
Abstract
The human glucocorticoid receptor (GR) gene expresses two splicing isoforms α and β through alternative use of specific exons 9α and 9β. In contrast to the classic receptor GRα, which mediates most of the known actions of glucocorticoids, the functions of GRβ have been largely unexplored. Owing to newly developed methods, for example microarrays and the jellyfish fluorescence proteins, we and others have recently revealed novel functions of GRβ. Indeed, this enigmatic GR isoform influences positively and negatively the transcriptional activity of large subsets of genes, most of which are not responsive to glucocorticoids, in addition to its well-known dominant negative effect against GRα-mediated transcriptional activity. A recent report suggested that the “ligand-binding domain” of GRβ is active, forming a functional ligand-binding pocket associated with the synthetic compound RU 486. In this review, we discuss the functions of GRβ, its mechanisms of action, and its pathologic implications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glucocorticoids, the end-products of the hypothalamic–pituitary adrenal axis, are steroid hormones crucial for the regulation of basal and stress-related homeostasis [1, 2]. Glucocorticoids are also essential for the proper functioning of virtually all organs and tissues of the organism, including the central nervous (CNS) and cardiovascular systems, metabolic organs, such as the liver and adipose tissue, and the immune/inflammatory response [3, 4]. In addition, glucocorticoids at “pharmacologic” or “stress-related” doses are irreplaceable therapeutic means of treatment of many allergic, inflammatory, autoimmune, and lymphoproliferative diseases [4].
The actions of glucocorticoids are mediated by a ubiquitous intracellular receptor protein, the glucocorticoid receptor (GR), which functions as a hormone-activated transcription factor of glucocorticoid target genes [5, 6]. The human GR gene is located in chromosome 5 and encodes two splicing variants GRα and GRβ by alternative use of different terminal exons 9α and 9β [5, 7]. GRα is the classic receptor, binding to glucocorticoids and mediating most of the known glucocorticoid actions [5]. In contrast, GRβ does not bind glucocorticoids but functions as a dominant negative inhibitor of GRα-induced transactivation of GRE-containing, glucocorticoid-responsive promoters; its physiologic/pathologic roles have not yet been well elucidated [8, 9].
Using the microarray technique, which enabled us to evaluate gene expression en masse, we and others recently found that the GRβ isoform has intrinsic, GRα-independent transcriptional activity, in addition to its well-known dominant negative effect on GRα [10, 11]. In this review article, we will summarize known GRβ activities and discuss newly identified actions of this GR isoform.
The human GR gene, splicing variants GRα and GRβ, and their multiple translational isoforms
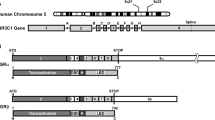
The GR, also known as nuclear receptor superfamily 3, group C, member 1 (NR3C1), belongs to the steroid/sterol/thyroid/retinoid/orphan nuclear receptor superfamily, which consists of over 130 members preserved from the early metazoans to humans [5, 12]. The human GR gene, located in the short arm of chromosome 5 (5q31.3), consists of nine exons, and its expression is regulated by at least three different promoters (A, B, and C) [7, 13], with promoter A alternatively used with three unique promoter fragments 1A1, 1A2, and 1A3 [13]. Thus the GR gene can produce five different transcripts from different promoters that encode the same GR proteins. In addition to alternative transcripts using the 5′ different promoters, the GR gene generates two 3′ splicing variant transcripts with alternative use of exon 9α and/or 9β (Fig. 1). Thus, the GR gene generates ten different transcripts that encode two protein molecules GRα and GRβ.

Genomic and complementary DNA and protein isoforms of the human GR and distribution of functional domains in its linearized molecule. The human GR gene consists of nine exons. Exon 1 is an untranslated region, exon 2 codes for the N-terminal “immunogenic” domain, exons 3 and 4 for the DNA-binding domain, and exons 5–9 for the hinge region and the ligand-binding domain. The GR gene contains two terminal exons 9 (9α and 9β), which produce the classic GRα (GRα-A) and GRβ (GRβ-A) through alternative splicing of these exons. C-terminal gray colored domains in GRα-A and GRβ-A show their specific portions. GRα N-terminal translational isoforms expressed from a single GRα transcript are shown in the middle panel of the figure. Similar N-terminal translational isoforms may also be produced from the GRβ-specific transcript using the same start sites (modified from Ref. [5]). AF-1 and -2 activation functions 1 and 2, DBD DNA-binding domain, HD hinge region, LBD Ligand-binding domain, NL1 and 2 Nuclear translocation signals 1 and 2, NTD N-terminal domain
Recently, it became evident that the GRα variant mRNA is translated from at least eight initiation sites into multiple GRα isoforms termed A through D (A, B, C1-C3 and D1-D3), producing different amino terminal isoforms with distinct specific transcriptional activities on glucocorticoid-responsive genes [14] (Fig. 1). These GR molecules are also differentially expressed in several different cell lines and tissues [14]. Given that GRα and GRβ share a common mRNA domain that contains the same translation initiation sites [15], it seems that the GRβ variant mRNA is also translated through the same initiation sites to a similar host of eight β isoforms [5] (Fig. 1).
The classic receptor GRα
GRα, the classic glucocorticoid receptor is ubiquitously expressed and mediates most of the known actions of glucocorticoids [3, 5]. The human GRα consists of 777 amino acids and has three major distinct functional domains, the N-terminal or immunogenic domain (NTD), the DNA-binding domain (DBD), and the ligand-binding domain (LBD) [6] (Fig. 1). The LBD of GRα consists of twelve α-helices and four β-sheets, among which helices 3, 4, 11, and 12 form the ligand-binding pocket for binding to glucocorticoids [16–18] (Fig. 2). GRα is located primarily in the cytoplasm in the absence of glucocorticoid ligand, as part of hetero-oligomeric complexes containing heat-shock proteins (HSPs) 90, 70, 50, 20 and, possibly, other proteins also [5, 6] (Fig. 3). After binding to its agonist ligand, GRα undergoes conformational changes, dissociates from HSPs, homo-dimerizes, and translocates as a monomer or dimer into the nucleus through the nuclear pore, via an active ATP-dependent process mediated by its nuclear localization signals (NL)-1 and 2 [12, 19]. NL-1 is located in the junction of DBD and the hinge region whereas NL-2 spans the entire LBD [19] (Fig. 1).

The three-dimensional structure of GRα associated with agonist dexamethasone (left) and antagonist RU 486 (right). Results from crystallographic analysis of the GRα associated with agonist dexamethasone (left) or with antagonist RU 486 (right) are shown [16, 75]. The LBD of GRα consists of twelve α-helices and four β-sheets, among which helices 3, 4, 11, and 12 form the ligand-binding pocket for binding to glucocorticoids. Helix 12 changes its localization dramatically upon binding to ligands, playing a critical role in the formation of a binding surface for the coactivator (LXXLL) motif. Image sources were downloaded from the RCSB Protein Data Bank (http://www.rcsb.org) whereas the images were created using the MacPyMOL software. Yellow bold arrow ligand-binding pocket, white arrow helix 12, white arrowhead the coactivator motif peptide fragment of the transcriptional intermediate factor 2

Nucleocytoplasmic shuttling and transcriptional regulation of GRα. Upon ligand binding, the activated GRα dissociates from the heat-shock proteins (HSPs) and translocates into the nucleus, where it binds as a homodimer to GREs in the promoter regions of target genes or interacts as a monomer with other transcription factors. GRα glucocorticoid receptor α, GRE glucocorticoid response element, HSPs heat-shock proteins, REs response elements, RNPII RNA polymerase II, TF transcription factor
Inside the nucleus, the ligand-activated GRα directly interacts as a dimer with specific DNA sequences, the glucocorticoid response elements (GREs), in the promoter regions of target genes, or as a monomer or dimer with other transcription factors via protein–protein interactions, indirectly influencing the activity of the latter on their own target genes [5, 12] (Fig. 3). GR contains two transactivation domains, activation functions (AF)-1 and 2, located at its NTD and LBD, respectively, through which the GR interacts with many proteins and protein complexes, for example the nuclear receptor coactivator (p160, p300/CREB-binding protein (CBP) and p300/CBP-associated factor (p/CAF)) complexes and the SWI/SNF and vitamin D receptor-interacting protein/thyroid hormone receptor-associated protein (DRIP/TRAP) chromatin-remodeling complexes, eventually influencing the activity of RNA polymerase II and its ancillary factors, altering the transcription rates of glucocorticoid-responsive genes [5, 6, 20] (Fig. 1).
GR also interacts with the nuclear receptor corepressor (NCoR) and its homolog silencing mediator of retinoic acid and thyroid hormone receptor (SMRT), which are macromolecular docking platforms for nuclear receptors and many transcription factors, repressing the transcriptional activity of the GR by attracting histone deacetylase/Sin3 complexes [20]. The p160 type coactivators and the NCoR/SMRT type corepressors establish equilibrium in their interaction with the GR to, respectively, facilitate or block its transcriptional activity [21]. Accumulation of coactivators and corepressors on the promoter-bound GR is dependent on the kind of ligands bound to the GR: agonist glucocorticoids attract the coactivator complexes to the promoter-bound GR whereas antagonists, for example RU 486, accumulate the corepressor complexes [22] (Fig. 2).
In addition to transactivation of the glucocorticoid-responsive genes explained above, GRα modulates other signal transduction cascades through mutual protein–protein interactions with specific transcription factors, by influencing their ability to stimulate or inhibit the transcription rates of their respective target genes (Fig. 3). This activity may be more important than the GRE-mediated one, granted that mice harboring a mutant GRα, which is active in terms of protein–protein interactions but inactive in terms of transactivation via DNA GREs, survive and procreate, in contrast to mice with a deletion of the entire GR gene that die immediately after birth from severe respiratory distress syndrome [23, 24]. The former mouse model and additional in vitro results indicate that GR interacts with and influences other transcription factors primarily as a monomer [23, 25].
The protein–protein interactions of GRα with other transcription factors may take place on promoters that do not contain GREs (tethering mechanism), and on promoters that have both GRE(s) and responsive element(s) of transcription factors that interact with GRα (“composite promoters”) [26]. Repression of transactivation of other transcription factors through protein–protein interactions may be particularly important in the suppression of immune function and inflammation by glucocorticoids [23, 25]. A substantial part of the effects of glucocorticoids on the immune system may be explained by the interaction between GRα with nuclear factor-κB (NF-κB), activator protein-1 (AP-1), and, probably, the signal transducers and activators of transcription (STATs) [27–30].
In addition to co-regulators and other transcription factors that modulate GR-induced transcriptional activity, several distinct signaling pathways regulate the transcriptional activity of the GR via post-translational modifications of the receptor protein [5]. These include methylation, acetylation, nitrosylation, sumoylation, and ubiquitination, as well as phosphorylation, which has been studied best. Indeed, several kinases, such as the cell-cycle-related kinases, mitogen-activated kinases, and the glycogen synthase kinases, phosphorylate specific serine or threonine residues of the GR. Interestingly, most of these residues are located in the AF-1 domain of the human GR NTD, thus phosphorylation of some or all of them modulates GR-induced transcriptional activity through alteration of co-regulator attraction to the promoter region of glucocorticoid-responsive genes, possibly by changing their affinity for the AF-1 domain of GR [31].
The splicing variant GRβ isoform
Similarly to the classic human GRα, the original human GRβ isoform is also ubiquitously expressed in most tissues. This isoform has been identified in both the zebrafish and humans, but not in mice [15, 32, 33]. The human (h) GRβ contains 742 amino acids and shares the first 727 amino acids from the N-terminus with hGRα [6, 15] (Fig. 1). hGRβ encodes an additional 15 nonhomologous amino acids in the C-terminus, whereas hGRα has an additional 50 amino acids forming a 777-amino-acid protein [6, 15] (Fig. 1). Therefore, hGRβ shares the same NTD and DBD with hGRα, but has a unique “LBD”. Because the divergence point (amino acid 727) is located at the C-terminal end of helix 10 in the hGRα LBD, the hGRβ “LBD” does not have helices 11 and 12 of the hGRα. As these helices are important for forming the ligand-binding pocket and for the creation of the AF-2 surface upon ligand binding [16] (Fig. 2), GRβ cannot form an active ligand-binding pocket, does not bind glucocorticoids, and, thus, does not directly regulate GRE-containing, glucocorticoid-responsive gene promoters. In the absence of the hGRβ “LBD”, the truncated hGR consisting of NTD and DBD is transcriptionally active on GRE-containing promoters [34], thus the hGRβ “LBD” somehow attenuates the transcriptional activity of the other subdomains of the molecule on GRE-driven promoters. Inside cells hGRβ can localize both in the cytoplasm and nucleus [9, 35].
Similarly to the human GR gene, the zebrafish (z) GR gene consists of nine exons and produces the zGRα and zGRβ proteins, which contain 746 and 737 amino acids, respectively [32] (Fig. 4). zGRα and zGRβ share the N-terminal 697 amino acids and have specific C-terminal portions which contain 47 and 40 amino acids, respectively. In contrast to hGRα and hGRβ, which are produced by alternative use of specific exons 9α and 9β, zGRα and zGRβ are formed as a result of intron retention [32]. zGRα and zGRβ use exon 1 to exon 8 for their common N-terminal 697 amino acids. zGRα uses exon 9 for its specific C-terminal portion whereas zGRβ continuously employs the rest of exon 8 and uses a stop codon located at the 3′ portion of this exon to express its specific C-terminal peptide [32] (Fig. 4). Protein alignment comparison of hGRα and zGRβ indicated that these two molecules have exactly the same divergence point and that their β isoform-specific C-terminal peptides show little sequence homology [32]. These pieces of molecular information indicate that hGRβ and zGRβ evolved independently. Nevertheless, zGRβ had the same functional properties as hGRβ, for example inability to bind glucocorticoids, a dominant negative activity on zGRα transcriptional activity on GRE-drive promoters, and strikingly similar tissue distribution [32]. Thus, hGRβ and zGRβ are produced by convergent evolution, most likely developed through strong requirement of this type of GR isoform in a physiologic situation.

Genomic and complementary DNA and protein isoforms of the zebrafish GR. The zebrafish (z) GR gene consists of nine exons. The zGR gene expresses zGRα and zGRβ splicing variants through intron retention [32]. C-terminal gray colored and shaded domains in zGRα and zGRβ show their specific portions. They are, respectively, encoded by exon 9 and the 3′ portion of exon 8, which are also shown in the same labeling. DBD DNA-binding domain, LBD Ligand-binding domain, NTD N-terminal domain, UTR untranslated region
The presence of nonligand-binding C-terminal variants is not unique to the GR. Similarly to the human and zebrafish GR, several other human steroid and nuclear receptors, for example the estrogen receptor β (ERβ), thyroid hormone receptor α (TRα), vitamin D receptor, constitutive androstane receptor (CAR), dosage-sensitive sex reversal-1 (DAX-1), nuclear receptor related 2 (Nurr2), neuron-derived orphan receptor-2 (NOR-2), peroxisome proliferator-activated receptor α (PPARα), and PPARγ, also have C-terminally truncated receptor isoforms defective in binding to cognate ligands with dominant negative activity on their corresponding classic receptors [36–45]. This suggests that evolution has allowed the development and retention of such alternative nuclear receptors, probably because they play useful biologic roles.
The dominant-negative effect of GRβ on GRα-induced transcriptional activity: physiologic and pathologic implications
In contrast to GRα, which has numerous and diverse actions [3], the functions of GRβ had not been revealed until we reported its dominant negative effect on GRα-induced transcriptional activity almost a decade after the original identification of this receptor isoform [8]. The dominant negative activity of GRβ was first demonstrated in transient transfection-based reporter assays using GRE-driven reporter genes, but was subsequently confirmed on endogenous, glucocorticoid-responsive genes, such as the mitogen-activated protein kinase phosphatase-1 (MPK-1), myocilin and fibronectin [46, 47]. Further, GRβ was shown to attenuate glucocorticoid-induced repression of the tumor necrosis factor (TNF) α and interleukin (IL)-6 genes [46]. We also confirmed this negative effect of GRβ on GRα-mediated transrepression using microarray analyses [10]. Several mechanisms explaining this GRβ function have been reported, including:
-
1
competition for GRE binding through their shared DBD;
-
2
heterodimerization with GRα; and
-
3
coactivator squelching through the preserved AF-1 domain [8, 34, 48].
All these different mechanisms of action seem to be functional, depending on the promoters and tissues affected by this GR isoform.
Several clinically oriented investigations suggest that GRβ is responsible for the development of tissue-specific insensitivity to glucocorticoids in various disorders, most of them associated with dysregulation of immune function. They include glucocorticoid-resistant asthma, rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), ankylosing spondylitis, chronic lymphocytic leukemia, and nasal polyps [49–55]. In these studies, various immune cells expressed elevated levels of GRβ, which correlated with reduced sensitivity to glucocorticoids. Elevated levels of pro-inflammatory cytokines, such as IL-1, 2, 4, 7, 8, and 18, TNFα, and interferons α and γ, might have been responsible for increased GRβ expression in cells from patients with these pathologic conditions, because these cytokines experimentally stimulated expression of GRβ in lymphocytes, neutrophils, or airway smooth-muscle cells [56–61]. Further, the presence of a single nucleotide polymorphism in the 3′ untranslated region of the hGRβ mRNA (rs6198G allele), which increases the stability of the mRNA, and thus, causes elevated expression of GRβ protein, was associated with increased incidence of RA, SLE, high blood pressure, ischemic heart diseases, and nasal carriage of Staphylococcus aureus [50, 62–64], possibly through inhibition of glucocorticoid actions by increased concentrations of GRβ. These pieces of clinical evidence further support the dominant negative activity of GRβ on GRα-induced transcription inside the human body, functioning as a negative regulator of glucocorticoid actions in local tissues.
GRβ has intrinsic, GRα-independent transcriptional activity
We and others recently performed transcriptome analyses using microarray techniques in cultured cells overexpressing GRβ, and found that these cells had a distinct mRNA expression profile compared with cells not overexpressing GRβ and those expressing GRα and treated with glucocorticoids [10, 11]. In a subsequent real-time PCR analysis, we also confirmed that GRβ regulates mRNA expression positively and negatively in a gene-specific fashion [10]. These results indicate that GRβ has intrinsic transcriptional activities independent of the activity of its isoform GRα. We have compared the microarray results obtained by us and those of others [10, 11], and found that the two studies share 78 genes modulated by overexpression of GRβ (Table 1). Specifically, 29 out of 78 genes were both down-regulated by GRβ overexpression, whereas only eight were up-regulated. Interestingly, 41 genes showed opposite response to GRβ between the two studies, suggesting that GRβ modulates mRNA expression of some of its responsive genes in a cell-specific and, possibly, cell culture condition-specific fashion.
Apparently, this intrinsic transcriptional activity of GRβ is not mediated by binding of the isoform to classic GREs, as GRβ does not affect the transcriptional activity of classic GRE-driven promoters, whereas the promoter regions of the genes, which we identified to be regulated by GRβ, do not contain GRE sequences [10]. Rather, GRβ directly modulates the transcriptional activity of its responsive genes, which are distinct from those responsive to glucocorticoids, possibly by altering the activity of transcriptional intermediate molecules or other transcription factors through physical protein–protein interactions. Indeed, we previously demonstrated that the AF-1 of GRβ, which presumably keeps the same protein structure and function as that of GRα, is transcriptionally active, contributing to its dominant negative activity against GRα-induced transactivation [34]. This transactivation domain of GRα interacts with numerous cofactor molecules, including CBP/p300 and p160-type histone acetyltransferase coactivators, components of the SWI/SNF chromatin modulators, DRIP150 of the DRIP/TRAP complex, and the steroid receptor RNA coactivator (SRA) [65–70]. Thus, it is possible that GRβ alters the transcriptional activity of its responsive genes by lodging into the transcriptional complexes formed on their promoter region through its AF-1 (Fig. 5). This mechanistic hypothesis is further supported by recent results from other groups, which showed GRβ repressed the transcriptional activity of AP-1 and NFκB, possibly through protein–protein interactions similar to those between GRα and these transcription factors [71].

Hypothetical models for GRβ-mediated modulation of the transcriptional activity of its responsive genes. a Through AF-1 located in the NTD, GRβ may interact with numerous transcriptional cofactors and transcriptional factors, lodge into the transcription intermediate complex formed on the promoter region of GRβ-responsive genes, and modulate their transcriptional activity. GRβ may attract histone deacetylases to the transcription intermediate complex formed on the promoter region of genes regulated by this GR isoform. b GRβ might also bind to hypothetical specific response elements located in the promoter region of responsive genes, directly modulating their transcriptional activity. GRβ glucocorticoid receptor β, HDACs histone deacetylases, REs response elements, RNPII RNA polymerase II, TF transcription factor
GRβ was also reported to suppress the transcriptional activity of the GATA3 transcription factor on its responsive IL-5 and 13 promoters by attracting histone deacetylases [72]. Alternatively, GRβ might bind DNA sequences unique to this isoform through its DBD, regulating transcription through hypothetical “GRβ REs” (Fig. 5). Because the subdomains of steroid hormone receptors affect each others’ activities [73, 74], the unique GRβ “LBD” might alter the binding specificity of its DBD to DNA and allow it to recognize a set of DNA sequences specific to GRβ and distinct from those of GRα.
The importance and exact roles of this intrinsic transcriptional activity of the GRβ isoform in physiology and pathophysiology have not yet been elucidated. We have performed a pathway analysis of our microarray results to define the biologic pathways where GRβ might play consistent roles [10], and found that this GR isoform may be involved in regulation of 43 distinct pathways recorded in the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Table 2). Among the pathways we found in this analysis, GRβ might strongly affect several cellular functions, such as cell communications (#13), focal adhesion (#26), ECM-receptor interaction (#27), expression of cell adhesion molecules (#28), and regulation of actin cytoskeleton (#34), and the metabolism of some amino acids and other bioactive molecules. Interestingly, GRβ might also play a role in the development/activity/apoptosis of cancer cells, as it also regulates mRNA expression of genes important for colorectal (#40), renal cell (#41), prostate (#42), and small cell lung cancer (#43) and apoptosis (#23). To further verify the biologic pathways, in which GRβ plays important roles, development of mice conditionally over-expressing human GRβ would be very helpful.
Issues on “ligand” and subcellular localization of GRβ
A previous publication demonstrated that only RU 486 among 57 native and synthetic steroids tested bound GRβ weakly at the “ligand-binding” pocket of the GRβ and slowly (over 6 h for completion) induced its nuclear translocation [11]. The results were supported by a nuclear translocation study using fluorescent protein-fused GRβ by scoring cellular localization of this fusion protein in different cells, by a whole-cell ligand-binding assay followed by the crude fractionation of radiolabeled ligand-associated receptors with a Sephadex column and by computer-based modeling of the GRβ “ligand-binding domain” associated with several steroids [11]. This report also demonstrated that RU 486 modulated GRβ-mediated transcriptional activity in microarray analysis [11]. Although the hypothesis presented in this publication is interesting, there are several points to be resolved. Yet undiscovered endogenous steroids or other related compounds with structures similar to that of RU 486 would be expected to be the endogenous ligands of GRβ. Crystallographic structural analysis of the GRβ “LBD” might help identifying a “ligand-binding pocket” in the GRβ “LBD” and hence its binding to RU 486. The cytoplasmic to nuclear translocation of GRβ demonstrated by the previous work was quite slow compared with that of GRα: in the former, the receptor took 6 h to complete its translocation whereas in the latter it did this within minutes [11]. GRβ and GRα share NL-1, which mediates the rapid nuclear translocation of GRα, whereas GRβ does not appear to have NL-2, which is dependent on the entire LBD of GRα, and causes slower nuclear translocation of the receptor [19]. Thus, the presence of yet unknown regulators specific to GRβ might be involved in the nuclear translocation of this isoform.
We independently performed several experiments addressing the potential activation of GRβ by RU 486, its subcellular localization, and cytoplasmic to nuclear translocation. In contrast to the previously reported findings [11], the green fluorescent protein-fused GRβ was mainly located in the nuclei of HeLa cells stably expressing this fusion protein, whereas it was heterogeneously distributed both in the cytoplasm and the nucleus in HCT116 cells that expressed the GRβ fusion protein transiently: some cells mainly expressed GRβ in the nucleus whereas others had it in the cytoplasm [10]. Addition of RU 486 did not stimulate the transcriptional activity of glucocorticoid-responsive and GRE-containing mouse mammary tumor virus promoter in transiently GRβ-expressing HCT116 cells, and did not induce cytoplasmic to nuclear translocation of this isoform [10]. The inconsistency of our results with those previously reported may have been caused by use of different experimental systems, for example cell lines and plasmids. This discrepancy suggests that the mechanisms of the regulatory actions of GRβ on the transcription of responsive genes inside the cells are quite complex.
Summary
In 1995, ten years after the original identification of the human GRβ by R. Evans’ group [15], we reported that GRβ had a dominant negative effect on GRα-induced transcriptional activity, an effect that was replicated a year later [8, 35]. After another decade, a new activity of GRβ, namely an intrinsic, GRα-independent transcriptional activity, was discovered by employing microarray-based transcriptome analyses [10, 11]. Despite continuous effort spanning 20 years, the molecular mechanisms of action and the roles of GRβ in physiology are still largely unknown, in contrast to those of the classic, glucocorticoid action-mediating GRα. The β isoform cannot modulate the transcriptional activity of GRE-containing promoters in the absence of GRα, even though it shares a perfect DBD with GRα [10]. Lack of GRβ in rodents stands against elucidation of its in vivo activity [33]. We hope that physiologic and pathologic roles of GRβ will be further clarified with future technical progress, for example development of mice conditionally expressing human GRβ, sophisticated transcriptome/promoter/proteome analyses with array techniques, evaluation of GRβ subcellular circulation/localization through fusion with fluorescent proteins, and crystallography-based structural analyses.
References
Chrousos GP (1995) The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med 332:1351–1362
Kino T, Chrousos GP (2001) Glucocorticoid and mineralocorticoid resistance/hypersensitivity syndromes. J Endocrinol 169:437–445
Kino T, Chrousos GP (2005) Glucocorticoid effect on gene expression. In: Steckler T, Kalin NH, Reul JMHM (eds) Handbook on stress and the brain. Elsevier, Amsterdam, pp 295–312
Chrousos GP (2001) Glucocorticoid therapy. In: Felig P, Frohman LA (eds) Endocrinology & metabolism. McGraw–Hill, New York, pp 609–632
Chrousos GP, Kino T (2005) Intracellular glucocorticoid signaling: a formerly simple system turns stochastic. Sci STKE 2005, pe48
Kino T, Chrousos GP (2004) Glucocorticoid and mineralocorticoid receptors and associated diseases. Essays Biochem 40:137–155
Chrousos GP, Kino T, Charmandari E (2008) Generalized glucocorticoid resistance. Eur J Endocrinol 2:93–99
Bamberger CM, Bamberger AM, de Castro M, Chrousos GP (1995) Glucocorticoid receptor β, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest 95:2435–2441
de Castro M, Elliot S, Kino T, Bamberger C, Karl M, Webster E, Chrousos GP (1996) The non-ligand binding β-isoform of the human glucocorticoid receptor (hGRβ): tissue levels, mechanism of action, and potential physiologic role. Mol Med 2:597–607
Kino T, Manoli I, Kelkar S, Wang Y, Su YA, Chrousos GP (2009) Glucocorticoid receptor (GR) β has intrinsic, GRα-independent transcriptional activity. Biochem Biophys Res Commun 381:671–675
Lewis-Tuffin LJ, Jewell CM, Bienstock RJ, Collins JB, Cidlowski JA (2007) Human glucocorticoid receptor β binds RU-486 and is transcriptionally active. Mol Cell Biol 27:2266–2282
Kino T, De Martino MU, Charmandari E, Mirani M, Chrousos GP (2003) Tissue glucocorticoid resistance/hypersensitivity syndromes. J Steroid Biochem Mol Biol 85:457–467
Breslin MB, Geng CD, Vedeckis WV (2001) Multiple promoters exist in the human GR gene, one of which is activated by glucocorticoids. Mol Endocrinol 15:1381–1395
Lu NZ, Cidlowski JA (2005) Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell 18:331–342
Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, Thompson EB, Rosenfeld MG, Evans RM (1985) Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 318:635–641
Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, Consler TG, Parks DJ, Stewart EL, Willson TM, Lambert MH, Moore JT, Pearce KH, Xu HE (2002) Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell 110:93–105
Tanenbaum DM, Wang Y, Williams SP, Sigler PB (1998) Crystallographic comparison of the estrogen and progesterone receptor’s ligand binding domains. Proc Natl Acad Sci USA 95:5998–6003
Williams SP, Sigler PB (1998) Atomic structure of progesterone complexed with its receptor. Nature 393:392–396
Savory JG, Hsu B, Laquian IR, Giffin W, Reich T, Hache RJ, Lefebvre YA (1999) Discrimination between NL1- and NL2-mediated nuclear localization of the glucocorticoid receptor. Mol Cell Biol 19:1025–1037
Rosenfeld MG, Lunyak VV, Glass CK (2006) Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev 20:1405–1428
Wang Q, Blackford JA Jr, Song LN, Huang Y, Cho S, Simons SS Jr (2004) Equilibrium interactions of corepressors and coactivators with agonist and antagonist complexes of glucocorticoid receptors. Mol Endocrinol 18:1376–1395
Schulz M, Eggert M, Baniahmad A, Dostert A, Heinzel T, Renkawitz R (2002) RU486-induced glucocorticoid receptor agonism is controlled by the receptor N terminus and by corepressor binding. J Biol Chem 277:26238–26243
Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, Schmid W, Herrlich P, Angel P, Schutz G (1998) DNA binding of the glucocorticoid receptor is not essential for survival. Cell 93:531–541
Cole TJ, Blendy JA, Monaghan AP, Krieglstein K, Schmid W, Aguzzi A, Fantuzzi G, Hummler E, Unsicker K, Schutz G (1995) Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev 9:1608–1621
Reichardt HM, Tuckermann JP, Gottlicher M, Vujic M, Weih F, Angel P, Herrlich P, Schutz G (2001) Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J 20:7168–7173
Miner JN, Yamamoto KR (1991) Regulatory crosstalk at composite response elements. Trends Biochem Sci 16:423–426
De Bosscher K, Haegeman G (2009) Minireview: latest perspectives on antiinflammatory actions of glucocorticoids. Mol Endocrinol 23:281–291
Karin M, Chang L (2001) AP-1-glucocorticoid receptor crosstalk taken to a higher level. J Endocrinol 169:447–451
Barnes PJ, Karin M (1997) Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med 336:1066–1071
Didonato JA, Saatcioglu F, Karin M (1996) Molecular mechanisms of immunosuppression and anti-inflammatory activities by glucocorticoids. Am J Respir Crit Care Med 154:S11–S15
Kino T, Ichijo T, Amin ND, Kesavapany S, Wang Y, Kim N, Rao S, Player A, Zheng YL, Garabedian MJ, Kawasaki E, Pant HC, Chrousos GP (2007) Cyclin-dependent kinase 5 differentially regulates the transcriptional activity of the glucocorticoid receptor through phosphorylation: clinical implications for the nervous system response to glucocorticoids and stress. Mol Endocrinol 21:1552–1568
Schaaf MJ, Champagne D, van Laanen IH, van Wijk DC, Meijer AH, Meijer OC, Spaink HP, Richardson MK (2008) Discovery of a functional glucocorticoid receptor β-isoform in zebrafish. Endocrinology 149:1591–1599
Otto C, Reichardt HM, Schutz G (1997) Absence of glucocorticoid receptor-β in mice. J Biol Chem 272:26665–26668
Charmandari E, Chrousos GP, Ichijo T, Bhattacharyya N, Vottero A, Souvatzoglou E, Kino T (2005) The human glucocorticoid receptor (hGR) β isoform suppresses the transcriptional activity of hGRα by interfering with formation of active coactivator complexes. Mol Endocrinol 19:52–64
Oakley RH, Sar M, Cidlowski JA (1996) The human glucocorticoid receptor β isoform. Expression, biochemical properties, and putative function. J Biol Chem 271:9550–9559
van der Vaart M, Schaaf MJ (2009) Naturally occurring C-terminal splice variants of nuclear receptors. Nucl Recept Signal 7:e007
Ogawa S, Inoue S, Watanabe T, Orimo A, Hosoi T, Ouchi Y, Muramatsu M (1998) Molecular cloning and characterization of human estrogen receptor βcx: a potential inhibitor of estrogen action in human. Nucleic Acids Res 26:3505–3512
Benbrook D, Pfahl M (1987) A novel thyroid hormone receptor encoded by a cDNA clone from a human testis library. Science 238:788–791
Ebihara K, Masuhiro Y, Kitamoto T, Suzawa M, Uematsu Y, Yoshizawa T, Ono T, Harada H, Matsuda K, Hasegawa T, Masushige S, Kato S (1996) Intron retention generates a novel isoform of the murine vitamin D receptor that acts in a dominant negative way on the vitamin D signaling pathway. Mol Cell Biol 16:3393–3400
Arnold KA, Eichelbaum M, Burk O (2004) Alternative splicing affects the function and tissue-specific expression of the human constitutive androstane receptor. Nucl Recept 2:1
Hossain A, Li C, Saunders GF (2004) Generation of two distinct functional isoforms of dosage-sensitive sex reversal-adrenal hypoplasia congenita-critical region on the X chromosome gene 1 (DAX-1) by alternative splicing. Mol Endocrinol 18:1428–1437
Ohkura N, Hosono T, Maruyama K, Tsukada T, Yamaguchi K (1999) An isoform of Nurr1 functions as a negative inhibitor of the NGFI-B family signaling. Biochim Biophys Acta 1444:69–79
Petropoulos I, Part D, Ochoa A, Zakin MM, Lamas E (1995) NOR-2 (neuron-derived orphan receptor), a brain zinc finger protein, is highly induced during liver regeneration. FEBS Lett 372:273–278
Gervois P, Torra IP, Chinetti G, Grotzinger T, Dubois G, Fruchart JC, Fruchart-Najib J, Leitersdorf E, Staels B (1999) A truncated human peroxisome proliferator-activated receptor α splice variant with dominant negative activity. Mol Endocrinol 13:1535–1549
Sabatino L, Casamassimi A, Peluso G, Barone MV, Capaccio D, Migliore C, Bonelli P, Pedicini A, Febbraro A, Ciccodicola A, Colantuoni V (2005) A novel peroxisome proliferator-activated receptor γ isoform with dominant negative activity generated by alternative splicing. J Biol Chem 280:26517–26525
Li LB, Leung DY, Hall CF, Goleva E (2006) Divergent expression and function of glucocorticoid receptor β in human monocytes and T cells. J Leukoc Biol 79:818–827
Zhang X, Clark AF, Yorio T (2005) Regulation of glucocorticoid responsiveness in glaucomatous trabecular meshwork cells by glucocorticoid receptor-β. Invest Ophthalmol Vis Sci 46:4607–4616
Oakley RH, Jewell CM, Yudt MR, Bofetiado DM, Cidlowski JA (1999) The dominant negative activity of the human glucocorticoid receptor β isoform. Specificity and mechanisms of action. J Biol Chem 274:27857–27866
Goleva E, Li LB, Eves PT, Strand MJ, Martin RJ, Leung DY (2006) Increased glucocorticoid receptor β alters steroid response in glucocorticoid-insensitive asthma. Am J Respir Crit Care Med 173:607–616
Derijk RH, Schaaf MJ, Turner G, Datson NA, Vreugdenhil E, Cidlowski J, de Kloet ER, Emery P, Sternberg EM, Detera-Wadleigh SD (2001) A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor β-isoform mRNA is associated with rheumatoid arthritis. J Rheumatol 28:2383–2388
Lee CK, Lee EY, Cho YS, Moon KA, Yoo B, Moon HB (2005) Increased expression of glucocorticoid receptor β messenger RNA in patients with ankylosing spondylitis. Korean J Intern Med 20:146–151
Longui CA, Vottero A, Adamson PC, Cole DE, Kino T, Monte O, Chrousos GP (2000) Low glucocorticoid receptor α/β ratio in T-cell lymphoblastic leukemia. Horm Metab Res 32:401–406
Pujols L, Mullol J, Benitez P, Torrego A, Xaubet A, de Haro J, Picado C (2003) Expression of the glucocorticoid receptor α and β isoforms in human nasal mucosa and polyp epithelial cells. Respir Med 97:90–96
Shahidi H, Vottero A, Stratakis CA, Taymans SE, Karl M, Longui CA, Chrousos GP, Daughaday WH, Gregory SA, Plate JM (1999) Imbalanced expression of the glucocorticoid receptor isoforms in cultured lymphocytes from a patient with systemic glucocorticoid resistance and chronic lymphocytic leukemia. Biochem Biophys Res Commun 254:559–565
Piotrowski P, Burzynski M, Lianeri M, Mostowska M, Wudarski M, Chwalinska-Sadowska H, Jagodzinski PP (2007) Glucocorticoid receptor β splice variant expression in patients with high and low activity of systemic lupus erythematosus. Folia Histochem Cytobiol 45:339–342
Leung DY, Hamid Q, Vottero A, Szefler SJ, Surs W, Minshall E, Chrousos GP, Klemm DJ (1997) Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor β. J Exp Med 186:1567–1574
Webster JC, Oakley RH, Jewell CM, Cidlowski JA (2001) Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative β isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci USA 98:6865–6870
Xu Q, Leung DY, Kisich KO (2003) Serine-arginine-rich protein p30 directs alternative splicing of glucocorticoid receptor pre-mRNA to glucocorticoid receptor β in neutrophils. J Biol Chem 278:27112–27118
Strickland I, Kisich K, Hauk PJ, Vottero A, Chrousos GP, Klemm DJ, Leung DY (2001) High constitutive glucocorticoid receptor β in human neutrophils enables them to reduce their spontaneous rate of cell death in response to corticosteroids. J Exp Med 193:585–593
Orii F, Ashida T, Nomura M, Maemoto A, Fujiki T, Ayabe T, Imai S, Saitoh Y, Kohgo Y (2002) Quantitative analysis for human glucocorticoid receptor α/β mRNA in IBD. Biochem Biophys Res Commun 296:1286–1294
Tliba O, Damera G, Banerjee A, Gu S, Baidouri H, Keslacy S, Amrani Y (2008) Cytokines induce an early steroid resistance in airway smooth muscle cells: novel role of interferon regulatory factor-1. Am J Respir Cell Mol Biol 38:463–472
Chung CC, Shimmin L, Natarajan S, Hanis CL, Boerwinkle E, Hixson JE (2009) Glucocorticoid receptor gene variant in the 3′ untranslated region is associated with multiple measures of blood pressure. J Clin Endocrinol Metab 94:268–276
van den Akker EL, Koper JW, van Rossum EF, Dekker MJ, Russcher H, de Jong FH, Uitterlinden AG, Hofman A, Pols HA, Witteman JC, Lamberts SW (2008) Glucocorticoid receptor gene and risk of cardiovascular disease. Arch Intern Med 168:33–39
van den Akker EL, Nouwen JL, Melles DC, van Rossum EF, Koper JW, Uitterlinden AG, Hofman A, Verbrugh HA, Pols HA, Lamberts SW, van Belkum A (2006) Staphylococcus aureus nasal carriage is associated with glucocorticoid receptor gene polymorphisms. J Infect Dis 194:814–818
Wallberg AE, Neely KE, Hassan AH, Gustafsson JA, Workman JL, Wright AP (2000) Recruitment of the SWI-SNF chromatin remodeling complex as a mechanism of gene activation by the glucocorticoid receptor tau1 activation domain. Mol Cell Biol 20:2004–2013
Ma H, Hong H, Huang SM, Irvine RA, Webb P, Kushner PJ, Coetzee GA, Stallcup MR (1999) Multiple signal input and output domains of the 160-kilodalton nuclear receptor coactivator proteins. Mol Cell Biol 19:6164–6173
Webb P, Nguyen P, Shinsako J, Anderson C, Feng W, Nguyen MP, Chen D, Huang SM, Subramanian S, McKinerney E, Katzenellenbogen BS, Stallcup MR, Kushner PJ (1998) Estrogen receptor activation function 1 works by binding p160 coactivator proteins. Mol Endocrinol 12:1605–1618
Wallberg AE, Neely KE, Gustafsson JA, Workman JL, Wright AP, Grant PA (1999) Histone acetyltransferase complexes can mediate transcriptional activation by the major glucocorticoid receptor activation domain. Mol Cell Biol 19:5952–5959
Hittelman AB, Burakov D, Iniguez-Lluhi JA, Freedman LP, Garabedian MJ (1999) Differential regulation of glucocorticoid receptor transcriptional activation via AF-1-associated proteins. EMBO J 18:5380–5388
Lanz RB, McKenna NJ, Onate SA, Albrecht U, Wong J, Tsai SY, Tsai MJ, O’Malley BW (1999) A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 97:17–27
Gougat C, Jaffuel D, Gagliardo R, Henriquet C, Bousquet J, Demoly P, Mathieu M (2002) Overexpression of the human glucocorticoid receptor α and β isoforms inhibits AP-1 and NF-κB activities hormone independently. J Mol Med 80:309–318
Kelly A, Bowen H, Jee YK, Mahfiche N, Soh C, Lee T, Hawrylowicz C, Lavender P (2008) The glucocorticoid receptor β isoform can mediate transcriptional repression by recruiting histone deacetylases. J Allergy Clin Immunol 121:203–208e1
Kumar R, Thompson EB (2005) Gene regulation by the glucocorticoid receptor: structure: function relationship. J Steroid Biochem Mol Biol 94:383–394
He B, Bowen NT, Minges JT, Wilson EM (2001) Androgen-induced NH2- and COOH-terminal Interaction Inhibits p160 coactivator recruitment by activation function 2. J Biol Chem 276:42293–42301
Kauppi B, Jakob C, Farnegardh M, Yang J, Ahola H, Alarcon M, Calles K, Engstrom O, Harlan J, Muchmore S, Ramqvist AK, Thorell S, Ohman L, Greer J, Gustafsson JA, Carlstedt-Duke J, Carlquist M (2003) The three-dimensional structures of antagonistic and agonistic forms of the glucocorticoid receptor ligand-binding domain: RU-486 induces a transconformation that leads to active antagonism. J Biol Chem 278:22748–22754
Acknowledgment
Literary work of this article was funded partly by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kino, T., Su, Y.A. & Chrousos, G.P. Human glucocorticoid receptor isoform β: recent understanding of its potential implications in physiology and pathophysiology. Cell. Mol. Life Sci. 66, 3435–3448 (2009). https://doi.org/10.1007/s00018-009-0098-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-009-0098-z