
Abstract
The development and function of blood cells are regulated by specific growth factors/cytokines and their receptors’ signaling pathways. In this way, these factors influence cell survival, proliferation and differentiation of hematopoietic cells. Central to this positive and/or negative control are the adaptor proteins. Since their identification 10 years ago, members of the Lnk adaptor protein family have proved to be important activators and/or inhibitors in the hematopoietic, immune and vascular system. In particular, the generation of animal and cellular models for the Lnk and APS proteins has helped establish the physiological role of these molecules through the identification of their specific signaling pathways and the characterization of their binding partners. Moreover, the recent identification of mutations in the LNK gene in myeloproliferative disorders, as well as the correlation of a single nucleotide polymorphism on LNK with hematological, immune and vascular diseases have suggested its involvement in the pathophysiology of these malignancies. The latter findings have thus raised the possibility of addressing Lnk signaling for the treatment of certain human diseases. This review therefore describes the pathophysiological role of this adaptor protein in hematological malignancies and the potential benefits of Lnk therapeutic targeting.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hematopoietic stem cells (HSCs) are responsible for the generation of all blood cells throughout the lifespan of an organism, a process called hematopoiesis. HSCs stay quiescent or proliferate toward either self-renewal or differentiation into distinct lineage-committed lymphoid and myeloid mature cells. Under steady-state conditions, most HSCs remain quiescent and only a small number proliferate to maintain a supply of mature blood cells without exhausting the HSC pool of an individual (Enver et al. 1998). However, in response to hematopoietic stress such as blood loss, HSCs exit quiescence and rapidly expand and differentiate to repopulate the peripheral hematopoietic compartments. These cellular events are tightly controlled by a number of growth factors and cytokines that influence cell survival, proliferation, differentiation and function of blood cells (Watowich et al. 1996).
Members of the cytokine receptor superfamily function through receptor oligomerization and subsequent conformational change resulting in transphosphorylation and activation of either an intrinsic kinase domain or specific receptor-associated kinases. One of the main families of cytoplasmic tyrosine kinases involved in cytokine signaling is the Janus kinase (JAK) family (Ortmann et al. 2000). Upon cytokine binding, the activated JAKs phosphorylate tyrosine residues in the receptor and subsequently downstream substrates, such as the signal transducers and activators of transcription (STAT) proteins. Once recruited to the receptor complex, STAT proteins are themselves phosphorylated on tyrosine, dimerize and translocate into the nucleus, where they activate the transcription of genes mediating cytokine-induced responses.
However, other signaling cascades are as well activated by cytokines, such as the Ras/mitogen-activated protein kinase (MAPK) and the phosphoinositide 3-kinase (PI3K)/Akt pathways. These pathways participate in the proliferation, survival, and differentiation of several cell types in the hematopoietic system (Geest and Coffer 2009; Leevers et al. 1999). Interestingly, deregulation of all these signaling pathways has been implicated in hematopoietic and autoimmune disorders, chronic inflammatory diseases and cancer (Ward et al. 2000; Schade et al. 2006; Khwaja 2006), underscoring the importance of multiple levels and mechanisms of control of the specific responses elicited by cytokine stimulation.
Role of Adaptor Proteins in Cytokine Signaling Regulation
Upon ligand binding, cytokine receptors undergo tyrosine autophosphorylation of the associated tyrosine kinase and of the receptor cytoplasmic domain at specific tyrosine (Y) residues. In this way, the cytoplasmic domain of these signaling molecules serves to initially localize the signaling response to the plasma membrane. Recruitment of particular targets to the receptor then determines the quality of the generated response. Indeed, the location of the proteins inside the cell and the kinetics of their activation are important features of signal-transduction pathways. How the signaling molecules are localized in the cell and how the strength and quality of the signal is regulated is an area of intense research. Central to this regulation are the so-called adaptor proteins, featuring as key molecules for the control of signaling complexes.
Adaptors are proteins lacking an enzymatic activity or other direct effector function. They can be transmembrane proteins, reside in the cytoplasm under resting conditions and be recruited to the membrane upon activation, or be localized by specific interactions in intracellular compartments such as the endoplasmic reticulum (ER) or lipid rafts. Regardless of their cellular localization, adaptor proteins possess an array of binding sites and modules that allow them to mediate specific protein–protein and protein–lipid interactions. Some representative binding domains present in adaptors include Src homology 2 (SH2) and phosphotyrosine-binding (PTB) domains, both binding to phosphotyrosine (pTyr) motifs, SH3 domains that bind to proline-rich sequences and pleckstrin homology (PH) domain that recognizes phospholipids (Pawson and Scott 1996). Moreover, there are specific features that distinguish a membrane adaptor from a cytoplasmic one. Membrane adaptors organize macromolecular complexes by mainly forming novel docking sites through the phosphorylation of multiple tyrosine residues. In contrast, cytoplasmic adaptors have a more varied array of domains and motifs that allow them to act both in a non-phosphotyrosine or phosphotyrosine-dependent manner, bringing proteins to the sites where these adaptors reside or are recruited. Interestingly, some enzymatically active molecules, such as tyrosine phosphatases and ubiquitin ligases contain in their structure the domains and motifs found in adaptors. This particularity allows these proteins to mediate true adaptor-like functions and also orchestrate signaling complex formation. Therefore, a single adaptor possessing different modules and binding sequences can serve as a scaffold protein for the recruitment of multiple proteins into complexes, thus bringing the effectors into close proximity with their targets. However, the general ability of adaptor proteins to amplify or inhibit signaling depends on the combination of different cell parameters such as the cell-specific expression and levels of the adaptors and that of their binding partners, their cell location, the stability of the interactions between the adaptor and its targets and in certain conditions, on the basal kinase/phosphatase activity in the cell.
In the last decades, the use of innovative biochemical, cellular and imaging techniques, as well as in vivo genetic approaches have provided new insights into the biology of the adaptor proteins function. These techniques have helped establish that adaptor proteins can affect the thresholds and the dynamics of signaling reactions by coordinating positive and negative feedback signals. To date, the majority of investigations on cytokine signaling pathways have mainly focused on the mechanisms of cytokine-receptor activation. However, recent research has now also focused on the mechanisms by which cytokine signals are attenuated or terminated. Indeed, stringent mechanisms of signal attenuation are essential for ensuring an appropriate, controlled cellular response following cytokine stimulation (Yasukawa et al. 2000). It is now easy to conceive how the aberrant assembly of macromolecular active signaling complexes can lead to disease: excess positive signaling or insufficient negative signaling may lead to autoimmunity, chronic inflammation or malignant transformation, while excess negative signaling or insufficient positive signaling may lead to immunodeficiency or certain hematological disorders.
The initiation, duration, magnitude and specificity of cytokine signaling is regulated at multiple levels by different mechanisms: (1) receptor internalization and inhibition mediated by soluble receptor antagonists and/or specific inhibitors; (2) tyrosine dephosphorylation of the receptor and signaling intermediates mediated by tyrosine phosphatases; (3) proteosomal degradation of signaling molecules and lastly (4) transcriptional suppression mediated by specific inhibitors such as the protein inhibitors of activated STATs (PIAS) proteins.
In this review, we address what is currently known about the function and regulation of the Lnk inhibitory adaptor protein, a key player in the control of cytokine signaling during normal and pathological hematopoiesis. However, the role of the other members of the family in the hematopoietic system will be mentioned when appropriate.
Structure, Origin and Cell Expression of the Lnk/SH2B Family
The Lnk (also known as SH2B) family of adaptor proteins comprises 3 members, Lnk (SH2B3), APS (for Adaptor protein with PH and SH2 domain, also known as SH2B2) and SH2-B [also known as PSM (proline-rich, PH and SH2 domain-containing signaling mediator) or SH2B1] (Rudd 2001). All members possess a dimerization (DD) domain and proline-rich motifs at the N-terminus, followed by a PH and SH2 domains, and several potential tyrosine phosphorylation sites (Fig. 1).

Structural representation of the Lnk family of adaptor proteins. Scheme presenting the functional domains and motifs present in all members of the Lnk family. The different isoforms of APS and SH2-B are also shown. DD dimerization domain, P proline-rich motif, PH pleckstrin domain, SH2 Src homology domain, Y tyrosine residue
The Lnk adaptor protein was the first member of this family identified as a 38 kDa protein expressed from a rat lymph node or mouse cDNA libraries (Huang et al. 1995; Takaki et al. 1997). However, it was found that the protein initially reported was partial. The complete Lnk protein was later being double in size (~70 kDa) (Li et al. 2000; Takaki et al. 2000; Velazquez et al. 2002). In contrast to SH2-B and APS, only one form of Lnk has been identified so far in mammalians and it is the only member with an invertebrate orthologue in Drosophila melanogaster (D-Lnk) to date (Werz et al. 2009). The Lnk adaptor protein is mainly expressed in hematopoietic cells and tissues, notably in HSC, and hematopoietic (lymphoid and myeloid) progenitors (HP) (Table 1). Interestingly, Lnk is not only an important regulator of cytokine signaling, but its expression is itself up-regulated by some of the cytokines essential for the development and function of hematopoietic cells, such as stem cell factor (SCF), thrombopoietin (TPO), and erythropoietin (EPO) (Kent et al. 2008; Buza-Vidas et al. 2006; Gery et al. 2009a, b; Baran-Marszak et al. 2010). Moreover, it was recently shown that Lnk is also highly expressed in endothelial progenitor (EPC) and mature (EC) cells and neurons with its expression being induced by tumor necrosis factor (TNF)-α and nerve growth factor (NGF), respectively (Fitau et al. 2006; Kwon et al. 2009; Wan et al. 2006; Wang et al. 2011). These findings implicate the Lnk adaptor as a key molecule in the negative feedback loop regulation of cytokine and growth factor pathways.
The APS protein was initially identified in a two-hybrid system screening of human B cells or adipocytes using as bait either an oncogenic form of the tyrosine kinase receptor c-Kit or the human insulin receptor (IR) cytoplasmic domain, respectively (Yokouchi et al. 1997; Moodie et al. 1999; Ahmed et al. 1999). Contrary to Lnk, there are two APS isoforms, SH2B2α and the recently identified SH2B2β (Fig. 1); the latter contains the N-terminal region and PH domain but lacks the SH2 domain (Li et al. 2007). APS adaptor protein is highly expressed in insulin-responsive tissues, especially in adipocytes and in the nervous system. However, it is also present in hematopoietic cells, notably in mature B cells and mast cells.
As for SH2-B, its gene encodes four isoforms (α, β, γ, δ) in the mouse and in the human genome. They result from alternative mRNA splicing at their 3′ terminus giving rise to proteins differing at their C-terminus [Fig. 1] (Nelms et al. 1999; Yousaf et al. 2001; Nishi et al. 2005). SH2-Bα and β isoforms were originally cloned from yeast tribrid and two-hybrid systems screening, respectively, using different proteins as baits: the tyrosyl-phosphorylated gamma subunit of the high-affinity immunoglobulin E (IgE), the cytoplasmic domain of the human IR and the JAK2 kinase domain (Osborne et al. 1995; Riedel et al. 1997; Rui et al. 1997). Although the SH2-B isoforms were initially identified in immune cells, they are mainly expressed and functional, as shown by gene inactivation in mice, in the adipose tissue, muscle, and brain with almost no role in the hematopoietic system. Indeed, deletion of the SH2B1 gene resulted in severe obesity, hyperphagia and both leptin and insulin resistance as well as infertility, which might be a consequence of resistance to insulin-like growth factor (IGF-1) (Ohtsuka et al. 2002; Duan et al. 2004; Ren et al. 2005). Therefore, SH2-B-deficient mice phenotype support a role for this adaptor as a positive regulator of JAK2 signaling pathways initiated by leptin, insulin and potentially by IGF-1.
Lnk Signaling Partners in Hematopoietic Cells
Since their identification, much effort has been focused in understanding the role of the Lnk family as regulators through the identification of their specific signaling pathways and the characterization of the molecules binding to their functional domains and motifs (Table 2). In this respect, crystallographic studies have revealed important structural differences among the Lnk adaptor family members that may explain their specific association with certain partners/effectors and the resulting cellular function.
The N-terminal region of the Lnk adaptor family is the least conserved region among all members. Crystal structure analysis of SH2-B and APS region revealed a novel dimerization (DD) domain containing at its core, a new structural motif, a phenylalanine zipper. This domain was shown to mediate SH2-B and APS homo and heterodimerization, which together with classical SH2 interactions, appears critical to their cellular functions (Dhe-Paganon et al. 2004; Nishi et al. 2005). As for Lnk, its DD domain is fairly different, having a leucine and tyrosine residues in the zipper. This difference seems to affect Lnk heterodimerization with APS or SH2-B only in yeast (Nishi et al. 2005), as it can still form heterodimers with SH2-B in PC12 neuronal cells (Wang et al. 2011). Conversely, Lnk homodimerization has only been shown in an over-expressed system and therefore its functional relevance has not been confirmed (Takizawa et al. 2006).
All members of the Lnk family and their isoforms possess a PH domain in their structure, suggesting an important role for this domain in the localization or translocation of these adaptor proteins to cellular membranes. Indeed, previous reports showed that the Lnk PHW191A mutant protein moderately affected Lnk modulation of TPO- and EPO-dependent biological responses (Tong and Lodish 2004; Tong et al. 2005). In agreement, a different PH mutant, W270A, exerted a similar effect on Kit-dependent signaling (Simon et al. 2008). Moreover, genetic modifications in the LNK gene, notably in exon 2 coding for the PH domain, have been recently identified in hematological, inflammatory and vascular diseases (see further in this review). This discovery suggests that Lnk PH domain plays a more significant role in the inhibitory function of the adaptor than originally believed. On the other hand, it has been reported that the Lnk PH domain displayed moderate affinity and little specificity to phosphoinositides in vitro. It is therefore possible that the Lnk PH domain may down-regulate membrane targeting of Lnk in the absence of docking site for the SH2 domain and increase binding stability to membrane receptors when the SH2 domain is engaged.
The best characterized domain of the Lnk adaptor proteins is the SH2 domain. This signaling module mediates most of the key biological functions and molecular interactions between this adaptor family and their partners/effectors described up to date. Although the sequence identity of the SH2 domains among all three members is fairly high (72–80 %), structural and biochemical studies have revealed important differences in this domain that determine their specificity and function. Indeed, crystallographic studies have demonstrated that the SH2 domain of APS is dimeric, with a rearranged C-terminal half of the molecule. This configuration favors a binding specificity for turn-containing phosphopeptide segments, such as the phosphorylated activation loop of the insulin receptor (Hu et al. 2003). In contrast, the SH2-B SH2 domain is monomeric, with a canonical SH2 domain architecture that preferentially binds conventional phosphotyrosine sequences, such as the one displayed in JAK2 pY813 (pYELL) (Hu and Hubbard 2006). As for Lnk, less is known on how its SH2 domain binds to its partners. Based on SH2-B data, Lnk SH2 domain is predicted to function as a monomer, since it lacks tryptophan 475 (Trp475), shown to be a critical residue for APS SH2 dimerization. However, one potentially important difference between Lnk and SH2-B is their binding preference. Lnk contains a cysteine instead of serine in SH2B at position 613 (Ser613) that can result in loss of selectivity for glutamate at the P + 1 position of the phosphotyrosine partner (Hu and Hubbard 2006). Indeed, a peptide library screening with Lnk SH2 domain allowed predicting a binding sequence, pY-[I/F/V/L]-X-[L/R/F/I] for this domain (Laura Velazquez, unpublished data). This binding motif has been found in some of Lnk’s specific partners, such as its first identified binding partner, the SCF receptor, the Kit protein. The primary Kit-binding site for Lnk SH2 domain was identified as pTyr567 [pYVYI], which resides in the juxtamembrane region of the receptor (Simon et al. 2008; Gueller et al. 2008). Similarly, the SH2 domain of APS was reported to bind to pTyr568 and pTyr936 in the human c-Kit receptor (Wollberg et al. 2003). Interestingly, the juxtamembrane region of Kit contains critical tyrosine residues 567/569 for the recruitment of positive and negative regulatory signaling molecules (Chan et al. 2003). In this system, a proposed mode of action of Lnk is that once bound to the juxtamembrane region of Kit, it will then block the association of activators with the receptor, resulting in down-regulation of SCF-mediated pathways. Indeed, expression of an SH2-inactive Lnk protein abolishes Lnk-mediated negative regulation of SCF-induced cell proliferation and migration (Simon et al. 2008). Since both Lnk and Kit proteins are expressed in the same hematopoietic cells, their association therefore plays an essential role in the regulation of biological responses important in these cell systems. Lnk has been also reported to bind through its SH2 domain to other tyrosine kinase receptors of the class III family, such as the platelet-derived growth factor receptor (PDGFR) and the macrophage-colony stimulating factor (M-CSF) receptor, the c-Fms protein, as well as the class VII NGF receptor, the TrkA protein. However, the physiological implications of these associations are not yet clear (Gueller et al. 2010, 2011; Wang et al. 2011).
The cytokine receptor-associated kinase, JAK2, was first characterized as binding partner of SH2-B and APS and it was later demonstrated for Lnk. This association results in activation of the kinase in the case of SH2-B and APS or in its inhibition when bound to Lnk. Different biochemical studies have shown that the interaction of the SH2 domains of SH2-B and APS occurs preferentially with kinase-active, tyrosyl-phosphorylated JAK2 (Rui et al. 1997; Nishi et al. 2005). The primary JAK2-binding site for the SH2 domain of the Lnk family members is pTyr813, which resides in an YXXL motif within the regulatory JAK homology 2 (JH2) pseudokinase domain of JAK2 (Kurzer et al. 2004, 2006; Bersenev et al. 2008). Interestingly, a secondary binding site for Lnk SH2 domain in JAK2 was identified as pTyr613 also present in the JH2 domain. However, this site seems to display lower affinity for Lnk SH2 domain than pTyr813 (Bersenev et al. 2008). Moreover, it was shown that Lnk is capable of binding the constitutive active JAK2-V617F form present in myeloproliferative neoplasms (MPNs) with higher affinity than JAK2 wild-type form (Bersenev et al. 2008; Gery et al. 2009a; Baran-Marszak et al. 2010). Together, these results show that in addition to the SH2-dependent interaction of the adaptor proteins of the Lnk family with phosphorylated JAK2, there is also a low-affinity interaction involving amino acids outside the SH2 domain in the adaptors and inactive JAK2 that may prevent abnormal activation of the kinase (Rui et al. 2000; Kurzer et al. 2006; Baran-Marszak et al. 2010).
Other Lnk binding partners have been reported using its SH2 domain in glutathione-S-transferase (GST) pulldown assays in T cells (Huang et al. 1995; Li et al. 2000). However, since the Lnk-deficient mice do not display a phenotype in this lineage, the physiological relevance of these interactions is uncertain.
Association of Lnk, APS and SH2-B with growth factor, cytokine and immunoreceptors or the JAK2 kinase allows phosphorylation of the adaptors and their proper localization at the signaling complex. These two events are necessary for the regulatory functions of the Lnk family and are primarily SH2-mediated. The C-terminal tyrosine residue is conserved in all members of this family, strongly suggesting it as a main site of phosphorylation after growth factor or cytokine stimulation and a potential docking site for SH2-containing molecules. The Y536 residue of Lnk was suggested to be phosphorylated upon SCF stimulation in a mast cell line (Takaki et al. 2002). However, mutation of this residue still allows this Lnk mutant form to be phosphorylated upon Kit activation in bone marrow-derived mast cells (BMMCs) (Simon et al. 2008). This result suggested that Lnk could be phosphorylated at sites other than Y536. Indeed, a similar result was reported with human Lnk mutated at this residue in Jurkat cells after T cell activation (Li et al. 2000). On the other hand, it seems that Lnk Y536 may play different biological roles depending on the signaling pathway: it is dispensable for lymphoid development, TPO- or SCF-dependent signaling pathways (Takaki et al. 2003; Tong and Lodish 2004; Simon et al. 2008), while it may have a regulatory role in IL3- and EPO-mediated proliferation (Tong et al. 2005; Simon et al. 2008). However, no specific signaling molecule has been identified as binding partner of Lnk at this site.
In contrast, it has been shown that the C-terminal Y618 of APS can be phosphorylated by activated growth factor (IR), cytokine (EPO) and immune (BCR) receptors and thus serves as binding site for the E3 ligase protein Cbl (Moodie et al. 1999; Yokouchi et al. 1997; Wakioka et al. 1999). The APS/Cbl association plays an important role in down-regulation of signaling through the ubiquitination, internalization and eventual degradation of the receptor complex, as has been shown for the IR (Ahn et al. 2004; Kishi et al. 2007). However, a similar biological significance of this association in cytokine or immune signaling has not been shown.
Of great interest is the identification of a group of binding partners of this adaptor family that are mainly involved in the regulation of actin polymerization, which suggests the implication of these proteins in the regulation of cell shape and motility. These signaling molecules associate with domains in Lnk, APS and SH2-B, other than the SH2 domain or phosphorylated tyrosines. In particular, SH2-Bβ has been shown to regulate growth hormone and prolactin-dependent actin reorganization and cell motility. This is partly due to the presence in SH2-Bβ of two actin-binding domains, one at the N-terminal region (amino acids 150–200) and the second at the C-terminus (amino acids 615–670) and of one filamin-binding domain also at the N-terminus (amino acids 200–260) (Rider et al. 2009; Rider and Diakonova 2011). Moreover, SH2-Bβ is capable of binding to Rac GTPase, a major actin regulating protein, through a proline-rich sequence in the N-terminal region, resulting in enhanced GH-induced actin reorganization and cell migration (Herrington et al. 2000; Diakonova et al. 2002). A similar sequence in APS can associate with Enigma, an actin-associated cytoskeleton protein (Barrès et al. 2005), while APS PH domain was shown to associate with Vav3, a guanine exchange factor (GEF) protein for Rac when over-expressed in NIH3T3 cells (Yabana and Shibuya 2002). In the case of Lnk, it was shown that the adaptor can down-regulate SCF-mediated Rac and p38MAPK signaling pathways, leading to control of mast cell migration and spreading (Simon et al. 2008; Elisabetta Dondi, personal communication). Interestingly, Rac is known to play an essential role in regulating HSCs/HPs actin cytoskeleton, which affects the migration, adhesion, and cell-cycle progression of these cells (Gu et al. 2003). Whether Lnk may be involved in the modulation of Rac-mediated actin cytoskeletal regulation and cell movement in HSCs/HPs remains to be addressed. Together, all these data suggest Rac as an important common effector of the Lnk family in cytoskeleton rearrangement and cell migration.
In the same context, several studies have recently demonstrated the key role of Lnk in the regulation of integrin-mediated adhesion and migration of various hematopoietic cell types. In HSC/HPs, Lnk seems to regulate their interaction and trapping by vascular cell adhesion molecule 1 (VCAM), which results in the modulation of their motility and proper lodging to niche-forming cells in the BM and spleen (Takizawa et al. 2006). As for the megakaryocytic lineage, Lnk was shown to modulate thrombopoiesis at two levels: (1) during megakaryocytic maturation and platelet release processes by regulating the crosstalk between integrin and TPO-mediated signaling pathways and (2) it contributes to platelet cytoskeleton rearrangement and fibrinogen spreading by facilitating the recruitment of the Fyn kinase to the β3 integrin tail, which results in the stabilization of thrombus formation in vivo (Takizawa et al. 2008, 2010). On the other hand, it was also shown that human Lnk interacts with the actin-binding protein ABP-280 (or Filamin A) in Cos7 and Jurkat T cells in a yeast two-hybrid system (He et al. 2000). This interaction is mediated by the PH and SH2 inter-domain sequence (56 amino acids) of human Lnk. Interestingly, increased filamin binding to the β-integrin subunit cytoplasmic tail was shown to affect cell migration on VCAM-1 (Calderwood et al. 2001). One can then speculate that Lnk may participate in the regulation of integrin-mediated cell adhesion and/or migration through its interaction with filamin in hematopoietic cells. All together, these findings underlie the important role of the Lnk family members in the regulation of actin cytoskeleton and cell motility.
Lnk-Deficient Mice as Animal Model to Study Hematopoiesis
The initial in vitro biochemical analysis on the Lnk family members, SH2-B and APS, showed that these adaptors functioned as substrates and/or activators of growth factor signaling pathways. However, APS was later shown to also serve as negative regulator of platelet-derived growth factor (PDGF)-induced mitogenesis (Yokouchi et al. 1999), the BCR receptor (Yokouchi et al. 1997) and of EPO-stimulated JAK2 (Wakioka et al. 1999). Conversely, Lnk has been mainly considered as a negative regulator of growth factor and cytokine-receptor signaling controlling cytokine-induced proliferation and migration. However, there are so far two signaling pathways where Lnk seems to also play a positive role: in the integrin αIIbβ3 outside-in signaling in mouse platelets and in the TNF-α-mediated PI3K/Akt pathway in human vascular endothelial cells (Takizawa et al. 2010; Fitau et al. 2006). These findings point out a dual positive/negative role for these adaptor proteins in growth factor and cytokine-receptor pathways. However, these data also raise questions on how do these adaptors with such a conserved structure can mediate opposite cellular functions. A possible explanation is that each adaptor displays a specific cell expression, signaling and effectors’ patterns that together with intrinsic structural differences, will determine their function as either cellular activators or inhibitors. In this context, the presence of “adaptor-specific inhibitory/activating” sequences could also explain the mechanistic differences exhibited by the Lnk family members. Detailed analysis of their sequence will certainly shed light on these adaptors’ cellular functioning.
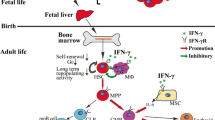
Nonetheless, the generation of mice and cell lines deficient for members of this family has helped establish the physiological role, as activators or inhibitors, for Lnk and APS in the hematopoietic system. Two of the main signaling pathways where Lnk has proved to be a key negative regulator are the SCF/Kit and TPO/Mpl pathways. These cytokines are crucial for the development, proliferation, migration and survival of progenitors (HSC, hematopoietic and endothelial) and mature (mast cells and megakaryocytes/platelets) cells. Interestingly, Lnk-deficient mice display phenotypes in these same cell types, which confirmed Lnk inhibitory role in Kit and Mpl signaling pathways. In HSC and hematopoietic progenitors, Lnk has been shown to regulate cell self-renewal, proliferation and apoptosis, as shown by studies on Lnk −/−-derived HSC. Further analysis of these cells demonstrated an enhanced TPO hypersensibility, resulting in abnormal TPO signaling with increased JAK2/STAT5, Akt and Bcl-xL activation, while down-regulating p38MAPK (Ema et al. 2005; Buza-Vidas et al. 2006; Seita et al. 2007; Bersenev et al. 2008; Suzuki et al. 2012). These findings therefore confirm that Lnk controls TPO-induced self-renewal, quiescence, proliferation and apoptosis of HSC. Moreover, Lnk deficiency enhances the ability of HSC and hematopoietic progenitors to reconstitute the hematopoietic system in irradiated hosts (Takaki et al. 2002; Ema et al. 2005). Indeed, transient inhibition of endogenous Lnk significantly increased the repopulating capacity of the transduced cells and thereby, engraftment (Takizawa et al. 2006). Conversely, analysis of Lnk −/−-derived hematopoietic progenitors show an hypersensibility to several cytokines (SCF, IL-7, IL-3, TPO, EPO) resulting in sustained MAPK, JAK/STAT activation and therefore, cell proliferation (Takaki et al. 2000; Velazquez et al. 2002; Tong et al. 2005; Takizawa et al. 2008). Interestingly, this function cannot be restored by APS expression in Lnk −/− hematopoietic progenitors, thus demonstrating Lnk-specific function in these cells (Simon et al. 2008).
In mature cells like megakaryocytes/platelets, Lnk has been shown to control TPO-dependent megakaryopoiesis and thrombopoiesis. Studies on primary Lnk −/− megakaryocytes indicated an abnormal proliferation due to the absence of negative regulation of TPO signaling pathways (Fig. 2). Indeed, Lnk through its SH2 domain negatively modulates MPL signaling by attenuating three major signaling pathways: JAK2/STAT, MAPK and Akt (Tong and Lodish 2004; Takizawa et al. 2008). Moreover, Lnk is capable of binding and regulating the mutated forms of the Mpl receptor, MPL-W515L, and of the JAK2 kinase, JAK2-V617F, both expressed in MPNs in primary megakaryocytes and megakaryocytic cell lines (Gery et al. 2007, 2009a; Bersenev et al. 2008; Baran-Marszak et al. 2010).

Schematic model of Lnk and APS-mediated signaling pathways in hematopoietic cells. Lnk and APS can interact with tyrosine kinase (a), cytokine (b) receptors or (c) integrins, either directly (a) or via an associated kinase (b and c) inhibiting in this way, their downstream substrates. GF growth factor, GFR growth factor receptor, CR cytokine receptor, KD kinase domain, ECM extracellular matrix, FA focal adhesion, FAK focal adhesion kinase, ILK integrin-linked kinase, GSK3β glycogen synthase kinase 3β
As in TPO, a similar regulatory role has been attributed to Lnk during erythropoiesis. Lnk −/− mice exhibited a recovery after erythopoietic stress superior to that of wild-type animals. In addition, primary Lnk −/− erythroblasts displayed an increase in cell proliferation and survival, due to the deregulated activation of EPOR-mediated signaling pathways: MAPK, JAK2/STAT, and Akt (Tong et al. 2005). This function is mediated by Lnk SH2 domain, also responsible for Lnk phosphorylation following EPO stimulation.
The second hematopoietic mature cell line where Lnk plays an important role is in mastocytes. This is not surprising considering that the SCF/Kit pathway is essential for several biological responses in mast cells. Indeed, studies on Lnk −/− mast cells demonstrated the physiological role of this adaptor in SCF-mediated signaling pathways controlling proliferation (MAPK and JNK) and migration (Rac and p38MAPK) (Takizawa et al. 2006; Simon et al. 2008). These functions are mainly mediated by Lnk SH2 domain which binds first to regulatory tyrosines in Kit receptor and then to specific effectors involved in the different SCF-dependent biological responses (Fig. 2). On the other hand, APS has been also reported to play a role in mast cells, notably through the FcεRI receptor. Indeed, studies on APS −/− mastocytes showed that APS controls actin rearrangement and the magnitude of degranulation induced by FcεRI receptor cross-linking (Kubo-Akashi et al. 2004).
Recently, Lnk was also reported to participate as negative regulator in M-CSF-mediated signaling pathways in macrophages (Gueller et al. 2010). Lnk seems to bind to the c-Fms receptor through its SH2 domain and regulate M-CSF-induced activation of Akt and Erk pathways, as well as macrophage migration. These data suggest that Lnk is involved in the regulation of macrophage biological functions, which may imply a role for this adaptor in innate immunity.
The first physiological role for Lnk revealed by analysis of Lnk-deficient mice was in B cell lymphopoiesis. These animals exhibited the selective expansion of pro-/pre-B and immature B cells in bone marrow and spleen. Indeed, the histological analysis of spleen of Lnk-deficient mice showed dramatic changes in the lymphoid follicles in the white pulp, which resulted from an increase in B220+ lymphocytes (Velazquez et al. 2002). Moreover, Lnk −/− lymph nodes were often enlarged and showed increased B cell numbers. This abnormal proliferation of B cell clonogenic progenitors was partly due to hypersensitivity to SCF and IL-7, two cytokines that synergize and support proliferation of B cell precursors (Takaki et al. 2000; Velazquez et al. 2002). The regulatory role of Lnk in the Kit signaling pathway in the B cell lineage was further demonstrated in competitive repopulation assays. In these studies, Lnk −/− mice on a Kit W/+ background exhibited partial, but significant normalization of B cell overproduction displayed by Lnk-deficient animals, confirming the negative role of Lnk in SCF/Kit signaling in these cells (Takaki et al. 2002). Alternatively, Lnk over-expression in transgenic mice show impaired B cell production in an Lnk dose-dependent manner confirming the negative control mediated by this adaptor in B-lineage cell production (Takaki et al. 2003). However, no effect on mature B cells was observed in the absence of Lnk, suggesting either a lack of role for Lnk in this population or a functional compensation by APS in these cells. Indeed, the expression of APS adaptor was mainly detected in mature B cells, with a weaker expression in immature cells and absent in B cell clonogenic progenitors and pre/pro-B cells (Yokouchi et al. 1997; Elisabetta Dondi, personal communication). In mature B cells, APS has been shown to play a role in B cell development and function, as shown by APS-deficient animals. Ablation of APS in mice caused an increase in B-1 cell number and an enhanced humoral immune response against a thymus-independent type 2 antigen, while B-2 cells exhibited normal proliferative responses and tyrosine phosphorylation upon BCR stimulation (Iseki et al. 2004). Moreover, APS −/− derived B-1 cells displayed lower filamentous actin (F-actin) content than wild-type cells. In contrast, APS transgenic mice showed reduced numbers of peritoneal B-1 and splenic B cells with impaired BCR-induced proliferation of mature B cells. Moreover, APS co-localized with F-actin accumulated during the capping of BCR complexes in these cells (Iseki et al. 2005). All together, these results suggest a negative regulatory role for APS in BCR signaling, cell proliferation and actin reorganization in mature B-1 cells. However, little is known, besides the Kit receptor, on the signaling partners of Lnk and APS in the B cell lineage, that could explain the regulatory role of these two adaptors in the proliferation, survival and migration of B cell progenitors (IL-7/SCF signaling pathways) and mature (BCR signaling) cells. Certainly, this is still an open research area in the Lnk adaptor family to explore.
A few years ago, three groups showed that besides its specific expression in hematopoietic cells, the Lnk adaptor was also highly expressed in the vascular system, notably in endothelial cells (Nobuhisa et al. 2003; Fitau et al. 2006; Wan et al. 2006). Lnk was first detected in the endothelial cells lining the dorsal aorta in the aorta–gonad–mesonephros (AGM) region at embryonic day 11.5, where it inhibits SCF-induced hematopoietic cell development via its SH2 domain (Nobuhisa et al. 2003). Moreover, its expression and function was subsequently reported in endothelial mature cells (EC) (Fitau et al. 2006). Indeed, studies by Charreau’s group showed that the pro-inflammatory cytokine TNF-α rapidly up-regulates Lnk at mRNA and protein level, with its subsequent phosphorylation (Boulday et al. 2002; Fitau et al. 2006). Furthermore, they demonstrated that Lnk down-regulates VCAM-1 and E-selectin expression in activated vascular ECs via modulation of TNF-α signaling pathways: on one hand inhibiting the ERK1/2 cascade, and on the other hand activating the PI3-kinase/Akt pathway with phosphorylation of the endothelial nitric oxide synthase (eNOS) protein. Recently, Lnk was also shown to regulate the activation of integrin signaling pathways in ECs. In this system, Lnk interacts directly with the integrin-linked kinase (ILK), resulting in the phosphorylation of Akt, GSK3β, paxillin and FAK (Devallière et al. 2012). This leads to the control of focal adhesion (FA) formation and EC migration. Moreover, the α-parvin protein was identified as one of the molecular targets of Lnk responsible for these integrin-mediated biological functions. All these data have allowed proposing a model in which Lnk down-regulates α-parvin expression through its interaction with ILK in response to cell adhesion to extracellular matrix (ECM) (Fig. 2). As a result, Lnk blocks FAs and delayed migration. Together, these findings confirm Lnk as a key adaptor in ECs via two major signaling pathways, TNF-α and β-integrin.
Recently, it was demonstrated that Lnk-deficient mice also exhibited an increase in endothelial progenitor cells (EPC) numbers that display an enhanced capacity for colony formation. Through the use of different molecular, physiological and morphological approaches, it was shown that Lnk deficiency promotes vasculogenesis/angiogenesis and osteogenesis through the mobilization and recruitment of HSCs/EPCs via activation of the SCF/Kit signaling pathway in the ischemic and perifracture zone, respectively, thereby establishing an optimal environment for neovascularisation, bone healing and remodeling (Kwon et al. 2009; Matsumoto et al. 2010). Therefore, these findings strongly suggest that Lnk regulates bone marrow EPC kinetics during vascular and bone regeneration.
Lastly, it seems that Lnk might play an important function in systems other than the hematopoietic one. In a recent study, Wang et al. showed that Lnk is expressed in the cortex of embryonic rat brain, notably in cortical cells. Using PC12 cells, they demonstrated that Lnk represses NGF-induced activation of PLCγ, MEK-ERK1/2, PI3K/Akt pathways and the expression of Egr-1, resulting in inhibition of neurite differentiation and outgrowth (Wang et al. 2011). Lnk exercises its inhibitory function by binding to the phosphorylated NGF receptor, TrkA, via its SH2 domain. In this way, it blocks further binding of the other two members of the Lnk family, SH2B and APS that act as activators of the TrkA receptor in this system.
Role of the Lnk Adaptor Family in Human Diseases
The generation of mice deficient for adaptor proteins has been central to understanding the physiological role of these molecules in the hematopoietic and immune systems. As described previously, animal models for the Lnk and APS members of this family have been invaluable in demonstrating the important roles played by these adaptor proteins in fine-tuning signaling in hematopoietic and immune cells at specific stages of development. The strong resemblance between these in vivo models and certain human hematological and immune diseases has allowed to identify signaling effectors and pathways implicated in the initiation and progression of some of these malignancies (Table 3). However, in some cases, these animal models have also raised important questions regarding the mechanisms of action of these regulators and their potential therapeutic application.
Lnk-deficient mice display a phenotype reminiscent of human BCR-ABL negative MPNs: hypersensitivity to cytokines, increased number of hematopoietic progenitors, high platelet counts, splenomegaly together with fibrosis and extramedullary hematopoiesis (Velazquez et al. 2002; Campbell and Green 2008). These clonal disorders are characterized by the abnormal proliferation, cytokine regulation and/or absence of negative feedback regulation of hematopoietic cells. In this sense, it is possible that Lnk has a key role in the development of these diseases, notably in essential thrombocythemia (ET) and primary myelofibrosis (PMF); these MPNs are characterized by the excessive proliferation of the megakaryocytic/platelet lineage and the megakaryocyte/granulocytic lineage, respectively.
In the last 5 years, the identification of mutations in major signaling molecules in MPNs has changed the way these diseases are now approached and studied. One of the most valuable discoveries in the field is the Val617Phe acquired mutation in the JAK2 gene (JAK2-V617F), as it represents the first reliable molecular marker of Ph− MPN (Campbell and Green 2008). However, the precise mechanism implicated in the pathogenic role of JAK2-V617F constitutive active kinase is still unclear. It is possible that this mutant kinase causes the abnormal activation of downstream signaling molecules, among which Lnk likely plays an important negative regulatory role, as shown by its binding to and inhibition of the JAK2-V617F form (Gery et al. 2009a; Bersenev et al. 2008; Baran-Marszak et al. 2010). Moreover, a differential level of expression of Lnk was demonstrated in megakaryocyte/platelets and CD34+ cells from MPNs patients (Baran-Marszak et al. 2010), suggesting its implication in the development of these diseases. This was further confirmed by the recent identification of different LNK mutations in JAK2-V617F-negative and positive MPN patients (Fig. 3). These LNK mutations occur at low frequency (5–7 %) in MPNs and therefore are considered rare events. However, other LNK mutations have been identified in leukemic transformation of MNPs at a higher frequency (13 %), all except one, targeting a “hot spot” located between codons 208 and 234 in the PH domain (Pardanani et al. 2010). Certainly, the first mutations identified were mainly on exon 2 coding for the PH domain (Oh et al. 2010; Lasho et al. 2010). Nonetheless, mutations in other exons of the LNK gene have been recently reported (Ha and Jeon 2011; Hurtado et al. 2011). In addition, LNK mutations are not exclusive events, as they can be found in conjunction with mutations in other genes, like JAK2, TET2 and CBL (Tefferi 2010). These findings suggest that LNK mutations induce an MPN phenotype that may depend on different parameters, such as the presence of other mutations (Lasho et al. 2011). In this context, it was reported that loss of Lnk cooperates with oncogenes, such as JAK2 and BCR-ABL, to induce MPN in mice. These animals exhibit a disproportionate expansion of myeloid progenitors and immature precursors in vitro and in vivo (Bersenev et al. 2010). Moreover, aged Lnk −/− mice seem to spontaneously develop a chronic myeloid leukemia (CML)-like MPN, suggesting a role for Lnk in myeloid expansion in vivo. However, this myeloid cell hyperproliferation fails to trigger blast crises, supporting the need of Lnk deficiency for additional oncogenic events to promote blast transformation.

Point mutations and polymorphism found in the Lnk/SH2B3 gene in human diseases. Scheme showing the localization of the point mutations in LNK gene identified in MPNs (bottom). The non-synonymous polymorphism R262W has been correlated to certain human disease and hematological parameters (top). MPNs myeloproliferative neoplasms, nsSNP non-synonymous single nucleotide polymorphism
On the other hand, recent studies have shown that LNK mRNA levels were also elevated in myelodysplastic syndromes and acute myeloid leukemia cells. Moreover, Lnk protein was shown to bind to certain oncogenic tyrosine kinases, such as JAK2-V617F, KIT-D816V, and FIP1L1-PDGFRA, and inhibit proliferation induced by these oncogenes (Gery et al. 2009a, b; Gueller et al. 2011). Together with data on MPNs, these results suggest a relevant role for Lnk in these proliferative hemopathies that can be exploited for therapeutic purposes.
Of great interest is the recent publication of several genome-wide association studies (GWAS) revealing that different diseases may share a LNK susceptibility variant. The LNK/SH2B3 gene maps on chromosome 12 at 12q24 and single nucleotide polymorphisms (SNP) in this gene has been reported in exon 2, 3 and 5, which implicate the PH and SH2 domains of the Lnk protein. In particular, the non-synonymous (ns) SNP rs3184504 results in a missense mutation at position 262 in exon 2 leading to a R262W amino acid substitution in the PH domain (Fig. 3). Surprisingly, this nsSNP has recently been associated with inflammatory disorders, such as celiac disease (Hunt et al. 2008; Zhernalova et al. 2010), type 1 diabetes (Lavrikova et al. 2011), asthma (Gudbjartsson et al. 2009) and multiple sclerosis (Alcina et al. 2010), and also to eight clinically relevant hematological parameters (Soranzo et al. 2009; Ganesh et al. 2009) in different populations. Furthermore, the LNK R262W variant has also been associated to cardiovascular diseases such as myocardial infarction, coronary heart disease and hypertension (Gudbjartsson et al. 2009; Ikram et al. 2010). All these data suggest that the LNK nsSNP rs3184504 could be a risk variant for these diseases contributing to their pathogenesis, and in consequence, providing a useful diagnostic marker.
On the other hand, APS-deficient mice display an increase in CD5+ B-1 cells in the peritoneal cavity and humoral responses to type-2 antigen, indicating a negative regulatory role for APS in BCR-mediated cell proliferation and cytoskeletal regulation in these cells. Interestingly, this B-1 cell subtype is greatly increased in patients with chronic lymphocytic leukemia (CLL), which suggests a role for APS in the proliferation of B-1 cells in this lymphoproliferative disease.
Potential Application of the Lnk Adaptor in Regenerative Medicine
Given Lnk-restricted expression to the hematopoietic and vascular system and the important role it plays in the expansion of HSC, hematopoietic and endothelial progenitor cells (EPCs), Lnk protein seems an excellent new target for the treatment of hematopoietic and vascular diseases. Indeed, recent reports have shown that Lnk plays a key role in vascular regeneration in hindlimb ischemia, retinopathy and spinal cord injury. In these in vivo mouse models, loss of Lnk or its inhibition leads to SCF-mediated growth upregulation of HSCs and endothelial progenitor cells (EPCs), resulting in enhanced endothelial commitment into endothelial lineage cell types, mobilization from bone marrow into peripheral blood, and recruitment to ischemic sites for neovascularisation (Kwon et al. 2009; Kamei et al. 2010). Moreover, Lnk was shown to increase astrocyte network maturation in the retinopathy model, while it promotes repair of injured spinal cord through the acceleration of angiogenesis with nonpathogenic effect and astrogliosis. Taken together, these results provide strong evidence that Lnk regulates the commitment of bone marrow cells to EPCs in vascular regeneration as shown by Lnk deletion.
Furthermore, Lnk seems also to participate in the regenerative response during bone fracture healing. This was demonstrated in Lnk-deficient mice where vasculogenesis/angiogenesis and osteogenesis were promoted by the mobilization and recruitment of HSCs/EPCs via activation of the SCF signaling pathway in the perifracture zone. This condition established a favorable environment for bone healing and remodeling (Matsumoto et al. 2010).
On the other hand, it was shown that Lnk loss or its inhibition causes the abnormal expansion and enhanced ability of HSCs for engraftment (Takizawa et al. 2006). The relevance of this Lnk feature is that these cells can then be used for bone marrow transplantation, whose limiting factor is the availability of HSC in sufficient numbers. Furthermore, APS expression in these cells does not restore Lnk function, demonstrating the unique role of Lnk in HSC/HP proliferation and self-renewal. Lastly, of great interest is the recent report on the expression of human Lnk in cultured porcine aortic endothelial cells (PAEC) and its functional significance in protecting these cells from inflammation and apoptosis (Chatelais et al. 2011). These results open up the possibility of using Lnk to protect vascular endothelial cells from inflammation in xenotransplantation.
Overall, these findings suggest that selective targeting of Lnk may be a novel and effective therapeutic approach for hematopoietic, vascular and bone diseases and injuries. However, Lnk lacks any enzymatic activity for drug targeting. An alternative possibility is therefore to block Lnk protein–protein interactions for targeting specific signaling pathways. In some pathological contexts, the association of the Lnk adaptor with mutated or oncogenic forms of its targets is modified from that engaged with its normal counterpart (Baran-Marszak et al. 2010). These findings suggest the use of the binding sequence in the adaptor to exclusively inhibit the oncogenic protein and signaling pathway, while sparing the normal cell signaling cascades. Conversely, the use of dominant negative forms of the Lnk protein can be advantageous, notably where this adaptor displays dual functions, as positive and negative regulator, as it allows modulate specifically their function depending on the cell type and biological response to be addressed.
Conclusion
Over the last years, the pivotal role that Lnk plays in maintaining hematopoietic homeostasis by preventing inappropriate cellular activation has become clear. For this, Lnk acts upon three key signaling intermediates: the receptor, JAK kinases, and STATs/downstream effectors, to switch off the signal completely, as part of a negative feedback loop. It is therefore not surprising that the Lnk protein is also tightly regulated, pointing out the complexity of Lnk regulatory mechanisms.
While the initial analysis of the Lnk mouse model highlighted the critical roles for this inhibitory adaptor in immune function and hematological malignancy, recent studies on Lnk −/− animals show its implication in vascular and bone regeneration, as well as its potential use in bone marrow and xenotransplantation. All these findings therefore point out the possibility of selective targeting of Lnk as a novel, safe and effective therapeutic approach for hematopoietic, vascular and bone diseases and/or injuries. In this context, a better understanding of the spectrum of signaling alterations provoked by mutant forms of Lnk identified in human pathologies will likely reveal therapeutic strategies for patients with these mutations. Moreover, the association of an nsSNP in the Lnk gene with different inflammatory, myeloproliferative and vascular diseases suggests the implication of this molecule as risk factor and its potential use as biomarker in these diseases.
Thus, one of the future research lines is to understand how the Lnk adaptor orchestrates the functional activity and fate of its partners to produce the desired intensity of a signaling response. Although a great deal of research remains to be done to clarify the roles of this inhibitory adaptor and its mutant forms in human diseases such as MPNs and inflammation, we can predict that it will lead to the development of Lnk adaptor-based strategies for therapeutic purposes.
Abbreviations
- HSC:
-
Hematopoietic stem cells
- HP:
-
Hematopoietic progenitors
- JAK:
-
Janus kinase
- STAT:
-
Signal transducer and activator of transcription
- MAPK:
-
Mitogen-activated protein kinase
- PI3K:
-
Phosphoinositide 3-kinase
- SH2:
-
Src homology 2
- PH:
-
Pleckstrin homology
- DD:
-
Dimerization domain
- APS:
-
Adaptor protein with PH and SH2 domain
- SCF:
-
Stem cell factor
- TPO:
-
Thrombopoietin
- EPO:
-
Erythropoietin
- TNF-α:
-
Tumor necrosis factor-alpha
- MPN:
-
Myeloproliferative neoplasms
- BCR:
-
B cell receptor
- AGM:
-
Aorta–gonad–mesonephros
- EPC:
-
Endothelial progenitor cells
- EC:
-
Endothelial cells
- SNP:
-
Single nucleotide polymorphism
References
Ahmed Z, Smith BJ, Kotani K et al (1999) APS, an adapter protein with a PH and SH2 domain, is a substrate for the insulin receptor kinase. Biochem J 341(Pt 3):665–668
Ahn MY, Katsanakis KD, Bheda F, Pillay TS (2004) Primary and essential role of the adaptor protein APS for recruitment of both c-Cbl and its associated protein CAP in insulin signaling. J Biol Chem 279(20):21526–21532
Alcina A, Vandenbroeck K, Otaegui D et al (2010) The autoimmune disease-associated KIF5A, CD226 and SH2B3 gene variants confer susceptibility for multiple sclerosis. Genes Immun 11:439–445
Baran-Marszak F, Magdoud H, Desterke C et al (2010) Expression level and differential JAK2-V617F-binding of the adaptor protein Lnk regulates JAK2-mediated signals in myeloproliferative neoplasms. Blood 116(26):5961–5971
Barrès R, Gonzalez T, Le Marchand-Brustel Y, Tanti JF (2005) The interaction between the adaptor protein APS and Enigma is involved in actin organization. Exp Cell Res 308:334–344
Bersenev A, Wu C, Balcerek J, Tong W (2008) Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J. Clin. Invest. 118(8):2832–2844
Bersenev A, Wu C, Balcerek J et al (2010) Lnk constraints myeloproliferative diseases in mice. J. Clin. Investig 120(6):2058–2069
Boulday G, Coulon F, Fraser CC et al (2002) Transcriptional up-regulation of the signaling regulatory protein LNK in activated endothelial cells. Transplantation 74(9):1352–1354
Buza-Vidas N, Antonchuk J, Qian H et al (2006) Cytokines regulate postnatal hematopoietic stem cell expansion: opposing roles of thrombopoietin and LNK. Genes Dev 20:2018–2023
Calderwood DA, Huttenlocher A, Kiosses WB et al (2001) Increased filamin binding to beta-integrin cytoplasmic domains inhibits cell migration. Nat Cell Biol 3:1060–1068
Campbell PJ, Green AR (2008) The myeloproliferative disorders. N Engl J Med 355:2452–2466
Chan MP, Ilangumaran S, La Rose J et al (2003) Autoinhibition of the Kit receptor tyrosine kinase by the cytosolic juxtamembrane region. Mol Cell Biol 23:3067–3078
Chatelais M, Devallière J, Galli C, Charreau B (2011) Gene transfer of the adaptor Lnk (SH2B3) prevents porcine endothelial cell activation and apoptosis: implication for xenograft's cytoprotection. Xenotransplantation 18:108–120
Devallière J, Chatelais M, Fitau J et al (2012) LNK (SH2B3) is a key regulator of integrin signaling in endothelial cells and targets α-parvin to control cell adhesion and migration. FASEB. doi:10.1096/fj.11-193383
Dhe-Paganon S, Werner ED, Nishi M et al (2004) A phenylalanine zipper mediates APS dimerization. Nat Struct Mol Biol 11(10):968–974
Diakonova M, Gunter DR, Herrington J, Carter-Su C (2002) SH2-Bβ is a Rac-binding protein that regulates cell motility. J Biol Chem 277(12):10669–10677
Duan C, Yang H, White MF, Rui L (2004) Disruption of the SH2-B gene causes age-dependent insulin resistance and glucose intolerance. Mol Cell Biol 24:7435–7443
Ema H, Sudo K, Seita J et al (2005) Quantification of self-renewal capacity in single hematopoietic stem cells from normal and Lnk-deficient mice. Dev Cell 8:907–914
Enver T, Heyworth CM, Dexter TM (1998) Do stem cells play dice? Blood 92:348–351
Fitau J, Boulday G, Coulon F et al (2006) The adaptor molecule Lnk negatively regulates TNFα-dependent VCAM-1 expression in endothelial cells through inhibition of the ERK1 and 2 pathways. J Biol Chem 281(29):20148–20159
Ganesh SK, Zakai NA, Van Rooij FJA, Soranzo N et al (2009) Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat Genet 41(11):1191–1198
Geest CR, Coffer PJ (2009) MAPK signaling pathways in the regulation of hematopoiesis. J. Leuk. Biol. 86:237–250
Gery S, Gueller S, Chumakova K et al (2007) Adaptor protein Lnk negatively regulates the mutant MPL, MPLW515L associated with myeloproliferative disorders. Blood 110(9):3360–3364
Gery S, Qi C, Gueller S et al (2009a) Lnk inhibits myeloproliferative disorder-associated JAK2 mutant, JAK2V617F. J Leuk Biol. 85:957–965
Gery S, Gueller S, Nowak V et al (2009b) Expression of the adaptor protein Lnk in leukemia cells. Exp Hematol 37:585–592
Gu Y, Filippi MD, Cancelas JA et al (2003) Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science 302(5644):445–449
Gudbjartsson DF, Bjornsdottir US, Halapi E et al (2009) Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat Genet 41(3):342–347
Gueller S, Gery S, Nowak V et al (2008) Adaptor protein Lnk associates with Y568 in c-Kit. Biochem. J. 415:241–245
Gueller S, Goodrigde HS, Niebuhr B et al (2010) Adaptor protein Lnk inhibits c-fms-mediated macrophage function. J. Leuk. Biol. 88:699–706
Gueller S, Hehn S, Nowak V et al (2011) Adaptor protein Lnk binds to PDGF receptor and inhibits PDGF-dependent signaling. Exp Hematol. doi:10.1016/j.exphem.2011.02.001
Ha JS, Jeon DS (2011) Possible new LNK mutations in myeloproliferative neoplasms. Am J Hematol 86(10):866–868
He X, Li Y, Schembri-King J et al (2000) Identification of actin binding protein, ABP-280, as a binding partner of human Lnk adaptor protein. Mol Immunol 37:603–612
Herrington J, Diakonova M, Rui L et al (2000) SH2-B is required for Growth hormone-induced actin reorganization. J Biol Chem 275(17):13126–13133
Hu J, Hubbard SR (2006) Structural basis for phosphotyrosine recognition by the Src Homology-2 domains of the adapter proteins SH2-B and APS. J Mol Biol 361:69–79
Hu J, Liu J, Ghirlando R et al (2003) Structural basis for recruitment of the adaptor protein APS to the activated Insulin receptor. Mol Cell 12:1379–1389
Huang X, Li Y, Tanaka K et al (1995) Cloning and characterization of Lnk, a signal transduction protein that links T-cell receptor activation signal to phospholipase C1, Grb2, and phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 92:11618–11622
Hunt K, Zhernakova A, Turner G et al (2008) Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet 40(4):395–402
Hurtado C, Erquiaga I, Aranaz P et al (2011) LNK can also be mutated outside PH and SH2 domains in myeloproliferative neoplasms with and without V617FJAK2 mutation. Leuk Res 35(11):1537–1539
Ikram MK, Xueking S, Jensen RA et al (2010) Four novel loci (19q13, 6q24, 12q24, and 5q14) influence the microcirculation in vivo. PLoS Genet 6(10):e1001184
Iseki M, Kubo C, Kwon SM et al (2004) Increased numbers of B-1 cells and enhanced responses against TI-2 antigen in mice lacking APS and adaptor molecule containing PH and SH2 domains. Mol Cell Biol 24:2243–2250
Iseki M, Kubo-Akashi C, Kwon SM et al (2005) APS, an adaptor molecule containing PH and SH2 domains, has a negative regulatory role in B cell proliferation. Biochem. Biophys. Research. Commun. 330:1005–1013
Kamei N, Kwon SM, Alev C et al (2010) Lnk deletion reinforces the function of bone marrow progenitors in promoting neovascularization and astrogliosis following spinal cord injury. Stem Cells. 28(2):365–375
Kent DG, Dykstra BJ, Cheyne J et al (2008) Steel factor coordinately regulates the molecular signature and biologic function of hematopoietic stem cells. Blood 112:560–567
Khwaja A (2006) The role of Janus kinases in haemopoiesis and haematological malignancy. Br J Haematol 134:366–384
Kishi K, Mawatari K, Sakai-Wakamatsu K et al (2007) APS-mediated ubiquitination of the insulin receptor enhances its internalization, but does not induce its degradation. Endocrinology 54(1):77–88
Kubo-Akashi C, Seki M, Kwon SM et al (2004) Roles of a conserved family of adaptor proteins, Lnk, SH2-B, and APS, for mast cell development, growth, and functions: APS-deficiency causes augmented degranulation and reduced actin assembly. Biochem. Biophys. Research. Commun. 315:356–362
Kurzer JH, Argetsinger LS, Zhou YJ et al (2004) Tyrosine 813 is a site of JAK2 autophosphorylation critical for activation of JAK2 by SH2-Bβ. Mol Cell Biol 24(10):4557–4570
Kurzer JH, Saharinen P, Silvennoinen O, Carter-Su C (2006) Binding of SH2-B family members within a potential negative regulatory region maintains JAK2 in an active state. Mol Cell Biol 26(17):6381–6394
Kwon SM, Suzuki T, Kawamoto A et al (2009) Pivotal role of Lnk adaptor protein in endothelial progenitor cell biology for vascular regeneration. Circ Res 104:969–977
Lasho TL, Pardanani A, Tefferi A (2010) LNK mutations in JAK2 mutation-negative erythrocytosis. N Engl J Med 363(12):1189–1190
Lasho TL, Tefferi A, Finke C, Pardanani A (2011) Clonal hierarchy and allelic mutation segregation in a myelofibrosis patient with two distinct LNK mutations. Leukemia. doi:10.1038/leu.2011.45
Lavrikova EY, Nikitin AG, Kuraeva TL et al (2011) The carriage of the type I diabetes-associated R262W variant of human LNK correlates with increased proliferation of peripheral blood monocytes in diabetic patients. Pediatr Diabetes 12(2):127–132
Leevers SJ, Vanhaesebroeck B, Waterfield MD (1999) Signalling through phosphoinositide 3-kinases: the lipids take centre stage. Curr Opin Cell Biol 11:219–225
Li Y, He X, Schembri-King J et al (2000) Cloning and characterization of human Lnk, an adaptor protein with pleckstrin homology and Src homology 2 domains that can inhibit T cell activation. J. Immunol. 164:5199–5206
Li M, Li Z, Morris DL, Rui L (2007) Identification of SH2B2β as an inhibitor for SH2B1- and SH2B2α-promoted Janus kinase-2 activation and insulin signaling. Endocrinology 148:1615–1621
Matsumoto T, Li M, Nishimura H et al (2010) Lnk-dependent axis of SCF-cKit signal for osteogenesis in bone fracture healing. J Exp Med 207:2207–2223
Moodie SA, Alleman-Sposeto J, Gustafson TA (1999) Identification of the APS protein as a novel insulin receptor substrate. J Biol Chem 274:11186–11193
Nelms K, O’Neill TJ, Li S et al (1999) Alternative splicing, gene localization, and binding of SH2-B to the insulin receptor kinase domain. Mamm Genome 10:1160–1167
Nishi M, Werner ED, Oh BC et al (2005) Kinase activation through dimerization by human SH2-B. Mol Cell Biol 25:2607–2621
Nobuhisa I, Takizawa M, Takaki S et al (2003) Regulation of hematopoietic development in the aorta–gonad–mesonephros region mediated by Lnk adaptor protein. Mol Cell Biol 23(23):8486–8494
Oh ST, Simonds EF, Jones C et al (2010) Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood 116(6):988–992
Ohtsuka S, Takaki S, Iseki M et al (2002) SH2-B is required for both male and female reproduction. Mol Cell Biol 22:3066–3077
Ortmann RA, Cheng T, Visconti R et al (2000) Janus kinases and signal transducers and activators of transcription: their roles in cytokine signaling, development and immunoregulation. Arthritis Res 2:16–32
Osborne MA, Dalton S, Kochan JP (1995) The yeast tribrid system: genetic detection of trans-phosphorylated ITAM-SH2-interactions. Biotechnology 13:1474–14781
Pardanani A, Lasho T, Finke C et al (2010) LNK mutation studies in blast-phase myeloproliferative neoplasms, and in chronic-phase disease with TET2, IDH, JAK2 or MPL mutations. Leukemia 24(10):1713–1718
Pawson T, Scott JD (1996) Signaling through scaffold, anchoring, and adaptor proteins. Science 278:2075–2080
Ren D, Li M, Duan C, Rui L (2005) Identification of SH2-B as a key regulator of leptin sensitivity, energy balance, and body weight in mice. Cell Metab 2:95–104
Rider L, Diakonova M (2011) Adapter protein SH2B1β binds filamin A to regulate prolactin-dependent cytoskeletal reorganization and cell motility. Mol Endocrinol 25(7):1231–1243
Rider L, Tao J, Snyder S et al (2009) Adapter protein SH2B1β cross-links actin filaments and regulates actin cytoskeleton. Mol Endocrinol 23(7):1065–1076
Riedel H, Wang J, Hansen H, Yousaf N (1997) PSM, an insulin-dependent, pro-rich, PH, SH2 domain containing partner of the insulin receptor. J Biochem 122:1105–1113
Rudd EC (2001) Lnk adaptor: novel negative regulator of B cell lymphopoiesis. Sci STKE 2001:PE1
Rui L, Mathews LS, Hotta K et al (1997) Identification of SH2-Bβ as a substrate of the tyrosine kinase JAK2 involved in growth hormone signaling. Mol Cell Biol 17(11):6633–6644
Rui L, Gunter DR, Herrington J, Carter-Su C (2000) Differential binding to and regulation of JAK2 by the SH2 domain and N-terminal region of SH2-Bβ. Mol Cell Biol 20(9):3168–3177
Schade AE, Wlodarski MW, Maciejewski JP (2006) Pathophysiology defined by altered signal transduction pathways. the role of JAK-STAT and PI3K signaling in leukemic large granular lymphocytes. Cell Cycle 5(22):2571–2574
Seita J, Ema H, Ooehara J et al (2007) Lnk negatively regulates self-renewal of hematopoietic stem cells by modifying thrombopoietin-mediated signal transduction. Proc. Natl. Acad. Sci. USA 104(7):2349–2354
Simon C, Dondi E, Chaix A et al (2008) Lnk adaptor protein down-regulates specific Kit-induced signaling pathways in primary mast cells. Blood 112:4039–4047
Soranzo N, Spector TD, Mangino M et al (2009) A genome-wide meta-analysis identifies 22 loci associated with eight haematological parameters in the HaemGen consortium. Nat Genet 41(11):1182–1190
Suzuki N, Yamazaki S, Ema H et al (2012): Homeostasis of hematopoietic stem cells regulated by the myeloproliferative disease associated-gene product Lnk/Sh2b3 via Bcl-xL. Exp Hematol 40:166–174.e3
Takaki S, Watts JD, Forbush KA et al (1997) Characterization of Lnk. An adaptor protein expressed in lymphocytes. J Biol Chem 272:14562–14570
Takaki S, Sauer K, Iritani BM et al (2000) Control of B cell production by the adaptor protein Lnk: definition of a conserved family of signal-modulating proteins. Immunity 13:599–609
Takaki S, Morita H, Tezuka Y, Takatsu K (2002) Enhanced hematopoiesis by hematopoietic progenitor cells lacking intracellular adaptor protein, Lnk. J Exp Med. 195:151–160
Takaki S, Tezuka Y, Sauer K et al (2003) Impaired lymphopoiesis and altered B cell subpopulations in mice overexpressing Lnk adaptor protein. J Immunol 170:703–710
Takizawa H, Kubo-Akashi C, Nobuhisa I et al (2006) Enhanced engraftment of hematopoietic stem/progenitor cells by the transient inhibition of an adaptor protein, Lnk. Blood 107(7):2968–2975
Takizawa H, Eto K, Yoshikawa A et al (2008) Growth and maturation of megakaryocytes is regulated by Lnk/SH2B3 adaptor protein through crosstalk between cytokine- and integrin-mediated signals. Exp Hematol 36(7):897–906
Takizawa H, Nishimura S, Takayama N et al (2010) Lnk regulates integrin aIIbβ3 outside-in signaling in mouse platelets, leading to stabilization of thrombus development in vivo. J. Clin. Investig 120(1):179–190
Tefferi A (2010) Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia 24:1128–1138
Tong W, Lodish FH (2004) Lnk inhibits Tpo-mpl signaling and Tpo-mediated megakaryocytopoiesis. J Exp Med 200:569–580
Tong W, Zhang J, Lodish FH (2005) Lnk inhibits erythropoiesis and Epo-dependent JAK2 activation and downstream signaling pathways. Blood 105:4604–4612
Velazquez L, Cheng AM, Fleming HE et al (2002) Cytokine signaling and hematopoietic homeostasis are disrupted in Lnk-deficient mice. J Exp Med 195(12):1599–1611
Wakioka T, Sasaki A, Mitsui K et al (1999) APS, an adaptor protein containing pleckstrin homology (PH) and Src homology-2 (SH2) domains inhibits the JAK-STAT pathway in collaboration with c-Cbl. Leukemia 13:760–767
Wan M, Li Y, Xue H, Li Q, Li J (2006) TNF-α induces Lnk expression through PI3K-dependent signaling pathway in human umbilical vein endothelial cells. J. Surg. Res. 136:53–57
Wang TC, Chiu H, Chang YJ et al (2011) The adaptor protein SH2B3 (Lnk) negatively regulates neurite outgrowth of PC12 cells and cortical neurons. PLoS ONE 6(10):e26433
Ward AC, Touw I, Yoshimura A et al (2000) The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood 95:19–29
Watowich SS, Wu H, Socolovsky M et al (1996) Cytokine receptor signal transduction and the control of hematopoietic cell development. Annu Rev Cell Dev Biol 12:91–128
Werz C, Köhler K, Hafen E, Stocker H (2009) The Drosophila SH2B family adaptor Lnk acts in parallel to Chico in the insulin signaling pathway. PLoS Genet 5(8):e1000596. doi:10.1371/journal.pgen.1000596
Wollberg P, Kennartsson J, Gottridsson E et al (2003) The adapter protein APS associates with the multifunctional docking sites Tyr-568 and Tyr-936 in c-Kit. Biochem J. 370:1033–1038
Yabana N, Shibuya M (2002) Adaptor protein APS binds the NH2-terminal autoinhibitory domain of guanine exchange factor Vav3 and augments its activity. Oncogene 21:7720–7729
Yasukawa H, Sasaki A, Yoshimura A (2000) Negative regulation of cytokine signaling pathways. Annu Rev Immunol 18:143–164
Yokouchi M, Suzuki R, Masuhara M et al (1997) Cloning and characterization of APS, an adaptor molecule containing PH and SH2 domains that is tyrosine phosphorylated upon B-cell receptor stimulation. Oncogene 15:7–15
Yokouchi M, Wakioka T, Sakamoto H et al (1999) APS, an adaptor protein containing PH and SH2 domains, is associated with the PDGF receptor and c-Cbl and inhibits PDGF-induced mitogenesis. Oncogene 18:759–767
Yousaf N, Deng Y, Kang Y, Riedel H (2001) Four PSM/SH2.B alternative splice variants and their differential roles in mitogenesis. J Biol Chem 276:40940–40948
Zhernalova A, Elbers CC, Ferwerda B, Romanos J et al (2010) Evolutionary and functional analysis of celiac risk loci reveals SH2B3 as a protective factor against bacterial infection. Am J Hum Genet 86:970–997
Acknowledgments
I want to thank the members of my laboratory and colleagues from the U978 INSERM research unit for their support and for sharing their data. I apologize for not citing all the articles published on the Lnk family due to space limitation. This work was supported by grants from INSERM and the Fondation ARC pour la Recherche sur le Cancer.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Velazquez, L. The Lnk Adaptor Protein: A Key Regulator of Normal and Pathological Hematopoiesis. Arch. Immunol. Ther. Exp. 60, 415–429 (2012). https://doi.org/10.1007/s00005-012-0194-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-012-0194-x