
Abstract
Objective
α-Lipoic acid (LA) exerts beneficial effects in cardiovascular diseases though its antioxidant and/or anti-inflammatory functions. It is postulated that the anti-inflammatory function of LA results from its antioxidant function. In this study we tested whether inhibition of NF-κB by LA is dependent on its antioxidant function.
Methods
Human umbilical vein endothelial cells (HUVECs) were treated with tumor necrosis factor-α (TNFα) in the presence of various antioxidants, including LA, tiron, apocynin, and tempol. The activation of the nuclear factor-κB (NF-κB) signaling pathway was then analyzed.
Results
LA, but not other tested antioxidants, inhibited TNFα-induced inhibitor-kappaB-α (IκBα) degradation and VCAM-1 and COX2 expression in HUVECs. Although LA activated the phosphatidylinositol-3-kinase (PI3-kinase)/Akt pathway in HUVECs, inhibition of Akt by LY294002 did not affect inhibition of TNFα-induced IκBα degradation by LA. In transient co-transfection assays of a constitutively active mutant of IκB kinase-2 (IKK2), IKK2(EE), and a NF-κB luciferase reporter construct, LA dose-dependently inhibited IKK2(EE)-induced NF-κB activation in addition to inhibiting IKK activity in in vitro assays. Consistent with the effect on luciferase expression, LA inhibited IKK2(EE)-induced cyclo-oxygenase-2 (COX2) expression, suggesting that IKK2 inhibition by LA may be a relevant mechanism that explains its anti-inflammatory effects.
Conclusions
LA inhibits NF-κB activation through antioxidant-independent and probably IKK-dependent mechanisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
α-Lipoic acid (LA) was first identified as a cofactor for mitochondrial α-ketoacid dehydrogenases in mammals [1]. A number of studies have demonstrated that LA exerts beneficial effects on various diseases, such as hypertension [2–4], atherosclerosis [5, 6], diabetes mellitus [7], hyperlipomia [6, 8], and age-associated cognitive and vascular decline [9]. However, the cellular and molecular mechanisms whereby LA exerts its beneficial effects on these diseases are only partially understood.
One mechanism that has received considerable attention is its effects in attenuating oxidative stress and inflammation. LA is a potent antioxidant in vitro and acts as a scavenger of various free radicals, including hydroxyl radical, singlet oxygen, peroxynitrite, and hypochlorous acid. [1] LA may also activate Phase II antioxidant enzymes including rate-limiting enzymes involved in glutathione synthesis. The effects of LA on reactive oxygen species (ROS) pathways is probably important in its anti-inflammatory function as ROS could serve as second messengers mediating inflammatory cytokine production through nuclear factor-kappaB (NF-κB) [10]. However, some studies have challenged the cause–effect relationship between ROS and NF-κB activation with evidence that ROS do not mediate NF-κB activation induced by pro-inflammatory cytokines [11].
In the present study, we examine whether LA inhibits NF-κB through its antioxidant function, and show that LA but not other tested antioxidants inhibited inhibitor-kappaB-α (IκBα) degradation and inflammatory gene expression induced by tumor necrosis factor-α (TNFα). This was paralleled by an inhibitory effect of LA but not other tested antioxidants on inhibitor IκB kinase-2 (IKK2), offering evidence that LA inhibits NF-κB activation independent of its antioxidant function.
Methods
Cell treatment
Human umbilical vein endothelial cells (HUVECs) were obtained from ATCC (Manassas, VA, USA), and maintained in endothelial cell basic medium supplemented with 10% FBS (Promocell). Before treatment with TNFα, HUVECs had been starved in endothelial cell basic medium supplemented with 0.5% FBS overnight. To measure IκBα degradation and the inflammatory gene expression, HUVECs were treated with 10 ng/ml TNFα for 10 min and 8 h, respectively.
Western blotting
This was performed using standard techniques with primary antibodies as follows: monoclonal anti-β-actin (Sigma), rabbit anti-IκBα (Upstate Biotechnology), monoclonal anti-vascular cell adhesion molecule-1 (VCAM-1) (R&D Systems), monoclonal anti-cyclo-oxygenase-2 (COX2) (Cayman Chemical), goat anti-Akt1 (Santa Cruz), and rabbit anti-phospho-Akt (Cell Signaling Technology). Signals were detected by chemiluminescence and analyzed by densitometry.
Lucigenin chemiluminescence assay for superoxide detection
Mouse liver was homogenized in PBS, and the protein concentration was adjusted to 5 µg/µl. Cell lysates, 20 µl/well, were plated on 96-well microplates in Kreps–HEPES buffer (pH = 7.4; initially gassed with 95% O2 and 5% CO2. Antioxidants, including tiron (20 mM), LA (2 mM), apocynin (0.3 mM) and tempol (0.1 mM) were added prior starting the measurement. Lucigenin at 5 μM and NADPH at 100 μM were then added to start measurement. Experiments were performed with a Berthold Luminometer LB 960, with settings at 1 s measurement and 30 s kinetics.
IKK2 inhibition assay
A K-LISA™ IKK2 Inhibitor Screening Kit (Calbiochem) was used to examine the inhibitory effect of LA and other antioxidants on IKK2. Phosphorylation of glutathione S-transferase (GST)-IkBα by IKK2 was performed in a total of 50 μl solution containing 100 ng IKK2, 0.75 μM GST-IkBα, 60 mM HEPES–NaOH (pH 7.5), 3 mM MgCl2, 3 mM MnCl2, 3 μM Na-orthovanadate, 1.2 mM DTT, 10 μM ATP, and various concentration of LA or other antioxidants. The reaction solution was incubated at 30°C for 30 min. The phospho-IkBα was then measured with anti-phospho IκBα (Ser32/Ser36). Experiments were performed in duplicate.
Transfection
The plasmid expressing a constitutively active IKK2 mutant IKK2(EE) (Plasmid 11105) was obtained from Addgene (Cambridge, MA, USA). Transfection was performed with Attractene (Qiagen) according to the manufacturer’s instructions. Briefly, DNA/Attractene complex was prepared at a ratio of 4 μg/15 μl, and then added to 90% confluent HEK293 cells. These cells were incubated with the complex overnight and then treated with LA for 16–24 h.
NF-κB activation assays
HUVECs had been starved in endothelial cell basic medium supplemented with 0.5% FBS overnight, and treated with 10 ng/ml TNFα for 30 min in the presence of antioxidants. Nuclear proteins were then extracted with NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce, Rockford, IL, USA) and subject to electrophoretic mobility shift assay (EMSA) with lightshift Chemiluminescent EMSA Kit (Pierce) according to the manufacturer’s instructions. Oligonucleotides (sequences: NF-κB sense, AgT TgA ggg gAC TTT CC Cag gC; NF-κB antisense, gCC Tgg gAA AgT CCC CTC AAC T) were synthesized by Invitrogen, and labeled with Biotin 3′ end DNA Labeling Kit (Pierce). Binding reactions were performed in 20 μl solution containing 10× binding buffer (2 μl), glycerol (2.5%), MgCl2 (5 mM), poly(dIdC) (50 ng/μl), NP-40 (0.05%), nuclear extract (8 μg proteins), and biotin–DNA (20 fmol). The binding action products were then resolved by 6% native polyacrylamide gel and transferred to nylon membrane. After cross-linking, biotin–DNA was visualized by chemiluminescence.
Statistical analysis
All data are expressed as mean ± SE. Statistical comparisons of dose–response curves were performed with two-way repeated-measures analysis of variance (ANOVA) using GraphPad Prism version 4.0b (Graphpad Software, La Jolla, CA, USA). Otherwise, statistical comparisons were performed with Student’s t-test and Bonferroni post hoc tests of selected comparisons where appropriate. Statistical significance was set at P < 0.05.
Results
LA inhibits TNFα-induced NF-κB activation independent of its antioxidant function
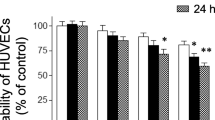
IκBα degradation is a critical step for NF-κB activation in response to TNFα stimulation. Consistent with previous studies, LA dose-dependently inhibited TNFα-induced IκBα degradation in HUVECs (Fig. 1a, b). To test whether LA inhibits NF-κB activation through its antioxidant function, we compared the effect of LA on TNFα-induced IκBα degradation to other well-known antioxidants. Figure 1c and d show that LA, but not other tested antioxidants, inhibited TNFα-induced IκBα degradation. Next, we investigated if this effect of LA on IκBα resulted in altered expression of two classic NF-κB-dependent genes, VCAM-1 and COX2 in HUVECs. As shown in Fig. 1e and f, LA, but not other tested antioxidants, inhibited TNFα-induced expression of VCAM-1 and COX2, further confirming that the antioxidant function of LA is not a major mechanism for its inhibitory effect on NF-κB activation in response to TNFα. Figure 1g shows that LA and other antioxidants at the doses used in the previously described experiments efficiently scavenged superoxide generated by mouse liver lysate in the presence of NADPH, verifying the quality of all the used antioxidants.

The antioxidant function of LA is not involved in its inhibition of TNFα-induced NF-κB activation. a, b HUVECs were starved overnight and pre-treated with various concentration of LA for 20 min, followed by treatment with TNFα (10 ng/ml) for 10 min. Cells were then lyzed in RIPA buffer, and IκBα was analyzed by Western blotting. A representative picture (a) and the summary (b) of three independent experiments are presented. *P < 0.05 versus TNFα only, Student’s t-test with Bonferoni correction. c, d HUVECs were starved overnight and pre-treated with LA (1 mM), tiron (20 mM), apocynin (0.3 mM), PEG-SOD (100 U/ml) and tempol (0.1 mM) for 20 min, followed by treatment with TNFα (10 ng/ml) for 10 min. Cells were then lysed in RIPA buffer, and IκBα was analyzed by Western blotting. A representative picture (c) and the summary (d) of four independent experiments are presented. *P < 0.05 versus vehicle, Student’s t-test with Bonferoni correction. e, f HUVECs were starved overnight and pre-treated with LA (1 mM), tiron (20 mM), apocynin (0.3 mM), PEG-SOD (100 U/ml) and tempol (0.1 mM) for 20 min, followed by treatment with TNFα (10 ng/ml) for 30 min (NF-κB DNA binding activity assay) or 8 h (all other measurements). Nuclear proteins or total proteins were prepared. The NF-κB DNA binding activity was assessed by EMSA, and a representative results of two independent experiments is presented. The expression of COX2 and VCAM-1 were examined by Western blot. A representative picture (e) and the summary (f) of three independent experiments were presented. *P < 0.05 versus vehicle, Student’s t-test with Bonferoni correction. g In the presence of the indicated antioxidants, the superoxide production by NADPH oxidase in mouse liver lysate was measured by lucigenin chemiluminescence assay, n = 6, *P < 0.05 versus control, one way-ANOVA
LA inhibits TNFα-induced NF-κB activation independent of Akt activation
LA inhibits lipopolysaccharide (LPS)-induced NF-κB activation via activating the phosphoinositide 3-kinase (PI3 kinase)/Akt signaling pathway in THP-1 cells [12]. To test if LA inhibits NF-κB activation in HUVECs through activation of the PI3 kinase/Akt signaling pathway, we examined the effect of LA and other antioxidants on Akt activation in HUVECs. Figure 2a, b showed that LA, but not other antioxidants, increased Akt phosphorylation, indicating that the activation of the PI3-kinase/Akt signaling pathway by LA in HUVECs is also independent of its antioxidant function. To test if PI3-kinase/Akt pathway activation by LA contributes to the inhibition of NF-κB activation, a PI3-kinase inhibitor, LY294002, was used to block PI3-kinase/Akt activation. As shown in Fig. 2c–e, 50 µM LY294002 significantly inhibited Akt phosphorylation by approximately 85%. However, this treatment with LY294002 did not inhibit the effect of LA on TNFα-induced IκBα degradation, suggesting that Akt activation by LA in HUVECs is not involved in its effects on NF-κB.

Akt activation by LA does not contribute to its inhibiting NF-κB signaling. a, b HUVECs were starved overnight and pre-treated with LA (1 mM), tiron (20 mM), apocynin (0.3 mM), PEG-SOD (100 U/ml) and tempol (0.1 mM) for 20 min, followed by treatment with TNFα (10 ng/ml) for 10 min. Akt phosphorylation was then analyzed by Western blotting. A representative picture (a) and the summary (b) of four independent experiments are presented. *P < 0.05 versus vehicle, # P < 0.05 versus TNFα, Student’s t-test with Bonferoni correction. c, d, HUVECs were starved overnight and pre-treated with LA (1 mM), or LA (1 mM) plus LY294002 (30 µM) for 20 min, followed by treatment with TNFα (10 ng/ml) for 10 min. IκBα degradation and Akt phosphorylation were then analyzed by Western blotting. A representative picture (c) and the summary (d, e) of four independent experiments are presented. *P < 0.05 versus vehicle, # P < 0.05 versus TNFα, $ P < 0.05 versus TNFα + LA, Student’s t-test with Bonferoni correction
LA inhibits IKK2
Given that IκBα degradation is regulated by IKK2-dependent phosphorylation, we tested whether LA inhibits IKK2. Figure 3a shows that LA inhibited IKK2 activity concentration-dependently, with a half maximal inhibitory concentration (IC50) of 0.15 mM (95% CI: 0.09–0.25 mM). We next examined if this effect is unique to LA. As shown in Fig. 3b, other tested antioxidants did not inhibit IKK2 activity, indicating that LA inhibits NF-κB activation in endothelial cells through inhibition of IKK2.

LA inhibits IKK2 in vitro. a The effect of the indicated concentration of LA on IKK2 activity was measured with IKK2 screening kit (Calbiochem). Results were expressed as a percentage of vehicle control, n = 5. b The effect of antioxidants was analyzed with IKK2 screening kit (Calbiochem), n = 5. *P < 0.05 versus IKK−, # P < 0.05 versus IKK+ , $ P < 0.05 versus IKK+ LA, Student’s t-test with Bonferoni correction
LA inhibits constitutively active IKK2-induced COX2 expression
To verify that LA can inhibit IKK2 in living cells, we tested the effect of LA on a constitutively active IKK2 mutant IKK2(EE)-induced NF-κB-dependent gene expression. However, HUVECs is a hard-to-transfect cell line. Therefore, we over-expressed IKK2(EE) and a NF-κB reporter construct in HEK293 cells. Figure 4a shows that LA inhibited IKK2(EE)-induced luciferase expression dose-dependently, with an IC50 of 0.3 mM (95% CI: 0.1–0.6 mM). Consistent with the effect on luciferase expression, LA inhibited IKK2(EE)-induced COX2 expression (95% CI of IC50: 0.1–0.9 mM; Fig. 4b, c), strongly supporting that LA may inhibit NF-κB activation in living cells through inhibition of IKK2.

LA inhibits IKK2 in living cells. a HEK293 cells were transfected with a constitutively active mutant of IKK2, IKK2(EE), and a NF-κB reporter construct. Twenty-four hours later, these cells were treated with the indicated concentration of LA for 24 h, and the activity of the reporter gene, luciferase, was analyzed by chemiluminescence assay, n = 3. b and c HEK293 cells were transfected with a constitutively active mutant of IKK2, IKK2(EE), and a NF-κB reporter construct. Twenty-four hours later, these cells were treated with the indicated concentration of LA for 24 h, and the expression of COX2 was analyzed by Western blot, n = 3. *P < 0.05 versus IKK2(EE)−/LA−, # P < 0.05 versus IKK2(EE)+/LA−, Student’s t-test with Bonferoni correction
Discussion
In the present study, we show that (1) LA, but not other tested antioxidants, inhibits NF-κB activation, (2) LA-induced activation of the PI3-kinase/Akt pathway in HUVECs does not contribute to its effects on NF-κB inhibition, (3) inhibition of IKK2 by LA may represent an important mechanism by which it mediates inhibitory effects on NF-κB activation.
Previous studies have provided important insights into the therapeutic effects of LA in inflammatory conditions [2–4, 6–9]. Principal amongst these are its potent free-radical scavenging, activation of anti-inflammatory pathways, prevention of NF-κB activation, and induction of Phase II antioxidant defense mechanisms resulting in alteration of cellular redox status. Since oxidative stress can lead to direct NF-κB activation, it has been presumed that LA may prevent NF-κB activation through direct ROS scavenging. An alternative mechanism has been demonstrated in elegant studies by Zhang et al. [12] whereby LA through activation of PI3K-Akt may prevent NF-κB activation. Inhibition of PI3K/Akt signaling enhances LPS-induced activation of NF-κB and AP-1 and expression of pro-inflammatory mediators in cultured THP-1 cells.
Our data demonstrate that LA, but not other tested antioxidants, inhibits IκBα degradation and NF-κB-dependent gene expression, strongly indicating that LA inhibits NF-κB activation independent of its antioxidant function. This is consistent with previous studies showing that LA but not N-acetyl cysteine (NAC) inhibited TNFα-induced IKK/NF-κB signaling in human aortic endothelial cells [13]. However, in contrast to previous studies where the inhibition of NF-κB activation appeared to require activation of the PI3-kinase/Akt pathway, our results indicate that inhibition of PI3K by up to 85% had no effect on prevention of IκBα degradation by LA in response to TNFα. These results indicate that the effects of PI3K/Akt on NF-κB may be dependent on cell type and context (LPS vs. TNFα) [14, 15]. However, since the Akt activation was not completely blocked, we cannot rule out the possibility that Akt activation at a low level may be enough to inhibit NF-κB signaling. Our results are consistent with previous reports that PI3-kinase/Akt does not affect TNFα-induced NF-κB activation in HUVECs [15] and demonstrate that Akt activation by LA is independent of its antioxidant function, because other tested antioxidants did not activate Akt.
IKK2 is essential for IκBα degradation and subsequent NF-κB activation in response to TNFα. Our data demonstrate that LA inhibited IKK2 in vitro, providing a potential molecular mechanism for the inhibitory effect of LA on NF-κB signaling. Although we could not rule out the possibility that there are other mechanisms whereby LA inhibits NF-κB activation in living cells, the comparable IC50s of IKK2 inhibition in vitro and inhibition of NF-κB-dependent luciferase expression in living cells indicates that IKK2 inhibition may be a critical mechanism for LA inhibiting NF-κB.
In conclusion, our data demonstrate that LA inhibits NF-κB through inhibition of IKK2 rather than its antioxidant function.
References
Smith AR, et al. Lipoic acid as a potential therapy for chronic diseases associated with oxidative stress. Curr Med Chem. 2004;11(9):1135–46.
Louhelainen M, et al. Lipoic acid supplementation prevents cyclosporine-induced hypertension and nephrotoxicity in spontaneously hypertensive rats. J Hypertens. 2006;24(5):947–56.
Thirunavukkarasu V, Anitha Nandhini AT, Anuradha CV. Lipoic acid attenuates hypertension and improves insulin sensitivity, kallikrein activity and nitrite levels in high fructose-fed rats. J Comp Physiol. 2004;174(8):587–92.
Vasdev S, et al. Salt-induced hypertension in WKY rats: prevention by alpha-lipoic acid supplementation. Mol Cell Biochem. 2003;254(1–2):319–26.
Zhang WJ, et al. Dietary alpha-lipoic acid supplementation inhibits atherosclerotic lesion development in apolipoprotein E-deficient and apolipoprotein E/low-density lipoprotein receptor-deficient mice. Circulation. 2008;117(3):421–8.
Zulkhairi A. et al. Alpha lipoic acid possess dual antioxidant and lipid lowering properties in atherosclerotic-induced New Zealand white rabbit. Biomed Pharmacother. 2008;62(10):716–22.
Kamenova P. Improvement of insulin sensitivity in patients with type 2 diabetes mellitus after oral administration of alpha-lipoic acid. Hormones (Athens). 2006;5(4):251–8.
Yang RL, et al. Lipoic acid prevents high-fat diet-induced dyslipidemia and oxidative stress: a microarray analysis. Nutrition. 2008;24(6):582–8.
Milgram NW, et al. Acetyl-l-carnitine and alpha-lipoic acid supplementation of aged beagle dogs improves learning in two landmark discrimination tests. FASEB J. 2007;21(13):3756–62.
Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991;10(8):2247–58.
Hayakawa M, et al. Evidence that reactive oxygen species do not mediate NF-kappaB activation. EMBO J. 2003;22(13):3356–66.
Zhang WJ, et al. Alpha-lipoic acid attenuates LPS-induced inflammatory responses by activating the phosphoinositide 3-kinase/Akt signaling pathway. Proc Natl Acad Sci USA. 2007;104(10):4077–82.
Zhang WJ, Frei B. Alpha-lipoic acid inhibits TNF-alpha-induced NF-kappaB activation and adhesion molecule expression in human aortic endothelial cells. FASEB J. 2001;15(13):2423–32.
Marone R, et al. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008;1784(1):159–85.
Gustin JA, et al. Cell type-specific expression of the IkappaB kinases determines the significance of phosphatidylinositol 3-kinase/Akt signaling to NF-kappa B activation. J Biol Chem. 2004;279(3):1615–20.
Acknowledgments
This study was supported by grants from National Institutes of Health (NIH) R01ES013406 and R01ES015146 to Dr. Rajagopalan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Liwu Li.
Rights and permissions
About this article
Cite this article
Ying, Z., Kampfrath, T., Sun, Q. et al. Evidence that α-lipoic acid inhibits NF-κB activation independent of its antioxidant function. Inflamm. Res. 60, 219–225 (2011). https://doi.org/10.1007/s00011-010-0256-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-010-0256-7