
Abstract
Background
Multiple sclerosis (MS) is a chronic and autoimmune disease of the central nervous system (CNS), mainly characterized by inflammatory demyelination, which manifests as relapses and diffuse damage and brain volume loss, both accounting for neurodegeneration, and therefore, physical disability. MS typically affects young adults and is commonly diagnosed in the early years by acute relapses, which then followed through partial or complete remission period. The clinical course of MS is characterized as four major classifications, including relapsing–remitting (RRMS), primary progressive (PPMS), progressive relapsing (PRMS), and secondary progressive (SPMS).
Purpose
This review provides comprehensive overview of the current treatments and future innovative approaches in the treatment of MS.
Results
Currently, there is no definite cure for MS. The treatment of MS has mainly been based on the prescription of immunosuppressive and immune-modulating agents. However, a number of disease-modifying treatments (DMTs) have been designed that reduce the attack rate and delay progression and mainly target inflammation settings in these patients. Although remarkable advancements have occurred in the therapy of MS, the rate of progressive disability and early mortality is still worrisome. Recently, a monoclonal antibody (ocrelizumab) was demonstrated to be beneficial in a clinical trial of primary progressive MS. Furthermore, novel treatment strategies concentrating on the remyelination or neuroprotection are under evaluation.
Conclusions
In spite of prosperous experiences in MS therapy, the future research, hopefully, will bring substantial improvements in the understanding and approaches of MS therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is characterized as chronic and autoinflammatory setting causing neurodegeneration and inflammatory immune responses in the central nervous system (CNS), including brain and the spinal cord. MS is the most prevalent non-traumatic cause of CNS complications in young people worldwide. The immune system has been the main culprit of MS [1,2,3,4,5]. In most cases, initial disease course of MS is relapsing–remitting (RRMS) with periods of relapses followed by periods of remission. In most of the MS patients, the relapsing course is further advanced towards a secondary progressive phase (SPMS) [6]. However, in about 15% of cases, MS is progressed from the initial phase with a primary progressive phase (PPMS) lacking superimposed relapse periods [6]. Contemporary classification guidelines concentrate on the inflammatory image of MS, which can be manifested at all phases of the disease and can be targeted using disease-modifying treatments (DMTs) [7]. Currently, there are a number of DMTs available for the therapy of RRMS and their primary purpose is to reduce the relapse level and the inflammation severity in CNS [8].
During last decades, several advancements have occurred in the treatment of MS. To date, new much efficient treatments for MS therapy are available, after several years of treatment with DMTs such as interferon beta (IFNβ) and glatiramer acetate (GA) as the main treatment options, fingolimod was the first oral DMT, which was approved in 2010 in United States. Since then, a number of other oral medications has been validated or are in phase III trials currently [9]. Currently, three monoclonal antibodies are approved for MS therapy and some other agents are in the final phase of development. Although a promising progression in treating MS has occurred, the currently available medications are unable to respond the future needs raised by the complicated nature of MS. Hence, in this review article, we have tried to summarize the current available treatments for MS as well as draw the novel landscape in MS therapy with prospects on the future of disease treatment development.
Previously approved therapies in MS
Injectable drugs
Three major IFNβ products are available for administration as first-line DMTs to treat relapsing MS. All of these products were approved after relevant process of single double-blind, placebo-controlled, phase III clinical trials [10]. Of three these products, two of them are administered subcutaneously and the other one is injected intramuscularly. Copaxone, which is a four amino acid synthetic copolymer [11], was approved after a single phase III randomized clinical trial [12] and was shown to be effective in treatment of RRMS. Copaxone and IFNβ possess various immunomodulatory effects but with almost similar function in reducing the relapse rate up until about 30% [13].
A large observational cohort study demonstrated that IFNβ and Copaxone therapy ameliorates progression of disability as evaluated by Expanded Disability Status Scale (EDSS) scores after 6 years of medication [14]. IFNβ and Copaxone therapy have been known to be generally safe and favorably tolerated. However, both IFNβ and Copaxone need periodical and long-term self-injections. Among the side effects of IFNβ product are increased levels of liver enzymes, flu-like symptoms, and injection-site unwanted reactions. On the other side, the side effects of Copaxone are injection-site unwanted reactions as well as post-injection reactions which are seen in approximately 15% of patients [15]. Of the approved medications for MS therapy, the humanized antibodies such as daclizumab, natalizumab, alemtuzumab [16], and mitoxantrone have been associated with promising effects but have drawn safety issues. Parenteral administration of these drugs is much prevalent that may have severe side effects, such as autoimmune-associated complications by alemtuzumab, progressive multifocal leukoencephalopathy (PML) by natalizumab, liver injury, skin reactions, and colitis by daclizumab, and finally cardiotoxicity and acute leukemia by mitoxantrone. Natalizumab, which is a humanized recombinant monoclonal antibody, targets α4-integrin [16]. This biological medication interrupts the leukocyte migration from the peripheral blood into the CNS through inhibiting the binding of leukocytes by α4-integrin to the vascular cell adhesion molecule (VCAM) located on the endothelial cell [16].
This interference possesses a beneficial influence on CNS inflammation by blocking the binding and later diapedesis of lymphocytes through the blood–brain barrier (BBB). In a placebo-controlled phase III trial, resulting in approval of natalizumab, intravenous administration in the dose of 300 mg monthly decreased RR up to 68% and interrupted disability progression up to 42% by 2 years [17] and decreased the MRI activity up to 92% [18]. After that, natalizumab was reintroduced in 2006 with description of risk management programs [19]. The risk of PML stratification in cases with MS on natalizumab underlies duration of treatment, prior immunosuppressant utilization, and the anti-JC virus (JCV) antibody conditions imply to JVC infection [20, 21]. This issue permits increased risk stratification in treatment with natalizumab [22]. Natalizumab therapy may trigger the production of persistent neutralizing antibodies (NABs) in 4–6% of cases, typically occurring within the first 12 months. It has been shown that NABs are related with increased rates of infusion-related adverse responses and can decrease the treatment efficacy [23].
Alemtuzumab, which is a humanized monoclonal antibody, targets CD52 molecule expressed on natural killer (NK) cells, lymphocytes, monocytes, and some other granulocytes [24, 25]. Alemtuzumab causes quick lymphopenia, which takes years to last, through antibody-dependent cellular cytotoxicity (ADCC) [16]. The subcutaneous administration of alemtuzumab was compared to IFNβ-1 injection that was conducted three times a week in two phase III trials of RRMS [26, 27]. It was observed that alemtuzumab decreased the annualized relapse rate (ARR) up to 49–55%, reduced the rate of progression disability up to 30–42%, and attenuated MRI gadolinium-enhancing lesions up to 61–63% [27, 28]. In Europeans, alemtuzumab has been prescribed as a first-line therapy in active RRMS, although some neurologists would administer it as a second-line medication due to the risk of secondary autoimmunity following the therapy period [23].
Daclizumab, which is a humanized monoclonal antibody, targets against the interleukin (IL)-2 receptor subunit CD25 expressed on T cells [29]. Although daclizumab’s influence on decreasing CD25+ T cells is short and small, it causes expansion of CD56bright NK cells, correlating with the clinical efficiency of the drug [29]. Randomized double-blind trials (phase II and III trial) demonstrated that daclizumab had promising influence as observed by MRI manifestations [30,31,32] either as supplementary therapy to IFNΒ-β1a or placebo. Daclizumab did not show indications of rebound effects following treatment stop. Unique side effect of daclizumab is cutaneous complications. Most of the skin complications are patches of eczema, which usually needs no medications [33], although mild-to-severe rashes occurred in 19% of cases require interrupting the treatment. The skin lesions demonstrated nonspecific characteristics of eczematous dermatitis, with infiltration of CD56+ lymphocytes, which were not associated with the clinical manifestations [33]. Recently, FDA approved daclizumab for treatment of RRMS [34]. Daclizumab should be prescribed to patients with insufficient response to two or more conventional treatments to MS. It is mandatory for patients to be evaluated for liver function before initiating daclizumab therapy as well as monthly before each dose, and, afterwards, for up to 6 months after the last dose of administration [34].
Mitoxantrone functions through inhibiting type II topoisomerase and disruption of DNA synthesis. Mitoxantrone transmits through the disrupted blood brain barrier (BBB) and may stimulate microglial death [35]. It was approved by the FDA for rapidly improving SPMS and RRMS after a number of clinical trials [36, 37]. Mitoxantrone is administered through infusions monthly at doses of 12 mg/m2, although the cumulative dose is restricted because of hematologic and cardiologic side effects. The prescription of mitoxantrone was quickly decreased because of severe complications like acute leukemia [38] as well as due to advent of alternative more efficient and less toxic medications [23] (Table 1).

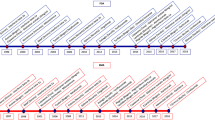
Therapeutic targets of monoclonal antibodies in treatment of MS. The illustration depicts schematic view of an intracranial blood vessel that goes into the CNS. Most of the targets for the currently available monoclonal antibody to treat MS patients are located in the blood vessel, or better put, the peripheral players in mechanobiology of the immune system. Anti-CD20 monoclonal antibodies, including rituximab, ocrelizumab, and ofatumumab deplete B cells. The α4β1 integrin (VLA-4) receptors on B and T cells are blocked by natalizumab, leading to inhibition of these cells to bind vascular cell adhesion protein (VCAM) and cross through blood brain barrier (BBB) into the CNS. Alemtuzumab causes depletion of CD52-expressing B and T cells, and therefore, memory and regulatory T (Treg) cells are substituted. Through targeting the CD25 subunit of the IL2Rα on T cells, daclizumab, instead of depleting T cells, functions in increasing the number of CD56bright NK cells, triggering tolerogenic or immunoregulatory response. By blocking LINGO-1, opicinumab stimulates differentiation of oligodendrocyte (OG) precursor and, therefore, remyelination occurs. Opicinumab has been beneficial during the damage phase in the CNS, including axonal damage, demyelination, microgliosis, and reactive astrocytosis
Drugs with oral administration
Teriflunomide has been approved for treatment of mild-to-moderate rheumatoid arthritis (RA). This drug interrupts the mitochondrial enzyme involved in de novo pyrimidine synthesis dihydroorotate dehydrogenase (DHODH) [39]. Studies in two phase III trials in RRMS demonstrated that teriflunomide decreased the ARR in comparison to placebo up to 31–36%, the level of disability progression up to 26–27%, and the MRI gadolinium-enhancing lesions displayed 80% reduction [40, 41]. Studies showed that teriflunomide demonstrated same effects on the ARR and treatment interruption compared with subcutaneous IFNβ-1a administration [42]. Teriflunomide has been evaluated in a double-blind, randomized, placebo trial on patients with clinically isolated syndrome (CIS) having silent MRI lesions and it led to procrastination in the time to a second relapse and amelioration in the recent MRI lesions [43]. Among the side effects of teriflunomide are alanine aminotransferase (ALT) increase, diarrhea, headache, nausea, and hair thinning [44]. The most usual reason for stopping the treatment with teriflunomide is ALT elevation; hence a periodical ALT evaluation within the first 6 months of treatment and afterwards every second month is suggested [44].
The recently approved oral DMT for treatment of RRMS is delayed-release dimethyl fumarate (DMF), which is administered in a 240 mg dose capsule twice a day. Although its mechanism of action has not fully characterized yet, it has been proposed that DMF activates the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) pathway [45].
DMF was examined in two phase III trials in RRMS, demonstrating a reduction of ARR up to 44–53%, the rate of disability progression up to 22–32%, and the MRI gadolinium-enhancing lesions up to approximately 75–94% [46, 47]. Moreover, the phase III trials indicated that DMF therapy led to decreased clinical and MRI disease activity [48]. The prevalent side effects of DMF are nausea, diarrhea, flushing, and abdominal pain [47]. Moreover, DMF may induce leucopenia and elevate liver transaminases.
Fingolimod, approved by FDA in 2010, was the first line of oral treatment for relapsing forms of MS. The drug is administered as 0.5 mg dose capsule once daily. Fingolimod is sphingosine-1-phosphate (S1P) receptor antagonist and functions non-selectively in degrading the S1P1 receptor on lymphocytes [49, 50]. The drug captures T lymphocytes in secondary lymphatic tissues, and therefore, leads to amelioration of inflammation in MS [51].
Fingolimod was evaluated in two phase III trials in RRMS and demonstrated a reduction in ARR up to 48–55%, the rate of disability progression up to 25–30%, and the MRI gadolinium-enhancing lesions over 80% [52]. In comparison to IFNβ-1a, intramuscular injection of fingolimod once in a week caused a decline in ARR up to 52%, the rate of disability progression up to 25%, and the MRI gadolinium-enhancing lesions over 50% [53]. A fingolimod phase III trial in patients with PPMS resulted in no postponement of disability progression [54]. Most prevalent side effects of fingolimod are cough, diarrhea, headache, back pain, and upper respiratory tract infection [55]. It is suggested to perform electrocardiogram monitoring steadily for 6 h after the first dose of fingolimod due to the possibility of bradycardia and atrioventricular block upon first administration. In one of the phase III trials, a death case was observed because of a fulminant primary varicella zoster. Afterwards, examination for varicella zoster infection is recommended in cases under therapy with fingolimod. Moreover, it is advised to perform vaccination in case with no infection history [53] (Table 1).
New drugs in trial
Monoclonal antibodies
There are three anti-CD20 agents, namely, ocrelizumab, rituximab, and ofatumumab, which function to deplete pre-B cells and mature B cells, that have been evaluated for MS therapy [25]. Rituximab targets CD20 and has been utilized to treat MS and patients with neuromyelitis optica. Rituximab in a phase II trial was accompanied with reduction in recent MRI gadolinium-enhancing lesions up to 91%. Moreover 78% of cases manifested an infusion-related side effect. Occurrence of infection had same rate in both groups [56]. A small phase II trial of intrathecal rituximab demonstrated low efficacy as observed with evaluation of CSF biomarkers [57]. While treatment with rituximab caused a transient and incomplete depletion of B cells in the CSF samples, the impression on peripheral B cells was favorable and lasting [57]. It has been suggested that rituximab can be a choice in RRMS patients with no respond to first- and second-line medications, in patients with other autoimmune disorders [58], and in cases with stable RRMS who change their therapies from natalizumab to other DMT because of high PML risk [59].
Ocrelizumab in a phase II RRMS trial reduced the rate of MRI enhancing lesions [60]. Although infection side effects were equal between RRMS and placebo groups, infusion-related adverse effects were seen in the ocrelizumab group more frequently than in the placebo group. Studies have indicated that ocrelizumab reduced the annual relapse rate up to 46 and 47%, in OPERA I and II phase III trials, respectively [61]. Furthermore, a reduction of clinical disability by 40% was reported. As well, ocrelizumab caused a reduction of the count of T1 gadolinium-enhancing lesions in the brain up to 94% [61].
It was also observed that 47.9 and 47.5% of patients with ocrelizumab therapy in OPERA I and OPERA II, respectively, after 96 weeks demonstrated no evidence of MS relapses, disability progression, and T2 or gadolinium-enhancing T1 lesions [62]. A phase III clinical trial, named ORATORIO, evaluated intravenous ocrelizumab 600 mg every 6 months for PPMS therapy and compared it to placebo group [63]. The first drug in trial, namely, ocrelizumab, demonstrated primary and secondary efficacy results in a phase III PPMS survey. Ocrelizumab displayed significant decrease in the relative risk of 12-week confirmed disability progression (CDP) up to 24% and 24-week CDP up to 25% [63]; as well, ocrelizumab therapy reduced the volume of T2 hyperintense lesions and decreased the loss of whole brain volume in comparison to placebo group [63].
Ofatumumab is a monoclonal antibody, which is also prescribed for lymphocytic leukemia. It interrupts the early activation of the B lymphocyte and demonstrates lower antigenicity. Ofatumumab was evaluated in a small phase II clinical trial and demonstrated promising results, resulting in a 99% decrease in MRI activity and no significant side effects [64]. The effectiveness and safety of ofatumumab were measured in a phase II trial in 232 patients with RRMS in comparison to placebo, demonstrating a 90% decrease of MRI lesions after 12 weeks of therapy initiation. Therapy with 60 mg dose displayed five serious side effects, while no cases of opportunistic infections or PML were observed [65] (Fig. 1).
Laquinimod
Laquinimod is a carboxamide derivative, which was validated to decrease disease activity in RRMS patients, but with intense side effects [66]. Laquinimod has been assessed for treatment of neurodegenerative disease like Huntington’s disease and progressive RRMS [67]. Studies on the EAE (experimental autoimmune encephalomyelitis) mice, the animal model of MS, indicated that laquinimod caused a decline in overall inflammation, axonal injury, and demyelination [68]. Apparently, laquinimod interrupts CD4+ T cells and macrophage infiltration into the CNS. The drug also causes an increase in the serum level of brain-derived neurotrophic factor, which may have protective effects against neuronal damages [69]. Two phase III trials of ALLEGRO (Assessment of Oral Laquinimod in Preventing Progression in Multiple Sclerosis) [70] and BRAVO (Benefit–Risk Assessment of Avonex and Laquinimod) [71] have evaluated the efficacy of laquinimod in RRMS patients. ALLEGRO indicated that laquinimod led to a mild, but significant decrease of relapse rate and disease progression [70].
At first, BRAVO did not support the beneficial effect of laquinimod in reducing the annual relapse rate [71]. However, later by adjusting the groups for the number of patients having frequent lesions and increased T2 lesion volume, the relapse rate was observed. Overall laquinimod demonstrated more marked influences on the disease disability progression as well as brain atrophy in relation to influence on relapses rate and recent MRI lesion occurrence [72]. The safety and efficacy of laquinimod have been investigated via a phase III of RRMS (CONCERTO) and a phase II of PPMS (ARPEGGIO). Both of these studies intended to evaluate the efficacy of two doses of laquinimod, namely, 0.6 and 1.5 mg/day, in comparison to placebo. Nonetheless, the evaluation of higher doses of laquinimod was stopped following the occurrence of cardiovascular complications in eight cases in 2016 [1] (Table 1).
Cladribine
Cladribine, an adenosine deaminase-resistant purine nucleoside, is prescribed as chemotherapeutic agent primarily for the treatment of hairy cell leukemia as well as other neoplasms [73]. Cladribine preferentially acts on monocytes lymphocytes and integrates into the DNA of dividing cells leading to apoptosis [74]. Although cladribine selectively causes depletion of circulating T- and B-cell numbers, it shows little effect on NK cell count [75]. In two double-blind randomized trials, the cladribine intravenous administration was examined for PPMS, SPMS therapy and indicated promising outcomes [76, 77]. Moreover, it was evaluated as an oral agent for RRMS [75]. In a placebo-controlled trial, cladribine was evaluated for RRMS therapy, which decreased both the severity and prevalence of relapses and reduced the MRI-enhancing lesions [78]. The 120-week evaluation indicated that the clinical favorable effects of 3.5 mg/kg cladribine used in the initial 2 years of the trial on disability, relapses, and MRI outcome can last for at least 4 years in most of the patients [79, 80].
A randomized double-blind phase IIb trial of cladribine tablets in 3.5 mg/kg dose as supplementary to IFN-β therapy in 2-year evaluation on relapsing MS patients indicated that number of relapses was decreased (23%) in patients treated with cladribine plus IFNβ in comparison to 56% relapse rate in patients with placebo plus IFNβ therapy [81]. On the other hand, the average numbers of recent enhancing T1 and active T2 lesions were decreased after cladribine plus IFNβ therapy in comparison to placebo plus IFNβ regimen treatment [81]. Another study demonstrated that both doses of cladribine significantly caused a delay in MS diagnosis in comparison to placebo [82].
Using the 2010 McDonald criteria, it was demonstrated that cladribine 3.5 mg/kg significantly decreased the risk of more disability and relapse exacerbation in comparison to placebo [83]. Cladribine was prescribed for the treatment of RRMS in Russia and Australia, but was stopped after a while. Nonetheless, a meta-analysis of performed phase III trials of approved DMTs for RRMS as well as the CLARITY trial did not confirmed an elevated risk of cancer due to cladribine [84]. It seems that a long-term screening is needed to evaluate the safety of cladribine and approved DMDs for the risk of cancer [84].
Siponimod and ozanimod
Siponimod, also called BAF312, is a regulator of sphingosine pathway and is available for oral administrations. It acts more selectively in comparison to fingolimod by targeting S1P-1 and S1P-5 [85]. Siponimod was evaluated in a phase II trial for RRMS treatment and resulted in amelioration of brain MRI lesions and relapses rate [86].
Ozanimod is another oral drug that selectively modulates S1P receptor. Ozanimod was successfully evaluated in a phase II trial with promising ameliorative effects as measured through MRI manifestations [26].
Transplantation of autologous bone marrow
A number of studies using animal models have demonstrated that syngeneic bone marrow transplantation leads to immunosuppression which can exert antigen-specific tolerance [87]. Over the course of past few years, because of the high rate of adverse occurrences related to bone marrow transplantation, it was set aside as a therapy for MS patients only in cases that failed to respond to all other treatments and represented a poor prognosis [88]. Studies show that high-dose immune ablation and autologous hematopoietic stem cell transplantation (HSCT) could regenerate the cells of immune system repertoire and weaken the immune tolerance [89], and therefore, negatively impress the final outcome with clinical complication. Phase I clinical trials have demonstrated that autologous HSCT may ameliorate the disease activity, and therefore, improve the life quality in MS patients [88, 90]. A phase II trial evaluating the efficacy of HSCT versus mitoxantrone in treatment of RRMS and SPMS revealed that HSCT decreased new T2 count, enhancing lesions, and the AAR in comparison to mitoxantrone medication [91]. In a phase II trial during 6–7 years of screening, it was observed that 70% of the patients receiving an aggressive immune-ablative treatment after a HSCT with graft depletion of autoreactive lymphocytes did not represent any signs of disease activity, manifested through relapses, new MRI lesions, and EDSS progression [92]. Despite these observations are promising, and attempts and advancements has decreased the risks and side effects, there are many uncertainties about the exertion of HSCT as a potential second-line treatment for MS refractory [93, 94] (Table 1).
By removing the mature lymphocytes from the graft before transplantation, it was observed that graft-mediated immune responses were eliminated accompanied by beneficial effects on decreasing disease activity [92]. However, it seems that therapeutic approaches by exerting HSCT in MS need to be reconsidered alongside with the contribution of ever-growing array of available therapeutic approaches and designing more sophisticated trials.
Strategies to restore myelination
Remyelination happens during initial phases of MS development, and the repair mechanism in the CNS face impairments over time, particularly when the disease tends to be chronic [95]. During the repair steps, it is vital for oligodendrocyte precursor cells (OPC) to be differentiated into mature cells [95]. The crosstalk between the immune system and OPC possesses particular specifications in MS setting [96, 97]. The process of neurodegeneration is seen early in the course of the MS and repeated demyelination episodes can result in local elimination of myelin producing OPCs [98]. Most data on the demyelination procedure have been achieved from investigations performed on animal models of MS [99].
It has been shown that tocopherol derivative TFA-12, a member of the vitamin E family, that displays anti-inflammatory properties induces differentiation of OPC, and therefore, leads to repair of myelin in EAE mice [100]. Expression of myelin gene in oligodendrocytes is induced by lithium chloride through exerting Akt/CREB and Wnt/β-catenin signaling pathways [101]. Indomethacin, which is a non-steroidal anti-inflammatory drug (NSAID), can cross through the blood brain barrier (BBB) and promote OPC differentiation into mature cells. As a result, indomethacin can trigger remyelination through Wnt/β-catenin pathway [102]. Furthermore, nuclear retinoid X receptor (RXR)-γ, by ligation to receptors inside the OPC, positively modulates the remyelination via stimulating the differentiation of OPC [103, 104].
In vitro experiments have indicated that miconazole and clobetasol can increase differentiation of oligodendrocytes from human OPC by glucocorticoid receptor and mitogen-activated protein kinase signaling pathways [105].
As a specific transmembrane protein of nervous system, leucine-rich repeat and immunoglobulin domain containing 1 (LINGO-1) can possibly be a therapeutic target with respect to remyelination. Oligodendrocytes express LINGO-1, which inhibits the potential of these cells to differentiate and myelinate. Moreover, LINGO-1 is expressed on axons and confers regeneration of axons [106, 107]. Antibodies against LINGO-1 lead to differentiation of OPC in lesions displaying demyelination and alleviate axonal injuries [108, 109].
Opicinumab, a human monoclonal antibody, binds to LINGO-1 and promotes remyelination and retrieves [110]. As the first remyelinating therapeutics, anti-LINGO-1 antibodies have been tried in humans. In a phase I randomized trial of opicinumab, one or two doses over 100 mg/kg were tolerated and did not demonstrate intense side effects with low immunogenicity [111].
In a study, human induced pluripotent stem cells (iPS) were differentiated into OPCs and transferred to a mouse with myelin-deficiency [112]. These OPCs were differentiated into astrocytes and oligodendrocytes and caused myelination of the brains of the host animal, and ultimately led to increased survival rate [112]. Nonetheless, with respect to the multifocal nature of MS, repeated transplantation of the OPC is needed in all the lesions with demyelination presentations [98].
On the other hand, there is declined recruitment rate of OPC in MS lesions; hence chemoattractants like Sema3A receptor neuropilin-1 can be regarded as a new class of therapies to ameliorate remyelination process [113]. Nowadays, it is not fully determined that remyelination could hinder the neurodegeneration process. However, it seems to have the potential of restoring neuronal function or limitation of degeneration of nerve components at least [98]. As a result, new much more sophisticated approaches of remyelination therapies could possibly be part of MS therapy in the future implementations (Table 1).
Mesenchymal stem cells
Mesenchymal stem cells (MSC) can be obtained from bone marrow and then be engrafted lacking the necessity for immunosuppressive interventions. It seems that the therapeutic potential of MSC is systemic [114]. The therapeutic mechanisms of action by MSC transplantation are due to anti-inflammatory modulations of the environment. In a phase II trial evaluating the potential of MSC for MS therapy, no remarkable adverse events were reported [115]. Another trial in MS patients with amyotrophic lateral sclerosis indicated that intrathecal and intravenous injection of autologous MSCs was almost safe; its implementation raised quick immunomodulatory outcomes [116].
Strategies targeting T cell
Despite varieties in the activation circumstances or subpopulation of MS T cells, healthy subjects may also develop immune responses against MS related antigens such as myelin oligodendrocyte protein (MOG), myelin proteins including basic protein (MBP), and proteolipid protein (PLP) [117,118,119,120]. Nonetheless, it has proposed that possible mechanisms of disease initiation and perpetuation might be epitope spreading, molecular mimicry, and bystander activation. As a result, it seems that an alternative therapeutic strategy in MS could be restoring self-tolerance toward autoantigens by means of immunization approaches [121]. Furthermore, stimulation of tolerance in T cells [122], particularly regulatory T cells (Treg) signaling could be promising in restoring self-tolerance [123]. A phase I trial disclosed that it is possible to implement antigen-coupled cell tolerization in MS subjects [124]. Animal studies as well as human trials in MS patients by vaccination with T-cells have been performed [125, 126]. Phase II, placebo-controlled clinical trials through preventing the encephalitogenicity of myelin-reactive T cells in MS patients have been established [126, 127].
It was indicated that targeting MOG, MBP and PLP antigens through myelin-reactive T-cells was accompanied with safety in RRMS patients, although 44% of cases were previously receiving DMTs [127]. A placebo-controlled, double-blind autologous T cell vaccine (TCV) trial was carried out in progressive MS, in which 19 RPMS patients were under therapy with attenuated autologous T cells against various antigens derived from MBP, MOG, and PLP [128]. It was observed that the TCV trial on these patients was safe representing no serious side effects. Moreover, it was associated with promising effects on relapses and disability of the patients [128]. It seems that exertion of various peptides and several anti-myelin T-cell lines was responsible for the desirable effects [128] (Table 1).
Epigenetic therapy
Epigenetics is defined as heritable modifications in gene expression occurring without alterations in the nucleotide sequence of DNA. DNA methylation, histone modifications, and microRNA-associated gene expression regulation are the main mechanisms for epigenetic regulations [129,130,131]. Several studies have demonstrated that an array of aberrant regulation of these mechanisms lead to pathogenesis of autoimmune disease such as MS [29, 132,133,134]. Acknowledging the aberrant epigenetic regulations involved in the etiopathology of autoimmune diseases has drawn the attention towards an array of researches to dissect the tissue-/cell-specific epigenetic clinical markers for early diagnosis as well to develop novel medications. The novel therapeutic trend, called ‘epigenetic therapy’, includes medications with the ability to regulate methylation patterns, histones tails, and expression pattern of miRNAs. Among these medications are currently compounds like histone deacetylase (HDAC) inhibitors and DNA methyltransferase (DNMT) inhibitors. In patients with MS, studies have been carried out in animal models and in vitro trials, which seem promising to pass clinical trials in the future.
Therapy with trichostatin A (TSA), a HDAC inhibitor, led to ameliorated spinal cord inflammation, demyelination, neuronal and axonal loss, and improvement of disability in the relapsing period of EAE C57BL/6 mice through decreased inflammatory cell infiltration into the CNS [135]. Vorinostat, a HDAC inhibitor, interrupted dendritic cell (DC)-directed Th1 and Th17-polarizing production of cytokine and improved EAE by decreasing CNS inflammation and demyelination, mediated by Th1 and Th17 cells. Moreover, vorinostat therapy in EAE mice, led to downregulation of costimulatory molecules of DCs, including CD80 and CD86, and HLA-DR. As a consequence, vorinostat conferred favorable therapeutic effects in EAE mice and seems promising for the treatment of human MS in the future [136]. Valproic acid (VPA) is a HDAC inhibitor and has been accompanied with beneficial therapeutic effects in EAE mice, as represented with alleviated disease severity and duration. The beneficial outcomes were manifested by diverging the development of CD4+ T cells from Th1 and Th17 to Th2 and Treg profile; thus, downregulation of the proinflammatory cytokines such as tumor necrosis factor (TNF)-α, IFN-γ, IL-1β, and IL-17 in the spinal cord was manifested. Furthermore, VPA treatment led to increased expression of IL-4, which is an anti-inflammatory cytokine. Treatment with VPA reduced the infiltration and accumulation of lymphocytes and macrophages in spinal cords of EAE mice [137]. All of these outcomes are in line with positive therapeutic effects for MS.
Curcumin is a naturally polyphenolic phytochemical compound that inhibits the function of lysine acetyltransferase enzymes. Curcumin decreases the severity of EAE in rats through suppressing the infiltration of inflammatory cells into the spinal cord. Curcumin therapy in animal models resulted in down-expression of IL-6, IL-17, IL-21, STAT3, TGFβ, and RORγ, suggesting that curcumin prevents differentiation of CD4+ T cells to Th17 cell, a main player in MS [138]. Oral administration of Resveratrol activates Sirt1 (deacetylase enzyme), resulting in limited neuronal damage in EAE female SJL/J mice [139]. In EAE mice, 5-aza-2′-deoxycytidine (Decitabine, a DNMT inhibitor) caused both the count and immunosuppressive activity of Foxp3+ Tregs. Moreover, effector cells in the periphery were suppressed [140]. By suppressing the CNS inflammation, 5-aza-2′-deoxycytidine seems promising in MS therapy. It was also revealed that suppressing the miR-326 expression led to reduced count of Th17 cells, and thus, improved disease severity, proposing the miR-326 potential in Th17 polarization and MS therapy [141]. Although the exertion of epigenetic therapy has just started, there are convincing observations in animal models of MS. If epigenetic drugs are prosperous in other disorders like cancer and autoimmune diseases, this new trend of therapeutic approach will hopefully be used in treatment of MS patients in the future.
Future directions
It appears that monoclonal antibodies, relative to other approaches, could win the completion of best practice therapy for MS patients; a great deal of research has been diverged to this path. Therapeutics by monoclonal antibodies has conferred the opportunity to targeting pathogenesis mechanisms of the immune system, including CD20 (rituximab), CD25 (daclizumab), CD52 (alemtuzumab), and VLA-4 (natalizumab). Regarding such approaches, the final challenge for physicians will be to recognize the well-suited drug for patients. In recent years, assessing monoclonal antibodies targeting IL-17A and anti-LINGO-1 has gained attentions and been exciting future approach in treatment of MS patients. Modulation of IL-17A signaling could be a promising strategy for designing therapeutics in several autoinflammatory settings. The function of anti-LINGO-1 therapy through evaluations by phase II proof-of-concept trial could be interesting. Despite seemingly high efficacy of these new drugs, potential adverse effects of them on immune surveillance of the CNS and host’s immune system are still not well known. Therefore, we will have to carefully monitor the patients that are receiving such medications to disclose the potential side effects precisely. Other than that, identification of new molecular pathogenesis aspects of MS will hopefully open new horizons for designing novel therapeutics for these patients.
Concluding points
The well-suited treatment approach for MS therapy is still controversial. Choosing the treatment approach should consider the circumstance of the inflammation. Drugs like alemtuzumab and natalizumab are preferred in individuals with active MS. Over the course of past years, the marked impression on increasing life expectancy in the population underlies in primary prevention. MS has been speculated to be the result of the interactions between the genetic factors and environmental agents. Among the factors present in environment, infection with the Epstein–Barr virus (EBV), vitamin D deficiency, smoking, obesity, and lack of exposure to intestinal parasites are contributing agents of the MS risk. Giving that researches about preventing MS are of high priority nowadays, developments of prevention approaches seem to be on the fast track. Treating the right patient with the suitable drug soon in the initiation steps of the disease course, before disability would occur, could be accompanied with long-term positive results. It is not far from expectations that over the next decades, further information from techniques interested in preventing MS and on methodologies to prevent neurons or promote remyelination would change the image of MS therapy. Finally, the future of MS therapy would largely be contingent upon a comprehensive understanding of the immunopathogenesis of MS.
References
Coclitu C, Constantinescu CS, Tanasescu R. The future of multiple sclerosis treatments. Expert Rev Neurother. 2016;16(12):1341–56.
Javan M-R, Seyfizadeh N, Aslani S, Farhoodi M, Babaloo Z. Molecular analysis of interleukin-25 exons 1 and 2 and its serum levels in Iranian patients with multiple sclerosis. Am J Clin Exp Immunol. 2014;3(2):91.
Javan MR, Shahraki S, Safa A, Zamani MR, Salmaninejad A, Aslani S. An interleukin 12 B single nucleotide polymorphism increases IL-12p40 production and is associated with increased disease susceptibility in patients with relapsing-remitting multiple sclerosis. Neurol Res. 2017;39(5):435–41.
Javan MR, Aslani S, Zamani MR, Rostamnejad J, Asadi M, Farhoodi M, et al. Downregulation of immunosuppressive molecules, PD-1 and PD-L1 but not PD-L2, in the patients with multiple sclerosis. Iran J Allerg Asthma Immunol. 2016;15(4):296.
Azimi M, Ghabaee M, Moghadasi AN, Noorbakhsh F, Izad M. Immunomodulatory function of Treg-derived exosomes is impaired in patients with relapsing-remitting multiple sclerosis. Immunol Res. 2018. 1–8.
Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15(9):545–58.
Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ, et al. Defining the clinical course of multiple sclerosis The 2013 revisions. Neurology. 2014;83(3):278–86.
Tanasescu R, Ionete C, Chou I-J, Constantinescu C. Advances in the treatment of relapsing-remitting multiple sclerosis. Biomed J. 2014;37(2):41.
Thomas RH, Wakefield RA. Oral disease-modifying therapies for relapsing-remitting multiple sclerosis. Am J Health Syst Pharm. 2015;72(1).
Ali R, Nicholas RSJ, Muraro PA. Drugs in development for relapsing multiple sclerosis. Drugs. 2013;73(7):625–50.
Boster AL, Ford CC, Neudorfer O, Gilgun-Sherki Y. Glatiramer acetate: long-term safety and efficacy in relapsing-remitting multiple sclerosis. Expert Rev Neurother. 2015;15(6):575–86.
Johnson KP, Brooks B, Cohen J, Ford C, Goldstein J, Lisak R, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis results of a phase III multicenter, double-blind, placebo-controlled trial. Neurology. 1995;45(7):1268–76.
Lugaresi A, Di Ioia M, Travaglini D, Pietrolongo E, Pucci E, Onofrj M. Risk–benefit considerations in the treatment of relapsing-remitting multiple sclerosis. Neuropsychiatr Dis Treat. 2013;9:893.
Palace J, Duddy M, Bregenzer T, Lawton M, Zhu F, Boggild M, et al. Effectiveness and cost-effectiveness of interferon beta and glatiramer acetate in the UK multiple sclerosis risk sharing scheme at 6 years: a clinical cohort study with natural history comparator. Lancet Neurol. 2015;14(5):497–505.
Ford C, Goodman A, Johnson K, Kachuck N, Lindsey J, Lisak R, et al. Continuous long-term immunomodulatory therapy in relapsing multiple sclerosis: results from the 15-year analysis of the US prospective open-label study of glatiramer acetate. Mult Scler J. 2010;16(3):342–50.
Craddock J, Markovic-Plese S. Immunomodulatory therapies for relapsing-remitting multiple sclerosis: monoclonal antibodies, currently approved and in testing. Expert Rev Clin Pharmacol. 2015;8(3):283–96.
Polman CH, O’connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910.
Miller D, Soon D, Fernando K, MacManus D, Barker G, Yousry T, et al. MRI outcomes in a placebo-controlled trial of natalizumab in relapsing MS. Neurology. 2007;68(17):1390–401.
Rommer P, Zettl U, Kieseier B, Hartung HP, Menge T, Frohman E, et al. Requirement for safety monitoring for approved multiple sclerosis therapies: an overview. Clin Exp Immunol. 2014;175(3):397–407.
McGuigan C, Craner M, Guadagno J, Kapoor R, Mazibrada G, Molyneux P, et al. Stratification and monitoring of natalizumab-associated progressive multifocal leukoencephalopathy risk: recommendations from an expert group. J Neurol Neurosurg Psychiatry. 2015: jnnp-2015-311100.
Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366(20):1870–80.
Plavina T, Subramanyam M, Bloomgren G, Richman S, Pace A, Lee S, et al. Anti-JC virus antibody levels in serum or plasma further define risk of natalizumab-associated progressive multifocal leukoencephalopathy. Ann Neurol. 2014;76(6):802–12.
Torkildsen Ø, Myhr KM, Bø L. Disease-modifying treatments for multiple sclerosis—a review of approved medications. Eur J Neurol. 2016;23(S1):18–27.
Jones JL, Coles AJ. Mode of action and clinical studies with alemtuzumab. Exp Neurol. 2014;262:37–43.
Singer BA, editor. Parenteral treatment of multiple sclerosis: the advent of monoclonal antibodies. Seminars in neurology; 2016. Thieme Medical Publishers.
Cohen JA, Arnold DL, Comi G, Bar-Or A, Gujrathi S, Hartung JP, et al. Safety and efficacy of the selective sphingosine 1-phosphate receptor modulator ozanimod in relapsing multiple sclerosis (RADIANCE): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(4):373–81.
Coles AJ, Twyman CL, Arnold DL, Cohen JA, Confavreux C, Fox EJ, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1829–39.
Cohen JA, Coles AJ, Arnold DL, Confavreux C, Fox EJ, Hartung H-P, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1819–28.
Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, et al. Regulatory CD56bright natural killer cells mediate immunomodulatory effects of IL-2Rα-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci. 2006;103(15):5941–6.
Wynn D, Kaufman M, Montalban X, Vollmer T, Simon J, Elkins J, et al. Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta. Lancet Neurol. 2010;9(4):381–90.
Gold R, Giovannoni G, Selmaj K, Havrdova E, Montalban X, Radue E-W, et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECT): a randomised, double-blind, placebo-controlled trial. The Lancet. 2013;381(9884):2167–75.
Giovannoni G, Gold R, Selmaj K, Havrdova E, Montalban X, Radue E-W, et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECTION): a multicentre, randomised, double-blind extension trial. Lancet Neurol. 2014;13(5):472–81.
Cortese I, Ohayon J, Fenton K, Lee C-C, Raffeld M, Cowen EW, et al. Cutaneous adverse events in multiple sclerosis patients treated with daclizumab. Neurology. 2016;86(9):847–55.
Baldassari LE, Rose JW. Daclizumab: development, clinical trials, and practical aspects of use in multiple sclerosis. Neurotherapeutics. 2017:1–17.
Li J-M, Yang Y, Zhu P, Zheng F, Gong F-L, Mei Y-W. Mitoxantrone exerts both cytotoxic and immunoregulatory effects on activated microglial cells. Immunopharmacol Immunotoxicol. 2012;34(1):36–41.
Millefiorini E, Gasperini C, Pozzilli C, D’andrea F, Bastianello S, Trojano M, et al. Randomized placebo-controlled trial of mitoxantrone in relapsing-remitting multiple sclerosis: 24-month clinical and MRI outcome. J Neurol. 1997;244(3):153–9.
Hartung H-P, Gonsette R, Konig N, Kwiecinski H, Guseo A, Morrissey SP, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360(9350):2018–25.
Tanasescu R, Debouverie M, Pittion S, Anxionnat R, Vespignani H. Acute myeloid leukaemia induced by mitoxantrone in a multiple sclerosis patient. J Neurol. 2004;251(6):762–3.
Tanasescu R, Evangelou N, Constantinescu CS. Role of oral teriflunomide in the management of multiple sclerosis. Neuropsychiatr Dis Treat. 2013;9:539.
O’connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, Olsson TP, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365(14):1293–303.
Confavreux C, O’Connor P, Comi G, Freedman MS, Miller AE, Olsson TP, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(3):247–56.
Vermersch P, Czlonkowska A, Grimaldi LM, Confavreux C, Comi G, Kappos L, et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler J. 2014;20(6):705–16.
Miller AE, Wolinsky JS, Kappos L, Comi G, Freedman MS, Olsson TP, et al. Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis (TOPIC): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(10):977–86.
Comi G, Freedman MS, Kappos L, Olsson TP, Miller AE, Wolinsky JS, et al. Pooled safety and tolerability data from four placebo-controlled teriflunomide studies and extensions. Mult Scler Relat Disord. 2016;5:97–104.
Linker RA, Gold R. Dimethyl fumarate for treatment of multiple sclerosis: mechanism of action, effectiveness, and side effects. Curr Neurol Neurosci Rep. 2013;13(11):394.
Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–107.
Havrdova E, Hutchinson M, Kurukulasuriya NC, Raghupathi K, Sweetser MT, Dawson KT et al. Oral BG-12 (dimethyl fumarate) for relapsing–remitting multiple sclerosis: a review of DEFINE and CONFIRM: Evaluation of: Gold R, Kappos L, Arnold D, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med 2012; 367: 1098–107; and Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med 2012; 367: 1087–97. Expert Opin Pharmacother. 2013;14(15):2145–56.
Gold R, Arnold DL, Bar-Or A, Hutchinson M, Kappos L, Havrdova E, et al. Long-term effects of delayed-release dimethyl fumarate in multiple sclerosis: Interim analysis of ENDORSE, a randomized extension study. Mult Scler J. 2017;23(2):253–65.
Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277(24):21453–7.
Tanasescu R, Constantinescu CS. Pharmacokinetic evaluation of fingolimod for the treatment of multiple sclerosis. Expert Opin Drug Metab Toxicol. 2014;10(4):621–30.
Groves A, Kihara Y, Chun J. Fingolimod: direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J Neurol Sci. 2013;328(1):9–18.
Kappos L, Radue E-W, O’connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401.
Cohen JA, Barkhof F, Comi G, Hartung H-P, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–15.
Lublin F, Miller DH, Freedman MS, Cree BA, Wolinsky JS, Weiner H, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387(10023):1075–84.
Ayzenberg I, Hoepner R, Kleiter I. Fingolimod for multiple sclerosis and emerging indications: appropriate patient selection, safety precautions, and special considerations. Ther Clin Risk Manag. 2016;12:261.
Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing–remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–88.
Komori M, Lin YC, Cortese I, Blake A, Ohayon J, Cherup J, et al. Insufficient disease inhibition by intrathecal rituximab in progressive multiple sclerosis. Ann Clin Transl Neurol. 2016;3(3):166–79.
Berenguer-Ruiz L, Sempere AP, Gimenez-Martinez J, Gabaldon-Torres L, Tahoces L, Sanchez-Perez R, et al. Rescue therapy using rituximab for multiple sclerosis. Clin Neuropharmacol. 2016;39(4):178–81.
Alping P, Frisell T, Novakova L, Islam-Jakobsson P, Salzer J, Björck A, et al. Rituximab versus fingolimod after natalizumab in multiple sclerosis patients. Ann Neurol. 2016;79(6):950–8.
Kappos L, Li D, Calabresi PA, O’Connor P, Bar-Or A, Barkhof F, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378(9805):1779–87.
Hauser S, Comi G, Hartung H, Selmaj K, Traboulsee A, Bar-Or A. on behalf of the OPERA I and II clinical investigators. Efficacy and safety of ocrelizumab in relapsing multiple sclerosis—results of the interferon-beta-1a-controlled, double-blind, Phase III OPERA I and II studies ECTRIMS Online Libr. 2015;116634.
Traboulsee A, Arnold D, Bar-Or A, Comi G, Hartung H-P, Kappos L, et al. Ocrelizumab no evidence of disease activity (NEDA) status at 96 weeks in patients with relapsing multiple sclerosis: analysis of the phase III double-blind, double-dummy, interferon beta-1a-controlled OPERA I and OPERA II studies (PL02. 004). Neurology. 2016;86(16 Supplement):PL02.004.
Montalban X, Hemmer B, Rammohan K, Giovannoni G, De Seze J, Bar-Or A. Efficacy and safety of ocrelizumab in primary progressive multiple sclerosis-results of the placebo-controlled, double-blind, Phase III ORATORIO study. Mult Scler. 2015;21(S1):781–2.
Sorensen PS, Lisby S, Grove R, Derosier F, Shackelford S, Havrdova E, et al. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis A phase 2 study. Neurology. 2014;82(7):573–81.
Bar-Or A, Grove R, Austin D, Tolson J, Vanmeter S, Lewis E, et al. The MIRROR study: a randomized, double-blind, placebo-controlled, parallel-group, dose-ranging study to investigate the safety and MRI efficacy of subcutaneous ofatumumab in subjects with Relapsing-Remitting Multiple Sclerosis (RRMS)(I7-1.007). Neurology. 2014;82(10 Supplement):I7-1.007.
Brück W, Wegner C. Insight into the mechanism of laquinimod action. J Neurol Sci. 2011;306(1):173–9.
Kim W, Zandoná ME, Kim S-H, Kim HJ. Oral disease-modifying therapies for multiple sclerosis. J Clin Neurol. 2015;11(1):9–19.
Yang J-S, Xu L-Y, Xiao B-G, Hedlund G, Link H. Laquinimod (ABR-215062) suppresses the development of experimental autoimmune encephalomyelitis, modulates the Th1/Th2 balance and induces the Th3 cytokine TGF-β in Lewis rats. J Neuroimmunol. 2004;156(1):3–9.
Thöne J, Gold R. Laquinimod: a promising oral medication for the treatment of relapsing-remitting multiple sclerosis. Expert Opin Drug Metab Toxicol. 2011;7(3):365–70.
Filippi M, Rocca MA, Pagani E, De Stefano N, Jeffery D, Kappos L, et al. Placebo-controlled trial of oral laquinimod in multiple sclerosis: MRI evidence of an effect on brain tissue damage. J Neurol Neurosurg Psychiatry. 2013:jnnp-2013-306132.
Vollmer T, Sorensen P, Selmaj K, Zipp F, Havrdova E, Cohen J, et al. A randomized placebo-controlled phase III trial of oral laquinimod for multiple sclerosis. J Neurol. 2014;261(4):773–83.
Varrin-Doyer M, Zamvil SS, Schulze-Topphoff U. Laquinimod, an up-and-coming immunomodulatory agent for treatment of multiple sclerosis. Exp Neurol. 2014;262:66–71.
Huynh E, Sigal D, Saven A. Cladribine in the treatment of hairy cell leukemia: initial and subsequent results. Leuk Lymphoma. 2009;50(sup1):12–7.
Beutler E. Cladribine (2-chlorodeoxyadenosine). Lancet. 1992;340(8825):952–6.
Thöne J, Ellrichmann G. Oral available agents in the treatment of relapsing remitting multiple sclerosis: an overview of merits and culprits. Drug Healthc Patient Saf. 2013;5:37.
Beutler E, Sipe J, Romine J, Koziol J, McMillan R, Zyroff J. The treatment of chronic progressive multiple sclerosis with cladribine. Proc Natl Acad Sci. 1996;93(4):1716–20.
Sipe J, Romine J, Koziol J, McMillan R, Beutler E, Zyroff J. Cladribine in treatment of chronic progressive multiple sclerosis. Lancet. 1994;344(8914):9–13.
Cook S, Vermersch P, Comi G, Giovannoni G, Rammohan K, Rieckmann P, et al. Safety and tolerability of cladribine tablets in multiple sclerosis: the CLARITY (CLAdRIbine tablets treating multiple sclerosis orallY) study. Mult Scler Jo. 2011;17(5):578–93.
Giovannoni G, Comi G, Cook S, Rieckmann P, Rammohan K, Soelberg-Soerensenn P, et al. Clinical efficacy of cladribine tablets in patients with relapsing-remitting multiple sclerosis (RRMS): final results from the 120-week phase IIIb extension trial to the CLARITY study (P3. 028). Neurology. 2016;86(16 Supplement):P3.028.
Comi G, Cook S, Giovannoni G, Rammohan K, Rieckmann P, Soelberg-Sorensen P et al, editors. Magnetic resonance imaging (MRI) outcomes in patients with relapsing-remitting multiple sclerosis (RRMS) treated with cladribine tablets: results from the CLARITY study, a 96-week, phase III, double-blind, placebo-controlled trial. J Neurol; 2009 (Dr Dietrich Steinkopff Verlag).
Montalban X, Cohen B, Leist T, Moses H, Hicking C, Dangond F. Efficacy of cladribine tablets as add-on to IFN-beta therapy in patients with active relapsing MS: final results from the phase II ONWARD study (P3. 029). Neurology. 2016;86(16 Supplement):P3.029.
Leist TP, Comi G, Cree BA, Coyle PK, Freedman MS, Hartung H-P, et al. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): a phase 3 randomised trial. Lancet Neurol. 2014;13(3):257–67.
Freedman M, Leist T, Comi G, Cree B, Coyle P, Hartung H-P, et al. Efficacy of cladribine tablets in ORACLE study patients who retrospectively met 2010 McDonald multiple sclerosis (MS) criteria at baseline (P3. 035). Neurology. 2016;86(16 Supplement):P3.035.
Pakpoor J, Disanto G, Altmann DR, Pavitt S, Turner BP, Marta M, et al. No evidence for higher risk of cancer in patients with multiple sclerosis taking cladribine. Neurol Neuroimmunol Neuroinflamm. 2015;2(6):e158.
Gonzalez-Cabrera PJ, Brown S, Studer SM, Rosen H. S1P signaling: new therapies and opportunities. F1000 prime reports. 2014;6.
Selmaj K, Li DK, Hartung H-P, Hemmer B, Kappos L, Freedman MS, et al. Siponimod for patients with relapsing-remitting multiple sclerosis (BOLD): an adaptive, dose-ranging, randomised, phase 2 study. Lancet Neurol. 2013;12(8):756–67.
Karussis D, Vourka-Karussis U, Mizrachi-Koll R, Abramsky O. Acute/relapsing experimental autoimmune encephalomyelitis: induction of long lasting, antigen-specific tolerance by syngeneic bone marrow transplantation. Mult Scler J. 1999;5(1):017–21.
Radaelli M, Merlini A, Greco R, Sangalli F, Comi G, Ciceri F, et al. Autologous bone marrow transplantation for the treatment of multiple sclerosis. Curr Neurol Neurosci Rep. 2014;14(9):478.
Muraro PA, Douek DC. Renewing the T cell repertoire to arrest autoimmune aggression. Trends Immunol. 2006;27(2):61–7.
Karussis D, Petrou P, Vourka-Karussis U, Kassis I. Hematopoietic stem cell transplantation in multiple sclerosis. Expert Rev Neurother. 2013;13(5):567–78.
Mancardi GL, Sormani MP, Gualandi F, Saiz A, Carreras E, Merelli E, et al. Autologous hematopoietic stem cell transplantation in multiple sclerosis A phase II trial. Neurology. 2015;84(10):981–8.
Atkins HL, Bowman M, Allan D, Anstee G, Arnold DL, Bar-Or A, et al. Immunoablation and autologous haemopoietic stem-cell transplantation for aggressive multiple sclerosis: a multicentre single-group phase 2 trial. Lancet. 2016;388(10044):576–85.
Freedman M, Atkins HL. Haematopoietic stem cell transplants should be a second-line therapy for highly active MS–YES. Mult Scler J. 2016:1352458516654311.
Soelberg Sorensen P. Haematopoietic stem cell transplants should be a second-line therapy for highly active MS–NO. Mult Scler J. 2016;22(10):1260–3.
Franklin RJ, Kotter MR. The biology of CNS remyelination. J Neurol. 2008;255:19–25.
Peferoen L, Kipp M, Valk P, Noort JM, Amor S. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology. 2014;141(3):302–13.
Foote A, Blakemore W. Inflammation stimulates remyelination in areas of chronic demyelination. Brain. 2005;128(3):528–39.
Münzel EJ, Williams A. Promoting remyelination in multiple sclerosis—recent advances. Drugs. 2013;73(18):2017–29.
Zendedel A, Beyer C, Kipp M. Cuprizone-induced demyelination as a tool to study remyelination and axonal protection. J Mol Neurosci. 2013;51(2):567–72.
Blanchard B, Heurtaux T, Garcia C, Moll NM, Caillava C, Grandbarbe L, et al. Tocopherol derivative TFA-12 promotes myelin repair in experimental models of multiple sclerosis. J Neurosci. 2013;33(28):11633–42.
Meffre D, Massaad C, Grenier J. Lithium chloride stimulates PLP and MBP expression in oligodendrocytes via Wnt/β-catenin and Akt/CREB pathways. Neuroscience. 2015;284:962–71.
Preisner A, Albrecht S, Cui Q-L, Hucke S, Ghelman J, Hartmann C, et al. Non-steroidal anti-inflammatory drug indometacin enhances endogenous remyelination. Acta Neuropathol. 2015;130(2):247–61.
Huang JK, Jarjour AA, Oumesmar BN, Kerninon C, Williams A, Krezel W, et al. Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat Neurosci. 2011;14(1):45–53.
de la Fuente AG, Errea O, van Wijngaarden P, Gonzalez GA, Kerninon C, Jarjour AA, et al. Vitamin D receptor–retinoid X receptor heterodimer signaling regulates oligodendrocyte progenitor cell differentiation. J Cell Biol. 2015;211(5):975–85.
Najm FJ, Madhavan M, Zaremba A, Shick E, Karl RT, Factor DC, et al. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature. 2015;522(7555):216–20.
Trifunovski A, Josephson A, Ringman A, Brené S, Spenger C, Olson L. Neuronal activity-induced regulation of Lingo-1. Neuroreport. 2004;15(15):2397–400.
Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci. 2004;7(3):221–8.
Rudick RA, Mi S, Sandrock AW Jr. LINGO-1 antagonists as therapy for multiple sclerosis: in vitro and in vivo evidence. Expert Opin Biol Ther. 2008;8(10):1561–70.
Wang CJ, Qu CQ, Zhang J, Fu PC, Guo SG, Tang RH. Lingo-1 inhibited by RNA interference promotes functional recovery of experimental autoimmune encephalomyelitis. Anat Record. 2014;297(12):2356–63.
Pepinsky RB, Shao Z, Ji B, Wang Q, Walus L, Lee X, et al. Exposure levels of anti-LINGO-1 Li81 antibody in the central nervous system and dose-efficacy relationships in rat spinal cord remyelination models after systemic administration. J Pharmacol Exp Ther. 2011;339(2):519–29.
Tran JQ, Rana J, Barkhof F, Melamed I, Gevorkyan H, Wattjes MP, et al. Randomized phase I trials of the safety/tolerability of anti-LINGO-1 monoclonal antibody BIIB033. Neurol Neuroimmunol Neuroinflamm. 2014;1(2):e18.
Wang S, Bates J, Li X, Schanz S, Chandler-Militello D, Levine C, et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell stem cell. 2013;12(2):252–64.
Boyd A, Zhang H, Williams A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol. 2013;125(6):841–59.
Abramowski P, Krasemann S, Ernst T, Lange C, Ittrich H, Schweizer M, et al. Mesenchymal stromal/stem cells do not ameliorate experimental autoimmune encephalomyelitis and are not detectable in the central nervous system of transplanted mice. Stem Cells Dev. 2016;25(15):1134–48.
Meamar R, Nematollahi S, Dehghani L, Mirmosayyeb O, Shayegannejad V, Basiri K, et al. The role of stem cell therapy in multiple sclerosis: An overview of the current status of the clinical studies. Adv Biomed Res. 2016;5.
Karussis D, Karageorgiou C, Vaknin-Dembinsky A, Gowda-Kurkalli B, Gomori JM, Kassis I, et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch Neurol. 2010;67(10):1187–94.
Amor S, Groome N, Linington C, Morris MM, Dornmair K, Gardinier MV, et al. Identification of epitopes of myelin oligodendrocyte glycoprotein for the induction of experimental allergic encephalomyelitis in SJL and Biozzi AB/H mice. J Immunol. 1994;153(10):4349–56.
Fritz R, Chou C, McFarlin D. Relapsing murine experimental allergic encephalomyelitis induced by myelin basic protein. J Immunol. 1983;130(3):1024–6.
Tuohy V, Sobel R, Lees M. Myelin proteolipid protein-induced experimental allergic encephalomyelitis. Variations of disease expression in different strains of mice. J Immunol. 1988;140(6):1868–73.
Johns TG, de Rosbo NK, Menon KK, Abo S, Gonzales MF, Bernard C. Myelin oligodendrocyte glycoprotein induces a demyelinating encephalomyelitis resembling multiple sclerosis. J Immunol. 1995;154(10):5536–41.
Fissolo N, Montalban X, Comabella M. DNA-based vaccines for multiple sclerosis: current status and future directions. Clin Immunol. 2012;142(1):76–83.
Billetta R, Ghahramani N, Morrow O, Prakken B, de Jong H, Meschter C, et al. Epitope-specific immune tolerization ameliorates experimental autoimmune encephalomyelitis. Clin Immunol. 2012;145(2):94–101.
Spence A, Klementowicz JE, Bluestone JA, Tang Q. Targeting Treg signaling for the treatment of autoimmune diseases. Curr Opin Immunol. 2015;37:11–20.
Lutterotti A, Yousef S, Sputtek A, Stürner KH, Stellmann J-P, Breiden P, et al. Antigen-specific tolerance by autologous myelin peptide–coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med. 2013;5(188):188ra75-ra75.
Hellings N, Raus J, Stinissen P. T-cell vaccination in multiple sclerosis: update on clinical application and mode of action. Autoimmun Rev. 2004;3(4):267–75.
Vandenbark AA, Abulafia-Lapid R. Autologous T-cell vaccination for multiple sclerosis. Biodrugs. 2008;22(4):265–73.
Fox E, Wynn D, Cohan S, Rill D, McGuire D, Markowitz C. A randomized clinical trial of autologous T-cell therapy in multiple sclerosis: subset analysis and implications for trial design. Mult Scler J. 2012;18(6):843–52.
Karussis D, Shor H, Yachnin J, Lanxner N, Amiel M, Baruch K, et al. T cell vaccination benefits relapsing progressive multiple sclerosis patients: a randomized, double-blind clinical trial. PLoS One. 2012;7(12):e50478.
Aslani S, Mahmoudi M, Garshasbi M, Jamshidi AR, Karami J, Nicknam MH. Evaluation of DNMT1 gene expression profile and methylation of its promoter region in patients with ankylosing spondylitis. Clin Rheumatol. 2016;35(11):2723–31.
Rezaei R, Mahmoudi M, Gharibdoost F, Kavosi H, Dashti N, Imeni V, et al. IRF7 gene expression profile and methylation of its promoter region in patients with systemic sclerosis. Int J Rheum Dis. 2017;20(10):1551–61.
Karami J, Mahmoudi M, Amirzargar A, Gharshasbi M, Jamshidi A, Aslani S, et al. Promoter hypermethylation of BCL11B gene correlates with downregulation of gene transcription in ankylosing spondylitis patients. Genes Immun. 2017;18(3):170–5.
Mahmoudi M, Aslani S, Nicknam MH, Karami J, Jamshidi AR. New insights toward the pathogenesis of ankylosing spondylitis; genetic variations and epigenetic modifications. Modern Rheumatol. 2017;27(2):198–209.
Aslani S, Mahmoudi M, Karami J, Jamshidi AR, Malekshahi Z, Nicknam MH. Epigenetic alterations underlying autoimmune diseases. Autoimmunity. 2016;49(2):69–83.
Foma AM, Aslani S, Karami J, Jamshidi A, Mahmoudi M. Epigenetic involvement in etiopathogenesis and implications in treatment of systemic lupus erythematous. Inflamm Res. 2017:1–17.
Camelo S, Iglesias AH, Hwang D, Due B, Ryu H, Smith K, et al. Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;164(1):10–21.
Ge Z, Da Y, Xue Z, Zhang K, Zhuang H, Peng M, et al. Vorinostat, a histone deacetylase inhibitor, suppresses dendritic cell function and ameliorates experimental autoimmune encephalomyelitis. Exp Neurol. 2013;241:56–66.
Zhang Z, Zhang Z-Y, Wu Y, Schluesener H. Valproic acid ameliorates inflammation in experimental autoimmune encephalomyelitis rats. Neuroscience. 2012;221:140–50.
Xie L, Li X-K, Funeshima-Fuji N, Kimura H, Matsumoto Y, Isaka Y, et al. Amelioration of experimental autoimmune encephalomyelitis by curcumin treatment through inhibition of IL-17 production. Int Immunopharmacol. 2009;9(5):575–81.
Shindler KS, Ventura E, Dutt M, Elliott P, Fitzgerald DC, Rostami A. Oral resveratrol reduces neuronal damage in a model of multiple sclerosis. J Neuroophthalmol. 2010;30(4):328.
Chan MW, Chang C-B, Tung C-H, Sun J, Suen J-L, Wu S-F. Low-dose 5-aza-2′-deoxycytidine pretreatment inhibits experimental autoimmune encephalomyelitis by induction of regulatory T cells. Mol Med. 2014;20(1):248.
Du C, Liu C, Kang J, Zhao G, Ye Z, Huang S, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol. 2009;10(12):1252–9.
Acknowledgements
We are thankful of the Department of Microbiology and Immunology facilities at Islamic Azad University-Tehran Medical Branch.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Responsible Editor: John Di Battista.
Rights and permissions
About this article

Cite this article
Gholamzad, M., Ebtekar, M., Ardestani, M.S. et al. A comprehensive review on the treatment approaches of multiple sclerosis: currently and in the future. Inflamm. Res. 68, 25–38 (2019). https://doi.org/10.1007/s00011-018-1185-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-018-1185-0