
Abstract
Objective
The aim of this study was to investigate the molecular mechanism of human bone marrow mesenchymal stem cells (hMSCs) secreting miR-26a exosomes on the function of skin fibroblasts.
Methods
Exosomes from hMSCs were extracted and identified by transmission electron microscopy, particle size was analyzed and protein markers were detected. Then, the exosomes were co-cultured with human skin fibroblasts (BJ). CCK-8, Annexin V/P and Transwell assays were used to detect the proliferation, apoptosis, and migration of BJ cells. In addition, the expressions of miR-26a, related proteins, and related inflammatory factors were detected by qRT-PCR, western blotting, and ELISA.
Results
Compared with the high glucose group, the proliferation rate, migration rate, and the expression of α-SMA, bcl-2, TLR4, NF-κB, TNF-α, IL-6, IL- and IL-1 were significantly decreased in the high glucose + MSC-Exo-miR-26a mimics group, while the apoptosis rate and the expression of miR-26a, cleaved-caspase 3, cleaved-caspase 9 and Bax were significantly increased. The results of the high glucose + MSC-Exo-miR-26a inhibitor group were the opposite.
Conclusion
These results suggest that hMSCs cells secreting miR-26a exosomes inhibited the proliferation, migration, and transdifferentiation of high glucose-induced BJ cells, and promoted cell apoptosis, which may be related to the TLR4/NF-κB signaling pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetes, burns, trauma, and surgery are common clinical problems. How to effectively promote wound healing and avoid scar formation is the key issue in wound treatment. Skin wound healing is a complex, multifactorial, and precisely regulated physiological process, which mainly includes four stages: the coagulation stage, inflammatory reaction stage, proliferation stage, and mature stage [1, 2]. It involves inflammation, granulation tissue formation, reepithelization, extracellular matrix remodeling, cell migration, proliferation, differentiation, and apoptosis.
Studies have shown that the proliferation, migration, and transdifferentiation of fibroblasts participate in the formation of granulation tissue and wound contraction, which is one of the decisive factors in determining the speed and quality of wound healing [3, 4]. In the early stage of wound healing, a large number of myofibroblasts rapidly shrink the wound through self-contraction. However, in the late stage, if myofibroblasts can’t undergo apoptosis and regression in time, it will lead to scar formation. Fibroblasts are one of the main sources of myofibroblasts in skin wounds [5, 6]. Therefore, elucidating the cellular function of fibroblasts and the mechanism of their transdifferentiation into myofibroblasts is one of the research hotspots in the field of wound healing.
The toll-like receptor 4 (TLR4) pathway plays an important role in wound healing. It mainly activates the nuclear factor kappa B (NF-κB) through the My D88 dependent pathway, and finally releases proinflammatory cytokines to play a regulatory role [7]. Bone marrow mesenchymal stem cells (BMSCs) are one of the main precursor cells in the bone marrow. They are not only the core cellular components of the hematopoietic system but also participate in the regeneration and repair of various tissues. Substantial evidence shows that BMSCs not only promote wound healing by increasing angiogenesis, granulation tissue formation, and paracrine effect, but also participate in the wound healing process by differentiating into keratinocytes [8,9,10]. Previous studies have shown that the wound microenvironment is the main factor influencing and regulating the participation of BMSCs in chronic wound epithelialization and healing. In chronic skin ulcer wounds, TNF-α induces fibroblast dysfunction by activating the NF-κB signaling pathway. Therefore, the TLR4/NF-κB signaling pathway, as an important pathway of skin wound healing, is worthy of further study.
Exosomes are small membranous vesicles with a diameter of 30–100 nm, which can be secreted by a variety of cells. Functional proteins, mRNA, microRNAs, and tRNAs play an important role in intercellular communication [11]. MiRNA is a kind of endogenous noncoding RNA with about 22 nucleotides in length. It can inhibit the expression of the target gene by recognizing the 3ʹ-noncoding region (3ʹ UTR) of the target gene mRNA. It has been proposed that miRNAs play a crucial role in diverse pathophysiological processes including differentiation of skin fibroblasts. MiR-26a is a highly conserved posttranscriptional regulatory factor, which plays an important role in the process of wound healing. Zhong et al. found that miR-26a attenuated ox-LDL-induced apoptosis of human endothelial cells through the TLR4/NF-κB pathway [12]. Duan et al. reported that adipose mesenchymal stem cell-derived extracellular vesicles containing microRNA-26a-5p target TLR4 and protect against diabetic nephropathy [13]. There is also evidence that overexpression of miR-26a can reduce the transdifferentiation of fibroblasts [14] but whether the mechanism is related to the TLR4/NF-κB pathway has not been reported yet. Therefore, the aim of this study was to investigate the regulatory effect of exon miR-26a secreted by BMSCs on the function of skin fibroblasts induced by glucose and further explore its mechanism.
Methods
Cell culture and grouping
Human skin fibroblasts (BJ) and human BMSCs (hMSCs) were obtained from the American Type Culture Collection (Manassas, VA). The cells were cultured at 37 ℃ in a 5% CO2 environment in DEME (Invitrogen, Life Technologies, NY) supplemented with 10% fetal bovine serum (FBS). BJ cells were divided into the control group (5.5 mmol/l glucose), high glucose group (15 mmol/l glucose), high glucose + MSC-Exo-miR-NC group (15 mmol/l glucose + MSC-Exo-miR-NC), high glucose + MSC-Exo-miR-26a mimics group (15 mmol/l glucose + MSC-Exo-miR-26a mimics) and high glucose + MSC-Exo-miR-26a inhibitor group (15 mmol/l glucose + MSC-Exo-miR-26a inhibitor) [15].
Transfection of miRNA
The negative control, miR-26a mimics (miR-26a mimics: UUCAAGUAAUCCAGGAUAGGCU) and miR-26a inhibitor (miR-26a inhibitor: AGCCUAUCCUGGAUUACUUGAA) were purchased from Ribobio (Guangzhou, China) and transfected into hMSCs. Before transfection, 1 × 105 cells were seeded into 96-well plants for 24 h. When the cells reached 80% confluence, they were transfected with negative control, miR-26a mimics or miR-26a inhibitor using Lipofectamine 2000 reagent (Invitrogen, CA) as described by the manufacturer’s protocol.
Extraction and identification of exosomes from hMSCs
The hMSCs were cultured in DMEM containing 10% FBS at 37 ℃ with 5% CO2 for 48 h. When the cell density reached 1 × 106–1 × 107/ml, the culture supernatant was collected. Then, the exosomes were extracted by SBI kit. The kit describes the specific extraction steps. The morphology of the exosomes was observed and photographed under an electron microscope, and the size of the exosomes was analyzed by particle size instrument. The expression of miR-26a was detected by RT-PCR and the expression of CD63 was detected by Western blot.
Fluorescence confocal experiment
The uptake of exosomes from hMSCs by BJ cells was detected by fluorescence confocal assay. PKH67 staining was added into exosome solution, which was fully mixed and incubated for 10 min. The labeled exosomes and BJ cells with a density of 1 × 105/ml were co-culture into a six-well plate containing a cover glass for a period of time. The cover glass was gently cleaned with phosphate buffer (PBS) and fixed with 4% paraformaldehyde solution at room temperature for 15 min. After wash with PBS for 3 times, the nuclei were stained with 4ʹ, 6-diamidino-2-phenylindole (DAPI), and then the anti-fluorescence quenching agent was added to cover the slide, and the images were taken under a confocal laser scanning microscope.
Cell proliferation was detected by Cell Counting Kit-8 (CCK-8)
Cell viability was measured using the Cell Counting Kit-8 (CCK-8, Boster Biological, China). Each group of cells were inoculated in 96-well plates. Then, 10 μl CCK-8 reagent was added to each well at 0 h, 24 h, 48 h, and 72 h, respectively. After that, all 96-well plates were maintained in an incubator at 37 °C for 2 h, and OD values at 450 nm were measured using a spectrophotometer.
Cell cycle distribution was detected by flow cytometry
After 48 h of culture, the cells were treated with 0.25% trypsin for 24 h, and then washed with PBS for 3 times. We added 0.5 ml propidium iodide staining solution to each tube of sample, and the cells were vortexed and resuspended. The cells were placed in the dark at 37 ℃ for 30 min and detected by upflow cytometry immediately. CELL Quest was used to analyze cell cycle distribution.
Apoptosis analysis by Annexin V/PI
After 48 h of culture, the cells were treated with 0.25% trypsin for 24 h, and then washed with PBS for 3 times. The cells were resuspended with 300 μl binding buffer. Then 5 μl annexin V-FITC was added to mix gently, and finally 10 μl PI staining solution was added for mixing. Thereafter, the cells were placed in the dark for 15 min at room temperature. The apoptosis rate was detected by flow cytometry and analyzed by Cell Quest software.
Transwell was used to detect cell migration
The Transwell chamber was placed in a 24-well plate, and 50 μL of each well of Matrigel gel was quickly absorbed with a sterile gun head into the precooled Transwell upper chamber. The cell concentration was diluted to 5 × 105/ml, and 200 μL cell suspension was added in the upper layer of the chamber. Serum-free medium (the control group) and ICA containing 25 μg/ml, 50 μg/ml and 100 μg/ml were added, respectively. Then, 500 μl DMEM medium containing 10% FBS was added in the lower chamber. The chamber was placed in 1% crystal violet dye solution, removed after 30 min at room temperature, and washed twice with PBS. The number of cells in the field of vision was recorded by taking pictures under a microscope with 100× magnification. The average value of 5 different fields in each chamber was calculated. All experiments were repeated 3 times.
Quantitative real-time PCR
Total RNA was extracted through TRIzol® Reagent (CA, US). cDNA was synthesized from 0.5 μg mRNA using a cDNA synthesis kit (Applied Biosystems, Japan). Quantitative RT-PCR was performed using an ABI Prism 7500 instrument (Applied Biosystems). The expression fold change was calculated using the comparative Ct (ΔΔCt) method. The primers for miR-26a, TLR4, NF-κB, α-SMA, cleaved-caspase 3 and 9, cyclin D1, and Bax are presented in Table S1. The incubation was initiated at 37 ℃ for 15 min, followed by 95 ℃ for 2 min, 95 ℃ for 20 s, 60 ℃ for 34 s and 40 cycles at 95 ℃ for 15 s, 55 ℃ for 15 min and 95 ℃ for 15 s. Each sample was processed in triplicate, and the experiment was repeated 3 times.
Western blotting assays
We extracted total cellular protein and mixed the protein samples with the loading buffer after protein quantification; after SDS-PAGE electrophoresis, the proteins were transferred to PVDF membrane (Washington, NY). The following antibodies were used for Western blotting: anti-CD63, anti-TLR4, anti-NF-kB, anti-α-SMA, anti-cleaved-caspase 3 and 9, anti-cyclin D1, anti-Bax, anti-Bcl-2 (1:1200), and anti-GAPDH (1:2000). Then, protein bands were developed using ECL reagents, and images were acquired using the ChemiDoc Imaging system. The Western blot assays were repeated at least 3 times.
Enzyme-linked immunosorbent assay (ELISA)
The serum levels of TNF-α, IL-6, IL-8, and IL-1β were determined by ELISA kits (Abcam, USA) according to the manufacturer’s instructions.
Luciferase reporter assay
The 3ʹ-UTR of TLR4 was amplified by Takara PCR Amplification Kit (Dalian, China). The cells were cultured in 96-well plates. Plasmids and miR-26a mimics were co-transfected into BJ cells. The experimental groups were as follows: control, miR mimics, and miR inhibitor. Each group was done in triplicate. After 24 h, the cells’ reporter activity was determined by Luciferase Reporter Assay Kit (Promega, USA).
Statistical analysis
All data were expressed as mean ± SD and were analyzed using ANOVA. The significance threshold was set at p < 0.05. SPSS 20.0 software was used for statistical analyses.
Results
Extraction and identification of exosomes from hMSCs
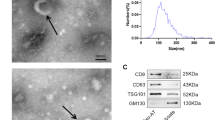
To identify the exosomes from hMSCs, we observed them by transmission electron microscope, analyzed their diameter by particle size analyzer, and detected the expression of exosome marker protein CD63 by Western blot. The results showed that round particles with a diameter of less than 200 nm (Fig. 1A), most of the extracted particles were about 82 nm in diameter (Fig. 1B), and CD63 was enriched in exosomes, but not in cells without exosomes (Fig. 1C). These results indicate that exosomes are successfully isolated. We detected the expression of miR-26 in hMSCs cells after transfection with miR-NC, miR-26a mimics and miR-26a inhibitor by RT-PCR. It was found that compared with the control group, the expression level of miR-26a in exosomes of the miR-NC group was not significantly different, the expression of miR-26a in exosomes of the miR-26a mimics group was significantly increased (P < 0.001), and the expression of miR-26a in exosomes of the miR-26a inhibitor group was significantly decreased (P < 0.001) (Fig. 1D). This indicated that miR-26a mimics and miR-26a inhibitor had significant effects on the expression of miR-26a in hMSCs. The exosomes from hMSCs were labeled with green fluorescence, and the nuclei of BJ cells were labeled with blue fluorescence. The uptake of exosomes by BJ cells was detected by fluorescence confocal assay. The results as shown in Fig. 1E, exosomes (green fluorescence) were clearly visible in BJ cells (blue fluorescence), indicating that exosomes from hMSCs could be transferred into BJ cells.

Identification of hMSCs derived exosomes and its transfer in BJ cells and the expression of miR-26a in exosomes. A Electron micrograph of exosomes extracted by ultracentrifugation (bar = 200 nm). B Nano tracking technique (NTA) was used to detect the diameter of exosomes. C The expression of CD63 in exosomes was extracted by western blotting. D The expression of miR-26a in exosomes was extracted by qRT-PCR. E Laser confocal detection of exosomes transfer to BJ cells
Effect of exosomes from hMSCs on the proliferation of BJ cells
To verify the effect of miR-26a in hMSCs derived exosomes on the proliferation ability of BJ cells, the proliferation of cells in each group was detected by CCK-8 assay in this study. It was found that miR-26a mimics significantly inhibited the proliferation of BJ cells induced by high glucose, whereas miR-26a inhibitor significantly promoted the proliferation of BJ cells induced by high glucose (Fig. 2).

The effect of miR-26a in hMSCs derived exosomes on the proliferation of BJ cells was detected by CCK-8 assay
Effect of exosomes from hMSCs on the cell cycle of BJ cells
To verify the effect of miR-26a in hMSCs derived exosomes on the BJ cell cycle, in this study, the cell cycle changes of each group were detected by flow cytometry, and the expression of cell cycle-related protein cycling D1 protein was detected by western blotting assays. It was found that compared with the control group, the proportion of cells in the G0-G1 phase in the high glucose group was decreased, and the proportion of cells in the S-phase and G2-M phase was increased. Compared with the high glucose group, the proportion of cells in the G0-G1 phase in the high glucose + MSC-Exo-miR-26a mimics group was increased, and the proportion of cells in the G2-M phase was decreased. Compared with the high glucose group, the proportion of cells in the G0-G1 phase in the high glucose + MSC-Exo-miR-26a inhibitor group was decreased, and the proportion of cells in the S-phase and G2-M phase was increased (Fig. 3A and B). The expression of cyclin D1 at mRNA and protein levels is shown in Fig. 3C and D. This result similarly demonstrated that miR-26a mimics suppressed the proliferation of BJ cells induced by high glucose, whereas miR-26a inhibitor had the opposite effect.

The effect of miR-26a in hMSCs derived exosomes on the cell cycle of BJ cells. A Cell cycle was detected by flow cytometry. B Statistical results of cell cycle distribution. C The mRNA expression of cyclin D1 was detected by qRT-qPCR. D The protein expression of cyclin D1 was detected by Western blotting. *P < 0.05 vs. the control group, **P < 0.01 vs. the control group, ***P < 0.001 vs. c the ontrol group, #P < 0.05 vs. the high glucose group, ##P < 0.01 vs. the high glucose group, ###P < 0.001 vs. the high glucose group
Effect of exosomes from hMSCs on the apoptosis of BJ cells
In this study, Annexin V/PI staining was used to detect the effect of miR-26a in hMSCs derived exosomes on the BJ cell apoptosis. qRT-PCR and Western blotting were used to detect the expression of apoptosis-related proteins cleaved-caspase 3, cleaved-caspase 9, Bax and Bcl-2 at mRNA and protein levels. The results showed that miR-26a mimics could increase the apoptosis rate of BJ cells induced by high glucose, while miR-26a inhibitor could decrease the apoptosis rate of BJ cells induced by high glucose (P < 0.05) (Fig. 4A and B). Compared with the high glucose group, cleaved-caspase 3, cleaved–caspase 9, and Bax expression were increased and Bcl-2 expression was decreased in the high glucose + MSC-Exo-miR-26a mimics group. However, in the high glucose + MSC-Exo-miR-26a inhibitor group, cleaved-caspase 3, cleaved-caspase 9, and Bax expression were decreased and Bcl-2 expression was increased (P < 0.05) (Fig. 4C and D).

The effect of miR-26a in hMSCs derived exosomes on the apoptosis of BJ cells. A Apoptosis was detected by Annexin V/PI staining. B Statistical results of apoptosis rate. C The mRNA expression of cleaved- caspase 3 and 9, Bax and Bcl-2 was detected by qRT-qPCR. D The protein expression of cleaved-caspase 3, cleaved-caspase 9, Bax and Bcl-2 was detected by western blotting. Q1 means Annexin-V negative/PI positive, normal cell damage caused by improper operation. Q2 represents Annexin-V positive/PI positive and apoptotic ratio of advanced cells. Q3 is the proportion of early apoptotic cells with Annexin-V positive/PI negative. Q4 is the proportion of living cells with Annexin-V negative/PI negative. *P < 0.05 vs. control group, **P < 0.01 vs. control group, ***P < 0.001 vs. the control group, #P < 0.05 vs. the high glucose group, ###P < 0.001 vs. the high glucose group
Effect of exosomes from hMSCs on the migration of BJ cells
Transwell assay was used to detect the effect of miR-26a in hMSCs derived exosomes on the BJ cell migration. Compared with the control group, the number of cell migration increased in the high glucose group (P < 0.05). Compared with the high glucose group, the number of cell migration in the high glucose + MSC-Exo-miR-26a mimics group was decreased (P < 0.05), while that in the high glucose + MSC-Exo-miR-26a inhibitor group was increased (P < 0.05) (Fig. 5).

The effect of miR-26a in hMSCs derived exosomes on migration of BJ cells. A Cell migration was detected by Transwell. B Statistical results of cell mobility. *P < 0.05 vs. control group, **P < 0.01 vs. the control group, ***P < 0.001 vs. the control group, #P < 0.05 vs. the high glucose group, ##P < 0.01 vs. the high glucose group, ###P < 0.001 vs. the high glucose group
Effect of exosomes from hMSCs on the transdifferentiation of BJ cells
α-Smooth muscle actin(α-SMA) is a marker protein used to determine transdifferentiation of fibroblasts, and its higher expression indicates a greater ability to transdifferentiate fibroblasts into myofibroblasts. In this study, we found that compared with the control group, the expression level of α-SMA in the high glucose group was significantly increased (P < 0.001). Compared with the high glucose group, the expression of α-SMA in the high glucose + MSC-Exo-miR-NC group was not significantly changed, the expression of α-SMA in the high glucose + MSC-Exo-miR-26a mimics group was significantly decreased (P < 0.001), and the expression of α-SMA in the high glucose + MSC-Exo-miR-26a inhibitor group was significantly increased (P < 0.01) (Fig. 6).

The effect of miR-26a in hMSCs derived exosomes on the transdifferentiation of BJ cells. A The mRNA expression of α-SMA was detected by RT-PCR. C The protein expression of α-SMA was detected by WB. *P < 0.05 vs. the control group, **P < 0.01 vs. the control group, ***P < 0.001 vs. the control group, #P < 0.05 vs. the high glucose group, ##P < 0.01 vs. the high glucose group, ###P < 0.001 vs. the high glucose group
Effect of exosomes from hMSCs on the miRNA-26a and TLR4 inflammatory pathway of BJ cells
In this study, the interaction between miR-26a and TLR4 was proved by luciferase reporting analysis (Fig. 7). Next, we verified the effect of miR-26a on the TLR4 inflammatory pathway through qRT-PCR, Western blotting, and ELISA assay. Compared with the control group, the expression level of miR-26a in the high glucose group was significantly decreased (P < 0.05). Compared with the high glucose group, the expression of the high glucose + MSC-Exo-miR-NC group was not significantly changed, the expression of miR-26a in the high glucose + MSC-Exo-miR-26a mimics group was significantly increased (P < 0.05), and the expression of miR-26a in the high glucose + MSC-Exo-miR-26a inhibitor group was significantly decreased (P < 0.05) (Fig. 8A.). However, the expression trend of TLR4, NF-κB, and related inflammatory factors TNF-α, IL-6, IL-8, and IL-1β in each group was opposite to that of miR-26a (Fig. 8B–D). This suggests that miR-26a has a negative regulatory relationship with TLR4/NF-κB pathway.

TLR4 is a target of miR-26a. A The 3-UTR of TLR4 contains the complementary sequences of miR-26a. B Luciferase reporter gene assay validated the interaction between TLR4 and miR-26a in BJ cells. *P < 0.05

The effect of miR-26a in hMSCs derived exosomes on the expression miR-26a and TLR4 related inflammatory factors in BJ cells. A The relative expression of miR-26a. B The mRNA expression of TLR4 and NF-κB was detected by qRT-PCR. C The protein expression of TLR4 and NF-κB was detected by western blotting. D Detection of TNF-α, IL-6, IL-8 and IL-1 β by ELISA. *P < 0.05 vs. the control group, **P < 0.01 vs. the control group, ***P < 0.001 vs. the control group, #P < 0.05 vs. the high glucose group, ##P < 0.01 vs. the high glucose group, ###P < 0.001 vs. the high glucose group
Discussion
Hypertrophic scar often occurs after trauma, which is a pathological healing phenomenon and not only affects the appearance but also may lead to body dysfunction. Histologically, the formation of hypertrophic scar is caused by excessive proliferation of fibroblasts and the transformation into myofibroblasts [16]. Therefore, it is of great significance to study the function of fibroblasts and the process of their transdifferentiation into myofibroblasts. MiRNAs account for about 3% of human genes, which not only regulate the formation of tissues and organs and the physiological process of cells but also affect the occurrence and development of many diseases [17]. It has been confirmed that abnormal miRNA expression may participate in the process of fibrosis and play a role in the pathogenesis of a variety of human diseases, including hypertrophic scar formation, heart failure, and cardiac hypertrophy.
MiR-26a was first cloned from HeLa cells. It contains 21 nucleotides (nt) and located on chromosome 3p [18, 19]. It is expressed in most human tissues. Sequence analysis showed that miR-26a is located at the unstable site of the chromosome [20]. Recent studies have shown that miR-26 family plays an important role in the process of fibrosis. Wang et al. found that overexpression of miR-26a in muscle prevented CKD-induced muscle atrophy through exosome-mediated miR-26a transfer [21]. Zhang et al. demonstrated that upregulation of miR-26a may play a role in myocardial fibrosis after acute myocardial infarction by inhibiting PTEN, enhancing the PI3K/Akt signaling pathway and MMP-9 level [14]. Qi et al. found that downregulation of miR-26a may be involved in the formation of hypertrophic scar [22]. The results of this study showed that when miR-26a was overexpressed in the exosomes secreted by hMSCs, the proliferation and migration ability of skin fibroblasts induced by 15 mmol/l glucose were decreased, and the apoptosis ability was increased. This study is consistent with the above results, indicating that upregulation of miR-26a can inhibit the proliferation and migration of skin fibroblasts cells, thus inhibiting the transdifferentiation process of skin fibroblasts.
Transdifferentiation of fibroblasts into myofibroblasts is a key event in the process of fibrosis response. The characteristics of myofibroblasts are between fibroblasts and smooth muscle cells. The ability of myofibroblasts to express α-smooth muscle actin (α-SMA) is significantly higher than that of fibroblasts, which is a marker to judge the transdifferentiation of fibroblasts. It has been proved that high glucose can promote the expression of α-SMA in skin fibroblasts and promote the transdifferentiation of BJ cells into myofibroblasts. The results of this study showed that hMSCs could inhibit the expression of α-SMA and transdifferentiation of skin fibroblasts induced by 15 mmol/l glucose by secreting exosomes encapsulated miR-26a.
NF-κB is widely distributed in mammals and is involved in the regulation of gene expression. It is closely related to inflammatory response, immune response, cell proliferation, apoptosis, and transdifferentiation. When cells are in the resting state, NF-κB binds to the inhibitory protein IκB and exists in the cytoplasm in an inactive form. When stimulated by cytokines, NF-κB is activated and enters the nucleus to combine with the corresponding target genes to initiate transcription of cytokines, chemokines and inflammatory mediators, resulting in the release of a large number of pro-inflammatory cytokines, free radicals, and other inflammatory mediators. At the same time, these cytokines stimulate NF-κB, which can further activate NF-κB, resulting in sustained or enlarged Inflammation. Ma et al. proved that high glucose can directly promote the activation of TLR4 in podocytes and renal tubular epithelial cells in vitro, resulting in the activation of NF-kB downstream of TLR4 and subsequent inflammatory reaction and fibrosis [23]. Jia et al. demonstrated that miR-26a can inhibit TLR4/NF-κB-mediated inflammatory response activation by targeting malat1, thereby regulating cardiomyocyte lipotoxic injury caused by increased saturated fatty acid levels [24]. Duan et al. found that secreted mir-26a exosomes from adipose tissue mesenchymal stem cells had a protective effect on diabetic nephropathy by regulating the TLR4/NF-κB signaling pathway [13]. However, whether miR-26a affects the function of BJ cells through the TLR4/NF-κB signaling pathway remains unclear. The results showed that upregulation of miR-26a could reduce the expression of TLR4 and NF-kB in skin fibroblasts. It is suggested that upregulation of miR-26a may inhibit the proliferation and transdifferentiation of skin fibroblasts and inhibit the occurrence and development of scar hyperplasia by inhibiting the TLR4/NF-κB signaling pathway and inhibiting the secretion of proinflammatory cytokines and the activation of inflammatory response.
Conclusion
In conclusion, upregulating the expression of miR-26a secreted by hMSCs can inhibit the proliferation, migration, and transdifferentiation of skin fibroblasts by inhibiting the NF-κB signaling pathway. This study provides a new idea for the treatment of posttraumatic scar hyperplasia.
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
References
Kondo T, Ishida Y. Molecular pathology of wound healing. Forensic Sci Int. 2010;203(1–3):93–8.
Childs DR, Murthy AS. Overview of wound healing and management. Surg Clin North Am. 2017;97(1):189–207.
Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526.
Darby IA, Zakuan N, Billet F, Desmoulière A. The myofibroblast, a key cell in normal and pathological tissue repair. Cell Mol Life Sci. 2015;73(6):1145–57.
Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmoulière A, Varga J, De Wever O, Mareel M, Gabbiani G. Recent developments in myofibroblast biology. Am J Pathol. 2012;180:1340–55.
Quaggin SE, Kapus A. Scar wars: mapping the fate of epithelial-mesenchymal-myofibroblast transition. Kidney Int. 2011;80(1):41–50.
Suga H, Sugaya M, Fujita H, Asano Y, Tada Y, Kadono T, et al. TLR4, rather than TLR2, regulates wound healing through TGF-β and CCL5 expression. J Dermatol Sci. 2014;73(2):117–24.
Badiavas AR, Badiavas EV. Potential benefits of allogeneic bone marrow mesenchymal stem cells for wound healing. Expert Opin Biol Ther. 2011;11(11):1447–54.
Jiang L, Zhang Y, Zhu ZM, Gao F, Yang HH, Zhao ZG. Bone marrow mesenchymal stem cells accelerate wound healing in diabetic mice via inhibiting the expression of microRNA-155 to up-regulate Sirt1 in endothelial cells. Biomed Pharmacother. 2016;9(3):2980–8.
Qi Y, Dong Z, Chu H, Zhao Q, Jiang D. Denatured acellular dermal matrix seeded with bone marrow mesenchymal stem cells for wound healing in mice. Burns. 2019;45(7):1685–94.
Donker RB, Mouillet JF, Chu T, Hubel CA, Stolz DB, Morelli AE, Sadovsky Y. The expression profile of C19MC microRNAs in primary human trophoblast cells and exosomes. Mol Hum Reprod. 2012;18(8):417.
Zhong X, Zhang L, Li Y, Li P, Li J, Cheng G. Kaempferol alleviates ox-LDL-induced apoptosis by up-regulation of miR-26a-5p via inhibiting TLR4/NF-κB pathway in human endothelial cells. Biomed Pharmacother. 2018;108:1783–9.
Duan Y, Luo Q, Wang Y, Ma Y, Shi J. Adipose mesenchymal stem cell-derived extracellular vesicles containing microRNA-26a-5p target TLR4 and protect against diabetic nephropathy. J Biol Chem. 2020;295(37):12868–84.
Zhang S, Cui R. The targeted regulation of miR-26a on PTEN-PI3K/AKT signaling pathway in myocardial fibrosis after myocardial infarction. Eur Rev Med Pharmacol Sci. 2018;22(2):523–31.
Song J. Glucose and amino acids affect mouse skin fibroblast growth through mTORC1 signaling pathway. Chongqing Med. 2013.
Ahn JY, Park S, Yun YS, Song JY. Inhibition of type III TGF-β receptor aggravates lung fibrotic process. Biomed Pharmacother. 2010;64(7):472–6.
Veksler-Lublinsky I, Shemer-Avni Y, Kedem K, Ziv-Ukelson M. Gene bi-targeting by viral and human miRNAs. BMC Bioinform. 2010;11(1):249.
Sorg RJH. Wound repair and regeneration. Eur Surg Res. 2012;25(1):79–92.
Eming SA, Krieg T, Davidson JM. Gene therapy and wound healing. Clin Dermatol. 2007;25(1):79–92.
Huse JT, Brennan C, Hambardzumyan D, Wee B, Pena J, Rouhanifard SH, et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009;23(11):1327–37.
Wang B, Zhang A, Wang H, Klein JD, Tan L, Wang ZM, Du J, Naqvi N, Liu BC, Wang XH. miR-26a limits muscle wasting and cardiac fibrosis through exosome-mediated microRNA transfer in chronic kidney disease. Theranostics. 2019;9(7):1864.
Qi J, Liu Y, Hu K, Zhang Y, Wu Y, Zhang X. MicroRNA-26a inhibits hyperplastic scar formation by targeting Smad2. Exp Ther Med. 2018;15(5):4332–8.
Jin M, Chadban SJ, Zhao CY, Xiaochen C, Tony K, Usha P, et al. TLR4 activation promotes podocyte injury and interstitial fibrosis in diabetic nephropathy. PLoS ONE. 2014;9(5):e97985.
Jia P, Wu N, Jia D, Sun Y. Downregulation of MALAT1 alleviates saturated fatty acid-induced myocardial inflammatory injury via the miR-26a/HMGB1/TLR4/NF-κB axis. Diabetes Metab Syndr Obes Targets Ther. 2019;12:655–65.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81602742).
Author information
Authors and Affiliations
Contributions
QL and PH performed the research; PH designed the research study; WC contributed essential reagents or tools; WC and JB analyzed the data; QL wrote the paper; All authors approved the final version of the paper.
Corresponding author
Ethics declarations
Conflict of interest
There is no competing interest.
Ethics approval
The study protocol was approved by the Ethics Committee of Tongji Hospital.
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, Q., Huang, P., Chen, W. et al. Mechanism of bone marrow mesenchymal stem cells secreting miR-26a exosomes affecting high glucose-induced skin fibroblasts function by regulating TLR4/NF-κB signaling. Inflamm. Res. 70, 811–821 (2021). https://doi.org/10.1007/s00011-021-01478-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-021-01478-7