
Abstract
Background
The present study was designed to explore the potential regulatory mechanism between mitophagy and pyroptosis during sepsis-associated acute lung injury (ALI).
Methods
In vitro or in vivo models of sepsis-associated ALI were established by administering lipopolysaccharide (LPS) or performing caecal ligation and puncture (CLP) surgery. Pyroptosis levels were detected by electron microscopy, immunofluorescence, flow cytometry, western blotting and immunohistochemistry. Dual-luciferase reporter gene assay was applied to verify the targeting relationship between miR-138-5p and NLRP3. Methylation-specific PCR and chromatin immunoprecipitation assays were used to determine methylation of the miR-138-5p promoter. Mitophagy levels were examined by transmission electron microscopy and western blotting.
Results
NLRP3 inflammasome silencing alleviated alveolar macrophage (AM) pyroptosis and septic lung injury. In addition, we confirmed the direct targeting relationship between miR-138-5p and NLRP3. Overexpressed miR-138-5p alleviated AM pyroptosis and the pulmonary inflammatory response. Moreover, the decreased expression of miR-138-5p was confirmed to depend on promoter methylation, while inhibition of miR-138-5p promoter methylation attenuated AM pyroptosis and pulmonary inflammation. Here, we discovered that an increased cytoplasmic mtDNA content in sepsis-induced ALI models induced the methylation of the miR-138-5p promoter, thereby decreasing miR-138-5p expression, which may activate the NLRP3 inflammasome and trigger AM pyroptosis. Mitophagy, a form of selective autophagy that clears damaged mitochondria, reduced cytoplasmic mtDNA levels. Furthermore, enhanced mitophagy might suppress miR-138-5p promoter methylation and relieve the pulmonary inflammatory response, changes that were reversed by treatment with isolated mtDNA.
Conclusions
In summary, our study indicated that mitophagy induced the demethylation of the miR-138-5p promoter, which may subsequently inhibit NLRP3 inflammasome, AM pyroptosis and inflammation in sepsis-induced lung injury. These findings may provide a promising therapeutic target for sepsis-associated ALI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sepsis is considered a lethal critical illness resulting from dysregulated host responses to infection, and sepsis-induced deaths account for 19.7% of deaths worldwide [1]. Sepsis is characterized by life-threatening organ dysfunction, and the lung is one of the most common and vulnerable organs [2]. Although the mechanism of acute lung injury (ALI) has been studied extensively for decades, the lack of effective therapeutic targets and strategies for ALI remains a significant challenge [3]. In addition, survivors of LPS-induced ALI have a poor prognosis and suffer long-term and irreversible physical impairment [4, 5]. Thus, the identification of novel therapeutic targets and exploration of effective measures for sepsis-associated ALI are very important to improve the prognosis and quality of life of patients.
As a type of selective autophagy, mitophagy is a targeted autophagy process that removes damaged or dysfunctional mitochondria and is very important for maintaining intracellular homeostasis [6]. Dysfunctional mitochondrial autophagy may lead to the accumulation of cytoplasmic mitochondrial DNA (mtDNA) [7]. In response to microbial infection, endogenous cell-derived molecules, known as damage-associated molecular patterns (DAMPs), have been reported to activate the immune response [8]. During LPS-induced ALI, LPS is one of the common pathogen-associated molecular patterns (PAMPs) serving as “signal 1” to activate nuclear factor-kappa B (NF-κB) in macrophages. Meanwhile, the cytosolic mtDNA released from impaired mitochondria acts as “signal 2” to activate the NOD-like receptor protein 3 (NLRP3) inflammasome in macrophages [9, 10]. The NLRP3 inflammasome has been reported to play an essential role in pyroptosis, a form of gasdermin D (GSDMD)-dependent programmed cell death mediated by inflammatory cysteinyl aspartate-specific proteases (caspases), which may contribute to ALI progression [9, 11,12,13]. However, the underlying mechanisms by which mtDNA activates the NLRP3 inflammasome in ALI have not been completely elucidated. miRNAs are short noncoding RNAs that suppress the expression of specific genes, such as NLRP3, by targeting their 3’ untranslated region [14, 15]. Moreover, a bioinformatics analysis identified NLRP3 as a putative target of miR-138-5p. miRNAs have been linked to the pathogenesis of respiratory diseases [16, 17]. Notably, miRNA promoter methylation may regulate miRNA expression and participate in the pathogenesis of several diseases [18, 19]. Ying Y et al. reported that miR-495 promoter methylation downregulates miR-495, which inhibits NLRP3 inflammasome activation to protect against ALI [20]. Interestingly, mtDNA released from damaged hepatocytes decreases the expression of miR-223 in neutrophils via Toll-like receptor 9 (TLR9)/NF-κB signalling [21]. Furthermore, an mtDNA deficiency might result in changes in the methylation of a number of genes, which might be reversed by restoring mtDNA in cells otherwise lacking the entire mitochondrial genome [22]. Besides, increased mtDNA copy numbers induce methylation of nuclear DNA cytosine–phosphate–guanine (CpG) [23]. Another study also reported that mitochondrial dysfunction increases miR-663 expression via hypermethylation of the miR-663 promoter [24]. miR-138-5p has been reported to be involved in several respiratory diseases, such as non-small cell lung cancer (NSCLC) [25] and ALI [26, 27]. However, the role of mtDNA in regulating miR-138-5p and NLRP3 expression during ALI has never been studied by performing in vivo and in vitro experiments.
As shown in our recent study, miR-138-5p suppresses NLRP3 inflammasome activation to alleviate pulmonary inflammatory reactions in ALI. This study aimed to explore whether mitophagy contributes to miR-138-5p promoter demethylation by regulating mtDNA and participates in alveolar macrophage (AM) pyroptosis during ALI.
Materials and methods
Cell culture and treatment
Rat AMs (NR8383 cells) purchased from the Chinese Academy of Sciences Cell Bank (Shanghai, China) were cultured in Ham’s F-12 K complete medium (Boster, PYG0036, Wuhan, China) containing 15% foetal bovine serum (Gibco, 10099-141, Australia) 37 °C with 5% CO2 in air. AMs were cultured in six-well plates at a density of 2 × 105 cells/well prior to transfection. AMs were transfected with short-hairpin-RNA (shRNA)-negative control (sh-NC) or shRNA-targeting NLRP3 (sh-NLRP3) (MOI = 80) purchased from GeneChem (Shanghai, China) to silence NLRP3. AMs were transfected with mimic-NC, miR-138-5p-mimic, inhibitor-NC, or miR-138-5p-inhibitor (RiboBio Co. Ltd, Guangzhou, Guangdong, China) mixed with riboFECTtm CP Reagent for 48 h to overexpress or knock down miR-138-5p, respectively. Furthermore, the working concentration was maintained at 100 nM. After transfection, the cells were treated with DMSO or 5-Aza (Sigma–Aldrich, USA, A3656) for 24 h. For mitophagy induction, AMs were treated with carbonyl cyanide m-chlorophenylhydrazone (CCCP, Med Chem Express, 100941, China) at a final concentration of 10 μM. AMs were transfected with isolated mtDNA for overexpression. AMs were treated with 1.0 μg/mL ethidium bromide (EtBr; Sigma Aldrich, E1510) to construct ρ0 cells lacking mtDNA. Macrophages used in our study were transfected using Lipofectamine 3000 (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s instructions. Subsequently, when the cells reached 70–80% confluence, AMs or ρ0 cells were treated with 1 μg/mL LPS (Escherichia coli 055:B5, Sigma–Aldrich) for 18 h to establish cell injury models. Experiments were performed at least 3 times in duplicate.
Establishment of a mouse model of sepsis-induced ALI
Eight-week-old male C57Bl/6 mice were purchased from Tianqin Biotechnology Co., Ltd. (Changsha, China). Beijing Viewsolid Biotech Co., Ltd. (Beijing, China) was commissioned to construct NLRP3-/- mice. Animals were randomly assigned to the sham, caecal ligation and puncture (CLP), NLRP3−/−, CLP + NLRP3−/−, CLP + AgomiR-NC, CLP + AgomiR-138-5p, CLP + 5-Aza, CLP + 5-Aza + AntagomiR-NC, CLP + 5-Aza + AntagomiR-138-5p, CLP + CCCP or CLP + CCCP + mtDNA groups. Animals were weighed and anaesthetized with 3% sodium pentobarbital. Ordinary mice were pretreated with PBS, AgomiR-NC (1 nmol in 50 μl of PBS, intratracheal injection, 72 h), AgomiR-138-5p (1 nmol in 50 μl of PBS, intratracheal injection, 72 h), 5-Aza (0.35 mg/kg, intraperitoneal injection, 3 times per week for 1 week), 5-Aza (0.35 mg/kg, intraperitoneal injection, 3 times per week for 1 week) + AntagomiR-NC (5 nmol in 50 μl of PBS, intratracheal injection, 72 h), 5-Aza (0.35 mg/kg, intraperitoneal injection, 3 times per week for 1 week) + AntagomiR-138-5p (5 nmol in 50 μl of PBS, intratracheal injection, 72 h), CCCP (4 mg/kg, intraperitoneal injection, 2 h), or CCCP (4 mg/kg, intraperitoneal injection, 2 h) + mtDNA (5 μg, intratracheal injection, 48 h). Mice in the sham and NLRP3-/- groups were subjected to laparotomy surgery without caecal ligation and perforation. At the same time, mice in other groups were subjected to CLP surgery. Briefly, the caecum was ligated with 3.0 silk thread at a location 1/2 of the blind end and punctured twice with an 18G needle. Then, a small amount of intestinal contents was extruded, and the abdomen was closed. The animals were euthanized 24 h after surgery. Experiments were performed at least 6 times in duplicate.
Scanning electron microscopy (SEM)
Cell membrane integrity was detected using SEM. Collected cells were fixed with 2.5% fresh glutaraldehyde (Solarbio, Shanghai, China, P1126) overnight at 4 °C and then rinsed three times for 45 min with 0.1 M PBS. Next, the cells were dehydrated in a graded series of ethanol solutions (30%, 50%, 70%, and 90%, 15–30 min each), 100% alcohol (15–30 min, twice), 100% ethanol:100% tert-butyl alcohol = 1:1 and 1:3 for 20 min each, and 100% tert-butyl alcohol for 20 min. The specimens were transferred to a critical point dryer for drying, and the dried samples were subsequently sputter-coated with gold–palladium. The images were collected with a scanning electron microscope (JSM-7001F, Japan) operating at 3.0 kV.
Transmission electron microscopy (TEM)
TEM was applied to determine the variations in cell membrane integrity and mitochondrial morphology. Cells or lung tissues were fixed overnight at 4 °C with 2.5% fresh glutaraldehyde (Solarbio, Shanghai, China, P1126) and washed with 0.1 M PBS. The fixed cells or lung tissues were treated with 1% osmium tetroxide for 2 h and then dehydrated in a graded series of ethanol solutions (30%, 50%, 70%, and 90%, 15–30 min each), followed by 100% alcohol (15–30 min, twice), and embedded in the embedding agent. Then, the samples were incubated at 70 °C for 12 h. Finally, 70–90 nm ultrathin sections were cut and sequentially stained with 2% uranium dioxide acetate for 15–30 min and lead citrate for 5–15 min. Finally, the cell membrane and mitochondria were visualized with a transmission electron microscope (JEM1200EX, Japan).
Haematoxylin and eosin (H&E) staining
The tissue samples were fixed with 4% paraformaldehyde, embedded in paraffin, and sliced into 4 µm thick sections. Lung tissue sections obtained after fixation were subjected to H&E staining to assess the lung injury severity. Images were captured using a microscope (Nikon), and the lung injury score was assessed by two pathologists, as previously described [28].
Immunohistochemistry
The expression levels of NLRP3, caspase-1 and cle-GSDMD levels in the lung tissues were detected using immunohistochemistry. Briefly, the lung tissue sections were immersed in citrate buffer at 100 °C (0.01 M, pH 6.0) for antigen recovery. Slices were incubated with a 3% hydrogen peroxide blocking solution for 10 min at room temperature to inhibit endogenous peroxidase activity. After blocking with 5% bovine serum albumin, the sections were incubated with specific primary antibodies against NLRP3 (Abcam, ab214185, 1:200, USA), caspase-1 (Proteintech, 22915-1-AP, 1:400, China), and cle-GSDMD (Affinity Biosciences, AF4012, 1:100) overnight at 4 °C. Next, the slices were washed with PBS three times and incubated with a goat anti-rabbit IgG secondary antibody (Boster, 1:200, Wuhan, China) at room temperature for 1 h. Afterward, the sections were visualized with a 3,3′-diaminobenzidine solution, counterstained with haematoxylin, and photographed and analysed with a light microscope (Olympus, Tokyo, Japan).
Immunofluorescence staining for FLICA caspase-1 and propidium iodide (PI)
Pyroptosis was detected by performing immunofluorescence staining for FLICA caspase-1 and PI using the FAM–FLICA Caspase Assay Kit (ImmunoChemistry Technologies, 98, USA).
Flow cytometry
Pyroptosis was detected using flow cytometry with the FAM–FLICA Caspase Assay Kit (ImmunoChemistry Technologies, 98, USA).
Western blot analysis
Cells were homogenized in radioimmunoprecipitation assay (RIPA) lysis buffer (Boster, AR0105, Wuhan, China) mixed with phenylmethylsulfonyl fluoride (PMSF), a protease inhibitor. After loading and separation on 10% SDS–PAGE gels, the proteins were subsequently transferred to polyvinylidene fluoride membranes. Then, 5% nonfat dry milk was applied to block the nonspecific protein binding sites for 2 h. Membranes containing target proteins were then incubated overnight at 4 °C on a shaker with the following primary antibodies: rabbit antibody against NLRP3 (Abcam, ab263899, 1:1000, USA), rabbit antibody against caspase-1 (Proteintech, 22915-1-AP, 1:1000, China), rabbit antibody against cle-GSDMD (Cell Signalling Technology, 39754, 1:1000, USA), rabbit antibody against LC3B (Abcam, ab192890, 1:2000, USA), and rabbit antibody against GAPDH (Proteintech, 10494-1-AP, 1:10,000, China). Afterward, the membranes were washed with TBST three times and incubated with HRP-conjugated Affinipure goat anti-rabbit IgG (H + L) (Proteintech, SA00001-2, 1:20,000, China) at room temperature for 1 h. Finally, the target proteins on the membranes were quantified using ImageJ software, with GAPDH serving as the internal reference.
ELISA quantification
The culture medium was collected and centrifuged to obtain the supernatant fractions. The BALF was collected from mice in each group 24 h after CLP. IL-1β (ab255730 and ab197742) and IL-18 (ab213909 and ab216165) levels in the cell supernatant and BALF were detected using ELISA kits purchased from Abcam.
Dual-luciferase reporter gene assay
The bioinformatics database http://www.miRNA.org/miRNA/home.do was used to analyse the interaction between miR-138-5p and NLRP3. According to the predicted binding site, the synthesized 3′ UTR sequence of the NLRP3 mRNA and its mutant sequence were integrated into the pmirGLO vector (Promega, CA, USA) to generate NLRP3-wild type (WT) and NLRP3-Mut reporter plasmids. Next, NLRP3-WT and NLRP3-Mut reporter plasmids with correct sequences were cotransfected with miR-138-5p-mimic or mimic-NC into cells. Forty-eight hours later, the luciferase activity in cells was detected using the dual-luciferase reporter assay system (E1910, Promega, CA, USA).
RNA extraction and qRT–PCR analysis
Total RNA was extracted from cells or lung tissues using a TransZol Up Plus RNA Kit (TransGen Biotech, ER501-01, China). Next, miRNAs were quantified using a Bulge-loop™ miRNA qRT–PCR Primer Set designed by RiboBio (RiboBio Co. Ltd., Guangdong, China). The U6 gene was used as the internal reference for miR-138-5p. The β-actin gene served as the internal reference for NLRP3 and caspase-1. qRT–PCR was performed with a LightCycler PCR instrument (Roche) using PerfectStart® Green qPCR SuperMix (TransGen Biotech, China) according to the manufacturer’s instructions. The primer sequences for qRT–PCR were as follows: U6, forward: CTCGCTTCGGCAGCACATATACTA, reverse: ACGAATTTGCGTGTCATCCTTGC; miR-138-5p, forward: GGCCGGACTAAGTGTTGT, reverse: GCAGGGTCCGAGGTATTC;; β-actin, forward: TACTGCCCTGGCTCCTAGCA, reverse: TGGACAGTGAGGCCAGGATAG; NLRP3, forward: AACTTGCAGAAGCTGGGGTT, reverse: GGTGCAGAAGTCCCTCACAG; and caspase-1, forward: AACACCCACTCGTACACGTC, reverse: TGAGGTCAACATCAGCTCCG.
Methylation-specific PCR (MSP) assay
The EZ DNA Methylation-Gold Kit (Zymo Research, D5005, Irvine, CA, USA) was used to measure the methylation of the miR-138-5p promoter region. In MSP amplification, the sequences of the primers for detecting promoter methylation were miR-138-5p-MF: TTTTAATCGATTGGAGTCGGA and miR-138-5p-MR: TATATCCTATAATCCCGCGAAA, and the sequences of the primers for detecting the unmethylated promoter were miR-138-5p-UF: TTAATTGATTGGAGTTGGAG and miR-138-5p-UR: TCCTATATCCTATAATCCCACAAAA. These primers were synthesized by BGI Genomics Co., Ltd. (China). The purified DNA sample and the CT conversion reagent were added to a tube for denaturation and bisulfite conversion. Then, the bisulfite-treated DNA was amplified using “hot start” polymerases (ZymoTaq™). Agarose gel electrophoresis was applied to detect the amplification products.
Chromatin immunoprecipitation (CHIP) assay
AMs treated with 1% formaldehyde (final concentration) were collected and ultrasonically crushed. Antibodies against DNA methyltransferase 1 (DNMT1) (Santa Cruz Biotechnology, sc-271729), DNA methyltransferase 3a (DNMT3a) (Santa Cruz Biotechnology, sc-373905), and DNA methyltransferase 3b (DNMT3b) (Santa Cruz Biotechnology, sc-376043) were bound to the miR-138-5p gene promoter and precipitated using Protein A agarose/salmon sperm DNA. Afterward, the immunoprecipitates were washed to remove nonspecifically bound DNA, followed by reverse cross-linking and purification, and the eluted products were subjected to qRT–PCR.
DNA isolation and mtDNA copy number analysis
The mtDNA copy number was assessed using qRT–PCR as previously described [29]. Total DNA was isolated from AMs or ρ0 cells using the M5 HiPer Universal DNA Mini Kit (Mei5bio), and 100 ng were detected using qRT–PCR analysis. The mtDNA and nuclear DNA contents were determined by amplifying Mt-cyb and Actb sequences. The mtDNA copy number was calculated as the mtDNA/nDNA ratio. The following primers for qRT–PCR were synthesized by Tsingke Biotechnology Co., Ltd. (China): mitochondrial (Mt-cyb), forward: GCAGCTTAACATTCCGCCCAATCA, reverse: TACTGGTTGGCCTCCGATTCATGT; and nuclear (Actb), forward: ATCATGTTTGAGACCTTCAACACCC, reverse: CATCTCTTGCTCGAAGTCTAGG.
Detection of mtDNA in cytosolic extracts
Digitonin cell extracts were generated as previously described [30]. Cells were divided into two equal parts; one was resuspended in 500 μL of 50 μM NaOH and boiled for 30 min to dissolve DNA. Fifty microlitres of 1 M Tris–HCl (pH 8) were added to neutralize the pH, and these extracts served as normalization controls for total mtDNA. The second equal part was resuspended in approximately 500 μL of buffer containing 150 mM NaCl, 50 mM HEPES (pH 7.4), and 15–25 µg/ml digitonin. The homogenates were incubated at room temperature for 10 min with end-over-end rotation to allow selective permeabilization of the plasma membrane and then centrifuged at 1000 g for 10 min three times to pellet intact cells. The cytosolic supernatant was transferred to fresh tubes and centrifuged at 17,000g for 10 min to eliminate any remaining cell debris. DNA was then isolated from these pure cytosolic fractions using the M5 HiPer Universal DNA Mini Kit (Mei5bio). mtDNA-specific primers were used to conduct qRT–PCR of both whole-cell extracts and cytosolic fractions. The obtained Ct values reflected the mtDNA abundance.
mtDNA isolation and transfection
According to the manufacturer’s instructions, mtDNA was isolated from the rat liver using the Mitochondrial DNA Isolation Kit (Biovision, K280-50). The mtDNA was resuspended in TE buffer and stored at − 20 °C for future use. The concentration and A260/280 value of mtDNA were determined using a Nanodrop 2000 ultramicroscopic spectrophotometer. Cells were transfected with 5 μg/mL mtDNA using Lipofectamine 3000 (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s instructions.
Immunofluorescence staining of lung tissues
OCT-embedded fresh mouse lung tissues were cut into 5 μm sections. Sections were blocked with PBS containing 2% bovine serum albumin and 0.2% Triton X-100 at room temperature for 30 min and coincubated with antibodies against CD68 (Immunoway, YM3050, 1:100, China), NLRP3 (Boster, BA3677, 1:200, Wuhan, China) and cle-GSDMD (Affinity Biosciences, AF4012, 1:100) at 4 °C overnight. Afterward, the slices were incubated with the corresponding secondary antibodies, AF488-conjugated goat anti-mouse IgG (H + L) or AF594-conjugated goat anti-rabbit IgG (H + L), at 25 °C in the dark. The nuclei were stained using DAPI staining solution. Images were obtained with an Olympus IX71 microscope (Olympus, Tokyo, Japan).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 7. Measurement data are presented as the means ± SD. An unpaired t test was used to analyse data between two groups that conformed to normal distribution and homogeneity of variance. Comparisons among multiple groups were assessed using one-way ANOVA and Tukey’s multiple comparisons test. A value of P < 0.05 was considered statistically significant.
Results
Effect of NLRP3 on sepsis-associated ALI induced by AM pyroptosis
Normal and LPS-treated AMs were assayed for cell membrane pore formation using SEM and TEM to verify the effect of LPS on AM pyroptosis. LPS promoted the formation of pores in AM membranes, indicating the oligomerization of activated GSDMD-N-terminal at the cell membrane (Fig. 1A, B). Previous studies have indicated an important role of the NLRP3 inflammasome in AM pyroptosis during sepsis-induced ALI. WT and NLRP3−/− mice were subjected to a sham operation or CLP to further verify this finding. Compared with the sham group, histology revealed that the CLP group exhibited significant alveolar structure destruction, accompanied by alveolar haemorrhage, pulmonary oedema, a thickened alveolar wall, neutrophil infiltration, and increased lung injury scores, which were markedly reversed by NLRP3 knockdown (Fig. 1C, D). Moreover, the expression of NLRP3, caspase-1, and cle-GSDMD in mouse lung tissues was evaluated using immunohistochemical staining. The expression levels of the aforementioned proteins in the CLP group were higher than those in the sham group, whereas NLRP3 knockdown decreased the expression levels of these proteins in mice subjected to CLP (Fig. 1E, F). Furthermore, LPS-treated AMs were transfected with sh-NC or sh-NLRP3. Next, immunofluorescence staining for FLICA caspase-1 and PI was applied to detect pyroptosis, and the results revealed increased pyroptosis of AMs after LPS treatment, which was rescued by NLRP3 silencing (Fig. 1G). The pyroptosis of AMs was further measured using flow cytometry, as illustrated in Fig. 1H, I; exposure to LPS indeed increased the rate of pyroptosis, while transfection with sh-NLRP3 blocked the pyroptosis of LPS-treated AMs. In addition, the increased expression levels of NLRP3, pro-caspase-1, P20, GSDMD, and cle-GSDMD induced by LPS were markedly reversed by sh-NLRP3 transfection, as determined using western blotting (Fig. 1J, K). Moreover, quantitative ELISA revealed that LPS increased the production of the inflammatory cytokines IL-Iβ and IL-18. However, the levels of the inflammatory cytokines IL-Iβ and IL-18 in the LPS + sh-NLRP3 group were significantly lower than those in the LPS + sh-NC group (Fig. 1L), suggesting that NLRP3 deficiency may relieve LPS-induced inflammation in AMs. In summary, our data indicate that NLRP3 deficiency significantly blocks AM pyroptosis and pulmonary inflammatory responses in sepsis-induced ALI models.

NLRP3 silencing alleviates sepsis-associated ALI induced by AM pyroptosis. A, B SEM and TEM show pore formation in the cell membrane after LPS treatment; scale bars, 100 μm. WT and NLRP3-/- mice underwent a sham operation or CLP. C, D Pathological alterations in mouse lung tissues were examined using H&E staining (200×); scale bars 100 μm. E, F Immunohistochemical staining showing the NLRP3, caspase-1, and cle-GSDMD contents in mouse lung tissues (200×); scale bars 100 μm. G Immunofluorescence staining for FLICA caspase-1 and PI indicates the pyroptosis of AMs (200 ×); scale bars 100 μm. H, I Pyroptosis levels in AMs were determined using flow cytometry. J, K Western blotting detected the levels of the NLRP3, pro-caspase-1, P20, GSDMD, and Cle-GSDMD proteins in AMs. L Expression levels of IL-1β and IL-18 in AMs were measured using ELISA. Measurement data are presented as the means ± SD (n = 3/6). *P < 0.05, **P < 0.01, ***P < 0.001; NS no statistically significant difference
miR-138-5p directly targets NLRP3 to suppress the pulmonary inflammatory response
Since an increasing number of studies have revealed that dysregulated miRNAs in macrophages participate in the occurrence and development of sepsis, RNAinter and miRDB were applied to predict miRNAs targeting NLRP3 in mice (Fig. 2A). A bioinformatics analysis was subsequently conducted to explore the relationship between miR-138-5p and NLRP3, and the results revealed that the complementary region in the 3′ UTR of the NLRP3 gene contains the potential target miRNA-138-5p sequence (Fig. 2B). Moreover, a dual-luciferase reporter gene assay revealed that miR-138-5p-mimic transfection significantly decreased the luciferase activity of the WT-NLRP3-3′ UTR construct but not the mutant NLRP3-3′ UTR in AMs (Fig. 2C). As determined using qRT–PCR analysis, LPS stimulation significantly diminished miR-138-5p expression in AMs, suggesting that miR-138-5p may play an essential role in the etiopathogenesis of sepsis-associated ALI (Fig. 2D). Then, we transfected LPS-treated AMs with miR-138-5p-mimic or mimic-NC. Immunofluorescence staining for FLICA caspase-1 and PI (Fig. 2E) and flow cytometry (Fig. 2F, G) were applied to detect the pyroptosis of AMs, and the results showed that miR-138-5p-mimic transfection protected LPS-treated AMs from pyroptosis. Similarly, western blot analysis showed higher levels of NLRP3, pro-caspase-1, P20, GSDMD, and cle-GSDMD in the LPS group than in the control group, changes that were markedly reversed by miR-138-5p-mimic transfection (Fig. 2H, I). Next, The levels of IL-1β and IL-18 were quantified using ELISA, and LPS increased the levels of these inflammatory cytokines. However, transfection of the miR-138-5p-mimic decreased the levels of these inflammatory cytokines in LPS-exposed AMs (Fig. 2J). In addition, H&E staining indicated that CLP-treated animals showed significant histopathological alterations and higher lung injury scores than sham mice, which were significantly reversed by AgomiR-138-5p treatment (Fig. 2K, L). As detected using immunohistochemistry, the expression levels of NLRP3, caspase-1, and cle-GSDMD in mouse lung tissues were significantly upregulated after CLP surgery, but these changes were ameliorated by the delivery of AgomiR-138-5p (Fig. 2M, N). In summary, miR-138-5p alleviates AM pyroptosis and pulmonary inflammatory responses by directly targeting the NLRP3 inflammasome and inhibiting its activation during sepsis-associated ALI.

miR-138-5p inhibits pulmonary inflammation by directly targeting NLRP3. A miRNAs targeting mouse NLRP3 were screened using RNAinter and miRDB. B Binding site between miR-138-5p and the NLRP3 3′ UTR was predicted using the bioinformatics database miRNA.org. C Dual-luciferase reporter gene assay detecting the luciferase activity of NLRP3-WT and NLRP3-Mut after mimic-NC or miR-138-5p-mimic cotransfection. D Expression of miR-138-5p in AMs treated with PBS or LPS, as detected using qRT–PCR. E Immunofluorescence staining for FLICA caspase-1 and PI indicates the pyroptosis of AMs (200×); scale bars 100 μm. F, G Pyroptosis levels in AMs determined using flow cytometry. H, I Western blots showing the levels of the NLRP3, pro-caspase-1, P20, GSDMD, and Cle-GSDMD proteins in AMs. J Levels of IL-1β and IL-18 in LPS-exposed AMs after mimic-NC and miR-138-5p-mimic transfection were detected using ELISA. K, L Representative images of the histology of lung tissues from mice subjected to a sham operation, CLP, CLP + AgomiR-NC, or CLP + AgomiR-138-5p (200 ×); scale bars 100 μm. M, N Immunohistochemical staining for NLRP3, caspase-1, and cle-GSDMD contents in mouse lung tissues (200×); scale bars 100 μm. Measurement data are presented as the means ± SD (n = 3/6). *P < 0.05, **P < 0.01, ***P < 0.001; NS no statistically significant difference
miR-138-5p expression was reduced in a methylation-dependent manner during sepsis-associated ALI
Notably, miRNA promoter methylation leads to decreased miRNA expression. Accordingly, we wondered whether the decreased expression of miR-138-5p in LPS-treated AMs was caused by miR-138-5p promoter methylation. Using the MethPrimer website, we confirmed the presence of a CpG island in the miR-138-5p promoter region (Fig. 3A) and subsequently performed an MSP assay using designed primers. LPS stimulation resulted in methylation of the miR-138-5p promoter region in AMs, which was significantly reversed by treatment with 5-Aza (Fig. 3B). Moreover, the CHIP assay revealed increased levels of DNMT1 and DNMT3a/b enrichment at the miR-138-5p promoter region in LPS-stimulated AMs compared to unstimulated AMs but were decreased in LPS-stimulated AMs treated with 5-Aza relative to the LPS-stimulated AMs administered DMSO (Fig. 3C). As evidenced by the qRT–PCR analysis, miR-138-5p expression was significantly decreased in LPS-treated AMs compared with untreated AMs, whereas 5-Aza stimulation upregulated the expression of miR-138-5p in LPS-exposed AMs (Fig. 3D). In addition, the findings of the MSP assay also suggested that the miR-138-5p promoter region was methylated in the lung tissues from mice subjected to CLP compared to sham mice but were rescued by the administration of the DNA methylation inhibitor 5-Aza (Fig. 3E). Moreover, the miR-138-5p expression level in lung tissues from the CLP group was decreased compared to the sham group; compared with the CLP group, the expression level of miR-138-5p in the CLP + 5-Aza group was increased (Fig. 3F). Thus, based on the aforementioned data, we inferred that miR-138-5p expression is diminished in a methylation-dependent manner during sepsis-associated ALI.

Methylation of the miR-138-5p promoter in AMs may result in decreased expression of miR-138-5p during sepsis-associated ALI. A CpG island in the miR-138-5p promoter region predicted by the MethPrimer database. B Methylation of the miR-138-5p promoter in AMs examined using the MSP assay. C Levels of DNMT1 and DNMT3a/b enrichment in miR-138-5p promoter of AMs, as detected using CHIP assay. D Expression of miR-138-5p in AMs, as measured using qRT–PCR analysis. E Methylation of the miR-138-5p promoter in lung tissues from mice subjected to a sham operation, CLP, CLP + 5-Aza, as determined using the MSP assay. F Expression of miR-138-5p in mouse lung tissues, as detected using qRT–PCR analysis. Measurement data are presented as the means ± SD (n = 3/6). *P < 0.05, **P < 0.01, ***P < 0.001; NS no statistically significant difference
miR-138-5p promoter demethylation attenuates the pyroptosis and inflammation of AMs in sepsis-associated ALI
LPS induced inflammatory responses and pyroptosis, especially methylation of the miR-138-5p promoter in AMs. We hypothesized that inhibition of miR-138-5p promoter methylation would block inflammation and pyroptosis in AMs. AM pyroptosis was detected by performing immunofluorescence staining for FLICA caspase-1 and PI to verify this hypothesis; 5-Aza treatment protected LPS-treated AMs from pyroptosis, while miR-138-5p-inhibitor transfection offset the protective effects of 5-Aza treatment (Fig. 4A). Similarly, flow cytometry was also applied to detect the pyroptosis of AMs, and the results showed that 5-Aza protected LPS-treated AMs from pyroptosis, while transfection of the miR-138-5p inhibitor offset the protective effect of 5-Aza (Fig. 4B, C). In addition, western blot analysis showed higher levels of NLRP3, pro-caspase-1, P20, GSDMD, and cle-GSDMD in the LPS group than in the control group, which were markedly rescued by the delivery of 5-Aza, suggesting that the delivery of 5-Aza blocked LPS-induced pulmonary inflammatory responses and pyroptosis. However, the anti-inflammatory effect of 5-Aza was reversed by miR-138-5p-inhibitor transfection (Fig. 4D, E). The levels of IL-1β and IL-18 were quantified ELISA, and LPS increased the levels of these inflammatory factors. However, the administration of 5-Aza diminished the levels of these inflammatory factors in LPS-exposed AMs, while the miR-138-5p inhibitor abolished the anti-inflammatory effects of 5-Aza (Fig. 4F). Besides, we also detected histopathological changes in lung tissues using H&E staining. As illustrated in Fig. 4G, H, 5-Aza significantly inhibited alveolar inflammatory damage in mice subjected to CLP; however, the anti-inflammatory effects of 5-Aza were weakened when AntagomiR-138-5p was administered. The expression levels of NLRP3, caspase-1 and cle-GSDMD in mouse lung tissues were evaluated using immunohistochemistry (Fig. 4I, J) and CLP surgery increased the expression levels of these proteins. Treatment with 5-Aza reduced the expression levels of these proteins, while a miR-138-5p inhibitor abolished the protective functions of 5-Aza. Briefly, inhibition of miR-138-5p promoter methylation suppresses the inflammatory response and pyroptosis of AMs during sepsis-induced ALI.

Suppression of miR-138-5p promoter methylation attenuates the pyroptosis and inflammation of AMs in response to sepsis-induced ALI. A Immunofluorescence staining for FLICA caspase-1 and PI represents the pyroptosis of AMs (200×); scale bars 100 μm. B, C Pyroptosis of AMs, as determined using flow cytometry. D, E Western blots showing the levels of the NLRP3, pro-caspase-1, P20, GSDMD, and Cle-GSDMD proteins in AMs. F Expression levels of IL-1β and IL-18 in AMs, as examined using ELISA. G, H Pathological alterations in mouse lung tissues measured using H&E staining (200 ×); scale bars 100 μm. I, J Immunohistochemical staining for NLRP3, caspase-1, and cle-GSDMD contents in mouse lung tissues (200×); scale bars 100 μm. Measurement data are presented as the means ± SD (n = 3/6). *P < 0.05, **P < 0.01, ***P < 0.001; NS no statistically significant difference
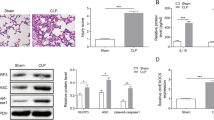
Increased mitophagy reduces cytoplasmic mtDNA levels, suppressing miR-138-5p promoter methylation
A retrograde pathway from mitochondria to the nucleus has been identified, and mitochondrial metabolism, including an altered mtDNA copy number, may affect epigenetic modifications in the nucleus, such as nuclear DNA methylation [24]. As determined using TEM, the formation of mitophagosomes was observed in mouse lung tissues after CLP surgery. In addition, CCCP treatment enhanced mitophagy and contributed to the formation of both mitophagosomes and mitolysosomes in the lung tissues of mice subjected to CLP (Fig. 5A). Consistent with the TEM results, western blot analysis of the autophagy biomarker LC3B also revealed activated autophagy in the CLP group that was further enhanced after CCCP treatment (Fig. 5B, C). A previous study revealed that mitochondria are damaged and cytoplasmic mtDNA levels are significantly increased in macrophages after LPS stimulation [9]. Thus, we speculated that the increase in mitophagy might suppress the methylation of the miR-138-5p promoter by reducing the cytoplasmic mtDNA level. AMs were treated with low concentrations or EtBr (0.1–2 μg/mL) to deplete mtDNA and to verify this hypothesis [31]. Treatment with 1.0 μg/mL EtBr significantly reduced the mtDNA copy number in AMs (Fig. 5D), suggesting that ρ0 cells were successfully constructed. Next, we detected cytoplasmic mtDNA levels in AMs or ρ0 cells using qRT–PCR analysis. As expected, the results revealed that LPS increased cytoplasmic mtDNA levels in AMs but not ρ0 cells. Interestingly, CCCP stimulation decreased cytoplasmic mtDNA levels to a similar extent as observed in LPS-treated ρ0 cells, which was reversed by the transfection of isolated mtDNA (Fig. 5E). MSP (Fig. 5F) and CHIP (Fig. 5G) assays indicated that LPS exposure induced miR-138-5p promoter methylation, whereas mtDNA deficiency induced by the mitophagy inducer CCCP markedly inhibited LPS-induced miR-138-5p promoter methylation, which was significantly rescued by the transfection of isolated mtDNA. Enhanced mitophagy increased miR-138-5p expression in LPS-treated AMs, which was reversed by the transfection of isolated mtDNA (Fig. 5H). As determined using the MSP assay, miR-138-5p promoter methylation levels were increased in the CLP group compared with the sham group, while enhanced mitophagy reduced methylation levels of the miR-138-5p promoter to a similar extent as observed in the CLP + 5-Aza group. In addition, mtDNA treatment increased miR-138-5p promoter methylation levels (Fig. 5I). The miR-138-5p expression level was reduced in lung tissues from the CLP group compared to that in the sham group. However, the mitophagy inducer CCCP increased the miR-138-5p expression level in lung tissues from the CLP group, which was reversed by mtDNA treatment (Fig. 5J). In summary, enhanced mitophagy may reduce cytoplasmic mtDNA levels to inhibit methylation of the miR-138-5p promoter.

Enhanced mitophagy may reduce cytoplasmic mtDNA levels to inhibit miR-138-5p promoter methylation. A Mitophagosomes and mitolysosomes in the lung tissues of mice. Purple arrow, nucleus; blue arrow, normal mitochondria; red arrow, mitophagosome; and double red arrow, mitolysosome. B, C Levels of LC3BI and LC3BII proteins in mouse lung tissues were detected using western blotting. D Ratio of mtDNA to genomic DNA in whole extracts of AMs, as determined using qRT–PCR analysis. E mtDNA levels in the cytosol of AMs treated with PBS, LPS, LPS + CCCP, LPS + CCCP + mtDNA or ρ0 cells treated with LPS. F Methylation of the miR-138-5p promoter in AMs or ρ0 cells, as determined using the MSP assay. G Levels of DNMT1 and DNMT3a/b enrichment in miR-138-5p promoter of AMs or ρ0 cells, as detected using the CHIP assay. H qRT–PCR analysis of the expression of miR-138-5p in AMs or ρ0 cells. I Methylation of the miR-138-5p promoter in mouse lung tissues from the sham, CLP, CLP + CCCP, CLP + CCCP + mtDNA, and CLP + 5-Aza groups, as measured using the MSP assay. J Expression of miR-138-5p in mouse lung tissue, as determined using qRT–PCR analysis. Measurement data are presented as the means ± SD (n = 3/6). *P < 0.05, **P < 0.01, ***P < 0.001; NS no statistically significant difference
Increased mitophagy alleviates sepsis-associated ALI induced by AM pyroptosis
Immunofluorescence staining for FLICA caspase-1 and PI was performed to examine the pyroptosis of AMs and determine the role of mitophagy in AM pyroptosis and ALI. The autophagy inducer CCCP protected LPS-treated AMs from pyroptosis, similar to the effect of ρ0 cells. Transfection with isolated mtDNA offset the protective effects of CCCP (Fig. 6A). Similarly, the pyroptosis of AMs was also detected by flow cytometry, and the result was consistent with that of immunofluorescence staining (Fig. 6B, C). Furthermore, the levels of NLRP3, pro-caspase-1, P20, GSDMD, and cle-GSDMD were examined using western blot analysis, and LPS increased the levels of these proteins, while the administration of CCCP decreased the levels of these proteins to a similar extent as observed in LPS-treated ρ0 cells. However, transfection with isolated mtDNA abolished the anti-inflammatory effects of CCCP (Fig. 6D, E). ELISA quantification revealed higher levels of IL-1β and IL-18 in the LPS group than in the control group, which were markedly rescued by increased mitophagy, suggesting that enhanced mitophagy blocked LPS-induced pulmonary inflammatory responses. In contrast, the anti-inflammatory effect of enhanced mitophagy was reversed by the transfection of isolated mtDNA (Fig. 6F). Moreover, H&E staining was applied to detect histopathological changes in lung tissues. As illustrated in Fig. 6G, H, enhanced mitophagy markedly suppressed alveolar destruction in mice subjected to CLP; however, the protective effects of enhanced mitophagy were weakened after isolated mtDNA transfection. As determined using immunohistochemistry (Fig. 6I, J), the levels of NLRP3, caspase-1 and cle-GSDMD in the CLP group were higher than those in the sham group, whereas treatment with CCCP decreased the levels of these proteins, which were reversed by the transfection of isolated mtDNA. Overall, these results substantiate the hypothesis that increased mitophagy may suppress AM pyroptosis to alleviate sepsis-associated ALI.

Enhanced mitophagy relieves sepsis-associated ALI induced by AM pyroptosis. A Immunofluorescence staining for FLICA caspase-1 and PI represents the pyroptosis of AMs treated with PBS, LPS, the combination of PBS and CCCP, or the combination of LPS and CCCP, or of ρ0 cells treated with LPS (200 ×); scale bars 100 μm. B, C Pyroptosis of AMs, as determined using flow cytometry. D, E Levels of the NLRP3, pro-caspase-1, P20, GSDMD, and Cle-GSDMD proteins in AMs were determined using western blotting. F Expression levels of IL-1β and IL-18 in AMs were detected using ELISA. G, H After establishing the sepsis-induced ALI model, lung tissue samples were collected to analyse the pathological alterations (200×); scale bars 100 μm. I, J Immunohistochemical staining for NLRP3, caspase-1, and cle-GSDMD contents in mouse lung tissues (200×); scale bars 100 μm. Measurement data are presented as the means ± SD (n = 3/6). *P < 0.05, **P < 0.01, ***P < 0.001; NS no statistically significant difference
Mitophagy-induced demethylation of the miR-138-5p promoter suppresses pyroptosis to protect against sepsis-associated ALI in vivo
Immunofluorescence staining of lung tissues was conducted to further verify the underlying mechanism by which mitophagy inhibits AM pyroptosis during sepsis-induced ALI. As illustrated by the number of CD68/NLRP3 (Fig. 7A, andC) or CD68/cle-GSDMD (Fig. 7B, andD) double-positive cells, the inflammation and pyroptosis of AMs in animals with sepsis-associated ALI was markedly inhibited by NLRP3 knockout, miR-138-5p overexpression, DNA demethylation, and enhanced mitophagy, but these changes were significantly reversed when additional AntagomiR-138-5p or mtDNA was administered. Quantitative ELISAs were also applied to examine the levels of proinflammatory factors in BALF from mice with sepsis-associated ALI, and the results revealed that NLRP3 knockout, AgomiR-138-5p, 5-Aza, and CCCP mitigated the inflammatory response of mice subjected to CLP, while miR-138-5p knockdown or mtDNA overexpression offset their protective effects (Fig. 7E). The qRT–PCR analysis showed reduced levels of NLRP3 and caspase-1 mRNAs following NLRP3 knockout, miR-138-5p overexpression, DNA demethylation, and enhanced mitophagy, which were reversed by miR-138-5p knockdown or mtDNA overexpression (Fig. 7F).

Mitophagy-induced demethylation of the miR-138-5p promoter blocks the pyroptosis and inflammation of AMs in vivo. A–D Immunofluorescence staining for CD68 (green) and NLRP3/cle-GSDMD (red) in AMs during sepsis-associated ALI. The inflammatory response and pyroptosis of AMs were suppressed by NLRP3 knockout, AgomiR-138-5p, 5-Aza and CCCP, while AntagomiR-138-5p or mtDNA treatment augmented the inflammatory response and pyroptosis of AMs in a model of sepsis-induced ALI (200 ×); scale bars, 100 μm. E Expression levels of IL-1β and IL-18 in BALF were determined using ELISA. F NLRP3 and caspase-1 mRNA expression levels were detected using qRT–PCR analysis. Measurement data are presented as the mean ± SD (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001; NS no statistically significant difference
Discussion
In the present study, LPS treatment contributed to NLRP3 inflammasome activation, while the overexpression of miR-138-5p restrained NLRP3 inflammasome activation, eventually inhibiting the progression of sepsis-associated ALI. In addition, methylation of the miR-138-5p promoter was observed in LPS-treated AMs and mice subjected to CLP, resulting in the downregulation of miR-138-5p. We also showed that LPS treatment in vitro and CLP surgery in vivo increased the level of cytoplasmic mtDNA, which subsequently mediated pyroptosis to aggravate pulmonary inflammatory responses. Moreover, the present study revealed that enhanced mitophagy inhibited the methylation of the miR-138-5p promoter during sepsis-associated ALI. Based on the aforementioned findings, we hypothesized that increasing mitophagy suppresses the methylation of the miR-138-5p promoter to alleviate pulmonary inflammation, which may be a promising therapeutic target for sepsis-associated ALI (Fig. 8).

Schematic diagram describing the mechanism by which mitophagy inhibits pyroptosis through miR-138-5p promoter demethylation in sepsis-induced ALI model. Cytoplasmic mtDNA downregulates miR-138-5p expression by promoting miR-138-5p promoter methylation, which activates the NLRP3 inflammasome, resulting in caspase-1 activation. Inflammatory caspases promote the cleavage and activation of the pore-forming effector protein GSDMD, which cleaves pro-IL-1β and pro-IL-18 into IL-1β and IL-18. Accordingly, AM pyroptosis levels were increased to aggravate ALI. However, mitophagy could induce miR-138-5p promoter demethylation by reducing cytoplasmic mtDNA level, thereby inhibiting AM pyroptosis and alleviating septic lung injury
As shown in our previous studies, NLRP3 inflammasome activation is involved in the inflammatory responses to LPS-induced ALI, the inhibition of which alleviates LPS-induced ALI [26, 27]. LPS treatment activates the NLRP3 inflammasome and increases the levels of caspase-1, cle-GSDMD, and inflammatory cytokines, such as IL-1β and IL-18 [20, 32], consistent with our findings. Furthermore, miRNAs have been shown to regulate inflammatory responses by inhibiting NLRP3 [33, 34]. For example, miR-7b overexpression reduces NLRP3 expression, thus downregulating caspase-1, IL-1β, and IL-18 levels [15]. Feng et al. [35, 36]. reported that miR-138-5p targets the NLRP3/Caspase-1 signalling pathway to regulate hippocampal neuroinflammation and cognitive dysfunction in rats. Notably, miR-138-5p functions as a tumour suppressor to repress the proliferation and invasion of lung cancer cells [37, 38] and regulate the development of ALI [26, 27]. Our recent study indicated that the lncRNA NLRP3 downregulated miR-138-5p to promote pyroptosis and exacerbate inflammatory responses by targeting NLRP3 during LPS-induced ALI [26]. Overexpression of miR-138-5p inhibited NLRP3 inflammasome activation, inflammatory injury, and pyroptosis in AMs in a sepsis-associated ALI model.
DNA methylation, a crucial component of the epigenetic modification of mammalian genomic DNA, plays an important role in mediating multiple disease processes. In addition, a high methylation level in promoters is likely to induce gene silencing by repressing transcription [39]. DNA methylation marks potentially provide biomarker information for patients with severe sepsis and even help differentiate between early and late-onset sepsis [40, 41]. A genome-wide analysis of DNA methylation has shown that abnormal DNA methylation in lung tissues may participate in the pathophysiological process of LPS-induced ALI [42]. Numerous findings have revealed that methylation of miRNA promoters participates in the transformation or progression of human diseases [43, 44]. According to a recent study, miR-19b-3P protects mice from inflammatory injury, while methylation of the miR-19b-3P promoter aggravates the inflammatory response in LPS-induced ALI [45]. Methylation-associated silencing of miR-495 inhibits the proliferation and invasion of human gastric cancer and breast cancer cells [46]. Moreover, Ying et al. [20] discovered that LPS stimulation leads to miR-495 promoter methylation, which may downregulate the expression level of miR-495, thus exacerbating lung injury, promoting neutrophil infiltration, and contributing to the progression of LPS-triggered ALI. The present studied identified a decreased expression level of miR-138-5p during sepsis-associated ALI as a result of promoter methylation that subsequently facilitated NLRP3 inflammasome activation and the pulmonary inflammatory response.
During microorganism infection, mtDNA, one of the essential mitochondrial DAMPs, is released from the mitochondria into the cytoplasm through BAK/BAX macropores [47, 48]. Huang et al. [49] has shown that the internalized bacterial endotoxin LPS activates the pore-forming protein GSDMD to form mitochondrial pores and enables mtDNA release into the cytoplasm. As a result, mtDNA suppresses endothelial cell proliferation programs to promote inflammatory injury. Cytosolic mtDNA triggers inflammasome assembly and subsequently induces Caspase-1-dependent pyroptosis [50, 51]. In addition, intraperitoneal mtDNA administration may provoke ALI and systemic inflammation [52]. Ning et al. [9] also found that LPS stimulation significantly increased the mtDNA level in the cytoplasm, subsequently promoting LPS-induced ALI by activating the NLRP3 inflammasome and pyroptosis of macrophages. Therefore, mtDNA has been increasingly considered a potent agonist of the innate immune system that affects antimicrobial responses and inflammatory pathology [53]. However, autophagy has been shown to alleviate mtDNA-induced ALI by inhibiting the NLRP3 inflammasome [54]. In the current study, LPS stimulation caused leakage of mtDNA into the cytoplasm, while mtDNA deficiency inhibited pyroptosis in AMs and relieved LPS-induced lung inflammation injury. In several diseases related to mitochondrial DNA, nuclear genomes undergo epigenetic modifications, such as chromatin remodelling and alterations in DNA methylation and miRNA expression [55, 56]. mtDNA may drive changes in nuclear DNA expression, which are associated with DNA methylation [57, 58]. Furthermore, mtDNA regulates the methylation of the miR-663 promoter, thus controlling mitochondrial to nuclear retrograde signalling [24]. Here, we showed that enhanced mitophagy might diminish the level of cytoplasmic mtDNA, thus inhibiting the methylation of the miR-138-5p promoter and pulmonary inflammation injury.
Conclusion
Overall, we found that a decrease in cytosolic mtDNA levels induced by mitophagy increases miR-138-5p expression levels in a demethylation-dependent manner, which subsequently attenuates NLRP3 inflammasome activation and pyroptosis of AMs during sepsis-associated ALI. Our study helped us better understand the underlying mechanisms by which mitophagy suppresses the activation of the NLRP3 inflammasome in individuals with ALI. In addition, the present study has provided promising evidence for targeting the mitophagy/miR-138-5p/NLRP3 axis to protect against sepsis-induced ALI.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
References
Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. The Lancet. 2020;395:200–11.
Kumar V. Pulmonary innate immune response determines the outcome of inflammation during pneumonia and sepsis-associated acute lung injury. Front Immunol. 2020;11:1722.
Hayes M, Curley G, Ansari B, Laffey JG. Clinical review: Stem cell therapies for acute lung injury/acute respiratory distress syndrome-hope or hype? Crit Care. 2012;16:1–14.
Needham DM, Wozniak AW, Hough CL, Morris PE, Dinglas VD, Jackson JC, et al. Risk factors for physical impairment after acute lung injury in a national, multicenter study. Am J Respir Crit Care Med. 2014;189:1214–24.
Zhang Y-c, Zuo W-q, Rong Q-f, Teng G-l, Zhang Y-m. Glucocorticoid receptor expression on acute lung injury induced by endotoxin in rats. World J Emerg Med. 2010;1:65.
Xian H, Liou Y-C. Functions of outer mitochondrial membrane proteins: mediating the crosstalk between mitochondrial dynamics and mitophagy. Cell Death Differ. 2021;28:827–42.
Gkirtzimanaki K, Kabrani E, Nikoleri D, Polyzos A, Blanas A, Sidiropoulos P, et al. IFNα impairs autophagic degradation of mtDNA promoting autoreactivity of SLE monocytes in a STING-dependent fashion. Cell Rep. 2018;25:921-33.e5.
Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20:95–112.
Ning L, Wei W, Wenyang J, Rui X, Qing G. Cytosolic DNA-STING-NLRP3 axis is involved in murine acute lung injury induced by lipopolysaccharide. Clin Transl Med. 2020;10: e228.
West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. 2017;17:363–75.
Liu B, He R, Zhang L, Hao B, Jiang W, Wang W, et al. Inflammatory caspases drive pyroptosis in acute lung injury. Front Pharmacol. 2021;12: 631256.
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8.
Luo D, Liu F, Zhang J, Shao Q, Tao W, Xiao R, et al. Functional crosstalk between long non-coding RNAs and the NLRP3 inflammasome in the regulation of diseases. Mol Immunol. 2021;131:191–200.
Davis BN, Hata A. Regulation of MicroRNA biogenesis: a miRiad of mechanisms. Cell Commun Signal. 2009;7:1–22.
Liao H, Zhang S, Qiao J. Silencing of long non-coding RNA MEG3 alleviates lipopolysaccharide-induced acute lung injury by acting as a molecular sponge of microRNA-7b to modulate NLRP3. Aging (Albany N Y). 2020;12:20198.
Pattarayan D, Thimmulappa RK, Ravikumar V, Rajasekaran S. Diagnostic potential of extracellular microRNA in respiratory diseases. Clin Rev Allergy Immunol. 2018;54:480–92.
Gon Y, Shimizu T, Mizumura K, Maruoka S, Hikichi M. Molecular techniques for respiratory diseases: microRNA and extracellular vesicles. Respirology. 2020;25:149–60.
Outeiro-Pinho G, Barros-Silva D, Aznar E, Sousa A-I, Vieira-Coimbra M, Oliveira J, et al. MicroRNA-30a-5p me: a novel diagnostic and prognostic biomarker for clear cell renal cell carcinoma in tissue and urine samples. J Exp Clin Cancer Res. 2020;39:1–11.
Du J, Zhang P, Luo J, Shen L, Zhang S, Gu H, et al. Dietary betaine prevents obesity through gut microbiota-drived microRNA-378a family. Gut Microbes. 2021;13:1–19.
Ying Y, Mao Y, Yao M. NLRP3 inflammasome activation by microRNA-495 promoter methylation may contribute to the progression of acute lung injury. Mol Therapy-Nucleic Acids. 2019;18:801–14.
He Y, Feng D, Li M, Gao Y, Ramirez T, Cao H, et al. Hepatic mitochondrial DNA/Toll-like receptor 9/MicroRNA-223 forms a negative feedback loop to limit neutrophil overactivation and acetaminophen hepatotoxicity in mice. Hepatology. 2017;66:220–34.
Smiraglia D, Kulawiec M, Bistulfi GL, Ghoshal S, Singh KK. A novel role for mitochondria in regulating epigenetic modifications in the nucleus. Cancer Biol Ther. 2008;7:1182–90.
Castellani CA, Longchamps RJ, Sumpter JA, Newcomb CE, Lane JA, Grove ML, et al. Mitochondrial DNA copy number can influence mortality and cardiovascular disease via methylation of nuclear DNA CpGs. Genome Med. 2020;12:1–17.
Carden T, Singh B, Mooga V, Bajpai P, Singh KK. Epigenetic modification of miR-663 controls mitochondria-to-nucleus retrograde signaling and tumor progression. J Biol Chem. 2017;292:20694–706.
Song N, Li P, Song P, Li Y, Zhou S, Su Q, et al. MicroRNA-138-5p suppresses non-small cell lung cancer cells by targeting PD-L1/PD-1 to regulate tumor microenvironment. Front Cell Dev Biol. 2020;8:540.
Luo D, Dai W, Feng X, Ding C, Shao Q, Xiao R, et al. Suppression of lncRNA NLRP3 inhibits NLRP3-triggered inflammatory responses in early acute lung injury. Cell Death Dis. 2021;12:1–15.
Luo D, Liu F, Zhang J, Shao Q, Tao W, Xiao R, et al. Comprehensive analysis of LncRNA-mRNA expression profiles and the ceRNA network associated with pyroptosis in LPS-induced acute lung injury. J Inflamm Res. 2021;14:413–28.
Samman Y, Masood I, Killampalli VV, Howell N, Alpar EK, Banerjee SK. The value of lung injury score in assessing the outcome of patients with Rib fracture. Eur J Trauma Emerg S. 2005;31:133–7.
Ishimoto Y, Inagi R, Yoshihara D, Kugita M, Nagao S, Shimizu A, et al. Mitochondrial abnormality facilitates cyst formation in autosomal dominant polycystic kidney disease. Mol Cell Biol. 2017;37:e00337-e417.
West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–7.
Hashiguchi K, Zhang-Akiyama Q-M. Establishment of human cell lines lacking mitochondrial DNA. In: Mitochondrial DNA. Springer; 2009. p. 383–91.
Zhang Y, Li X, Grailer JJ, Wang N, Wang M, Yao J, et al. Melatonin alleviates acute lung injury through inhibiting the NLRP3 inflammasome. J Pineal Res. 2016;60:405–14.
Wang D, Sun S, Xue Y, Qiu J, Ye T, Zhang R, et al. MicroRNA-223 negatively regulates LPS-induced inflammatory responses by targeting NLRP3 in human dental pulp fibroblasts. Int Endod J. 2021;54:241–54.
Qing Z, Kaixin Z, Yanfei H, Yiming Z, Hua X, Ling Z, et al. MicroRNA-223 triggers inflammation in porcine aorta by activating NLRP3 inflammasome under selenium deficiency. J Cell Physiol. 2021;236:4555–64.
Feng X, Hu J, Zhan F, Luo D, Hua F, Xu G. MicroRNA-138-5p regulates hippocampal neuroinflammation and cognitive impairment by NLRP3/Caspase-1 signaling pathway in rats. J Inflamm Res. 2021;14:1125–43.
Feng X, Zhan F, Luo D, Hu J, Wei G, Hua F, et al. LncRNA 4344 promotes NLRP3-related neuroinflammation and cognitive impairment by targeting miR-138-5p. Brain Behav Immun. 2021;98:283–98.
Bai X, Shao J, Zhou S, Zhao Z, Li F, Xiang R, et al. Inhibition of lung cancer growth and metastasis by DHA and its metabolite, RvD1, through miR-138-5p/FOXC1 pathway. J Exp Clin Cancer Res. 2019;38:1–13.
Cui D, Feng Y, Shi K, Zhang H, Qian R. Long non-coding RNA TRPM2-AS sponges microRNA-138-5p to activate epidermal growth factor receptor and PI3K/AKT signaling in non-small cell lung cancer. Ann Transl Med. 2020;8:1313.
Gu T, Goodell MA. The push and pull of DNA methylation. Science. 2021;372:128–9.
Lorente-Pozo S, Navarrete P, Garzón MJ, Lara-Cantón I, Beltrán-García J, Osca-Verdegal R, et al. DNA methylation analysis to unravel altered genetic pathways underlying early onset and late onset neonatal sepsis. A pilot study. Front Immunol. 2021;12: 622599.
Binnie A, Walsh CJ, Hu P, Dwivedi DJ, Fox-Robichaud A, Liaw PC, et al. Epigenetic profiling in severe sepsis: a pilot study of DNA methylation profiles in critical illness. Crit Care Med. 2020;48:142–50.
Zhang XQ, Lv CJ, Liu XY, Hao D, Qin J, Tian HH, et al. Genome-wide analysis of DNA methylation in rat lungs with lipopolysaccharide-induced acute lung injury. Mol Med Rep. 2013;7:1417–24.
Cai Q, Chen F, Xu F, Wang K, Zhang K, Li G, et al. Epigenetic silencing of microRNA-125b-5p promotes liver fibrosis in nonalcoholic fatty liver disease via integrin α8-mediated activation of RhoA signaling pathway. Metabolism. 2020;104: 154140.
Ma Y, Chai N, Jiang Q, Chang Z, Chai Y, Li X, et al. DNA methyltransferase mediates the hypermethylation of the microRNA 34a promoter and enhances the resistance of patient-derived pancreatic cancer cells to molecular targeting agents. Pharmacol Res. 2020;160: 105071.
Jiang L, Wang M, Sun R, Lin Z, Liu R, Cai H, et al. Methylation of miR-19b-3p promoter exacerbates inflammatory responses in sepsis-induced ALI via targeting KLF7. Cell Biol Int. 2021;45:1666–75.
Chen Y, Luo D, Tian W, Li Z, Zhang X. Demethylation of miR-495 inhibits cell proliferation, migration and promotes apoptosis by targeting STAT-3 in breast cancer. Oncol Rep. 2017;37:3581–9.
McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. 2018;359:eaao6047.
Faas M, De Vos P. Mitochondrial function in immune cells in health and disease. Biochim Biophys Acta (BBA) Mol Basis Dis. 2020;1866:165845.
Huang LS, Hong Z, Wu W, Xiong S, Malik AB. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity. 2020;52:475-86.e5.
Li Y, Shen Y, Jin K, Wen Z, Cao W, Wu B, et al. The DNA repair nuclease MRE11A functions as a mitochondrial protector and prevents T cell pyroptosis and tissue inflammation. Cell Metab. 2019;30:477-92.e6.
Wang X, Li X, Liu S, Brickell AN, Zhang J, Wu Z, et al. PCSK9 regulates pyroptosis via mtDNA damage in chronic myocardial ischemia. Basic Res Cardiol. 2020;115:1–14.
Zhang L, Deng S, Zhao S, Ai Y, Zhang L, Pan P, et al. Intra-peritoneal administration of mitochondrial DNA provokes acute lung injury and systemic inflammation via toll-like receptor 9. Int J Mol Sci. 2016;17:1425.
Riley JS, Tait SW. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020;21: e49799.
Peng W, Peng F, Lou Y, Li Y, Zhao N, Shao Q, et al. Autophagy alleviates mitochondrial DAMP-induced acute lung injury by inhibiting NLRP3 inflammasome. Life Sci. 2021;265: 118833.
Lopes AF. Mitochondrial metabolism and DNA methylation: a review of the interaction between two genomes. Clin Epigenet. 2020;12:1–13.
Chinnery PF, Elliott HR, Hudson G, Samuels DC, Relton CL. Epigenetics, epidemiology and mitochondrial DNA diseases. Int J Epidemiol. 2012;41:177–87.
Wang M, Zhao J, Wang Y, Mao Y, Zhao X, Huang P, et al. Genome-wide DNA methylation analysis reveals significant impact of long-term ambient air pollution exposure on biological functions related to mitochondria and immune response. Environ Pollut. 2020;264: 114707.
Vivian CJ, Brinker AE, Graw S, Koestler DC, Legendre C, Gooden GC, et al. Mitochondrial genomic backgrounds affect nuclear DNA methylation and gene expression. Cancer Res. 2017;77:6202–14.
Funding
This work was supported by the National Natural Science Foundation of China (81871548, 81560306, and 81460292).
Author information
Authors and Affiliations
Contributions
Kejian Qian and Fen Liu designed the study. Ying Yang performed experiments, analyzed the data and wrote the manuscript. Wei Peng and Ning Zhao made critical revisions to the article. Zeyao Xu, Jiaquan Chen and Yamei Cui conducted the immunohistochemical staining experiments. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, F., Yang, Y., Peng, W. et al. Mitophagy-promoting miR-138-5p promoter demethylation inhibits pyroptosis in sepsis-associated acute lung injury. Inflamm. Res. 72, 329–346 (2023). https://doi.org/10.1007/s00011-022-01675-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-022-01675-y