
Abstract
Hypoxia refers to environmental or clinical settings that potentially threaten tissue oxygen homeostasis. One unique aspect of skeletal muscle is that, in addition to hypoxia, oxygen balance in this tissue may be further compromised when exercise is superimposed on hypoxia. This review focuses on the cellular and molecular responses of human skeletal muscle to acute and chronic hypoxia, with emphasis on physical exercise and training. Based on published work, it is suggested that hypoxia does not appear to promote angiogenesis or to greatly alter oxidative enzymes in skeletal muscle at rest. Although the HIF-1 pathway in skeletal muscle is still poorly documented, emerging evidence suggests that muscle HIF-1 signaling is only activated to a minor degree by hypoxia. On the other hand, combining hypoxia with exercise appears to improve some aspects of muscle O2 transport and/or metabolism.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Exposure to high altitude refers to an environmental condition associated with whole body and tissue hypoxia, resulting from a drop in barometric pressure and hence a concomitant decrease in arterial oxygen availability. Such a condition is encountered by nearly 140 million people residing at high altitude worldwide [1] and also by numerous sea-level dwellers traveling or commuting to high altitude areas. Healthy humans studied at high altitude not only contribute to unravel the molecular and systemic mechanisms involved in O2 sensing and adaptive responses to the low oxygen environment, but also help in the understanding of various pathological situations associated with hypoxia. These pathologies include anemia and chronic obstructive pulmonary disease, as well as chronic heart failure. In addition, tumor growth and development can be associated with tissue hypoxia, since solid tumors become hypoxic as they grow larger [2]. Yet another situation that potentially leads to hypoxia is physical exercise. Even during normoxic conditions, oxygen availability can drop in the transition from rest to exercise, implying that exercising skeletal muscle must operate at a very low partial pressure of oxygen, estimated to be approximately ~3 mmHg [3].
Whatever the origin of the hypoxic stimulus may be (environmental or pathological), the ultimate consequence is an inadequate O2 delivery/availability at the tissue level, implying that tissue demand exceeds its O2 supply. All nucleated cells in the human body are able to sense O2 and to respond to O2 deficiency in order to maintain homeostasis. The main mediator of cellular hypoxia is the hypoxia inducible factor (HIF) pathway, discovered by the group of Semenza [4]. Like other tissues, resting skeletal muscle homeostasis is challenged during hypoxic exposure, either acutely or chronically. However, differently from other tissues, muscle function may be further compromised if exercise is superimposed during hypoxic exposure.
This review aims to characterize the particular response of human skeletal muscle tissue to hypoxia, both during acute and chronic exposure to hypoxia with emphasis on exercise. Special attention will be given to skeletal muscle gene expression and protein content, whereas the regulation of metabolism and oxygen delivery will not be discussed.
The HIF-1 pathway and its presence within human skeletal muscle
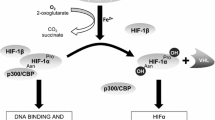
HIF-1 is a heterodimeric protein belonging to the basic helix-loop-helix-PAS family of transcription factors. This protein is composed of two subunits: HIF-1α, which has a short half-life (~5 min) and is highly sensitive to oxygen, and HIF-1β (or ARNT: aryl hydrocarbon nuclear receptor), which is constitutively expressed and remarkably insensitive to oxygen levels. Although the HIF family comprises two other members, HIF-2 and HIF-3, only HIF-1 is known to play a very general role in signaling hypoxia, whereas induction of HIF-2 with hypoxia is restricted to certain cells and HIF-3 function is still incompletely understood.
During normoxia, HIF-1α is degraded through hydroxylation. This process involves prolyl hydroxylases acting on HIF-1α oxygen-dependent degradation domain (ODDD). Once it has been hydroxylated, HIF-1α binds to von Hippel Lindau (VHL) protein, resulting in proteasomal degradation of HIF-1α.
Under hypoxic conditions, HIF-1α degradation is blocked because hydroxylation is inhibited. HIF-1α protein therefore accumulates, allowing for its binding to ARNT (HIF1-β) and hence the formation of a HIF-1 complex that can recognize hypoxia responsive elements (HRE) located in the nucleus of target genes (see also Table 1). The interaction between HIF-1 and HRE ultimately triggers the transcription of the target genes. To date, more than 100 HIF-1 downstream genes have been identified [5]. Those genes, which mainly enable the cells to cope with oxygen stress, are involved in erythropoiesis/iron metabolism, angiogenesis, glucose metabolism, and cell proliferation/survival and apoptosis, respectively. While this machinery helps normal cells to ameliorate local oxygen availability, it also contributes to the survival/development of tumor cells, thereby highlighting the dual role played by HIF-1.
The HIF-1 pathway can also be modulated under non-hypoxic conditions, either at the transcriptional or at the translational level [6, 7]. A variety of non-hypoxic environmental factors, including inflammation, reactive oxygen species, or nitric oxide can induce HIF-1α accumulation and target gene expression [5], which may further complicate our understanding of the cellular response to hypoxia. However, it is worth noting that the contribution of these regulators is only partially understood and that their influence on HIF-1 stabilization is far less than the dramatic induction caused by hypoxia.
HIF-1 in skeletal muscle tissue
Like in virtually all cells, skeletal muscle tissue is able to respond to hypoxia through the HIF-1 pathway. Animal studies have demonstrated that 1 h of systemic hypoxia is sufficient to increase HIF-1α protein expression in skeletal muscle [8]. In accordance with this, when leg oxygen delivery is impaired following blood flow restriction in humans, HIF-1α protein levels are known to be increased [9]. One particular aspect of skeletal muscle is that HIF-1α protein is highly expressed in this tissue even in normoxic conditions, suggesting that HIF-1 could have potential function in muscle homeostasis in normoxia [9, 10].
Of note is the fact that the HIF-1 response to hypoxia is time dependent. Previous data obtained in brain, liver, and kidney show that HIF-1 protein levels peak within the first hours of hypoxic exposure then progressively decline toward basal levels [8, 11]. The reason for HIF-1 to be down-modulated with “acclimatization” could be that local or systemic responses to hypoxia may attenuate the degree of cellular hypoxia in a tissue-dependent manner. That a similar response may occur in skeletal muscle tissue during sustained hypoxia could be hypothesized, but has not so far been verified. Our ongoing work, investigating the alterations of human skeletal muscle HIF-1α across time in hypoxia, provides the first indication that HIF-1α protein levels are barely modified in human skeletal muscle during environmental hypoxia (4,559 m of altitude), either acutely (0.5–8 h) or chronically (7–9 days) [12]. This preliminary finding questions the actual role of HIF-1α in vivo on the control of muscle adaptations during sustained hypoxia. In the same subjects, we concurrently observed permanently high levels of HIF-1α mRNAs at high altitude [13], suggesting that, in skeletal muscle, (1) factors other than hypoxia may be involved in the increase in HIF-1α mRNA, and (2) HIF-1α protein expression is probably not regulated at the transcriptional level. In summary, although HIF-1 has long been recognized as the master regulator of the cellular responses to hypoxia in numerous cell types, the few available data obtained from skeletal muscle tissue in vivo suggest that HIF-1 protein expression is only marginally altered during hypoxia. One reason could be the high pre-existing level of HIF-1 expression in skeletal muscle (aimed at maintaining homeostasis in a tissue operating at low physiological oxygen tension) that would leave only little room for additional changes when oxygenation is decreased.
Physical exercise per se may also challenge muscle oxygen homeostasis [3]. Accordingly, an alteration in HIF-1 signaling is expected. In support of this is the recent finding of increased HIF-1α protein expression in human skeletal muscle following an acute bout of normoxic exercise [9]. However, the exact pathway by which HIF-1 acts on muscle tissue in response to exercise-induced hypoxia and ultimately on exercise tolerance remains a complex issue. Recent studies using mice lacking skeletal muscle HIF-1α reveal that, surprisingly, endurance capacity is increased in these animals through an increase in oxidative metabolism [14]. However, these HIF-null mice are otherwise subject to increased muscle damage because of an impeded glycolytic metabolism, highlighting that HIF-1 is nevertheless essential in the metabolic control of muscle function.
If a single bout of exercise is associated with muscle hypoxia, it can be suggested that the repetition of exercise potentially challenges muscle oxygen homeostasis even more. The effect of chronic exercise on skeletal muscle function has been extensively investigated. Angiogenesis and alterations of metabolic control are well-known adaptations to endurance training [15]. It is thought that HIF-1 plays a central role in this setting, since HIF-1 modulates vascular endothelial growth factor, as well as several glycolytic enzymes. However, recent evidence shows that endurance training actually reduces muscle HIF-1α signaling [16]. Furthermore, HIF-1 null and wild-type muscles are shown to respond similarly to endurance training [17, 18], suggesting that the HIF-1 pathway is not essential for endurance training, while the latter could be useful during acute exercise.
Finally, how does skeletal muscle respond to hypoxia at the molecular level if exercise is superimposed and hence combines the effects of exercise and hypoxia on HIF-1 signaling. Although relevant because extremely challenging for local oxygen homeostasis, this issue is not well documented, making it difficult to understand the interaction between HIF-1 and its target genes during this condition. As mentioned above, combining acute hypoxia and acute exercise results in higher HIF-1 protein expression than with exercise alone [9]. If hypoxia is associated with endurance training, HIF-1α mRNA levels are found to be higher than after endurance training alone [19, 20]. Taken together, these data indicate that HIF-1 can be activated in skeletal muscle during exercise in hypoxia, highlighting its possible functional role on target genes involved in angiogenesis and/or energy metabolism. The early (acute) response of hypoxia on gene expression in human skeletal muscle is at present poorly described, and for this reason and the limited space available, this step will limited to that shown in Table 1.
Morphological and enzymatical adaptations in human skeletal muscle exposed to prolonged hypoxia
The first report describing the potential for human skeletal muscle to adapt to hypoxic exposure was published by Reynafarje in 1962 [21]. He showed that in miners permanently exposed to high altitude as compared to lowlanders cytocrome c reductase activity was increased by 78% and myoglobin content by 16%. Subsequently, it was for a long time believed that these adaptations had occurred to compensate for the lack of oxygen. Later, it has been argued that hypoxic stimulus is not sufficient in order to induce these responses alone, and that hypoxia has to be combined with either cold or exercise [22]. More recent data from our research group showed that capillary density was not increased in sea level residents exposed to 4,100 m in La Paz for 8 weeks [23], and also that 75 days exposure to 5,300 m in the base camp of Mount Everest did not cause the capillary number per muscle fiber to be altered (i.e., no neo-formation) in samples obtained from arm and leg skeletal muscle tissue [24]. In the later study, however, a decrease in fiber size resulted in more capillaries per area—a phenomenon often observed at this altitude or above [25, 26]. It has been speculated that the decrease in fiber size at altitude is due to a hypoxia-induced down-regulation of protein synthesis, because COPD patients have a reduced protein synthesis, and this has been hypothesized to be a direct effect of hypoxia (reviewed in [27]). While the muscle atrophy could be a consequence of hypoxic exposure per se, high altitude expeditions are also frequented by gastroenteritis, malnutrition, low physical activity levels, and low temperatures, and may very well be the reason for muscle atrophy [28].
Selected marker enzymes for oxidative metabolism (CS and HAD activity) were unchanged after 75 days at 5,300 m in the leg and arm muscles of active climbers and inactive base camp personnel [24]. Using the proteomic approach, it has recently been demonstrated that high altitude Sherpas have a slightly reduced HAD and lactate dehydrogenase protein content. Also, it seemed that, at least to some extent, high altitude residing Sherpas are protected from ROS-induced tissue damage and possess specific metabolic adaptations [29]. The protein density of lactate transport proteins MCT 1 and 4 do not seem to change in skeletal muscle in humans exposed to altitude, whereas proteins involved in acid–base regulation are increased [30]. Also, buffer capacity has been shown to increase in both arm and leg extremities following altitude exposure [24]. After a climbing expedition to Mt. Denali, a 13.8% down-regulation in muscle Na+/K+-ATPase has been reported [31]. In contrast to the climbing expedition to Mt. Denali, eight sea level natives exposed to 4,100 m altitude in the outskirts of La Paz (living in a modern apartment) did not experience any change in the three Na+/K+ pump subunits in muscle biopsies obtained after 2 and 8 weeks of high altitude exposure [30]. A decrease in Na+/K+-ATPase has been hypothesized to allow a given amount of work to be performed at lower ATP costs, and would thus seem a favorable adaptive response to high altitude exposure [32]. Since O2 consumption for a given workload seems unchanged following altitude exposure [33], this also argues against an altitude-induced down-regulation of skeletal muscle Na+/K+-ATPase. In summary, the current knowledge indicates that prolonged hypoxic exposure would not induce skeletal muscle angiogenesis, and also that oxidative enzymes in human skeletal muscle respond only marginally to long-term altitude exposure. This may come as a surprise, since one would think that both responses (angiogenesis and oxidative enzymes) should be increased knowing the HIF-1 pathway.
Human skeletal muscle gene response to exercise in hypoxia
As stated in the "Introduction", exercise may superimpose an already present hypoxic stimulus, and the gene response to hypoxic exercise has been investigated in at least two studies (and more if studies using ischemia are also regarded). In both studies, human subjects underwent a muscle biopsy before and after a 6-week training period, either involving two [20] or five [19] training sessions per week. The biopsies were obtained 24 h [19] or 48 h [20] following the last training session, and therefore it can unfortunately not be assessed whether the reported gene response is associated to the total 6-week stimulus or to the last hypoxic training session. Following exercise training, most genes peak their expression 2–8 h into recovery [34]. Since the biopsies in the above mentioned studies were obtained 24 and 48 h following the last exercise bout, (1) the potential increase in genes augmented as a consequence of the last exercise bout may not be represented in biopsies obtained at these time points, or (2) the augmented genes may be the result of the total training regimen. In order to be able to distinguish between the acute and chronic response, an additional biopsy should have been obtained following the very first training session. Regardless of origin, however, it may be assumed that the augmented mRNA levels may also induce increased protein contents. Although the degree of hypoxia, and also the training intensity was quite similar in both studies, the mRNA response was not similar. While mRNA levels of HIF-1 and myoglobin were augmented in both studies, VEGF was increased in [19] but not in [20]. On the other hand, the mRNA content of COX-1 and PFK did not increase more than in the normoxic control group in [19], whereas this was the case in [20]. In addition, Glut-4, PGC-1α, CS, MCT-1, and a few other genes were augmented in [20], but not investigated in [19]. To draw clear-cut conclusions regarding the effect of hypoxic exercise on gene expression, future studies could be conducted—if possible with subjects performing one-legged kicking in normoxia and hypoxia, with biopsies obtained in the early recovery period following the single exercise bout.
Morphological and enzymatical adaptations following hypoxic exercise training
The rationale of hypoxic exercise training relies on the hypothesis that such regimen may induce muscle adaptations that are beyond the responses triggered by chronic exercise alone. Different training models of hypoxic exercise may be used: living at sea level and training at altitude (as mentioned above) or training and living at altitude. Studies employing either model are discussed below.
In the above-mentioned studies (living at sea level and training in hypoxia), Vogt et al. [19] reported that total mitochondrial density was increased more in the hypoxic training groups as compared to those performing the training in normoxia. In addition, capillary length density was also increased as a consequence of the training performed in hypoxia (i.e., both training responses are different from those reported to occur with chronic altitude exposure). In agreement herewith, Ponsot et al. [35] reported that hypoxic training did not alter mitochondrial function but led to a better coupling between the energy utilization and production sites. They also reported that hypoxic training did not alter skeletal muscle fiber composition or the content of selected oxidative enzymes (as with chronic altitude exposure). Using a similar approach, Masuda and coworkers [36] investigated the effects of 8 weeks training in hypoxia (2,500 m) as compared to normoxia. Following training, there was no difference between groups in myoglobin, muscle fiber composition, capillarity, or citrate synthase activity. In yet another study, eight subjects were assigned to altitude (2,300 m) or sea level training (all living at sea level). Compared to the normoxic training group, the moderate altitude group experienced a decrease in muscle PFK activity but an increase in muscle capillary density [37]. Interestingly, the same research group conducted a study using one-legged training [38]. This model has the advantage that two situations with similar magnitudes of mitochondrial substrate flux but different blood oxygen contents can be compared. Ten subjects trained one leg under normoxic conditions and the other under hypoxic conditions. There was a greater increase of citrate synthase activity under hypoxic conditions than under normoxic conditions. In addition, the myoglobin content increased in the leg trained under hypobaric conditions, whereas it tended to decrease in the normoxia-trained leg. Capillary density did not respond to the addition of hypoxia. Using a somewhat similar model, Melissa and coworkers studied ten males before and after 8 weeks of unilateral cycle ergometry training so that one leg was trained while breathing an inspirate of 13.5% O2 and the other while breathing normal ambient air. Biopsies from quadriceps revealed an increase in CS, whereas succinate dehydrogenase and PFK activity, capillary density, fiber area, % fiber type, and mitochondrial and lipid volume density all remained unaltered between groups [39]. In line with this, six Scandinavian runners were taken to either Portugal (sea level) or Kenya (2,000 m) for a 14-day-long training camp. No differences were found between groups with regard to muscle fiber size, composition, or capillarization. Also, CS and HAD activity did not differ between the groups. In contrast to Scandinavian runners, local Kenyan runners (predominantly living and training at 2,000 m) were reported to have higher HAD activity levels, but the other characteristics were similar to those found in the Scandinavians [40]. Using an interesting model, Desplanches and coworkers [41] investigated the effects of supplementing high altitude natives with oxygen during training at altitude. The rationale for this was the belief that reduced muscle stress during endurance training in hypoxia could limit muscle adaptations. Compared to the non-oxygen-supplemented training group, the 6-week training program (5 weeks, 30 min/session at ~70% of max) with oxygen supplementation did not lead to differences in capillary-to-fiber ratio between groups, capillary density, volume density of total mitochondria, or CS-, PKF-, and HAD-activity.
In summary, it would seem that hypoxic training may increases CS more than training in normoxic conditions. Although such an adaptation theoretically improves muscle function and therefore exercise tolerance, the physiological significance of this enzymatic adaptation deserves further investigation. In addition, it appears that the response hereof likely depends on the degree of hypoxia and training duration. In contrast, it seems that structural changes—in addition to those already occurring with normoxic training—are less likely to occur when hypoxia is superimposed on chronic exercise.
New insights: interactions between iron metabolism, myoglobin and muscle function at high altitude
Iron plays a central role in a large number of essential cellular functions. Its pivotal role in oxygen transport has in recent years generated a considerable body of scientific work that is greatly improving our understanding of the interactions between tissue hypoxia and iron metabolism. However, reviewing the response of skeletal muscle tissue to hypoxia shows that the role of iron remains globally unknown while other cellular/molecular aspects have been documented.
The central role for HIF-1 signaling in oxygen homeostasis by regulating the glycoprotein hormone erythropoieitin (Epo) is well established. Erythropoiesis is a complex process that concurrently induces dramatic changes in iron metabolism (also influenced by HIF-1) in order to fulfill the high demand for iron within the bone marrow to synthesize hemoglobin. In response to prolonged hypoxia, the up-regulation of red blood cell production is associated with progressive systemic iron deficiency [42]. We recently investigated whether this high need for iron during enhanced erythropoiesis at high altitude could interact on skeletal muscle iron stores and ultimately on muscle oxygen homoeostasis in humans [13]. Our results indeed demonstrated that prolonged exposure to hypoxia induces a down-modulation in several iron proteins in skeletal muscle, and hence indicating muscle iron loss. The consequence of such muscle iron loss is a decrease in myoglobin protein expression at high altitude, suggesting an altered muscle oxygen homeostasis. Our results do not support previous evidence showing that prolonged hypoxia may enhance the synthesis of myoglobin [21, 38, 43]. Beyond the differences between the experimental approaches that may account for the divergent results, an important clue to our understanding of myoglobin biology is still lacking, namely the evidence that the transcriptional regulation of myoglobin is mediated by a HIF-1-dependent mechanism [44].
A question that remained unanswered is the physiological importance of myoglobin for exercise tolerance at high altitude. Although myoglobin is basically known as an oxygen-storage protein and a diffusion facilitator, its physiological role has been challenged by studies showing that myoglobin knockout mice are able to exercise normally, suggesting that muscle function can be preserved even in the absence of myoglobin [45]. Of note, however, is the fact that the myoglobin-null mice demonstrate a number of molecular adaptations/compensations that may explain their unexpected normal exercise capacity. Insights into the functional role of myoglobin may also be gained from previous physiological studies on altitude acclimatization indicating that exercise capacity at high altitude (which is permanently decreased in comparison to sea level) is immediately restored to sea level values when acute reoxygenation is applied to acclimatized lowlanders [46–48]. Such an observation leads to the speculation that myoglobin would not be a major determinant of exercise capacity in normoxia, otherwise any decrease in myoglobin content following altitude acclimatization [13] would be expected to impair the recovery of exercise capacity upon acute reoxygenation. Hence, the process of O2 diffusion from the capillaries to muscle mitochondria would be, to some extent, independent of myoglobin during normoxic conditions. During chronic hypoxia, leg O2 conductance (which is a measure of oxygen diffusing capacity in muscle tissue) is found to be reduced [47], suggesting that O2 conductance is a limiting factor for exercise tolerance at high altitude. Although such observation raises the possibility of a significant role played by myoglobin in maintaining muscle O2 homeostasis when O2 supply is restricted, to date, no evidence supports nor opposes this contention.
In summary, iron can be considered as an emerging issue for skeletal muscle function in hypoxia. However, how does high altitude exposure exactly alter muscle iron metabolism remains a complex issue since two stimuli, i.e., hypoxia and accelerated erythropoiesis, coexist during high altitude studies. In order to gain further insights into the interactions between oxygen, erythropoiesis, and iron, we recently investigated the effect of enhanced erythropoiesis on muscle iron metabolism under non-hypoxic conditions, by injecting healthy humans with recombinant erythropoietin. Surprisingly, we found that, under normoxic erythropoietin stimulation (1 month), muscle was not a source of iron for erythropoiesis. On the contrary, the changes in the expression of muscle iron proteins were indicative of skeletal muscle iron accumulation [49]. Such response differs from muscle iron loss occurring at high altitude [13] and highlights the potential role of hypoxia in triggering muscle iron mobilization. Accordingly, it can be speculated that skeletal muscle tissue would serve as an iron storage compartment only under massive stress, such as prolonged hypoxia.
Conclusion
The current published data suggest that skeletal muscle tissue is not remarkably altered with chronic exposure to hypoxia in resting humans. This contention is supported by two major aspects, which are first the absence of muscle angiogenesis, and second the marginal response of oxidative enzymes to long-term altitude exposure. Our understanding of the molecular events mediating muscle morphological and enzymatic responses to hypoxia is still fragmented. Nevertheless, emerging evidence suggests that HIF-1 signaling in skeletal muscle tissue displays a rather modest response to hypoxia, and thereby explains why muscle function is only barely modified in hypoxia. In contrast, the few data obtained in humans exercising in hypoxia raise the possibility that the combination of these two stimuli might result in structural and functional adaptations in skeletal muscle.
In conclusion, skeletal muscle tissue appears to be rather well adapted to the hypoxic environment, at least in healthy humans exposed to terrestrial hypoxia. That exercise training associated with hypoxia may improve muscle oxygen homoeostasis remains a relevant issue that deserves future work.
References
Niermeyer S, Zamudio S, Moore LG (2001) The people. In: Hornbein TF, Schoene RB (eds) High altitude, an exploration of human adaptation. Marcel Dekker, New York, pp 43–100
Harris AL (2002) Hypoxia—a key regulatory factor in tumour growth. Nat Rev Cancer 2:38–47
Richardson RS, Noyszewski EA, Kendrick KF, Leigh JS, Wagner PD (1995) Myoglobin O2 desaturation during exercise. Evidence of limited O2 transport. J Clin Invest 96:1916–1926
Wang GL, Semenza GL (1995) Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 270:1230–1237
Ke Q, Costa M (2006) Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol 70:1469–1480
Huang LE, Bunn HF (2003) Hypoxia-inducible factor and its biomedical relevance. J Biol Chem 278:19575–19578
Semenza GL (2002) Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol 64:993–998
Dm Stroka, Tobi Burkhardt, Isab Desbaillets, Rh Wenger, Dah Neil, Chri Bauer, Max Gassmann, Dani Candinas (2001) HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J 15:2445–2453
Ameln H, Gustafsson T, Sundberg CJ, Okamoto K, Jansson E, Poellinger L, and Makino Y (2005) Physiological activation of hypoxia inducible factor-1 in human skeletal muscle. FASEB J, 04-2304fje
Pisani DF, Dechesne CA (2005) Skeletal Muscle HIF-1α expression is dependent on muscle fiber type. J Gen Physiol 126:173–178
Chavez JC, Agani F, Pichiule P, LaManna JC (2000) Expression of hypoxia-inducible factor-1alpha in the brain of rats during chronic hypoxia. J Appl Physiol 89:1937–1942
Vigano A, Ripamonti M, De Palma S, Capitanio D, Vasso M, Wait R, Lundby C, Cerretelli P, Gelfi C (2008) Proteins modulation in human skeletal muscle in the early phase of adaptation to hypobaric hypoxia. Proteomics 8:4668–4679
Robach P, Cairo G, Gelfi C, Bernuzzi F, Pilegaard H, Vigano A, Santambrogio P, Cerretelli P, Calbet JAL, Mouterou S, Lundby C (2007) Strong iron demand during hypoxia-induced erythropoiesis is associated with down-regulation of iron-related proteins and myoglobin in human skeletal muscle. Blood 109:4724–4731
Mason SD, Howlett RA, Kim MJ, Olfert IM, Hogan MC, McNuly W, Hickey RP, Wagner PD, Kahn CR, Giordano FJ, Johnson RS (2004) Loss of skeletal muscle HIF-1alpha results in altered exercise endurance. PLoS Biol 2:e288
Hoppeler H, Howald H, Conley K, Lindstedt SL, Claassen H, Vock P, Weibel ER (1985) Endurance training in humans: aerobic capacity and structure of skeletal muscle. J Appl Physiol 59:320–327
Lundby C, Gassmann M, Pilegaard H (2005) Regular endurance training reduces the exercise induced HIF-1a and HIF-2a mRNA expression in human skeletal muscle in normoxic conditions. Eur J Appl Physiol 96:363–369
Mason SD, Johnson RS (2007) The role of HIF-1 in hypoxic response in the skeletal muscle. Adv Exp Med Biol 618:229–244
Mason SD, Rundqvist H, Papandreou I, Duh R, McNulty WJ, Howlett RA, Olfert IM, Sundberg CJ, Denko NC, Poellinger L, Johnson RS (2007) HIF-1α in endurance training: suppression of oxidative metabolism. Am J Physiol Regul Integr Comp Physiol 293:R2059–R2069
Vogt M, Puntschart A, Geiser J, Zuleger C, Billeter R, Hoppeler H (2001) Molecular adaptations in human skeletal muscle to endurance training under simulated hypoxic conditions. J Appl Physiol 91:173–182
Zoll J, Ponsot E, Dufour S, Doutreleau S, Ventura-Clapier R, Vogt M, Hoppeler H, Richard R, Fluck M (2006) Exercise training in normobaric hypoxia in endurance runners. III. Muscular adjustments of selected gene transcripts. J Appl Physiol 100:1258–1266
Reynafarje B (1962) Myoglobin content and enzymatic activity of muscle and altitude adaptation. J Appl Physiol 17:301–305
Jackson CG, Sillau AH, Banchero N (1987) Fiber composition and capillarity in growing guinea pigs acclimated to cold and cold plus hypoxia. Proc Soc Exp Biol Med 185:101–106
Lundby C, Pilegaard H, Andersen JL, van Hall G, Sander M, Calbet JAL (2004) Acclimatization to 4,100 m does not change capillary density or mRNA expression of potential angiogenesis regulatory factors in human skeletal muscle. J Exp Biol 207:3865–3871
Mizuno M, Savard GK, Areskog NH, Lundby C, Saltin B (2008) Skeletal muscle adaptations to prolonged exposure to extreme altitude: a role of physical activity? High Alt Med Biol 9:311–317
Hoppeler H, Kleinert E, Schlegel C, Claassen H, Howald H, Kayar SR, Cerretelli P (1991) Morphological adaptations of human skeletal muscle to chronic hypoxia. Int J Sports Med 11:S3–S9
MacDougall JD, Green HJ, Sutton JR, Coates G, Cymerman A, Young P, Houston CS (1991) Operation Everest II: structural adaptations in skeletal muscle in response to extreme simulated altitude. Acta Physiol Scand 142:427
Jagoe RT, Engelen MPKJ (2003) Muscle wasting and changes in muscle protein metabolism in chronic obstructive pulmonary disease. Eur Respir J 22:52s–563
Kayser B (1992) Nutrition and high altitude exposure. Int J Sports Med 13:S129–S132
Gelfi C, De Palma S, Ripamonti M, Wait R, Eberini I, Bajracharya A, Marconi C, Schneider A, Hoppeler H, and Cerretelli P (2004) New aspects of altitude adaptation in Tibetans: a proteomic approach. FASEB J, DOI:10.1096/fj.03-1077fje
Juel C, Lundby C, Sander M, Calbet JAL, van Hall G (2003) Human skeletal muscle and erythrocyte proteins involved in acid–base homeostasis: adaptations to chronic hypoxia. J Physiol 15:639–648
Green H, Roy B, Grant S, Burnett M, Tupling R, Otto C, Pipe A, McKenzie D (2000) Downregulation in muscle Na+-K+-ATPase following a 21-day expedition to 6,194 m. J Appl Physiol 88:634–640
Hochachka PW, Buck LT, Doll CJ, Land SC (1996) Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci USA 93:9493–9498
Lundby C, Calbet JAL, Sander M, van Hall G, Mazzeo RS, Stray-Gundersen J, Stager JM, Chapman RF, Saltin B, Levine BD (2007) Exercise economy does not change after acclimatization to moderate to very high altitude. Scand J Med Sci Sports 17:281–291
Pilegaard H, Saltin B, Neufer PD (2003) Exercise induces transient transcriptional activation of the PGC-1alpha gene in human skeletal muscle. J Physiol 546:851–858
Ponsot E, Dufour SP, Zoll J, Doutrelau S, N’Guessan B, Geny B, Hoppeler H, Lampert E, Mettauer B, Ventura-Clapier R, Richard R (2006) Exercise training in normobaric hypoxia in endurance runners. II. Improvement of mitochondrial properties in skeletal muscle. J Appl Physiol 100:1249–1257
Masuda KOK, Kuno S, Asano K, Shimojo H, Katsuna S (2001) Endurance training under 2,500-m hypoxia does not increase myoglobin content in human skeletal muscle. Eur J Appl Physiol 85:486–490
Terrados N, Melinchna J, Sylven C, Jansson E, Kaijser L (1988) Effects of training at simulated altitude on performance and muscle metabolic capacity in competitive road cyclists. Eur J Appl Physiol 57:203–209
Terrados N, Jansson E, Sylven C, Kaijser L (1990) Is hypoxia a stimulus for synthesis of oxidative enzymes and myoglobin? J Appl Physiol 68:2369–2372
Melissa L, Tarnopolsky M, Cipriano N, Green HJ (1997) Skeletal muscle adaptations to training under normobaric hypoxic versus normoxic conditions. Med Sci Sports Exerc 29:238–243
Saltin B, Kim CK, Tearrads N, Larsen H, Svedenhag J, Rolf CJ (1995) Morphology, enzyme activities and buffer capacity in leg muscles of Kenyan and Scandinavian runners. Scand J Med Sci Sports 5:222–230
Desplanches D, Hoppeler H, Tuscher L, Mayet MH, Spielvogel H, Ferretti G, Kayser B, Leuenberger M, Grunenfelder A, Favier R (1996) Muscle tissue adaptations of high altitude natives to training in chronic hypoxia or acute normoxia. J Appl Physiol 81:1946–1951
Robach P, Fulla Y, Westerterp KP, Richalet JP (2004) Comparative response of EPO and soluble transferrin receptor at high altitude. Med Sci Sports Exerc 36:1493–1498
Hoppeler H, Fluck M (2002) Normal mammalian skeletal muscle and its phenotypic plasticity. J Exp Biol 205:2143–2152
Ordway GA, Garry DJ (2004) Myoglobin: an essential hemoprotein in striated muscle. J Exp Biol 207:3441–3446
Garry DJ, Ordway GA, Lorenz JN, Radford NB, Chin ER, Grange RW, Bassel-Duby R, Williams RS (1998) Mice without myoglobin. Nature 395:905–908
Lundby C, Møller P, Kanstrup IL, Olsen NV (2001) Heart rate response to hypoxic exercise: role of dopamine D2-receptors and effect of oxygen supplementation. Clin Sci (Lond) 101:377–383
Lundby C, Sander M, van Hall G, Saltin B, Calbet JAL (2006) Maximal exercise and muscle oxygen extraction in acclimatizing lowlanders and high altitude natives. J Physiol 573:535–547
Calbet JAL, Boushel R, Radegran G, Sondergaard H, Wagner PD, Saltin B (2003) Why is VO2 max after altitude acclimatization still reduced despite normalization of arterial O2 content? Am J Physiol Regul Integr Comp Physiol 284:R304–R316
Robach P, Recalcati S, Girelli D, Gelfi C, achmann-Andersen NJ, Thomsen JJ, Norgaard AM, Alberghini A, Campostrini N, Castagna A, Vigano A, Santambrogio P, Kempf T, Wollert KC, Moutereau S, Lundby C, Cairo G (2009) Alterations of systemic and muscle iron metabolism in human subjects treated with low dose recombinant erythropoietin. Blood 113(26):6707–6715. doi:10.1182/blood_2008_09_178095
Dehne N, Kerkweg U, Otto T, Fandrey J (2007) The HIF-1 response to simulated ischemia in mouse skeletal muscle cells neither enhances glycolysis nor prevents myotube cell death. Am J Physiol Regul Integr Comp Physiol 293:R1693–R1701
Kubis HP, Hanke N, Scheibe RJ, Gros G (2005) Accumulation and nuclear import of HIF1 alpha during high and low oxygen concentration in skeletal muscle cells in primary culture. Biochim Biophys Acta Mol Cell Res 1745:187–195
Young AJ, Evans WJ, Fisher EC, Sharp RL, Costill DL, Maher JT (1984) Skeletal muscle metabolism of sea-level natives following short-term high altitude residence. Eur J Appl Physiol 52:463–466
Bigard AX, Brunet A, Guezennec CY, Monod H (1991) Skeletal muscle changes after endurance training at high altitude. J Appl Physiol 71:2114–2121
McClelland GB, Brooks GA (2002) Changes in MCT 1, MCT 4, and LDH expression are tissue specific in rats after long-term hypobaric hypoxia. J Appl Physiol 92:1573–1584
Green HJ, Sutton JR, Cymerman A, Young PM, Houston CS (1989) Operation Everest II: adaptations in human skeletal muscle. J Appl Physiol 66:2454–2461
Dill RP, Chadan SG, Li C, Parkhouse WS (2001) Aging and glucose transporter plasticity in response to hypobaric hypoxia. Mech Ageing Dev 122:533–545
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lundby, C., Calbet, J.A.L. & Robach, P. The response of human skeletal muscle tissue to hypoxia. Cell. Mol. Life Sci. 66, 3615–3623 (2009). https://doi.org/10.1007/s00018-009-0146-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-009-0146-8