
Abstract
Objective
Activated macrophages undergo a metabolic shift from oxidative phosphorylation (OXPHOS) to aerobic glycolysis, which plays a critical role in inflammation. Increasing evidence suggests the important role of propofol in the regulation of inflammatory response and metabolism, but the effect of propofol on the metabolic shift in macrophage, and the mechanisms involved remain unclear.
Methods
The effect of propofol on the metabolic switch was analyzed by extracellular acidification rate and oxygen consumption rate assays. The effect of propofol on glycolysis was analyzed by lactate and glucose uptake assay. The mRNA, protein, cell surface levels of glucose transporter 1 (GLUT1) and the silencing of GLUT1 were employed to understand the effects of GLUT1-mediated metabolism by propofol. Finally, to understand the antioxidation of propofol on the regulation of metabolism, the reactive oxygen species (ROS) production and NADPH activity were performed.
Results
We show that propofol can change the metabolic pathway switch from aerobic glycolysis to OXPHOS in LPS-activated macrophages. Moreover, propofol suppresses aerobic glycolysis via inhibited GLUT1-mediated glucose uptake. Furthermore, we show that propofol reduces ROS overproduction, which in turn inhibits GLUT1 expression. Finally, we find that propofol reduces ROS production via inhibits NADPH activity.
Conclusion
These findings shed light on the function and mechanism of propofol in the metabolic switch and highlight the importance of targeting metabolism by propofol in the clinical medication of inflammatory diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Macrophage is the front-line cell of innate immunity. They recognize and respond to pathogens or damaged cells via pathogen-associated molecular patterns (PAMP) or danger-associated molecular patterns (DAMP), leading to the inflammation response [1]. An excessive host response is associated with the pathogenesis of many diseases, such as Gram-negative bacilli-induced sepsis [2]. Depending on the type of stimuli, macrophages have two key roles: to respond to infection and to repair tissue damage, which requires the function of macrophages on host defense and inflammation. Recent studies have shown the critical role of metabolic reprogramming in host defense and inflammation [3, 4].
The reprogramming of metabolic pathways in immune cells has relevance in the regulation of innate immune functions and the pathogenesis of inflammatory [3,4,5]. A link between the inflammatory profile and the metabolic reprogramming of macrophages has become increasingly clear in the past few years [5, 6]. Growing evidence suggests that macrophages have the ability to switch from oxidative phosphorylation (OXPHOS) to aerobic glycolysis, which can lead to an accumulation of metabolic intermediates that serve as signaling molecules to regulate cellular processes, such as the production of inflammatory mediators [5,6,7]. One key mediator of aerobic glycolysis is the production of reactive oxygen species (ROS), which participate in normal physiological processes or contribute to a maladaptive response that leads to metabolic dysfunction and inflammatory signaling [8]. Researchers have reported ROS regulate metabolism via HIF-1α activity or glucose uptake [8, 9].
It is now recognized that anesthesia could influence metabolism, inflammation, and immunological changes in the perioperative period [10]. Propofol (2, 6-diisopropylphenol), a widely used intravenous anesthetic, has been used for the induction and maintenance of anesthesia and sedation in critical patient care. Recent pieces of evidence have shown that propofol has immunomodulatory effects, including suppression of inflammatory response [11, 12]. Also, several studies have shown that propofol is able to inhibit the expression of HIF-1α [13] or the overproduction of cytoplasmic or mitochondria ROS [14], which is high correlation with cellular metabolism [8]. Some studies demonstrated that propofol attenuates the adhesion of tumor and endothelial cells through inhibiting glycolysis in human umbilical vein endothelial cells [15]. A study has reported, in THP-1 cells, propofol suppressed lactate accumulation through the inhibition HIF-1α activation [13]. But, another study indicated that propofol induced the metabolic switch OXPHOS to glycolysis by suppressing mitochondrial function in human neuroblastoma SH-SY5Y cells [16]. The effect of propofol on the metabolic switch in macrophages, and the mechanisms involved remain unclear.
Therefore, in this study, we aim to explore the effect and mechanism of propofol on metabolism in LPS-induced activated macrophages. Here, we found that propofol can change the metabolic pathway switch from aerobic glycolysis to OXPHOS in LPS-induced macrophages. Besides, we demonstrated that propofol suppressed aerobic glycolysis and improved OXPHOS via regulating the production of nicotinamide adenine dinucleotide phosphate (NADPH)-derived ROS, which regulates glucose transporter 1 (GLUT1)-mediated glucose uptake. Overall, this article presents evidence for the metabolic regulation of propofol in activated macrophages.
Materials and methods
Animals
C57BL/6 male mice (6–8 weeks old, 25 g) were purchased from Southern Medical University Animal Center (Guangzhou, China). All the animal experimental protocols were reviewed and conformed to the committees of Southern Medical University Animal Center for care and committees of Guangdong Medical University.
Reagents
Propofol (56931), LPS (L3023), apocynin (178385) and NAC (A7250) were purchased from Sigma. Rabbit polyclonal antibody against ACTIN was from Cell Signaling Technology. Anti-Glucose Transporter GLUT1 antibody (ab115730), Alexa Fluor 488 Anti-Glucose Transporter GLUT1 antibody [EPR3915] (ab195359), H2DCFDA (ab113851) and 2NBDG (ab235976) were obtained from Abcam. Control siRNA (sc-37007), GLUT1 siRNA (sc-35494) were purchased from Santa Cruz Biotechnology.
Bone marrow-derived macrophages (BMDM) isolation and culture
BMDMs were, respectively, isolated from the tibia and femur of male C57BL/6 mice (6–8 week) and all the primary cells were cultured in the cell culture plates for 1 week in macrophage medium (RPMI-1640 medium with a supplement of 10% FBS, 100 μg/ml streptomycin, 100 U/ml penicillin, and 10 ng/ml MCSF).
CCK8 assay
Cell viability was assessed by Cell Counting Kit-8 (CCK8) assay following the manufacturer’s instructions (Dojindo, Japan).
Extracellular acidification rate and oxygen consumption rate assays
Extracellular acidification rate (ECAR) and cellular oxygen consumption rate (OCR) were measured with XF24 Extracellular Flux Analyzer (Seahorse Bioscience). ECAR and OCR were measured by Seahorse XF Glycolysis Stress Test Kit and Seahorse XF Cell Mito Stress Test Kit, respectively. In brief, cells (2 × 104) were seeded into a Seahorse XF 24 cell culture microplate. Cells were then switched into XF assay medium (containing 25 mM glucose, 2 mM sodium pyruvate, and 2 mM glutamine, and pH adjusted to 7.4) 1 h prior to assay. After baseline measurements, for ECAR, 10 mM glucose, 1 μM oligomycin, and 50 mM 2-DG were sequentially injected into each well at indicated time points. For OCR, 1 μM oligomycin, 0.5 μM FCCP, and 1 μM rotenone plus antimycin A (Rote/AA) were sequentially injected.
Mitochondrial membrane potential (JC-1) assay
BMDM cells were seeded in six-well plates at a density of 2 × 105 cells/well. After the treatments, the cells were collected and stained with JC-1 for 30 min at 37 ℃ in the dark and then washed twice with PBS. The cells were resuspended in 500 ml PBS and analyzed by flow cytometry.
RNA isolation and real‐time PCR analysis
TRIzol reagent (Invitrogen, Carlsbad, CA) was used to isolate total RNA. Total RNA was reverse transcribed to cDNA by a Reverse Transcription Kit (TaKaRa Bio-Technology, China). Quantitative real-time PCR was performed with the SYBR Green Mixture (TaKaRa Bio-Technology, China). The gene-specific primers as follow: GLUT1 (forward primer: GAA CCT GTT GGC CTT TGT GGC; reverse primer: GCT GGC GGT AGG CGG GTG AGC G), 18S (forward primer: GTA ACC CGT TGA ACC CC ATT; reverse primer: CCA TCC AAT CGG TAG TAG CG).
Western blotting
BMDM cells protein was lysed by RIPA buffer and the concentration was determined by BCA kit (ThermoFisher Scientific). The proteins were subjected to SDS-PAGE and transferred to PVDF membranes (Millipore). After blocking with 5% skimmed milk for 1 h at room temperature, the membranes were incubated with primary antibodies against GLUT1 (1:10,000, Abcam) or β-ACTIN (1:1000, Cell Signaling Technology) overnight at 4 ℃. The membranes were incubated with secondary antibodies for 1 h at room temperature, followed by visualization using ECL reagent (Millipore). The densitometric analysis of protein was performed by Image Pro Plus.
Cell transfection
For silence GLUT1 expression, cells were transfected with si-NC and si-GLUT1 by Lipofectamine™ 3000 Transfection Reagent (Invitrogen) according to the manufacturer’s instruction. After 24 h or 48 h, the silencing efficiency was measured by q-PCR and immunoblotting.
Flow cytometry assay
For measuring cell surface expression of GLUT1, after treatment, cells were incubated with AF488 Anti-Glucose Transporter GLUT1 antibody (Abcam, ab195359) for 30 min on ice, washed three times with PBS, and analyzed by flow cytometry.
NADPH activity and lactate assay
NADPH activity in cell lysate was measure by NADP/NADPH Quantitation Kit (Sigma-Aldrich, MAK038) following the manufacturer’s instructions. Lactate in culture medium was measured with the l-Lactate Assay Kit (Abcam, ab65331) according to the manufacturer’s instructions.
Reactive oxygen species detection
The intracellular ROS levels were examined using H2DCFDA (Abcam) by flow cytometry. In brief, after correspondingly treatments, 1.5 × 105 cells were collected and stained with 20 μM H2DCFDA for 30 min at room temperature. Cells were washed once in PBS and analyzed by flow cytometry. Data were analyzed using FlowJo (TreeStar, USA).
Glucose uptake assay
BMDM cells (2 × 105 cells/well) were cultured in a six-well plate. After treatments, cells were incubated 1 h in glucose-free media. And then, cells were stained with 300 μM 2NBDG for 30 min in 37 ℃ in the dark. Cells were then washed twice with PBS and analyzed by flow cytometry (BD Biosciences). Data were analyzed using FlowJo (TreeStar, USA).
Statistical analysis
Results are presented as mean ± SD of three independent experiments. Significance of differences between groups was determined using One-way ANOVA or t test. p < 0.05 was considered statistically significant.
Results
Propofol inhibits aerobic glycolysis and improves oxidative phosphorylation in LPS-induced macrophages
Macrophage activation elicits changes in metabolic profiles according to activation state. Activated macrophages (LPS stimulation) metabolic pathway shift from oxidative phosphorylation (OXPHOS) to glycolysis [5]. First, we examined the effect of propofol on LPS-induced lactate production. As shown in Fig. 1 a, 50 μM propofol significantly decreased LPS-induced lactate production, while 25 μM propofol did not affect lactate production. In addition, 50 μM propofol did not affect the BMDM cell viability (Fig. 1b). Therefore, we chose 50 μM propofol for the following experiments. For further investigated the effect of propofol on macrophages metabolic reprogramming. We measure the extracellular acidification rate (ECAR) and oxygen consumption rate (OCR), which reflects overall glycolytic flux and indicator of mitochondrial respiration, respectively. The difference between basal and maximal ECAR rates is known as the glycolytic reserve capacity. As shown in Fig. 1c and d, 50 μM propofol pretreatment showed a significant decrease in glycolytic reserve capacity in LPS-induced macrophages, indicating that propofol inhibits aerobic glycolysis. Besides, compare with LPS stimulation, pre-treated with propofol significantly increase basic state and maximal OCR (Fig. 1e, f), indicating that propofol improves OXPHOS. Furthermore, we found that propofol pretreatment maintained mitochondrial membrane potential at a higher level after LPS was stimulated (Fig. 1g, h), indicating that propofol protects mitochondrial function. Taken together, these results indicated propofol can change the metabolic switch from aerobic glycolysis to OXPHOS in LPS-induced macrophages.

Propofol is involved in regulating LPS-induced aerobic glycolysis in BMDM cells. a BMDM cells were pretreated with or without propofol (25 μM or 50 μM) for 1 h. Cells were stimulated with LPS (1 μg/ml) for 24 h. The concentration of lactate was determined in the supernatant. b BMDM cells were treated with indicated concentration of propofol for 24 h, cell viability was tested by CCK8 assay. c–h BMDM cells were treated with LPS (1 μg/ml, 24 h) in the presence or absence of 50 μM propofol for 1 h. ECAR and OCR rates were measured by extracellular flux analysis as a measurement for OXPHOS and glycolysis, respectively. During extracellular flux analysis, cells were sequentially treated with c, d glucose, oligomycin (OM), and 2-deoxyglucose (2-DG), to determine glycolysis parameters from the ECAR levels, or with e, f OM, FCCP, and rotenone (ROT) plus antimycin A (AA), to assess OXPHOS parameters from the OCR levels. g Mitochondrial membrane potential was tested by JC-1 assay. h JC-1 greed fluorescence-positive cells are shown. Error bars stand for the mean ± SD of at least triplicate experiments. *p < 0.05, **p < 0.01
Propofol suppresses aerobic glycolysis via inhibition of GLUT1 expression
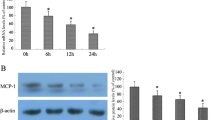
Glycolysis involves a series of reactions, including the transport of glucose across the plasma membranes of cells. We found that pre-treated propofol significantly decreased LPS-induced glucose uptake in macrophages (Fig. 2a, b), which indicating that propofol influences glucose transport. GLUT1, a glucose transporter, plays key roles in the activation of immune cells and glucose metabolism. We then investigated whether propofol might influence aerobic glycolysis via GLUT1 in macrophages; we examined the expression of GLUT1 at the mRNA, protein, and cell surface levels after pre-treated propofol. We found that propofol inhibited GLUT1 expression at both the mRNA and protein levels upon LPS stimulation (Fig. 2c–e). As GLUT1 locates in the cell plasma membrane, we tested the role of propofol in the GLUT1 cell surface expression. We found that propofol significantly decreased GLUT1 cell surface expression upon LPS stimulation (Fig. 2f, g). These results demonstrate that propofol can decrease GLUT1 cell surface expression via inhibiting GLUT1 mRNA and de novo generation of GLUT1 protein.

Propofol suppresses LPS-induced aerobic glycolysis through inhibition of GLUT1 expression. BMDM cells were pretreated with or without 50 μM propofol for 1 h, and then exposed to LPS (1 μg/ml) for 4 h (c) or 24 h (a, b, d–g). a, b Glucose uptake was measured by flow cytometry. c Gene transcript level of GLUT1 was analyzed by qPCR. d, e Protein level of GLUT1 was analyzed by western blot. f, g The cell surface expression of GLUT1 was analyzed by flow cytometry. BMDM cells were transfected with si-NC and si-GLUT1 for 24 h. h–j The efficiency of silencing was evaluated by qPCR and western blot. k BMDM cells transfected with si-NC or si-GLUT1 were treated with LPS (1 μg/ml) for 24 h to measure the concentration of lactate. Data shown represent three independent experiments. *p < 0.05, **p < 0.01
To further confirm the effect of GLUT1 on LPS-induced aerobic glycolysis, we silenced the expression of GLUT1 by GLUT1 siRNA. GLUT1 was significantly down-regulated at both the mRNA and protein levels (Fig. 2h-j). We found that production of lactate in macrophages upon LPS stimulation was significantly inhibited after GLUT1 silencing (Fig. 2k), indicating that GLUT1 contributes to LPS-induced aerobic glycolysis. Taken together, these results demonstrate that propofol dampens LPS-induced aerobic glycolysis via inhibiting GLUT1 expression in macrophages.
Propofol reduces ROS production in LPS-induced macrophage aerobic glycolysis
Experimental studies revealed the role of propofol in antioxidant activity [14]. In our experiment, we found that the increase of ROS production in LPS-induced macrophages aerobic glycolysis (Fig. 3a, b). We wondered whether ROS is involved in the mechanism underlying propofol inhibited aerobic glycolysis. To test this hypothesis, we monitored the intracellular ROS with or without propofol. We found that propofol significantly decreased intracellular ROS production upon LPS stimulation (Fig. 3a, b). Next, we tested the NADPH activity, which is essential for ROS production. We found that propofol pretreatment can inhibit NADPH activity (Fig. 3c). Besides, we found that pretreated with NAC or apocynin, which degrades intracellular ROS or inhibited NADPH oxidases respectively, can significantly decrease lactate (Fig. 3d, e). These results indicate propofol can inhibit LPS-induced aerobic glycolysis via reducing NADPH-induced ROS production.

Propofol inhibits LPS-induced aerobic glycolysis via reduces NADPH activity and ROS production. BMDM cells were treated with 1 μg/ml LPS for 4 h (a–c) or 24 h (d, e) in the presence or absence of 50 μM propofol, 10 mM NAC or 1 μM apocynin pretreatment for 1 h. a, b ROS production was analyzed by flow cytometry. c NADPH activity was measured. d, e The concentration of lactate was assessed in the supernatant. Data shown represent three independent experiments. *p < 0.05, **p < 0.01
ROS regulates GLUT1 expression
In our experiment, we have already confirmed that propofol dampens LPS-induced aerobic glycolysis via inhibiting GLUT1 expression and ROS production. We wondered the relationship between GLUT1 and ROS underlying LPS-induced aerobic glycolysis. We found that GLUT1 expression was decreased upon NAC or apocynin pretreatment at both the mRNA and protein levels (Fig. 4a–e). To further confirm the regulation of ROS to GLUT1, we measured the cell surface expression of GLUT1. As shown in Fig. 4f and g, NAC pretreatment can reduce the cell surface expression of GLUT1 upon LPS stimulation. Similarly, apocynin pretreatment decreased the cell surface expression of GLUT1 after LPS stimulation (Fig. 4h, i). we found that NAC pretreatment can reduce GLUT1-induced glucose uptake upon LPS stimulation (Fig. 4j, k). Taken together, these results suggest ROS reduce GLUT1 cell surface expression via inhibiting GLUT1 mRNA and de novo generation of GLUT1 protein.

ROS contributes to LPS-induced GLUT1 expression. BMDM cells were pretreated with or without 10 mM NAC or 1 μM apocynin for 1 h, and then exposed to LPS (1 μg/ml) for 4 h (a) or 24 h (b–k). a Gene transcript level of GLUT1 was analyzed by qPCR. b–e Protein level of GLUT1 was analyzed by western blot. f–i The cell surface expression of GLUT1 was analyzed by flow cytometry. j, k Glucose uptake was measured by flow cytometry. Data shown represent three independent experiments. *p < 0.05, **p < 0.01
Discussion
In response to invading pathogens, the innate immune system is the first line of defense. Macrophages play a critical role in host defense against infection and inflammatory diseases, when activated using TLR ligands (such as LPS), promote the production of inflammatory cytokines [1, 17]. Recent studies have shown the link between the inflammatory response and metabolic change, when macrophages activated, switch metabolism from OXPHOS to aerobic glycolysis, which promotes the production of inflammatory cytokines (such as il1b, HMGB1) [5, 6, 18]. Propofol, as a widely used intravenous anesthetic, has been demonstrated to protect tissue via the effect of anti-inflammatory [11, 14]. Also, some studies suggested that propofol regulate glucose metabolism in many cancer, including breast cancer [19], colon cancer [20], and pancreatic cancer [21]. But, the role of propofol in the metabolic switch in macrophages still remained unclear. In this study, we expounded the role of propofol in the metabolic switch between aerobic glyolysis and OXPHOS and demonstrated the mechanism of propofol in LPS-mediated metabolism.
Macrophages reprogram their metabolism and function according to environmental conditions and stimuli. In the normal resting macrophages, energy mainly from OXPHOS. Glucose entering glycolysis is converted to pyruvate, which enters the tricarboxylic acid (TCA) cycle where it becomes oxidized to generate ATP. However, once macrophages are activated (such as Toll-like receptor 4 (TLR4) agonist LPS), OXPHOS is suppressed, and cells switch to glycolysis as an alternative. The increase in glycolysis could be a mechanism to rapidly generate ATP, less energetically efficient when compared to the TCA cycle, but the mode of ATP generation, but it can be quickly induced and production more intermediate metabolites, which is necessary for inflammatory response. Stimulating macropaghes with LPS causes an increased glycolytic rate and glucose consumption, which are associated with enhanced expression of GLUT1 at both mRNA levels and protein levels as well as elevated lactate production [22, 23]. Furthermore, increased intracellular ROS production is observed in activated macrophages. In response to ROS, cells might upregulate glycolysis [24]. Similarly, we found that the expression of GLUT1, which mediates glucose uptake, is increased as well as the elevated ROS production. Interestingly, previous studies have reported that propofol has a positive effect on anti-inflammation via inhibiting ROS production [12, 14], which has a high correlation with cellular metabolism and infection disease [8]. Therefore, propofol has been a potential anesthetic for infection disease. However, the function of propofol on immunometabolism is unclear. Hence, we investigated the role of propofol on the metabolic switch.
Macrophages activation depends on cell metabolism reprogramming. In this study, we investigated the bioenergetic profiles of OXPHOS and aerobic glycolysis in LPS-induced macrophages and compared them with different concentrations of propofol pretreatment. Our results suggested that LPS stimulation leads to metabolic reprogramming via switching OXPHOS to aerobic glycolysis and pretreated with appropriate concentration of propofol (50 μM) could reverse this phenotype by normalizing its metabolic manner, switch the metabolism from aerobic glycolysis to OXPHOS. These results are similar to reports demonstrating a decrease in colorectal cancer cells [25] or umbilical vein endothelial cells [15] glycolysis after pretreatment with propofol. Conversely, some studies reported that propofol had cytotoxicity, even induced glycolysis and cell death in neuroblastoma SH-SY5Y cells [16] or Renal cell carcinoma cell lines [26], but these studies used a high concentration of propofol, which can lead the cell death. The study has shown, in macrophage cell line RAW264.7, the cells would be injured when the concentration of propofol up to 300 μM [27]. Similarly, in this study, we also found that a high dose of propofol (100 μM) tended to decrease the cell survival in BMDM. Therefore, our study demonstrated that an appropriate concentration of propofol can switch the metabolism from aerobic glycolysis to OXPHOS in activated macrophages. However, we should further consider the underlying mechanism of how propofol inhibits aerobic glycolysis and improve OXPHOS by LPS.
Previous studies have shown that glucose uptake plays a critical role in metabolism and that is the initial of glycolysis and oxidation phosphorylation [5, 6]. Cellular glucose uptake is mediated by glucose transporters (GLUTs), of which fourteen isoforms have been described with different kinetic properties and modes of regulation [28]. GLUT1, highly expressed in macrophages, regulates glucose metabolism, which drives the expression of proinflammatory mediators [22, 29]. Presently, we observed that propofol could inhibit glucose uptake via decrease the expression of GLUT1 at the mRNA, protein, and cell surface levels, indicating that propofol could regulate the de novo synthesis of GLUT1. Overexpression of GLUT1 in macrophages significantly increased glucose uptake, glycolysis, and the expression of proinflammatory cytokines associated with systemic metabolic dysfunction (e.g., TNF-α, IL-1β, and IL-6) [29]. In addition, adipose tissue macrophages displaying high surface GLUT1 contained greater cytokines, suggesting that GLUT1 correlates with in vivo inflammation [22]. GLUT1-overexpressing cells also contained less CoA, which limits excess pyruvate or fatty acids from entering the TCA cycle for oxidative metabolism [22]. Stimulation of cellular glucose uptake is frequently observed during conditions of oxidative stress. In muscle cells, endogenous ROS induced an increase in glucose uptake [30]. Similarly, in this study, we found that LPS-induced ROS production can upregulate GLUT1, which enhances glucose uptake. Propofol decreased GLUT1 expression by inhibiting ROS overproduction. Also, previous studies have shown that ROS-mediated HIF-1α stabilization enhanced GLUT1 expression and glucose uptake in Lewis lung carcinoma, colon, and breast cancer cells [9, 31, 32]. HIF-1α ubiquitinylation is inhibited allowing it to drive the transcription of various target genes, including GLUT1 [33]. Besides, we also found that propofol inhibited NADPH oxidases (NOX)-derived ROS production. One of the most well-known sources of intracellular ROS is the NOX family of enzymes [8, 34]. In the endothelial cell, NOX-dependent ROS production drives HIF1α-mediated GLUT1 expression. ROS-dependent aerobic glycolysis will enhance pentose phosphate pathway (PPP) activity, leading to elevated NADPH, which in turn, further enhances NOX-derived ROS production [34,35,36]. In addition, propofol can decrease the levels of cellular Ca2+, because of its antioxidation [14], leading to the activation of CAMK-ERK, which in turn inhibited HIF1α-mediated GLUT1 expression [25]. However, the exact mechanism of the relationship between propofol, ROS and GLUT1 needs to be further explored.
In conclusion, our study has indicated that an appropriate concentration of propofol switches the metabolism from aerobic glycolysis to OXPHOS. Furthermore, we also have identified the mechanisms of propofol in mediating LPS-induced metabolism through ROS-derived GLUT1-mediated glucose uptake. These findings suggest the metabolic regulation of propofol in activated macrophages proposes propofol as a promising candidate anesthetic for inflammatory diseases.
References
Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216.
Seymour CW, Liu VX, Iwashyna TJ, Brunkhorst FM, Rea TD, Scherag A, et al. Assessment of clinical criteria for sepsis: for the third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315:762–74.
O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16:553–65.
Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–43.
Van den Bossche J, O’Neill LA, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol. 2017;38:395–406.
Kelly B, O’Neill LAJ. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771–84.
Russell DG, Huang L, VanderVen BC. Immunometabolism at the interface between macrophages and pathogens. Nat Rev Immunol. 2019;19:291–304.
Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. 2018;122:877.
Jung KH, Lee JH, Quach CHT, Paik JY, Oh H, Park JW, et al. Resveratrol suppresses cancer cell glucose uptake by targeting reactive oxygen species-mediated hypoxia-inducible factor-1 alpha activation. J Nucl Med. 2013;54:2161–7.
Ciechanowicz SJ, Ma D. Anaesthesia for oncological surgery—can it really influence cancer recurrence? Anaesthesia. 2016;71:127–31.
Chen RM, Chen TG, Chen TL, Lin LL, Chang CC, Chang HC, et al. Anti-inflammatory and antioxidative effects of propofol on lipopolysaccharide-activated macrophages. In: Role of the mitochondria in human aging and disease: from genes to cell signalling, vol 1042. Hoboken: Wiley; 2005. p. 262–71.
Liu JH, Li YJ, Xia XH, Yang XY, Zhao RZ, Peer J, et al. Propofol reduces microglia activation and neurotoxicity through inhibition of extracellular vesicle release. J Neuroimmunol. 2019;333:476962.
Tanaka T, Takabuchi S, Nishi K, Oda S, Wakamatsu T, Daijo H, et al. The intravenous anesthetic propofol inhibits lipopolysaccharide-induced hypoxia-inducible factor 1 activation and suppresses the glucose metabolism in macrophages. J Anesth. 2010;24:54–60.
Zhong HH, Song R, Pang QN, Liu YW, Zhuang JL, Chen YM, et al. Propofol inhibits parthanatos via ROS-ER-calcium-mitochondria signal pathway in vivo and vitro. Cell Death Dis. 2018;9:932.
Qi J, Wu QC, Zhu XQ, Zhang S, Chen XY, Chen WK, et al. Propofol attenuates the adhesion of tumor and endothelial cells through inhibiting glycolysis in human umbilical vein endothelial cells. Acta Biochim Biophs Sin. 2019;51:1114–22.
Sumi C, Okamoto A, Tanaka H, Nishi K, Kusunoki M, Shoji T, et al. Propofol induces a metabolic switch to glycolysis and cell death in a mitochondrial electron transport chain-dependent manner. PLoS ONE. 2018;13:e0192796.
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. 2010;11:373–84.
Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. 2012;18:363–74.
Chen X, Lu P, Chen L, Yang SJ, Shen HY, Yu DD, et al. Perioperative propofol-paravertebral anesthesia decreases the metastasis and progression of breast cancer. Tumour Biol. 2015;36:8259–66.
Xu YJ, Li SY, Cheng Q, Chen WK, Wang SL, Ren Y, et al. Effects of anaesthesia on proliferation, invasion and apoptosis of LoVo colon cancer cells in vitro. Anaesthesia. 2016;71:147–54.
Chen XY, Wu QC, You L, Chen SS, Zhu MM, Miao CH. Propofol attenuates pancreatic cancer malignant potential via inhibition of NMDA receptor. Eur J Pharmacol. 2017;795:150–9.
Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem. 2014;289:7884–96.
Talwar H, Bouhamdan M, Bauerfeld C, Talreja TJ, Aoidi R, Houde N, et al. MEK2 negatively regulates lipopolysaccharide-mediated IL-1 beta production through HIF-1 alpha expression. J Immunol. 2019;202:1815–25.
Nishi K, Oda T, Takabuchi S, Oda S, Fukuda K, Adachi T, et al. LPS induces hypoxia-inducible factor 1 activation in macrophage-differentiated cells in a reactive oxygen species—dependent manner. Antioxid Redox Signal. 2008;10:983–95.
Chen X, Wu Q, Sun P, Zhao Y, Zhu M, Miao C. Propofol disrupts aerobic glycolysis in colorectal cancer cells via inactivation of the NMDAR-CAMKII-ERK pathway. Cell Physiol Biochem. 2018;46:492–504.
Sumi C, Okamoto A, Tanaka H, Kusunoki M, Shoji T, Uba T, et al. Suppression of mitochondrial oxygen metabolism mediated by the transcription factor HIF-1 alleviates propofol-induced cell toxicity. Sci Rep. 2018;8:8987.
Chen RM, Wu CH, Chang HC, Wu GJ, Lin YL, Sheu JR, et al. Propofol suppresses macrophage functions and modulates mitochondrial membrane potential and cellular adenosine triphosphate synthesis. Anesthesiology. 2003;98:1178–85.
Carruthers A, DeZutter J, Ganguly A, Devaskar SU. Will the original glucose transporter isoform please stand up! Am J Physiol Endocrinol Metab. 2009;297:E836–48.
Watanabe R, Hilhorst M, Zhang H, Zeisbrich M, Berry GJ, Wallis BB, et al. Glucose metabolism controls disease-specific signatures of macrophage effector functions. JCI Insight. 2018;3:e123047.
Pinheiro CH, Silveira LR, Nachbar RT, Vitzel KF, Curi R. Regulation of glycolysis and expression of glucose metabolism-related genes by reactive oxygen species in contracting skeletal muscle cells. Free Radical Biol Med. 2010;48:953–60.
Liemburg-Apers DC, Willems PHGM, Koopman WJH, Grefte S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch Toxicol. 2015;89:1209–26.
Lopez-Pascual A, Lorente-Cebrian S, Moreno-Aliaga MJ, Martinez JA, Gonzalez-Muniesa P. Inflammation stimulates hypoxia-inducible factor-1 alpha regulatory activity in 3T3-L1 adipocytes with conditioned medium from lipopolysaccharide-activated RAW 264.7 macrophages. J Cell Physiol. 2019;234:550–60.
Hayashi M, Sakata M, Takeda T, Yamamoto T, Okamoto Y, Sawada K, et al. Induction of glucose transporter 1 expression through hypoxia-inducible factor 1 alpha under hypoxic conditions in trophoblast-derived cells. J Endocrinol. 2004;183:145–54.
Ham M, Lee JW, Choi AH, Jang H, Choi G, Park J, et al. Macrophage glucose-6-phosphate dehydrogenase stimulates proinflammatory responses with oxidative stress. Mol Cell Biol. 2013;33:2425–35.
Xu KJ, He ZY, Chen M, Wang NF, Zhang DC, Yang L, et al. HIF-1 alpha regulates cellular metabolism, and Imatinib resistance by targeting phosphogluconate dehydrogenase in gastrointestinal stromal tumors. Cell Death Dis. 2020;11:586.
Freemerman AJ, Zhao LY, Pingili AK, Teng B, Cozzo AJ, Fuller AM, et al. Myeloid Slc2a1-deficient murine model revealed macrophage activation and metabolic phenotype are fueled by GLUT1. J Immunol. 2019;202:1265–86.
Acknowledgements
This study was supported by Pharmacy Research of Guangdong Provincial Hospital (No. 2020XC25). We thank Kexuan Liu, Chai Li (Southern Medical University) for their technical assistance.
Author information
Authors and Affiliations
Contributions
CZ devised the concept, designed the research and wrote the paper. WZ performed experiments, analyzed data and designed the figures. ZX, MT, and YW performed the experiments. The final version of the manuscript was approved by all the authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zeng, W., Xing, Z., Tan, M. et al. Propofol regulates activated macrophages metabolism through inhibition of ROS-mediated GLUT1 expression. Inflamm. Res. 70, 473–481 (2021). https://doi.org/10.1007/s00011-021-01449-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-021-01449-y