
Abstract
Objectives
To observe immune system changes in patients with secondary infection from severe acute pancreatitis (SAP).
Methods
Seventy-nine patients were recruited. The percentages of CD4+, CD8+, natural killer (NK), HLA-DR+ cells and B lymphocytes, and the CD4+/CD8+ ratio, were determined. In addition, tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-10 (IL-10), and interleukin-4 (IL-4) serum levels were determined on days 1, 7, 14, and 28.
Results
Fifteen patients had a secondary infection. The immune response of the infected group was quite different from the non-infected group, with a higher percentage of CD4+ and HLA-DR+ cells on days 1, 7, 14 and 28, a higher percentage of CD8+ and NK cells on days 14 and 28, a reduced CD4+/CD8+ ratio, and a reduction in B lymphocytes. The cytokine levels in the infected group were different from the non-infected group, with a rise in TNF-α and IL-6 through the first 2 weeks, but dropping at 1 month. IL-10 and IL-4 increased initially, but then dropped over the next 3 weeks.
Conclusions
An early excessive immune response followed by a subsequent immune deficiency is closely related to secondary SAP infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
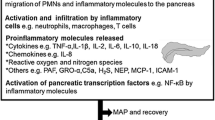
Severe acute pancreatitis (SAP) is a common emergency in abdominal surgery and has a substantial mortality risk of 10–30 % [1, 2]. The factors responsible for high patient mortality in SAP are immune dysfunction in the early stages (within the first week), and secondary infection with pancreatic necrosis in the later stages (usually after 10 days) [3–5]. The patient’s life is at risk due to local and systemic complications, which lead to multiple organ dysfunction syndrome (MODS) [6].
Clinical and experimental studies support the theory of excessive systemic inflammatory response syndrome (SIRS) fuelling SAP [7, 8]. Such an excessive inflammatory response can increase permeability, as represented by the systematic development of organ dysfunction, which can include the gut, lung, renal, and hepatic organs [9]. Increased permeability of intestinal mucosa can lead to intestinal bacterial translocation, and lead to secondary infection of the pancreatic and/or peripancreatic tissues [8].
SAP development and the marked change in the immune response is thought to play a central role in the development of SIRS, multiple organ dysfunction, secondary infection, and subsequent septic complications [10]. Recent studies have focused on the cytokine network and immune dysfunction as key pathological mechanisms of SAP. The focus of these studies was excessive inflammatory immune response-mediated pathological damage and cell-mediated immunity [11, 12]. However, few studies have investigated patient immunity with regard to secondary SAP infection in the clinic.
This study was undertaken to examine immune system changes in patients with SAP as indicated by plasma pro- and anti-inflammatory cytokines and the immune response of peripheral blood mononuclear cells (PBMC).
Materials and methods
Patients
Seventy-nine adult patients with SAP were recruited between July 1, 2007 and December 30, 2009. All patients were admitted directly to the National Medical Center of Biliopancreatic Diseases at the Nankai Clinical School of Tianjin Medical University. All patients met the severe disease criteria proposed at the International Symposium on Acute Pancreatitis [13]. The Balthazar CT severity index was applied to the initial contrast-enhanced CT. The initial severity of pancreatitis was assessed by using the Acute Physiology and Chronic Health Evaluation II (APACHE II) [14], Ranson scores, and Balthazar scores, and severe pancreatitis was diagnosed as an inclusion criterion with an APACHE II score of eight or higher, a Ranson score of four or higher, and Balthazar scores of four or higher. Patients who had autoimmune disease, immunodeficiency disease, malignancy, surgical interference, or pregnancy, or had received hormone or immunosuppressive agents within the last 3 months, were excluded from the study.
SAP secondary infections were confirmed by bacterial culture, blood culture, and/or computed tomography-guided fine needle aspiration (FNA) of pancreatic or peripancreatic tissue, and/or computed tomography-guided aspiration for suspected pancreatic or peripancreatic sepsis, or large localized fluid collections [2].
Inclusion and exclusion criteria
Inclusion criteria were: age between 18 and 60 years, more than 28 days of hospitalization, and signed informed consent. Exclusion criteria were: (1) patients diagnosed with concurrent pancreatic or peripancreatic infection at admission; (2) surgery during the study period; (3) pregnant or lactating females; (4) patients with cancer; (5) critically ill patients; (6) serious organ dysfunction; (7) immunodeficiency or autoimmune disease; and (8) patients participating in another research study.
Treatment
All patients were treated with standard Chinese protocols in our institution according to a standardized interdisciplinary management protocol, including intravenous fluids, organ system support, nutritional support, gastrointestinal decompression, and prophylactic antibiotics (ciprofloxacin/metronidazole or imipenem/cilastatin).
We used intragastric administration of traditional Chinese medicine twice each day after admission, over the course of the study. If the patient did not need a nasogastric tube, the drug was administrated orally.
Ethics
The study was approved by the hospital ethics committee. Patients or their relatives were given printed information, and signed informed consent was obtained before entry into the study.
Data collection
Data were collected from all patients prospectively. The data included gender, age, pancreatitis etiology, severity score, and demographic data. Biochemical data included the percentages of CD4+, CD8+, natural killer (NK), and HLA-DR+ cells and B lymphocytes, the CD4+/CD8+ ratio, and the serum levels of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-10 (IL-10), and interleukin-4 (IL-4). Peripheral venous blood samples (5 mL) were obtained from patients upon admission and on days 1, 7, 14, and 28 using a strict aseptic technique. One 2 mL aliquot of venous blood was obtained using BD Multitest IMK kit (BD Corporation, Franklin Lakes, NJ, USA), and stained with FITC anti-human CD14, PE anti-human HLA-DR, PE mouse IgG2b isotype control (all from eBioscience Corporation, San Diego, CA, USA), and examined using a FACSort flow cytometer (BD Corporation) and CellQuest software. Another 3 mL was used to detect inflammatory factors (TNF-α, IL-6, IL-10, and IL-4) using established enzyme-linked immunosorbent assay (ELISA) kits (Bio-Rad Laboratories, Hemel Hempstead, UK).
Statistical analysis
Comparisons of continuous and categorical variables between two groups used the nonparametric Mann–Whitney U test and the χ2 test with Yates correction, respectively. Data were analyzed using SPSS 11 (Chicago, IL, USA). Continuous data are presented as the mean ± standard error of the mean. Correlations were evaluated with the Spearman rank test. P < 0.05 was considered statistically significant.
Results
There were 79 patients in our study and 15 acquired a secondary infection. Data on gender, age, etiology, APACHE II score, and Balthazar scores at admission in the two groups are given in Table 1. There were no significant differences in age, gender, or etiology between the two groups. Based on the APACHE II and Balthazar scores, the severity at admission was different between the two groups.
Time required for SAP patients to acquire a secondary infection
Fifteen of the 79 patients had SAP with secondary infection. The time for secondary infection to appear is provided in Table 2. The time for secondary infection of peripancreatic or pancreatic necrosis was 7–20 days.
Comparison of PBMC
The PBMC levels in the infected group were quite different from the non-infected group, with a higher percentage of CD4+ and HLA-DR+ cells on days 1, 7, 14, and 28, higher than the percentage of CD8+ and NK cells on days 14 and 28, but exhibiting a lower CD4+/CD8+ ratio and B lymphocyte levels (Tables 3, 4, 5, 6, 7, 8). For SAP patients, the percentage of CD4+ cells increased significantly on day 7 and but was markedly depleted by 28 days in the infected group (P < 0.05, Table 3). The percentage of CD8+ cells was higher in the infected group at 28 days compared with the other time points (P < 0.05, Table 4). The CD4+/CD8+ ratios were elevated in infected patients on days 7, 14, and 28 compared with non-infected patients, and progressively decreased (P < 0.05, Table 7). The CD4+/CD8+ ratio was markedly depleted by 28 days in the infected group (P < 0.05, Table 7). The percentage of B lymphocytes progressively decreased in the infected group (P < 0.05, Table 6). In contrast, B lymphocyte levels were maintained at a relatively high and stable level in the non-infected group (P < 0.05, Table 6). HLA-DR levels were significantly higher in the infected group on days 7, 14, and 28 (P < 0.05, Table 8). In contrast, HLA-DR levels were markedly depleted by 28 days in the non-infected group (P < 0.05, Table 8). The HLA-DR+ cells were maintained at a relatively low and stable level in the non-infected group (P < 0.05, Table 8).
Comparison of inflammatory factors
Cytokine levels in the infected group were quite different from the non-infected group, with elevated TNF-α and IL-6 on days 1, 7, and 14, and decreased levels on day 28. IL-10 and IL-4 were elevated on day 1 but reduced on days 7, 14, 28 (P < 0.05; Tables 9, 10, 11, 12; Fig. 1). In infected patients, TNF-α peaked on day 7 but was markedly depleted by day 28, with similar levels on days 7 and 14 (P > 0.05, Table 9). In the infected group, IL-6 peaked on day 14 but was depleted by day 28, with similar levels on days 7 and 14 (P > 0.05, Table 10). The anti-inflammatory cytokines IL-10 and IL-4 increased slowly on days 7 and 14, peaking on day 14. In contrast, the pro-inflammatory and anti-inflammatory cytokines peaked on day 7 and gradually decreased until day 28 (Tables 11, 12).

Comparison of the changes in inflammatory factors between the two groups (pg/ml, mean ± SEM)
Discussion
SAP is one of the most serious pancreatic diseases and can follow two natural courses [1, 15]. The first course is characterized by SIRS, resulting from the release of inflammatory mediators. The second course appears 1–3 weeks following infection and is dominated by pancreatic necrosis, which can lead to sepsis-related complications [16], and occurs in 40–70 % of SAP patients [15, 17]. SAP pathogenesis is complicated, but immune damage resulting from the cytokine network is one of the key pathological mechanisms of SAP [18]. In early-stage SAP, hypercytokinemia plays a dominant role in SIRS development and triggers an excessive immune response [19, 20]. In later-stage SAP, the excessive inflammatory response is counteracted by a systemic release of anti-inflammatory mediators, followed by immunosuppression [19–21].
Researchers have found that inflammatory mediators contribute to early SAP causing systemic epithelial barrier dysfunction [22] and increased vascular endothelial permeability [23]. The gut fuels SAP [24], which activates development of a local or systemic inflammation response and secondary infection of pancreatic necrosis [22, 25].
TNF-α and IL-6 are the key proinflammatory cytokines mediating SAP. The excessive release of inflammatory mediators during SAP is the primary reason for secondary infection, which increases intestinal mucosa permeability, aggravates intestinal bacteria, and ultimately leads to death [26]. In contrast, the release of anti-inflammatory factors, such as IL-4 and IL-10, compensate for the inflammatory response, and increase during excessive inflammatory responses in SAP [27].
Cellular immunity may play a fundamental role in SAP and abnormal cellular immunity may be related to the outcome [28]. Immunological events are believed to be involved in SAP pathogenesis, although how immune dysfunction causes SAP is unclear [29]. Cell-mediated immunity is an important component of the immune system, and the percentages of T-helper (CD4+), T-suppressor (CD8+), NK, and HLA-DR+ cells and B lymphocytes, and the CD4+/CD8+ ratio in PBMC reflect immune status. The density of CD4+, CD8+, NK, and HLA-DR+ cells and B lymphocytes, and the ratio of CD4+/CD8+ cells in the periphery change in the course of SAP [30, 31]. There are at least two major CD4+ T-cell subsets, one which secretes IFN-α and IL-6, and one which produces IL-4 and IL-10 [32].
In summary, we describe a cohort of SAP patients with secondary infection treated by a combination of traditional Chinese medicine and Western medicine. The time to secondary infection for peripancreatic or pancreatic necrosis was 7–20 days. Although etiology, age, and gender were similar between the infection groups and the non-infected groups, the severity (based on APACHE II, Ranson scores, and Balthazar scores) and immunity at admission were quite different. The PBMC levels in the infected group were quite different from the non-infected group.
Our findings suggest that the severity of SAP is associated with bacterial contamination of pancreatic necrosis. SAP with necrosis infection is likely to show an aggressive course, during which severity increases. Our study showed that the excessive inflammatory response peak was 7 days and/or 14 days, whereas the time for secondary infection was 7–20 days.
Our results show that increased pro-inflammatory factors IL-6 and TNF-α and over-activation of CD4+, HLA-DR+, and NK cells and B lymphocytes were associated with secondary infection. The excessive release of anti-inflammatory cytokines IL-10 and IL-4 and CD8+ cells leads to a pro- and anti-inflammatory cytokine imbalance in late-phase SAP. Infection is caused mainly by an excessive immune response and subsequent immune dysregulation. Evidence has accumulated that an excessive immune response is counteracted by the systemic release of anti-inflammatory mediators, and an immunosuppression that is thought to contribute to secondary infections [33, 34]. Liu et al. [11] found that the levels of T-lymphocyte subsets in the combined traditional Chinese medicine and Western medicine treatment group were quite different from the conventional Western medicine treatment group. Shen et al. [12] reported that cytokine levels in the infected group were different from the non-infected group, with elevated TNF-α and IL-6 on days 1, and 7, and reduced levels on day 14. IL-10 and IL-4 increased on days 7 and 14.
In summary, an early excessive immune response and subsequent immune deficiencies are closely related to secondary infection from SAP. Future large-scale, high-quality, multicenter trials are required to clarify immune system dynamics throughout the course of SAP.
References
Beger HG, Rau B, Isenmann R. Natural history of necrotizing pancreatitis. Pancreatology. 2003;3:93–101.
Tsui NC, Zhao E, Li Z, et al. Microbiological findings in secondary infection of severe acute pancreatitis: a retrospective clinical study. Pancreas. 2009;38:499–502.
Bumbasirevic V, Radenkovic D, Jankovic Z, et al. Severe acute pancreatitis: overall and early versus late mortality in intensive care units. Pancreas. 2009;38:122–5.
Eckerwall GE, Axelsson JB, Andersson RG. Early nasogastric feeding in predicted severe acute pancreatitis: a clinical, randomized study. Ann Surg. 2006;244:959–65.
Mizuguchi T, Mukaiya M, Imaizumi H, et al. Successful management of severe acute pancreatitis with multiple organ failure. Pancreas. 2004;28:211–3.
Lytras D, Manes K, Triantopoulou C, et al. Persistent early organ failure: defining the high-risk group of patients with severe acute pancreatitis? Pancreas. 2008;36:249–54.
Zhang J, Yuan C, Hua G, et al. Early gut barrier dysfunction in patients with severe acute pancreatitis: attenuated by continuous blood purification treatment. Int J Artif Organs. 2010;33:706–15.
Zhang X, Chen L, Luo L, et al. Study of the protective effects of dexamethasone on ileum mucosa injury in rats with severe acute pancreatitis. Pancreas. 2008;37:e74–82.
Luan ZG, Zhang H, Ma XC, et al. Role of high-mobility group box 1 protein in the pathogenesis of intestinal barrier injury in rats with severe acute pancreatitis. Pancreas. 2010;39:216–23.
Takeyama Y. Significance of apoptotic cell death in systemic complications with severe acute pancreatitis. J Gastroenterol. 2005;40:1–10.
Liu Z, Shen Y, Cui N, et al. Clinical observation of immunity for severe acute pancreatitis. Inflammation. 2011;34:426–31.
Shen Y, Cui N, Miao B, et al. Immune dysregulation in patients with severe acute pancreatitis. Inflammation. 2010;34:36–42.
Bradley EL 3rd. A clinically based classification system for acute pancreatitis: summary of the International Symposium on Acute Pancreatitis, Atlanta, GA, September 11 through 13, 1992. Arch Surg. 1993;128:586–90.
Gravante G, Garcea G, Ong SL, et al. Prediction of mortality in acute pancreatitis: a systematic review of the published evidence. Pancreatology. 2009;9:601–14.
Beger HG, Rau B, Mayer J, Pralle U. Natural course of acute pancreatitis. World J Surg. 1997;21:130–5.
Halonen KI, Pettilä V, Leppäniemi AK, et al. Multiple organ dysfunction associated with severe acute pancreatitis. Crit Care Med. 2002;30:1274–9.
Wu XM, Ji KQ, Wang HY, et al. Total enteral nutrition in prevention of pancreatic necrotic infection in severe acute pancreatitis. Pancreas. 2010;39:248–51.
Zhang XP, Chen HQ, Liu F, et al. Advances in researches on the immune dysregulation and therapy of severe acute pancreatitis. J Zhejiang Univ Sci B. 2009;10:493–8.
Beger HG, Rau BM. Severe acute pancreatitis: clinical course and management. World J Gastroenterol. 2007;13:5043–51.
Shi CB, Zhao X, Lagergren A, et al. Immune status and inflammatory response differ locally and systemically in severe acute pancreatitis. Scand J Gastroenterol. 2006;41:472–80.
Bone R. Toward a theory regarding the pathogenesis of the systemic inflammatory response syndrome: what we do and what we do not know about cytokine regulation. Crit Care Med. 1996;24:163–72.
Liu H, Li W, Wang X, et al. Early gut mucosal dysfunction in patients with acute pancreatitis. Pancreas. 2008;36:192–6.
Liu HB, Cui NQ, Wang Q, et al. Sphingosine-1-phosphate and its analogue FTY720 diminish acute pulmonary injury in rats with acute necrotizing pancreatitis. Pancreas. 2008;36:e10–5.
Lu F, Huang H, Wang F, et al. Intestinal capillary endothelial barrier changes in severe acute pancreatitis. Hepatogastroenterology. 2011;58:1009–17.
Cicalese L, Sahai A, Sileri P, et al. Acute pancreatitis and bacterial translocation. Dig Dis Sci. 2001;46:1127–32.
Holmes CL, Russell JA, Walley KR. Genetic polymorphisms in sepsis and septic shock: role in prognosis and potential for therapy. Chest. 2003;124:1103–15.
de-Madaria E, Martínez J, Sempere L, et al. Cytokine genotypes in acute pancreatitis: association with etiology, severity, and cytokine levels in blood. Pancreas. 2008;37:295–301.
Curley PJ. Endotoxin, cellular immune dysfunction and acute pancreatitis. Ann R Coll Surg Engl. 1996;78:531–5.
Uehara S, Gothoh K, Handa H, et al. Immune function in patients with acute pancreatitis. J Gastroenterol Hepatol. 2003;18:363–70.
Curley PJ, McMahon MJ, Lancaster F, et al. Reduction in circulating levels of CD4-positive lymphocytes in acute pancreatitis: relationship to endotoxin, interleukin-6 and disease severity. Br J Surg. 1993;80:1312–5.
Yao W, Zhu Q, Yuan Y, et al. Thymosin alpha 1 improves severe acute pancreatitis in rats via regulation of peripheral T cell number and cytokine serum level. J Gastroenterol Hepatol. 2007;22:1866–71.
Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–93.
Kylänpää ML, Repo H, Puolakkainen PA. Inflammation and immunosuppression in severe acute pancreatitis. World J Gastroenterol. 2010;16:2867–72.
Bone R. Toward a theory regarding the pathogenesis of the systemic inflammatory response syndrome: what we do and what we do not know about cytokine regulation. Crit Care Med. 1996;24:163–72.
Acknowledgments
This work was supported by the National Science and Technology Support Program of China (grant no. 2006BAI04A15).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Artur Bauhofer.
Rights and permissions
About this article
Cite this article
Shen, Y., Cui, NQ. Clinical observation of immunity in patients with secondary infection from severe acute pancreatitis. Inflamm. Res. 61, 743–748 (2012). https://doi.org/10.1007/s00011-012-0467-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-012-0467-1