
Abstract
Occludin is a self-associating transmembrane tight junction protein affected in oxidative stress. However, its function is unknown. The cytosolic C-terminal tail contains a coiled coil-domain forming dimers contributing to the self-association. Studying the hypothesis that the self-association is redox-sensitive, we found that the dimerization of the domain depended on the sulfhydryl concentration of the environment in low-millimolar range. Under physiological conditions, monomers and dimers were detected. Masking the sulfhydryl residues in the domain prevented the dimerization but affected neither its helical structure nor cylindric shape. Incubation of cell extracts containing full-length occludin with sulfhydryl reagents prevented the dimerization; a cysteine/alanine exchange mutant also did not show dimer formation. This demonstrates, for the first time, that disulfide bridge formation of the domain is involved in the occludin dimerization. It is concluded that the redox-dependent dimerization of occludin may play a regulatory role in the tight junction assembly under physiological and pathological conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Occludin, tricellulin, claudins, and junctional adhesion molecules are transmembrane proteins of the tight junctions (TJ). Together with membrane-associated zonula occludens proteins (ZO), the multiprotein assembly regulates the paracellular permeability of endothelia and epithelia. Little is known about the oligomerization mechanism of TJ proteins. Defining the organization and control of the TJ, therefore, is a prerequisite for a better understanding of the TJ function at the molecular level.
The cytosolic C-terminal tail of occludin contains a coiled coil(CC)-domain [1], which is able to homodimerize [2] and to associate with ZO-1 [3]. Disruption of the barrier properties has been achieved through the transfection of occludin mutants without the intracellular part [4], supporting the observation that the CC-domain is functionally relevant for the TJ barrier [5]. ZO-1 may regulate the function of occludin, since occludin remains diffusely stained at the surface of fibroblasts lacking ZO-1 [6]. A regulatory role of occludin has been suggested as phosphorylated occludin is preferentially enriched at the TJ membrane [7]. Under pathological conditions, such as oxidative stress, occludin appears as an early and specific target for reactive species [8]. It is concentrated at cell contacts in fibroblasts as dots through homophilic oligomerization; cotransfection with claudins results in proper strand formation [9]. However, the regulation of occludin’s oligomerization is unknown.
Our aim was to test the hypothesis whether the self-association of the domain, involved in the oligomerization of occludin [2], is redox-sensitive, as disulfide bonds are assumed to contribute to the oligomerization [10]. Using a purified protein segment including the CC-domain and two cysteines, we are able to demonstrate that the dimerization is influenced by the ambient thiol concentration as well as by a cysteine residue in the segment.
Materials and methods
Cloning of occludin domain, protein preparation
Mouse occludin406–521 cDNA was obtained by RT-PCR, confirmed by sequencing [11], and recloned into pGEX-4T1 at the EcoRI and SalI restriction sites (GST; pGEX-4T1; Amersham) [3]. The vector was transfected into E. coli BL21(DE3). N-terminal GST-occludin406–521 was extracted (freezing/thawing, 6 × 15 s sonication) in TBS (20 mM Tris–HCl, pH 7.4, 150 mM NaCl) and protease inhibitor cocktail (Sigma) on ice. Nucleic acids were digested (25 U benzonase). The extract was filtered (0.2 μm), bound on glutathione-agarose affinity column (Sigma-Aldrich), and washed. Occludin406–521 was cleaved from GST (100 U thrombin; 1 ml TBS, 20 h) and eluted with TBS, resulting in occludin406–521 with additional N-terminal amino acids from the cloning site (GSPEFGS). Protein was estimated at 280 nm (DU 640; Beckman; ε = 18,910 M−1cm−1, www.expasy.ch). No mercaptoethanol was used in the buffer during the preparation of samples.
Masking cysteine residues, incubation with thiols
TBS containing 100 mM dithiothreitol (DTT; Roth) was added to purified occludin406–521 (Occl-Cys). The solution was covered with N2. Then, 2 μl 4-vinylpyridine/50 μg protein were added for 30 min at 37°C under N2 (Occl-Cys-R) [12]. The separation of the product was performed via HiPrep 26/10 desalting column (Amersham) by HPLC (LC-10AS pump, SPD-10A UV detector; Shimadzu) using TBS. For PAGE, the constructs were incubated with 0.2% SDS (w/v), 10% glycerine (v/v), 33 mM Tris/HCl pH 6.8, bromophenol blue, and 0.5–100 mM reduced glutathione (GSH; Sigma) or DTT for 5 min. Before size-exclusion chromatography, the protein was identified mass spectrometrically.
Mass spectrometry
Matrix-assisted laser desorption/ionization MS (MALDI-MS) was performed on MALDI-TOF (Perspective Biosystems) [2]. Then 1 μl freshly prepared eluate from affinity chromatography see above) was mixed with 1 μl of sinapic acid solution (10 mg matrix dissolved in 1 ml 0.3% trifluoroacetic acid in acetonitrile-water; 1:1, v/v) and measured in the linear mode at an acceleration voltage of 25 kV. Finally, 1 μl of the resulting mixture was applied to the sample plate. Samples were air-dried.
Gel electrophoresis, size-exclusion chromatography (SEC)
For SDS-PAGE (13% PAG) and immunoblotting [7], proteins were transferred to a polyvinylidendifluoride membrane (Amersham), blocked in 3% non-fat dried milk, and incubated with polyclonal rabbit anti-occludin antibodies (against 150 C-terminal amino acids; Zymed, #71-1500) and HRP (horse radish peroxidase)-conjugated goat anti-rabbit IgG (Zymed/Invitrogen). HRP was visualized by reagent I and II (1:1; Amersham) and chemiluminescence (Lumi-Imager F1; Boehringer). SEC was performed using HPLC (LC-10AS pump, degasser DGU-14A, SPD-10A UV detector, sampler FRC-10A; Shimadzu; and Superdex 75 10/300 GL; Amersham) with TBS or TBS with 20 mM DTT at 22°C. For calibration, Blue Dextran 2000, 2,000 kDa; bovine serum albumin, 67 kDa; ovalbumin, 43 kDa; chymotrypsinogen A, 25 kDa, and ribonuclease A, 13,7 kDa (LMW kit/Amersham) were used as globular markers. Proteins were centrifuged (20,000g, 4°C, 10 min) and buffers filtered (0.45 μm) before use; injection volume was 100 μl, flow-rate 0.5 ml/min. Protein absorption was monitored at 214 and 280 nm.
Light scattering, circular dichroism (CD)
Static and dynamic light scattering were performed simultaneously (scattering angle, 90°; laboratory-built apparatus; diode-pumped, continuous wave laser Millennia IIs; Spectra-Physics; high quantum yield avalanche photodiode) [2]. Translational diffusion coefficients were obtained using program CONTIN and converted into Stokes radii (R S) via Stokes–Einstein equation. Molar masses were estimated from the relative scattering intensities (toluene reference), applying a refractive index increment of 0.19 ml/g. Proteins were centrifuged (90 min, 50,000g, 22°C) before use. Using CD, ellipticity of the proteins was measured on J-720 spectropolarimeter (Jasco; 1 mm-quartz cell, 100 mM phosphate buffer pH 7.5, 150 mM NaF, 25°C) [11]. We calculated molar mean residue ellipticitiy by θmr = Ψ/cdn (Ψ = ellipticity, c = concentration, d = length, n = amino acids), content of helical secondary structure by [13].
Generation of fluorescent human occludin and of the C409A mutant
Human occludin (hOcc) cDNA was subcloned from pCMV-SPORT6 (clone IRAT p970A0847D6, RZPD—Deutsches Resourcenzentrum für Genomforschung into the expression vector pE-YFPC1 (Takara Bio Europe/Clontech) N-terminally encoding yellow fluorescent protein (YFP). hOcc cDNA was amplified by PCR using the primers 5′-ACT ATG TCG ACA TGT CAT CCA GGC CTC CTG-3′ and 5′-CTA TAC CCG GGC TGT TTT CTG TCT ATC ATA GTC TCC-3′ flanked by XmaI and SalI recognition sites. After amplification, pE-YFPC1 and hOcc were digested with XmaI and SalI, electrophoretically purified in agarose gel and ligated using T4 ligase (Fermentas). The resulting fusion product YFP-hOcc was amplified in E. coli DH5α and purified. The cysteine 409 in YFP-hOcc was then substituted by alanine using site directed mutagenesis. YFP-hOccl was subjected to multistep PCR amplification using two simultaneous runs with forward 5′-TGG CGG CGA GTC CGC TGA TGA GCT G-3′ or reverse primer 5′-CTC CAG CTC ATC AGC GGA CTC GCC G-3′. Both reaction products were mixed followed by third PCR. Immediately afterwards, a DpnI digestion (Fermentas) was carried out at 37°C for 3 h followed by heat inactivation of the enzyme. Thereafter, the DNA was kept at 4°C for 2 h and electroporated into E. coli. Colonies were grown and propagated, and the ligated plasmid was purified (Plasmid Purification Kit; Quiagen) and verified by automated sequencing.
Electrophoretic separation of full-length occludin
Human embryonic kidney (HEK) 293 cells [2] were transfected with YFP-hOccl using Lipofectamine 2000 (Invitrogen) following manufacturer’s recommendations. After 48 h, the expression was assessed by laser scanning confocal microscopy (LSM) and the cells were scraped into ice-cold PBS, quickly frozen in liquid N2 and stored at −86°C. The samples were thawed on ice, centrifuged at 15,000g, 4°C for 5 min and the supernatant discarded; the pellet was resuspended with occludin extraction buffer (1% Triton X-100, 25 mM HEPES/NaOH, pH 7.4, 150 mM NaCl, 4 mM EDTA; protease inhibitor cocktail, Sigma) and sonicated 2 × 10 mW for 20 s followed by a 10 min on-ice incubation and 20,000g centrifugation for 15 min at 4°C. The resulting Triton X-100 resistant pellets were resuspended with 125 mM Tris/HCl, 5% glycerol (pH 7.5) without (20 min on ice) or with DTT (150 mM, 37°C for 20 min). Similarly, the Triton X-100 soluble fraction was treated. Afterwards, all samples were diluted with loading buffer (125 mM Tris/HCl, 20% glycerol, pH 6.8) with or without 2% SDS, for electrophoretic separation in continuous native and SDS (0.05%) containing polyacrylamide gels (6%). Electrophoresis was performed at 80 V and 4°C for 2 h using native (25 mM Tris/HCl, 192 mM glycine) or conventional (0.1% SDS, 25 mM Tris/HCl, 250 mM glycine) running buffer pH 6.8. The gels were scanned in a FLA-5000 fluoroimager (Fujifilm) using a 473-nm SHG laser line powered at 450 V and a Fuji-LPB Y510 filter. Digitized images were analyzed using software AIDA 3.52 (Raytest) and Image J (NIH).
Results
Figure 1 demonstrates that occludin406–521 forms redox-sensitive dimers. The apparent molecular weight was ~40 kDa. Monomers appeared at ~20 and ~15 kDa (Fig. 1a). The 40 kDa-dimer disappeared at >20 mM GSH (Fig. 1b, lanes 5, 6) and ≥10 mM DTT (Fig. 1c, lanes 6, 7), both in a concentration-dependent manner. Due to the thiol-dependent dissociation, the 20 kDa-monomer increased from 1 to 10 mM DTT (Fig. 1c, lanes 5, 6). In contrast, 0.2–2.0% SDS (Fig. 1b, lanes 3, 4, and c, lanes 2, 3) and boiling at 95°C for 5 min (Fig. 1c, lanes 1, 4) did not affect the dimerization. In addition to the strong bands at 40 and 20 kDa after Coomassie staining, weak bands were traced at 15 and 30 kDa (Fig. 1b, c). The faint dimer band (30 kDa) disappeared at ≥1 mM DTT (Fig. 1c, lanes 5–7) and ≥5 mM GSH (Fig. 1b, lanes 5–8). Inversely, the faint monomer band (15 kDa) decreased within 1–10 mM GSH or DTT (Fig. 1b, lanes 7–9, and c, lanes 5–6). The compact 15 kDa-monomer probably contains an intramolecular disulfide bond (C408–C499) and the 30 kDa-dimer is probably a parallel arrangement resulting in two disulfide bonds (C408–C408′, C499–C499′). Both are considered misfolded because C499 is not accessible as it is embedded within the interaction between two intramolecular helices (Fig. 6). On the other hand, degradation products cannot be ruled out.

Redox-sensitive dimerization of murine occludin406–521 in SDS-PAGE. a Immunoblotting of occludin showed monomers at 15 and 20 kDa, dimers at 30 and 40 kDa. b Reduced glutathione (GSH) diminished the dimer bands (lanes 5–10) concentration-dependently (Coomassie, 0.2% SDS). c Dithiothreitol (DTT) diminished the dimer bands (lanes 5–7) concentration-dependently (Coomassie, 2% SDS). *GST-mOccl406–521 in E. coli lysate, Mw Molecular weight, large arrowhead dimer, small arrowhead monomer (other bands are considered misfolded as described in “Results”)
In SEC of oclludin406–521, a dimer and monomer peak were observed and showed similar characteristics as reported recently [2]. The dimer peaked at an apparent molecular weight slightly below 43 kDa, and the monomer did so considerably above 25 kDa. Under oxidative, thiol-free conditions, similar peak areas were estimated for monomer and dimer (Fig. 2, black curve). Under reductive 20 mM DTT, less than 10% of the area belonged to the dimer (Fig. 2, dashed curve).

Redox-sensitive dimerization of murine occludin406–521 in SEC. Control Dimer and monomer peak were detected (black line) as known [2]. Dithiothreitol (DTT) diminished the dimer and increased the monomer peak (dashed line). Arrows Molecular weight marker proteins
To verify whether the dimer formation is due to the sulfhydryl groups of the construct, Occl-Cys, with free sulfhydryl groups, was compared with Occl-Cys-R where both sulfhydryl groups were simply masked by 4-vinylpyridine. Occl-Cys and Occl-Cys-R were verified mass spectrometrically (14,622 g/mol; 14,833 g/mol). The difference between both numbers meets the molecular mass of 4-vinylpyridine documenting that both cysteines were masked. In SEC, the dimer and monomer areas of Occl-Cys were of similar extent. For Occl-Cys-R, however, ~95% of the peak area was monomeric and 5% dimeric. In SDS–PAGE (inset of Fig. 3), dimer-fractions 1 showed a strong 40 kDa-band and no monomer bands. In contrast, the monomer-fractions 3 exhibited no dimer, but a 20-kDa band. Bands of 15 and 30 kDa are considered misfolded as smaller molecular weights would correspond to intramolecular (C408–C499) and intermolecular (C408–C408′, C499–C499′) disulfide formation, respectively; however, C499 is considered inaccessible (Fig. 6).

Masking sulfhydryl residues in murine occludin406–521 (Occl-Cys-R) prevented its dimerization shown with free Occl-Cys, using size-exclusion chromatography. Arrows, molecular weight marker proteins. Inset: In SDS-PAGE (0.2% SDS, silver staining), Occl-Cys only exhibited dimers in Fr.1 and only monomers in Fr.3. Occl-Cys-R was mainly found as monomer in Fr.3. Mw Molecular weight, large arrowhead dimer, small arrowhead monomer (as mentioned in Fig. 1)
Blockage of the cysteines did not affect the structure of the CC-domain as CD spectroscopy almost revealed the same spectra for Occl-Cys and Occl-Cys-R (Fig. 4). Both displayed a double minimum (209, 222 nm), which is typical for α-helical secondary-structure elements in the domain [11]. The content of helical structure was 80% in Occl-Cys and 77% in Occl-Cys-R.

Circular dichroism of murine occludin406–521 in its sulfhydryl (Occl-Cys) and sulfhydryl-masked form (Occl-Cys-R) showed the same helical properties
Table 1 shows apparent molecular masses and Stokes radii of the constructs. The experimental molecular mass of 14,800 ± 1,500 g/mol for Occl-Cys-R coincides with the calculated mass 14,838 g/mol. This provides strong evidence for the monomeric state of Occl-Cys-R. The apparent molecular mass of 23,100 ± 2,500 g/mol for Occl-Cys was between the expected values for monomers and dimers which were demonstrated in solution by means of SEC (Figs. 2, 3). Taking into account that static light scattering measures a weight-averaged molecular mass, we have calculated a ratio of the weight concentrations for dimers to monomers of 1.44. This is in agreement to the ratio1.3 which was obtained from SEC (peak size, Fig. 3).
To confirm that the CC-domain also dimerizes within the full-length protein and in its natural environment, HEK cells transfected with YFP-hOccl were analyzed by native (Fig. 5b) and SDS–PAGE (Fig. 5a) using the YFP fluorescence. In cell extracts of non-transfected cells, no bands were detectable (Fig. 5, lanes 3, 7). High and low molecular weight bands (asterisks) were visible in cells transfected with YFP-hOccl (lanes 1, 5). The higher band (**) can be prevented by the addition of DTT (Fig. 5, lanes 2, 6). To prove that, within the CC-domain, the “freely accessible” Cys409 (for explanation, see Fig. 6) is responsible for the loss of the high molecular weight band (**), caused by the reducing thiol, we studied YFP-hOccC409A. In cells transfected with this mutant lacking C409, the higher molecular band was completely absent; only the low molecular weight band (*) was exhibited (Fig. 5, lanes 4, 8). Since the molecular weight cannot be determined in native gels we performed the same experiments in SDS–PAGE and identified a dominating monomeric (*) and a dimeric (**) band (Fig 5a, lane 1) at 90 and 180 kDa, respectively. These bands were not registered when non-transfected cells were applied (Fig. 5a, lane 3). Under DTT, the dimer was suppressed (Fig. 5a, lane 2) in the gel showing the monomer fluorescence only (Fig. 5a, lane 2). The cysteine mutant were not able to form the dimer, only allowing the detection of the monomer (Fig. 5a, lane 4).

The dimerization of full-length human occludin is thiol sensitive and can be prevented by the replacement of its cysteine 409 by alanine in the cytosolic C-terminal coiled coil-domain. To identify dimeric and monomeric forms, lysates from HEK 293 cells transfected with YFP (yellow fluorescent protein) tagged human occludin were treated with 1% triton X-100 and the insoluble fraction was analysed by a 2% SDS-PAGE and b native PAGE, followed by YFP laser scanning. The samples were analysed in presence (+) or absence (−) of 150 mM dithiothreitiol (DTT). *, **bands representing monomer and dimer, respectively; NT non-transfected cells, M cells transfected with the mutant human YFP-occludinCys409Ala

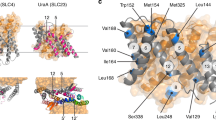
Structural scheme of murine occludin406−521 adapted from the x-ray structure of human occludin416−522 (http://molvis.sdsc.edu/fgij/fg.htm?mol=1wpa). C408 is freely accessible, but in contrast, C499 is shielded in the interface of two helices. In human occludin, the positions of the cysteines are 409 and 500
Discussion
We demonstrate that the homodimerization of the cytosolic C-terminal sequence of mouse occludin406–521 (mOccl406–521) is redox-sensitive due to a highly conserved cysteine. This matches earlier studies showing fluorescence resonance energy transfer between occludin molecules with intracellular fluorescence tags. The fluorescence was enriched in the contacts between two transfected cells [2], i.e., in the TJ area. These data are consistent with the view that the cytosolic part is involved in occludin’s self-association within the TJ. On the other hand, the crystal structure of the homologous, but shorter, human occludin416–522 (hOccl416–522) reveals a monomer [14]. This is explained by the fact that hOccl416–522 contains only one non-accessible cysteine (C500). In contrast, mOccl406–521 encloses two cysteines (C408, C499). Consequently, the N-terminal C408 is assumed as the binding area. This is supported by the observation that the N-terminal sequence (hOccl416–424) of hOccl416–522 is unstructured [14]. Thus, the assumption is supported that C408 in mOccl406–521 is flexible and accessible to form disulfide bonds (Fig. 6).
Immunoblotting of the sequence studied (molecular mass 14,628 g/mol) shows dimers and monomers simultaneously. The dimer, apparent relative molecular weight 40 kDa, is assumed as an antiparallel intermolecular interaction via disulfide bond formation (C408–C408′). This arrangement results in an elongated size of the dimer as determined by light scattering and, hence, in molecular weight shift as seen in SEC. The reducing agents GSH and DTT, known to abolish disulfide formation, lower the 40 kDa-dimer concentration-dependently (1–10 mM); conversely, the amount of the 20-kDa monomer increases. Cytosolic thiol, comprising primarily GSH, has the same concentration range [15]. Although the cytosol has a reducing environment [16] that should prohibit the generation of stable disulfide bridges numerous cytosolic proteins undergo disulfide bond formation [17]. Thus, the intracellular GSH level may regulate occludin’s dimerization and, as the occludin-sequence studied interacts with other proteins of the TJ [11], also the TJ assembly. As monomer and dimer coexist in similar amounts, the oligomerization should be sensitive to redox-changes. This matches the findings that occludin is particularly affected by oxidative stress [8].
Masking of the thiol groups prevents the dimerization, but does not influence the helical structure as measured by CD. The failure of dimer formation due to a blockage of the sulfhydryl groups confirms the conclusion that the dimerization occurs via HS-residues. It also means that the oxidative status controls the dimerization and not the secondary structure. Thus, changes in the dimerization of mOccl406–521 which is proposed to form a CC-domain can be of functional relevance. The structure of the CC-domain shows surface areas with local charge excess. As charged patches contribute to the association of the CC-domain with other TJ-proteins such as ZO-1 [3, 14], one can speculate that the disulfide-dimerization may modify the occludin-ZO-1 interaction and, in turn, the TJ-structure.
The detected Stokes radius for Occl-Cys-R (blocked cysteine residues) of R S = 2.49 nm is quite large for an apparent mass of 14,800 g/mol. This becomes evident if one calculates R S for a compactly folded globular protein [18], yielding 1.92 nm. The experimental R S, which is 29% larger, is consistent with the elongated structure that can be expected from the X-ray structure of the CC-domain [14]. This domain is composed of 3 helices including the C-terminal amino acid residues of occludin up to its C-terminus, while the N-terminal residues of the CC-domain are disordered. Based on the light scattering experiments, the structure is approximated by a cylinder (diameter 1.7 nm, axis length 7 nm).
To compute R S for the apparent molecular mass M app. = 23,100 g/mol, obtained for a monomer/dimer mixture of Occl-Cys, we calculate 2.27 nm. The corresponding experimental R S is by 28% larger. This tells us that the overall compactness of the dimer is similar or, at least, only slightly stronger than that of the monomer. We therefore expect a dimer consisting of two compact CC-domains joint by a flexible (disulfide bond) linker. It is interesting to note that we obtain a molecular mass of 29,700 g/mol for Occl-Cys-R if we used the experimentally observed R S of 2.49 nm and the inverse relationship M = 2505. R 2.71S [18]. This value of 29,700 g/mol explains the large apparent molecular weights determined by SEC primarily measuring the hydrodynamic volume of a particle.
The redox-sensitive dimerization of the CC-domain can also be demonstrated with the full-length molecule. Native and SDS-PAGE of cell extracts containing YFP-occludin show dimers and monomers. The dimeric bands are not observed when thiols have been added to the samples. This strongly underlines that occludin dimerization depends on intermolecular disulfide bridge formation. Moreover, the full-length occludin does not dimerize if the N-terminal cysteine of the CC-domain, assumed to be freely accessible (Fig. 6), has been substituted by alanine. This means that C409 of human occludin molecules (corresponding to murine C408) are directly involved in the dimerization of occludin in the cell and form intermolecular disulfide bonds. Our results are in agreement with those of others who suggested that occludin oligomerization may be mediated via disulfide bridges [10]. Others also found that oxidative stress, such as inflammation, reduces occludin oligomerization [19]. In addition, our study may explain the failure of oligomer detection in western blots [7] as the antibodies commercially available identify the cytosolic C-terminal amino acid sequence including the CC-domain. In the dimer, the sequence is partially covered and hence cannot be recognized by the antibody.
Occludin and GSH are highly sensitive to oxidative stress. In hypoxia, GSH is reduced [20], and cerebral endothelial cells down-regulate occludin 20]. The degradation of occludin is intensified in microvessel isolations after ischemia [8]. Reactive oxygen species diminish occludin in pigment epithelia TJ [21]. This would mean that attenuation of cytosolic thiol and, in turn, promotion of occludin dimers may disturb the integrity and function of the TJ as found after hypoxic injury [22]. This conclusion agrees with the notion that occludin is a specific target in the oxidative impairment of the TJ [8] and that disulfide bonds are involved in its oligomerization [10]. Taken together, one can assume that the redox-sensitivity of the CC-domain of occludin may play a role in the dysregulation of endothelial and epithelial barriers upon oxidative load.
Disulfide bridge formation in intracellular proteins often perturbs their function. Several enzymes are inactivated by oxidation of sulfhydryl residues in the active center [23]. The generation of disulfide bonds of redox-sensitive proteins can control multiple processes [17]. Our report demonstrates for the first time that occludin contains an intracellular redox-sensitive domain. Thus, occludin can be considered as a TJ-protein of which the dimerization of its cytosolic CC-domain and, hence, of the whole protein can be influenced by the redox-status.
References
Nusrat A, Chen JA, Foley CS, Liand TW, Tom J, Cromwell M, Quan C, Mrsny RJ (2000) The coiled-coil domain of occludin can act to organize structural and functional elements of the epithelial tight junction. J Biol Chem 275:29816–29822
Blasig IE, Winkler L, Lassowski B, Mueller SL, Zuleger N, Krause E, Krause G, Gast K, Kolbe M, Piontek J (2006) On the self-association potential of transmembrane tight junction proteins. Cell Mol Life Sci 63:505–514
Muller SL, Portwich M, Schmidt A, Utepbergenov DI, Huber O, Blasig IE, Krause G (2005) The tight junction protein occludin and the adherens junction protein alpha-catenin share a common interaction mechanism with ZO-1. J Biol Chem 280:3747–3756
Balda MS, Whitney JA, Flores C, Gonzalez S, Cereijido M, Matter K (1996) Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J Cell Biol 134:1031–1049
Chen YH, Merzdorf C, Paul DL, Goodenough DA (1997) C-terminus of occludin is required for tight junction barrier function in early Xenopus embryos. J Cell Biol 138:891–899
Van Itallie CM, Anderson JM (1997) Occludin confers adhesiveness when expressed in fibroblasts. J Cell Sci 110:1113–1121
Andreeva AY, Krause E, Muller EC, Blasig IE, Utepbergenov DI (2001) PKC regulates the phosphorylation and cellular localization of occludin. J Biol Chem 276:38480–38486
Maier CM, Hsieh L, Crandall T, Narasimhan P, Chan PH (2006) A new approach for the investigation of reperfusion-related brain injury. Biochem Soc Trans 34:1366–1369
Furuse M, Sasaki H, Fujimoto K, Tsukita S (1998) A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Ceel Biol 143:391–401
McCaffrey G, Staatz WD, Quigley CA, Nametz N, Seelbach MJ, Campos CR, Brooks TA, Egleton RD, Davis TP (2007) Tight junctions contain oligomeric protein assembly critical for maintaining blood–brain barrier integrity in vivo. J Neurochem 103:2540–2555
Schmidt A, Utepbergenov DI, Mueller SL, Beyermann M, Schneider-Mergener J, Krause G, Blasig IE (2004) Occludin binds to the SH3-hinge-GuK unit of ZO-1: potential mechanism of tight junction regulation. Cell Mol Life Sci 61:1354–1365
Andreu D, Albericio F, Solac NA, Munson MC, Ferrer M, Barany G (2002) Formation of disulfide bonds in synthetic peptides and proteins. In: Walker JM (ed) Protein protocols on CD-ROM v. 2.0, Sect. 17.7, Humana Press, Totowa
Chen Y-H, Yang JT, Martinez HM (1972) Determination of secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochem 11:4120–4131
Li Y, Fanning AS, Anderson JM, Lavie A (2005) Structure of the conserved C-terminal domain of occludin: identification of the ZO-1 binding surface. J Mol Biol 352:151–164
Reed MC, Thomas RL, Pavisic J, James SJ, Ulrich CM, Nijhout HF (2008) A mathematical model of glutathione metabolism. Theor Biol Med Model 5:8–24
Hwang C, Sinskey AJ, Lodish HF (1992) Oxidized redox-state of glutathione in the endoplasmic reticulum. Science 257:1496–1502
Cumming RC, Andon NL, Haynes PA, Park M, Fischer WH, Schubert D (2004) Protein disulfide bond formation in the cytoplasm during oxidative stress. J Biol Chem 279:21749–21758
Gast K, Modler AJ (2005) Studying protein folding and aggregation by laser light scattering. In: Buchner J, Kiefhaber, T (eds) Protein folding handbook, Part I, pp 673–709. Wiley–VCH, Weinheim
McCaffrey G, Seelbach MJ, Staatz WD, Nametz N, Quigley C, Campos CR, Brooks TA, Davis TP (2008) Occludin oligomeric assembly at tight junctions of the blood–brain barrier is disrupted by peripheral inflammatory hyperalgesia. J Neurochem 106:2395–2409
Guo S, Wharton W, Moseley P, Shi H (2007) HSP70 regulates cellular redox status by modulating glutathione-related enzyme activities. Cell Stress Chaperon 12:245–254
Bailey TA, Kanuga N, Romero IA, Greenwood J, Luthert PJ, Cheetham ME (2004) Oxidative stress affects the junctional integrity of retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 45:675–684
Krizbai IA, Bauer H, Bresgen N, Eckl PM, Farkas A, Szatmári E, Traweger A, Wejksza K, Bauer HC (2005) Effect of oxidative stress on the junctional proteins of cultured cerebral endothelial cells. Cell Mol Neurobiol 25:129–139
Rietsch A, Beckwith J (1998) The genetics of disulfide bond metabolism. Annu Rev Genet 32:163–184
Acknowledgment
Supported by the Deutsche Forschungsgemeinschaft DFG BL308/6-4.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Walter, J.K., Castro, V., Voss, M. et al. Redox-sensitivity of the dimerization of occludin. Cell. Mol. Life Sci. 66, 3655–3662 (2009). https://doi.org/10.1007/s00018-009-0150-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-009-0150-z