Interestingly, Tet(X) is an oxygen-dependent flavoprotein whereas the organism harboring it is an obligate anaerobe. This suggests that the gene must have migrated from some other genome. Moreover, it is borne by conjugative transposons Tn4351 and Tn4400 that also carry an erythromycin resistance gene (ermF). In fact, Tet(X) was discovered serendipitously, when Speer et al. [45] were trying to clone the associated erythromycin resistance gene ermF into E. coli and found the resulting recombinant strains to be tetracycline resistant as well. TetX orthologs, TetX1 and TetX2, have been identified on another transposon CTnDOT. Very recently, tet(X) has been discovered from Sphingobacterium sp. PM2-P1-29, on a mobilizable transposon-like element Tn6031 [48]. With a 99.8% nucleotide homology and 99.5% amino acid homology, this gene is almost identical to tetX2 of CTnDOT from Bacteriodes thetaiotaomicron. Although genes corresponding to tetX1 and ermF of CTnDOT were not homologous to Tn6031, the remainder of the transposon showed very high conservation. It is hypothesized that Sphingobacterium sp. could be the ancestral strain that originally harbored functional Tet(X) in nature and passed it on to Bacteriodes sp.
Several Tet(X)-like proteins have been identified in the chromosomes of bacteria like S. coelicolor and Cytophaga. In our investigations, we did not find that any of these proteins could inactivate tetracycline nor were they associated with tetracycline resistance (Thompson and Wright, unpublished). In another attempt to search for tetracycline inactivating determinants, 286 tetracycline resistant Streptomyces environmental isolates in our Streptomyces library were screened and not a single determinant was found to adopt an inactivation mechanism for tetracycline resistance (Thompson and Wright, unpublished).
Besides tet(X), there are two other genes that have been predicted to inactivate tetracycline. Tet(34), identified from Vibrio sp. present in the intestine of yellow tail tuna, shares significant homology to xanthine–guanine phosphoribosyltransferases (XGPRTs) that catalyze the synthesis of guanosine monophosphate and xanthine monophosphate, and thus supply purine nucleotides for translation [49]. Although it has been classified under determinants imparting tetracycline resistance by enzymatic action, the purified protein could not bring about any alteration in the antibiotic when assayed in our laboratory. In our view, Tet(34) needs to be revisited as a tetracycline resistance determinant. Another determinant in this group is tet(37) a small 327-bp gene, with proposed tetracycline inactivation product [50]. The predicted protein is not homologous to Tet(X) but shares homology with other flavoproteins, oxidoreductases, and NAD(P)-requiring enzymes. Protein functional analysis for Tet(37) and its exact mechanism of action on tetracycline remains to be determined.
Other mechanisms of tetracycline resistance
Self-resistance genes
Self-resistance genes usually are present as a part of the antibiotic biosynthesis gene cluster. Three resistance genes were found in the oxytetracycline producer Streptomyces rimosus: otrA, otrB, and otrC [51]. They are expected to impart resistance non-destructively for the obvious reason of avoiding a futile biosynthetic cycle. OtrA has a conserved GTPase domain and is a predicted ribosomal protection protein. The protein shares 50% sequence similarity with Tet(O) from C. jejuni as well as Tet(M) from Streptococcus sp, which impart resistance by similar mechanism in non-tetracycline-producing strains. OtrB resembles the efflux determinants of MFS proteins in Gram negative bacteria and imparts resistance by reduced intracellular accumulation of the drug [52, 53]. A similar gene, tcr3 has also been obtained from another tetracycline producer, S. aureofaciens, and is predicted to be an efflux pump [54]. OtrC is a putative ABC transporter, suggestive of its role in tetracycline efflux [19].
Reduced antibiotic permeability
Gram negative bacteria are naturally resistant to several antibiotics due to the presence of a lipopolysaccharide containing outer membrane layer. Tetracyclines can easily cross this barrier by forming a magnesium complex followed by a rapid passage via porin channels such as OmpF [10]. To date, mutants adapting such a mechanism of resistance are not prevalent in pathogenic isolates obtained from clinics. In the presence of antibiotic stress, E. coli overexpresses the global activator protein MarA, which besides inducing MDR efflux pump AcrAB, also down-regulates synthesis of OmpF, through the increased production of antisense RNA encoded by micF [55]. This has the combinatorial effect of reducing both tetracycline uptake and accumulation by the cell.
Target modification
Modification of drug target is often a prevalent mechanism by which pathogens escape antibiotic action. Though tetracycline resistance in isolates of Propionibacterium acnes has been reported since the 1980s from various parts of the world [56–58], no systematic study to characterize the mechanism was conducted until 1998, when Ross et al. [59] showed a point mutation (G → C) at position 1,058 in 16S-rRNA to be responsible for the resistant phenotype.
Different mutations in the 16S-rRNA of Helicobacter pylori strains showed variable resistance to tetracycline. More than one study has revealed substitutions (AGA → TTC) of nucleotides at positions 926, 927, and 928 in tetracycline resistant isolates of H. pylori [60, 61]. The mutations are located within the primary binding site of tetracycline. Further, it has been shown that each substitution has a cumulative effect in tetracycline resistance, as double (AGA → GTA/GGC) or single (AGA → GGA/AGC) base change could mediate low levels of tetracycline resistance [62, 63].
The impact of the tetracycline resistome
Since the identification of the first tetracycline resistance genes in the 1970s, numerous other genes with associated proteins of varying amino acid sequence have been identified. To keep track of the rapidly growing resistome, rules for nomenclature of new tetracycline resistance determinants were published in 1989 [64]. In a span of just one decade, the expanding resistome included all the available letters of the alphabet, which forced the introduction of a numerical pattern of nomenclature in 1999. tet(30) was the first gene to have been assigned a number. The emergence of mosaic genes has once again challenged existing nomenclature pattern. It has been suggested to incorporate the designations of the known tetracycline gene classes forming the hybrid in the order of their occurrence in the gene [65].
The diverse tetracycline resistome includes over 1,189 reported tetracycline genes, identified in more than 84 genera and 354 species of Gram positive and Gram negative bacteria [66]. Based on their sequence homology, these genes have been classified into 41 resistance determinant classes. Of these, 26 are efflux pumps, 11 ribosomal protection protein, and 2 are inactivators, while the mechanism of resistance by tet(34) remains unclear. In addition,, the resistome also hosts other resistance mechanisms such as MDR, point mutations, and mosaic genes that have not been included in these classes. This may make it the largest resistome against an individual class of antibiotics.
In a sampling for resistance determinants in diverse soils from various regions of the world, we showed a wide dispersion of antibiotic resistance, and the data on tetracyclines were informative [67]. Of 482 strains tested, 286 strains were found to be resistant to tetracyclines; ~60% of the strains in the sample. Even the resistance against semisynthetic second and third generation could be observed in a significant fraction of the sample (Fig. 9). Alarmingly, the incidence of resistance against tigecycline was more prevalent than its second generation precursor minocycline.

Venn diagram showing prevalence of resistance against first, second, and third generation tetracyclines from soil actinomycetes. Data are derived from [67]
Why is there such a plethora of tetracycline resistance genes in microbial populations? Baltz [68] has estimated that, out of 1,000 actinomycetes, one will produce tetracycline. In an average sample of soil, there are 106–108 bacteria, out of which 5–6% are actinobacteria. Therefore, if viewed on a global scale, the amount of naturally occurring tetracycline-like molecules in the environment is staggering. If we now add human application in agriculture and continued use in the community in particular as low cost treatment for disease, the planetary tetracycline burden in massive. Under such powerful selection pressure, it is not surprising in retrospect that the tetracycline resistome is so broad.
The impact of the tetracycline resistome has already been felt. Once a dominant class of antibiotic with desirable broad-spectrum activity against numerous pathogens and orally available, the widespread emergence of resistance has had a massive impact on these drugs. Preservation of the clinical utility of third generation tetracyclines such as tigecycline will require a thorough understanding of resistance at the molecule, population, and evolutionary levels along with vigilance and surveillance to identify and circumvent new resistance challenges as they emerge.
References
Fluit AC, Florijn A, Verhoef J, Milatovic D (2005) Presence of tetracycline resistance determinants and susceptibility to tigecycline and minocycline. Antimicrob Agents Chemother 49:1636–1638
Charest MG, Siegel DR, Myers AG (2005) Synthesis of minus-tetracycline. J Am Chem Soc 127:8292–8293
Charest MG, Lerner CD, Brubaker JD, Siegel DR, Myers AG (2005) A convergent enantioselective route to structurally diverse 6-deoxytetracycline antibiotics. Science 308:395–398
Lokeshwar BL, Selzer MG, Zhu BQ, Block NL, Golub LM (2002) Inhibition of cell proliferation, invasion, tumor growth and metastasis by an oral non-antimicrobial tetracycline analog (COL-3) in a metastatic prostate cancer model. Int J Cancer 98:297–309
Dezube BJ, Krown SE, Lee JY, Bauer KS, Aboulafia DM (2006) Randomized phase II trial of matrix metalloproteinase inhibitor COL-3 in AIDS-related Kaposi’s sarcoma: an AIDS Malignancy Consortium Study. J Clin Oncol 24:1389–1394
Anokhina MM, Barta A, Nierhaus KH, Spiridonova VA, Kopylov AM (2004) Mapping of the second tetracycline binding site on the ribosomal small subunit of E. coli. Nucleic Acids Res 32:2594–2597
Pioletti M, Schlünzen F, Harms J, Zarivach R, Glühmann M, Avila H, Bashan A, Bartels H, Auerbach T, Jacobi C, Hartsch T, Yonath A, Franceschi F (2001) Crystal structures of complexes of the small ribosomal subunit with tetracycline, edeine and IF3. EMBO J 20:1829–1839
Brodersen DE, Clemons WM Jr, Carter AP, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V (2000) The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell 103:1143–1154
Aleksandrov A, Simonson T (2008) Molecular dynamics simulations of the 30S ribosomal subunit reveal a preferred tetracycline binding site. J Am Chem Soc 130:1114–1115
Schnappinger D, Hillen W (1996) Tetracyclines: antibiotic action, uptake, and resistance mechanisms. Arch Microbiol 165:359–369
Wright GD (2007) The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol 5:175–186
D’Costa VM, Griffiths E, Wright GD (2007) Expanding the soil antibiotic resistome: exploring environmental diversity. Curr Opin Microbiol 10:481–489
Davies J, Wright GD (1997) Bacterial resistance to aminoglycoside antibiotics. Trends Microbiol 5:234–240
Liou GF, Yoshizawa S, Courvalin P, Galimand M (2006) Aminoglycoside resistance by ArmA-mediated ribosomal 16S methylation in human bacterial pathogens. J Mol Biol 359:358–364
Marshall CG, Lessard IA, Park I, Wright GD (1998) Glycopeptide antibiotic resistance genes in glycopeptide-producing organisms. Antimicrob Agents Chemother 42:2215–2220
Canton R, Coque TM (2006) The CTX-M beta-lactamase pandemic. Curr Opin Microbiol 9:466–475
Roberts MC (1996) Tetracycline resistance determinants: mechanisms of action, regulation of expression, genetic mobility, and distribution. FEMS Microbiol Rev 19:1–24
Chopra I, Roberts M (2001) Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol Mol Biol Rev 65:232–260 (second page, table of contents)
Roberts MC (2005) Update on acquired tetracycline resistance genes. FEMS Microbiol Lett 245:195–203
Guillaume G, Ledent V, Moens W, Collard JM (2004) Phylogeny of efflux-mediated tetracycline resistance genes and related proteins revisited. Microb Drug Resist 10:11–26
Agerso Y, Guardabassi L (2005) Identification of Tet 39, a novel class of tetracycline resistance determinant in Acinetobacter spp. of environmental and clinical origin. J Antimicrob Chemother 55:566–569
Thompson SA, Maani EV, Lindell AH, King CJ, McArthur JV (2007) Novel tetracycline resistance determinant isolated from an environmental strain of Serratia marcescens. Appl Environ Microbiol 73:2199–2206
Brown MG, Mitchell EH, Balkwill DL (2008) Tet 42, a novel tetracycline resistance determinant isolated from deep terrestrial subsurface bacteria. Antimicrob Agents Chemother 52:4518–4521
Kazimierczak KA, Rincon MT, Patterson AJ, Martin JC, Young P, Flint HJ, Scott KP (2008) A new tetracycline efflux gene, tet(40), is located in tandem with tet(O/32/O) in a human gut firmicute bacterium and in metagenomic library clones. Antimicrob Agents Chemother 52:4001–4009
Truong-Bolduc QC, Dunman PM, Strahilevitz J, Projan SJ, Hooper DC (2005) MgrA is a multiple regulator of two new efflux pumps in Staphylococcus aureus. J Bacteriol 187:2395–2405
Paulsen IT, Brown MH, Skurray RA (1996) Proton-dependent multidrug efflux systems. Microbiol Rev 60:575–608
Tamura N, Konishi S, Yamaguchi A (2003) Mechanisms of drug/H+ antiport: complete cysteine-scanning mutagenesis and the protein engineering approach. Curr Opin Chem Biol 7:570–579
Yamaguchi A, Someya Y, Sawai T (1992) Metal-tetracycline/H+ antiporter of Escherichia coli encoded by transposon Tn10. The role of a conserved sequence motif, GXXXXRXGRR, in a putative cytoplasmic loop between helices 2 and 3. J Biol Chem 267:19155–19162
Yamaguchi A, Akasaka T, Ono N, Someya Y, Nakatani M, Sawai T (1992) Metal-tetracycline/H+ antiporter of Escherichia coli encoded by transposon Tn10. Roles of the aspartyl residues located in the putative transmembrane helices. J Biol Chem 267:7490–7498
Yamaguchi A, Samejima T, Kimura T, Sawai T (1996) His257 is a uniquely important histidine residue for tetracycline/H+ antiport function but not mandatory for full activity of the transposon Tn10-encoded metal-tetracycline/H+ antiporter. Biochemistry 35:4359–4364
Hillen W, Berens C (1994) Mechanisms underlying expression of Tn10 encoded tetracycline resistance. Annu Rev Microbiol 48:345–369
Saenger W, Orth P, Kisker C, Hillen W, Hinrichs W (2000) The tetracycline repressor-A paradigm for a biological switch. Angew Chem Int Ed Engl 39:2042–2052
Pang Y, Brown BA, Steingrube VA, Wallace RJ Jr, Roberts MC (1994) Tetracycline resistance determinants in mycobacterium and streptomyces species. Antimicrob Agents Chemother 38:1408–1412
Taylor DE, Chau A (1996) Tetracycline resistance mediated by ribosomal protection. Antimicrob Agents Chemother 40:1–5
Kobayashi T, Nonaka L, Maruyama F, Suzuki S (2007) Molecular evidence for the ancient origin of the ribosomal protection protein that mediates tetracycline resistance in bacteria. J Mol Evol 65:228–235
Burdett V (1996) Tet(M)-promoted release of tetracycline from ribosomes is GTP dependent. J Bacteriol 178:3246–3251
Connell SR, Tracz DM, Nierhaus KH, Taylor DE (2003) Ribosomal protection proteins and their mechanism of tetracycline resistance. Antimicrob Agents Chemother 47:3675–3681
Rasmussen BA, Gluzman Y, Tally FP (1994) Inhibition of protein synthesis occurring on tetracycline-resistant, TetM-protected ribosomes by a novel class of tetracyclines, the glycylcyclines. Antimicrob Agents Chemother 38:1658–1660
Melville CM, Scott KP, Mercer DK, Flint HJ (2001) Novel tetracycline resistance gene, tet(32), in the Clostridium-related human colonic anaerobe K10 and its transmission in vitro to the rumen anaerobe Butyrivibrio fibrisolvens. Antimicrob Agents Chemother 45:3246–3249
Patterson AJ, Rincon MT, Flint HJ, Scott KP (2007) Mosaic tetracycline resistance genes are widespread in human and animal fecal samples. Antimicrob Agents Chemother 51:1115–1118
Warburton P, Roberts AP, Allan E, Seville L, Lancaster H, Mullany P (2009) Characterization of tet(32) genes from the oral metagenome. Antimicrob Agents Chemother 53:273–276
Oggioni MR, Dowson CG, Smith JM, Provvedi R, Pozzi G (1996) The tetracycline resistance gene tet(M) exhibits mosaic structure. Plasmid 35:156–163
Stanton TB, McDowall JS, Rasmussen MA (2004) Diverse tetracycline resistance genotypes of Megasphaera elsdenii strains selectively cultured from swine feces. Appl Environ Microbiol 70:3754–3757
van Hoek AH, Mayrhofer S, Domig KJ, Florez AB, Ammor MS, Mayo B, Aarts HJ (2008) Mosaic tetracycline resistance genes and their flanking regions in Bifidobacterium thermophilum and Lactobacillus johnsonii. Antimicrob Agents Chemother 52:248–252
Speer BS, Salyers AA (1989) Novel aerobic tetracycline resistance gene that chemically modifies tetracycline. J Bacteriol 171:148–153
Yang W, Moore IF, Koteva KP, Bareich DC, Hughes DW, Wright GD (2004) TetX is a flavin-dependent monooxygenase conferring resistance to tetracycline antibiotics. J Biol Chem 279:52346–52352
Moore IF, Hughes DW, Wright GD (2005) Tigecycline is modified by the flavin-dependent monooxygenase TetX. Biochemistry 44:11829–11835
Ghosh S, Sadowsky MJ, Roberts MC, Gralnick JA, LaPara TM (2009) Sphingobacterium sp. strain PM2–P1-29 harbours a functional tet(X) gene encoding for the degradation of tetracycline. J Appl Microbiol 106:1336–1342
Nonaka L, Suzuki S (2002) New Mg2+-dependent oxytetracycline resistance determinant tet 34 in Vibrio isolates from marine fish intestinal contents. Antimicrob Agents Chemother 46:1550–1552
Diaz-Torres ML, McNab R, Spratt DA, Villedieu A, Hunt N, Wilson M, Mullany P (2003) Novel tetracycline resistance determinant from the oral metagenome. Antimicrob Agents Chemother 47:1430–1432
Petkovic H, Cullum J, Hranueli D, Hunter IS, Perić-Concha N, Pigac J, Thamchaipenet A, Vujaklija D, Long PF (2006) Genetics of Streptomyces rimosus, the oxytetracycline producer. Microbiol Mol Biol Rev 70:704–728
Ohnuki T, Katoh T, Imanaka T, Aiba S (1985) Molecular cloning of tetracycline resistance genes from Streptomyces rimosus in Streptomyces griseus and characterization of the cloned genes. J Bacteriol 161:1010–1016
Dittrich W, Schrempf H (1992) The unstable tetracycline resistance gene of Streptomyces lividans 1326 encodes a putative protein with similarities to translational elongation factors and Tet(M) and Tet(O) proteins. Antimicrob Agents Chemother 36:1119–1124
McMurry LM, Levy SB (1998) Revised sequence of OtrB (tet347) tetracycline efflux protein from Streptomyces rimosus. Antimicrob Agents Chemother 42:3050
Cohen SP, McMurry LM, Levy SB (1988) marA locus causes decreased expression of OmpF porin in multiple-antibiotic-resistant (Mar) mutants of Escherichia coli. J Bacteriol 170:5416–5422
Eady EA, Jones CE, Gardner KJ, Taylor JP, Cove JH, Cunliffe WJ (1993) Tetracycline-resistant propionibacteria from acne patients are cross-resistant to doxycycline, but sensitive to minocycline. Br J Dermatol 128:556–560
Kurokawa I, Nishijima S, Asada Y (1988) The antibiotic susceptibility of Propionibacterium acnes: a 15-year bacteriological study and retrospective evaluation. J Dermatol 15:149–154
Leyden JJ, McGinley KJ, Cavalieri S, Webster GF, Mills OH, Kligman AM (1983) Propionibacterium acnes resistance to antibiotics in acne patients. J Am Acad Dermatol 8:41–45
Ross JI, Eady EA, Cove JH, Cunliffe WJ (1998) 16S rRNA mutation associated with tetracycline resistance in a gram-positive bacterium. Antimicrob Agents Chemother 42:1702–1705
Gerrits MM, de Zoete MR, Arents NL, Kuipers EJ, Kusters JG (2002) 16S rRNA mutation-mediated tetracycline resistance in Helicobacter pylori. Antimicrob Agents Chemother 46:2996–3000
Trieber CA, Taylor DE (2002) Mutations in the 16S rRNA genes of Helicobacter pylori mediate resistance to tetracycline. J Bacteriol 184:2131–2140
Dailidiene D, Bertoli MT, Miciuleviciene J, Mukhopadhyay AK, Dailide G, Pascasio MA, Kupcinskas L, Berg DE (2002) Emergence of tetracycline resistance in Helicobacter pylori: multiple mutational changes in 16S ribosomal DNA and other genetic loci. Antimicrob Agents Chemother 46:3940–3946
Gerrits MM, Berning M, Van Vliet AH, Kuipers EJ, Kusters JG (2003) Effects of 16S rRNA gene mutations on tetracycline resistance in Helicobacter pylori. Antimicrob Agents Chemother 47:2984–2986
Levy SB, McMurry LM, Burdett V, Courvalin P, Hillen W, Roberts MC, Taylor DE (1989) Nomenclature for tetracycline resistance determinants. Antimicrob Agents Chemother 33:1373–1374
Levy SB, McMurry LM, Roberts MC (2005) Tet protein hybrids. Antimicrob Agents Chemother 49:3099
Liu B, Pop M (2009) ARDB—antibiotic resistance genes database. Nucleic Acids Res 37:D443–D447
D’Costa VM, McGrann KM, Hughes DW, Wright GD (2006) Sampling the antibiotic resistome. Science 311:374–377
Baltz RH (2007) Antimicrobials from actinomycetes: back to the future. Microbe 2:125–131
Hinrichs W, Kisker C, Duvel M, Muller A, Tovar K, Hillen W, Saenger W (1994) Structure of the Tet repressor–tetracycline complex and regulation of antibiotic resistance. Science 264:418–420
Chopra I (1994) Tetracycline analogs whose primary target is not the bacterial ribosome. Antimicrob Agents Chemother 38:637–640
Sun C, Wang Q, Brubaker JD, Wright PM, Lerner CD, Noson K, Carest M, Siegel DR, Wang YM, Myers AG (2008) A robust platform for the synthesis of new tetracycline antibiotics. J Am Chem Soc 130:17913–17927
Levy SB, McMurry LM, Brdett V, Courvalin P, Hillen W, Roberts MC, Taylor DE (1999) Nomenclature for new tetracycline resistance determinants. Antimicrob Agents Chemother 43:1523–1524
Acknowledgments
This work is supported by the Natural Sciences and Engineering Research Council of Canada and the Canadian Institutes of Health Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Thaker, M., Spanogiannopoulos, P. & Wright, G.D. The tetracycline resistome. Cell. Mol. Life Sci. 67, 419–431 (2010). https://doi.org/10.1007/s00018-009-0172-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-009-0172-6