
Abstract
The idea that vaccination can be used to fight cancer is not new. Approximately 100 years ago, researchers attempted to stimulate a tumor-specific, therapeutic immune response to tumors by injecting patients with cells and extracts from their own tumors, or tumors of the same type from different individuals. During the last decade, great efforts have been made to develop immunotherapeutic approaches for the treatment of malignant diseases as alternatives to traditional chemo- and radiotherapy. A quintessential goal of immunotherapy in cancer is treatment with vaccines that elicit potent anti-tumor immune responses without side effects. In this article, we have attempted to review some of the most problematic issues facing the development of cancer vaccines. With the prospect of immunosuppression, an ill-designed cancer vaccine can be more harmful than a no-benefit therapy. We have noted that “immunoediting” and “immunodominance” are the premier setbacks in peptide-based vaccines and therefore it appears necessary not only to manipulate the activity of a vast number of principal components but also to finely tune their concentrations in time and space. In the face of all these quandaries, it is at least doubtful that any reliable anti-cancer vaccine strategy will emerge in the near future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite multiple advances in preventive and therapeutic approaches, cancer remains one of the major causes of death worldwide. The American Cancer Society has estimated nearly 1.5 million new cases of cancer for 2009 (American Cancer Society 2009). The three standard cancer therapies are surgical resection, chemotherapy, and radiation. Immunotherapy, which includes cancer vaccines, is considered a fourth, still investigational therapy having shown encouraging results in some human clinical trials (Waldmann 2003).
The idea that vaccination can be used to fight cancer is not new. Approximately 100 years ago, researchers attempted to stimulate a tumor-specific, therapeutic immune response to tumors by injecting patients with cells and extracts from their own tumors, or tumors of the same type from other individuals (Paul et al. 2007).
While standard treatments that involve chemotherapy or ionizing radiation have proven effective, they have limitations. Standard approaches can significantly reduce tumor and, in less advanced stages, often result in complete remission. However, the prognosis for advanced tumors has changed little over the past 50 years. Moreover, the majority of cancer-related deaths are caused not by the primary tumor, but by metastases. Side effects are another limitation of chemo- and radiotherapy.
During the last decade, great efforts have been made to develop immunotherapies for the treatment of malignant disease as alternatives to traditional chemo- and radiotherapy (Pardoll 1998). The quintessential goals of immunotherapy in cancer are vaccines that elicit potent anti-tumor immune responses without side effects. The goal is to stimulate both innate and adaptive immunity and cause recognition of tumors with subsequent elimination.
Immunotherapy of cancer continues to be a field of intense research, especially the induction of active immunity. The existence of tumor-specific antigens (TSAs) that can be targets of an immune response is well established (Renkvist et al. 2001). Increasingly, advances in cancer genomics anticipate an ever greater array of potential TSA targets. To date, a variety of immunization preparations and technologies have been employed, including whole tumor cell vaccines, tumor lysate vaccines, specific tumor antigens, tumor peptides, heat shock proteins, DNA vaccines, dexosomes, and dendritic cell (DC)-based vaccines. As a result, several putative tumor vaccines are in clinical trials (Jäger et al. 2002).
Apart from vaccines for specific types of cancer, it is important to consider the overall status and development of cancer vaccines. Here, we intend to delineate the most important issues involved in the development of a potent cancer vaccine.
Cancer Antigens
The effective immunotherapy requires cancer cells to have antigens on their surface that are: (i) unique, distinctly different than on normal cells, (ii) constitutively expressed during the cell cycle and, (iii) their constitutive expression is essential for cell survival. Unfortunately, there is no evidence that cancer-unique components of the plasma membrane that fulfill all three criteria mentioned above are indeed present on cancer cells (Darzynkiewicz 2006).
The identification of relevant TSAs and tumor-associated antigens (TAAs) capable of mediating tumor cell elimination is a central issue in the development of immunotherapeutic strategies. Immunotherapeutic protocols based on vaccination with autologous antigen-presenting cells (APCs) sensitized with TAA have been used for almost a decade in the treatment of metastatic human melanoma (Nestle et al. 1998). Although a large proportion of patients showed a detectable immune response, clinical trials demonstrated only limited evidence of tumor regression.
In recent years, a large number of TSAs and TAAs (see Table 1) have been identified, which can be recognized by T cells. However, the poor reactivity of T cells against them is a major obstacle still to be overcome. Perhaps the greatest hurdle in identifying an immunogenic cancer antigen is that the majority of TAAs and TSAs are self-antigens, which either are over-expressed or vary only in small mutations or altered post-translational modifications (Novellino et al. 2005). Accordingly, these antigens tend to be unreliable in eliciting efficient immunity owing to preexisting tolerance. As a result, T cell receptors (TCRs) often exhibit low affinity for these TAAs, due to prior deletion of T cells with high-affinity TCRs during negative selection in the thymus.
Peptides and Proteins
One of the potential solutions to the problem of TSAs and TAAs is to use heteroclitic peptides. Alterations of the major histocompatibility complex (MHC)-peptide-binding affinity by conservative amino acid modifications at MHC-binding residues may result in a higher capacity to target the wild-type epitopes (Muller et al. 1991) and to reject both newly implanted and established tumors. Although in many instances both in vitro and in vivo heteroclitic analogs often activate T cells better than the native ligand, the modified self-peptides do not always induce T cells that “recognize” naturally processed antigen on tumor cells (Fay et al. 2006). In fact, peptide-induced T cells that “recognize” peptide-pulsed targets, but not tumor cells with naturally processed antigen, have been reported in both murine models and cancer patients (Zaks and Rosenberg 1998).
Even if heteroclitic analogs were successful in provoking an immune response to natural tumor antigen, they would not obviate the hazard of artificially accelerating the purported “immunoediting” phenomenon. Immunoediting describes an early robust immune response that initially arrests tumor growth, but may eventually elicit the development of highly aggressive phenotypes down the road due to selection (Ostrand-Rosenberg 2008). Consistent with this concern, a recent study suggests that a reduction in the expression of NKG2D ligands in colorectal cancer is associated with poorer patient prognosis (McGilvray et al. 2009). This decline in prognosis being apparently due to tumor expression loss of these immunogenic proteins. One answer to this dilemma may be to vaccinate with peptides expressed in the more aggressive phenotypes.
Another approach to avoid potential immunoediting has been to vaccinate with multiple relevant epitopes. For example, vaccines providing the immune system with complete proteins are favored over single peptide-epitope vaccines, since the latter have to be identified per HLA type and may not contain all important epitopes (van der Burg et al. 2006). Whole protein vaccines may overcome obstacles presented by tumors including the immune suppressive milieu (Gorelik and Flavell 2001), cellular heterogeneity, and poor reactivity of T cells for TAAs. Alternatively, whole tumor cell vaccines, administered in the presence of adjuvant and genetically engineered tumor cells that express cytokines (i.e., granulocyte macrophage colony-stimulating factor (GM-CSF) and interleukin-2 (IL-2)), are being studied extensively (Finn 2003). These vaccines have the advantage of expressing relevant TAAs shared by the patient’s cancer cells.
These methods also have their setbacks. Most notably, the majority of antigen cocktails result in immunodominance, a robust immune response to only the most immunogenic epitope of the cocktail. Immunodominance, in turn, reintroduces the peril of immunoediting. In response to this problem, some groups have reported success in overcoming immunodominance by varied sequential vaccinations. For instance, Durántez et al. (2009) report that synthetic peptides followed by complex antigen vaccinations (such as virus or DNA) may surmount the prospect of antigen loss variants.
Immunodominance aside, peptides of highest affinity may not be appropriate in the design of effective anti-tumor vaccines for another reason; as immunogens, these high-affinity mimotopes may be associated with an expansion of cells with defective cytokine profiles and subsequently with a suppression of relevant responding T cells (McMahan et al. 2006). It has also been documented that there is no direct correlation between the higher immunogenic potential of peptide-pulsed DCs with their resultant in vitro tumor-specific cytotoxic T lymphocyte (CTL) response and in vivo functional activity against a growing tumor (Bellone et al. 1999).
Furthermore, several additional obstacles limit the use of peptides in vaccine therapy, such as a short half-life, high elimination rate, susceptibility of enzyme degradation in body fluid, difficulty in passing through biological barriers (immune system tissue, cell membranes, etc.), poor bioavailability through intestinal administration, and the potentially strong side effects of injection. Finally, peptide vaccinations are conflictingly associated with either low memory immunity, or differentiation of naïve CD8 cells toward the memory rather than the effector phenotype (Speiser et al. 2002). One possible approach to overcome this hurdle is to use tumor lysate to pulse DCs.
Carbohydrate Antigens
In addition to TSAs and TAAs, glycosylated antigens may play an important role in carcinogenesis. Their role may be connected with changes in the expression of specific glycosyltransferases that have been documented in several reviews (Cazet et al. 2010; Kaczmarek 2010; Ko et al. 2003). The over-expression of Thomsen-Friedenreich (TF), Tn, sialyl-Tn and Lewisy antigens is associated with disease progression and metastasis, although decreased expression of particular Lewis type glycoconjugates may also occur. In prostate cancer, increased expression of α1,2-fucosylated antigens is associated with α1,2-fucosyltransferases and FUT3. Changes in the expression of Lewisy and Lewisb glycotopes are observed in several tumors as well as breast cancer cells, but depend on the stage of disease. The majority of studies on monoclonal antibodies against sialyl-Lewisa and sialyl-Lewisx preventing metastases have been performed using murine models. Studies on anti-sialyl Lewisy monoclonal antibodies conjugated to antibiotic were also effective in the prevention of metastases in animal models.
The over-expression of carbohydrate TAAs as well as glycosyltransferases could serve as a basis for anti-cancer vaccines. Such structures as Lewisy, TF, sialyl-Tn, GM2, sialyl-GM3 or GD3 including globo H may be considered as antigens in developing cancer vaccines. Unfortunately, the therapeutic potential of such vaccines is complicated by several factors. Possible therapeutic potential lies in the construction of vaccines against fucosyltransferases by RNAi techniques. Suppression of the genes FUT1 and FUT4 inhibits the growth of epidermal tumor in mice (Zhang et al. 2008). Most of the positive results in animal studies have failed to translate to clinical trials. Antibodies against TFα-Keyhole Limpet Hemocyanin (KLH) and the sialyl-Tn-KLH conjugate were developed but did not show clinical efficacy. The reported occurrence of sTn expression in breast cancer is highly variable due to differences in staining protocols, monoclonal antibodies and methods of scoring used (Cazet et al. 2010). Clustered sTn haptens conjugated to KLH have been used to immunize breast cancer patients as at least 25–30% of breast cancers are sTn positive. Anti-cancer vaccines against carbohydrate TAAs have been developed and used in breast cancer treatment. Although this approach was shown to be effective in an animal model, the results of phase III clinical trials failed to show any benefit in patients (Julien et al. 2009). Anti-carbohydrate-Ag vaccines seem to be promising in inducing immunological responses if used in combination with cyclophosphamide and stem cell rescue.
Changes in glycosylation are associated with several enzymes (Cazet et al. 2010). Unfortunately, the resultant heterogeneity lowers the applicability of such vaccines. To overcome this obstacle, better defined target structures and more patient-oriented treatments are needed. Such vaccines may reduce disease relapse and death.
DNA
Naked DNA vaccination is an efficient means of inducing Ag-specific immunity (Gurunathan et al. 2000). It presents several potential advantages when compared with more conventional vaccination strategies: a single vaccine may comprise multiple antigens or chimeric DNA encoding for a fusion protein. Moreover, its large-scale production and storage is cheaper and easier than protein-based vaccines (Gurunathan et al. 2000).
DNA can be administered either alone or in complex with different carriers, and by different routes of vaccination, such as intradermal, subcutaneous, or intramuscular (Gurunathan et al. 2000). Physical administration of DNA by gene gun- or aerosol-based systems appears to elicit consistent responses in mice (Gurunathan et al. 2000). For instance, gene gun delivery of the tyrosinase-related protein (TRP)-2 DNA resulted in activation of a TRP-2-specific CTL response and delayed outgrowth of B16 melanomas, although no animals were cured by such treatment (Tüting et al. 1999).
In fact, a reasonable target organ for DNA vaccination is the skin, where resident professional APCs may take up the plasmid DNA and migrate to lymph nodes (Banchereau and Steinman 1998). Furthermore, subcutaneous administration of naked DNA requires neither pretreatment of the tissue nor gene gun delivery (Gurunathan et al. 1997).
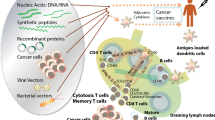
A flow chart summarizing selected cancer vaccine antigens is shown in Fig. 1.

Cancer vaccine antigens. Different types of antigens lead to different immune outcomes and have variable advantages and disadvantages
Antigen-Presenting Cells
An efficient anti-tumor immune response may require not only on antigen quality but also on a proper interaction with APCs. DCs represent unique APCs that are capable of sensitizing T cells to both new and recall antigens. In recent years, several groups have explored the use of DCs loaded in vitro with TAAs as therapeutic vaccines (Fong and Engleman 2000). DC vaccination has produced promising results primarily in patients with immunogenic tumors; with little success observed in tumors such as colorectal and lung cancers.
The problem is more complicated than just poor TAA immunogenicity. DC status also plays a role in processing and presenting peptides. It is generally accepted that DCs have to present the peptide–epitope in the presence of expressed co-stimulatory molecules; otherwise they may induce tolerance in CTL precursors (Carbone et al. 1998) resulting in tumor evasion. Additional problems with DC-based vaccinations include the failure of DCs to migrate from inoculation sites to the draining lymph nodes and the DC suppression associated with Tregs (Khazaie et al. 2009). It has also been reported that natural killer (NK) cells may inhibit antigen presentation by DCs thus suppressing subsequent CTL expansion, at least in tumors of low immunogenic potential such as lymphoma (Barber et al. 2007). One potential mode for this inhibition may be NK-mediated suppression of immature DCs via tumor necrosis factor (TNF) related apoptosis inducing ligands (Hayakawa et al. 2004). Consistent with this thought, elimination of NK cells has been demonstrated to increase “active” tumor-specific CD8+ T cells following immunization (Hayakawa et al. 2004). It is also reported that generation of tumor immunity by DCs correlates with their maturation stage. Due to low or absent expression of co-stimulatory molecules, immature DCs may be involved in inducing peripheral tolerance via T cell anergy (Banchereau and Palucka 2005). Interestingly, artificially matured (with lipopolysaccharide) DCs apparently lose some of this NK sensitivity. In addition, there is evidence that tumor onset and growth is slowed in tumor-challenged NK-depleted mice. Here again, mature DCs appear resistant to NK-mediated lysis (Hayakawa et al. 2004). Further, data suggest that mature DCs exhibit enhanced expression of co-stimulatory molecules and pro-migratory chemokine receptors while producing greater quantities of cytokines. As a result, mature DCs are increasingly used in cancer vaccine protocols; however, the method of maturation may be inconsequential (Han et al. 2009). Accordingly, mature antigen-loaded DCs have been demonstrated to induce peptide-specific CTL CD8 cells (Schuler-Thurner et al. 2000) and have been shown to induce a detectable TH1 cell response (the importance of the TH1 profile is discussed below) (Schuler-Thurner et al. 2002). In addition, mature DCs may also be more appropriate for reactivation of memory CD8 cells, as CD28 co-stimulation has been suggested to be requisite for this function (Borowski et al. 2007). Nevertheless, the advantages of mature DC-based vaccines have been mitigated by reports that mature DCs may expand CD25/CD4 T cell populations and induce peripheral tolerance (Yamazaki et al. 2003).
In addition to the NK inhibitory effect on DC antigen presentation, there are other recipient cell subsets, which may affect the function of APCs such as tumor cells themselves, stromal cells or myeloid derived suppressor cells (MDSCs).
Immunosuppressive features of tumor cells are a major obstacle for cancer immunotherapy. Cancer cells may evade T cell responses either avoiding immune “recognition” or “disabling” effector T cells. These include alterations in antigen presentation and secretion of immunosuppressive and proapoptotic factors. In one study, tumor cells were associated with an expansion of a population of NK1.1+/CD3+ cells, which was accompanied by elimination of mainly CD4+ lymphocytes, and production of a leukocyte-derived inhibitory factor (Banat et al. 2001; Rabinovich et al. 2007).
Researchers found that stromal cells, which form the connective tissue of tumors, may suppress tumor immunity. A stromal cell type that was first identified in human cancers expresses fibroblast activation protein-α (FAP). Depletion of FAP-expressing cells caused rapid hypoxic necrosis of both cancer and stromal cells in immunogenic tumors by a process involving interferon (IFN)-γ and TNF-α. Depleting FAP-expressing cells in a subcutaneous model of pancreatic ductal adenocarcinoma also permitted immunological control of growth. Therefore, FAP-expressing cells indicate that the tumor microenvironment may play an important role in modulating cancer immunotherapy (Kraman et al. 2010).
MDSC accumulate in response to pro-inflammatory mediators. It is thought that they play a role in obstructing immunotherapies that require the host’s cell-mediated and innate immunity. MDSC compete with APC for extracellular cysteine, and in the presence of MDSC, APC release of cysteine is reduced, thereby limiting the extracellular pool of cysteine and depriving T cells of cysteine (Ostrand-Rosenberg 2010; Srivastava et al. 2010). Given the complexity of the conditions that alter MDSC accumulation and the various modes by which MDSC “suppression” may occur, it is essential to understand which conditions and modes are dominant in order that MDSC accumulation and/or activity can be targeted in individual patients to minimize MDSC-associated immune suppression.
A further difficulty is the need to prime appropriate DCs. An inappropriate encounter, for example with dormant DCs or with the wrong subset of DCs, may lead to “silencing” of the immune response (Steinman et al. 2003). This problem may be alleviated to some extent be adding a strong Toll-like receptor (TLR) agonist to the peptide. It has been reported that patients vaccinated with Melan-A peptide and CpG 7909 in incomplete Freund’s adjuvant mounted CTL responses that were 1–3 orders of magnitude higher than in previous trials where this peptide was administered without CpG (Speiser et al. 2002). Thus, inclusion of proper TLR agonists drastically enhances the magnitude of the T cell response and promotes full effector function. Nevertheless, some TLR agonists may enhance tumor-mediated suppression of T cell proliferation and NK activity in vitro and in vivo again resulting in tumor evasion (Huang et al. 2005). However, in contrast to the dubious memorogenicity of peptide-only vaccines, DC-pulsed vaccines have shown the ability to induce peptide-specific memory T cells, though this appears to rely upon the route of vaccination (Banchereau and Palucka 2005).
Finally, using cell types other than APCs to present antigen may result in deletion of specific CD8+ T cells. It has been documented that the presentation of CTL peptide-epitopes on B cells induced a transient CTL response and subsequent deletion of these CD8+ T cells (Bennett et al. 1998).
Cytotoxic T Lymphocytes
CTL responses have been shown to be effective in the elimination of tumors in a number of experimental models (Ochsenbein 2002). In fact, it has been suggested that induced CTLs are most immediately responsible for observed tumor protection (Casares et al. 2001). CTLs can directly lyse tumor cells and also secrete cytokines such as IL-2, TNF, GM-CSF, and IFN-γ, which have direct anti-tumor effects. Additionally, CTLs constitute a major component of tumor-infiltrating lymphocytes. These cells have been associated with spontaneous tumor regression in humans (Zorn and Hercend 1999).
Hence, to generate systemic anti-tumor immune responses in cancer patients, vaccine strategies have focused on the induction of tumor-specific CD8+ CTLs, which recognize peptides of 8–12 residues presented by MHC class I molecules. CD8+ CTLs are the most important effector cells for anti-tumor immune responses and their proper activation leads to the killing of tumor cells (Rosenberg 2001). Although clinical trials in cancer patients have shown that epitope-specific CTLs can be induced in these patients, and that their induction correlated, in multiple instances, with partial or complete tumor responses (Rosenberg et al. 1998), the number of patients experiencing tumor regression has still been disappointingly low disallowing us to call the therapy successful (Parmiani et al. 2002).
The main problem here is to understand the key parameters needed to generate high quality anti-tumor CTLs. Robust anti-tumor CTLs can be developed by using DCs as a cellular adjuvant. It has been documented that the administration of Toxoplasma gondii-matured DCs as a therapeutic vaccine was able to induce pronounced CTL-mediated anti-tumor immunity which led to retarded tumor growth and the prolonged survival of tumor-bearing mice (Motamedi et al. 2009). However, the authors did not characterize the exact component of Toxoplasma which caused this significant effect on DC. According to the findings from different investigations T. gondii heat shock protein 70 is correlated with phenotypic differentiation and maturation of murine bone marrow-derived cells, an effect associated with TLR4 (Banchereau et al. 2000). The use of various exogenous or endogenous stimuli to mature DC followed by induction of robust CTL seems to be an effective approach and worthy of further investigation.
CD4+ T Lymphocytes
CD4+ T cells represent a highly heterogeneous population of cells that develop along different lineages depending upon polarizing cytokine signals during activation by antigens (DeNardo et al. 2010). Classically, CD4+ T lymphocyte subsets include TH1 and TH2 lineages. Through the production of these cytokines, TH1 cells regulate immune surveillance programs. In addition, following strong antigen-specific activation, TH1 cells may directly kill tumor cells by releasing high levels of IFN-γ, TNF-α and cytolytic granules. Thus, when present, TH1 cells may directly and indirectly restrain cancer development. Most recently it has been discovered that CD4+ T cells may not only be important for initial priming of helper-dependent CD8+ T cell responses, but may also play a critical role in programming CD8+ T cell memory development (Nakanishi et al. 2009).
Therefore, in anti-tumor immunotherapy CD4+ T cells are equally as important as CTLs. Accumulating evidence suggests that the CD4+ T cell response plays a key role in tumor immunity. This response is presumably required for expansion and persistence of CD8+ CTLs (Kalams and Walker 1998) and is generally believed to be necessary for the generation of both cellular and humoral anti-tumor immune responses (Toes et al. 1999). In addition, CD4+ T cells mediate the generation of CD8+ T memory cells (Janssen et al. 2003). Further, it has been suggested that the CD4+ T cell response must be particular as the TH1 helper cytokine profile appears most efficient in eliciting a strong CTL response (Casares et al. 2001). Secretion of effector cytokines such as IFN-γ by CD4+ T cells may sensitize tumor cells to CTL lysis via up-regulation of MHC class I molecules, stimulate the innate arm of the immune system at the tumor site, and prevent local angiogenesis (Qin and Blankenstein 2000). The importance of the CD4+ T cell response in tumor immunity was highlighted in murine studies showing that CD4+ T cells can eradicate tumor in the absence of CD8+ T cells (Levitsky et al. 1994). Therefore, the optimal anti-tumor immune response may consist in the concomitant activation of both CD4+ and CD8+ effector T cells.
Adding to this TH1 versus TH2 paradigm, studies of CD4 lineages have recently included a TH17 subset. TH17 cells are thought to play an important role in protection against some extracellular pathogens and in regulating auto-immune disease (Bettelli et al. 2008). As such, TH17 cell infiltration has been observed in patients with ovarian, prostate, and hepatocellular carcinoma where high numbers of IL-17-producing cells correlate with poor prognosis (Miyahara et al. 2008; Sfanos et al. 2008; Zhang et al. 2009). In mouse models of non-small cell lung cancer, IL-17 was associated with enhanced tumor growth and development of angiogenic vasculature (Numasaki et al. 2005). In contrast, in a B16 melanoma model, IL-17 depletion rendered mice more susceptible to metastasis (Martin-Orozco et al. 2009). Together, these experimental findings indicate that the role of TH17 cells in cancer development may also be context dependent and therefore it may be difficult to correlate their presence with outcome. Thus, at this stage of research the induction or repression of TH17 cells by cytokine cocktails or vaccines may cause more harm than benefit to cancer patients.
T Regulatory Cells
One of the basic problems in anti-tumor vaccination is the expansion of Treg cells which have been shown to inhibit specific CTL responses against tumor antigens (Somasundaram et al. 2002) and instead of eliminating tumor cells evoke tumor-related immune tolerance. In general, the capacity of Tregs to suppress effector cell expansion has been attributed to the intracellularly expressed cytotoxic T lymphocyte antigen 4 (CTLA-4, CD152) molecule (Kolar et al. 2009).
TAAs which could otherwise be good targets for anti-cancer immunotherapy, in some cases expand TAA specific Treg cells rather than stimulate effector T cells. Therefore, in the course of immunotherapy it is important to address this issue and strictly monitor the presence of Treg cells, Foxp3+ and effector T cells (IFN-γ+, IL-2+, IL-4+) in tumor and tumor draining lymph nodes. Moreover, inflammation enhances Treg function since prostaglandin E2 mediates differentiation of Tregs and increases their immune suppressive activity (Baratelli et al. 2005). In addition to an associated inhibitory effect on CD8+ T cells, CD4+ Tregs block killing by NK cells (Ghiringhelli et al. 2005) and thereby down-regulate both adaptive and innate anti-tumor immunity. Multiple vaccinations especially with the GM-CSF adjuvant have been shown to suppress immunity via Treg expansion rather than boost it (Eggermont 2009). Accordingly it has been suggested that vaccine/adjuvant administration be combined with a Treg suppression therapy such as anti-CTLA-4 or anti-CD25. Over the past few years, disruption of CD4+ Tregs through CTLA-4 has been a clinical focus of immunotherapy. Preliminary clinical trials of anti-CTLA-4 antibodies in melanoma and Renal Cell Carcinoma (RCC) patients suggest that this approach has a small therapeutic effect (Blansfield et al. 2005).
The presence of other regulatory cell populations in cancer patients has been studied in peripheral blood mononuclear cells from healthy donors and advanced RCC patients. The authors have shown a significantly higher percentage of “functioning” T suppressor cells in RCC patients (Ernstoff et al. 2007). Their contribution to tumor tolerance is still unclear, but they may play a significant role along with the other cell populations. Recent data suggest that DCs made tolerant by CD8+ T suppressor cells have reduced expression of co-stimulatory molecules (Chang et al. 2002). Thus, one approach to overcome CD8+ T cell suppression would be to increase co-stimulatory molecules by maturing DC ex vivo before clinical use.
Route of Vaccination
There are several major routes of vaccination: intramuscular, subcutaneous, intradermal and mucosal (intranasal, gastric, and via the genital tract). Since the main entry sites of most environmental antigens are mucosal surfaces this route of vaccination has drawn much attention. Mucosal vaccination elicits both humoral and cellular immune responses. It has been reported that a mucosal route of immunization is necessary for provision of long-term immunity and CTL-mediated protection against pathogens (Gallichan and Rosenthal 1996). Moreover, mucosally administered vaccines can stimulate serum IgG and mucosal IgA antibodies and induce CTL activity (Levine and Dougan 1998).
However, mucosal vaccination requires the co-administration of a toxin or an inflammatory adjuvant which has precluded their use in humans (Langermann 1996). Recently, oral immunization against 4T1 mammary adenocarcinoma has been reported. Several tumor antigens were expressed on the surface of the T7 phage and shown to trigger specific immune responses in BALB/c mice (Shadidi et al. 2008). If this approach works in other cancer models it may become an effective strategy for mucosal cancer vaccines.
Figure 2 inexhaustibly summarizes cancer vaccine approaches and possible outcomes. Serious consideration of cancer antigens used for vaccination, route of vaccination, in vitro APC loading and immune response modulators is needed to forestall deleterious effects of cancer vaccination.

Cancer vaccine approaches and possible outcome. I.M. Intramuscular, S.C. Subcutaneous, I.D. Intradermal
Adjuvants and Delivery Systems
Adjuvants are substances administered along with an antigen to enhance the humoral and/or cell-mediated immune response to the antigen. More than 100 adjuvant preparations have been described (Vogel and Powell 1995). Adjuvants may have up to five of the following modes of action: the “depot” effect, an antigen presentation effect and an antigen distribution or targeting effect, an immune activation/modulation effect, and a CTL induction effect (Cox and Coulter 1997). In addition to carrying and protecting the antigen from proteolytic destruction, an adjuvant should preserve the conformational integrity of an antigen and present the antigen to the appropriate effector cells, usually professional APCs and macrophages (Stills 2005). Adjuvants containing amphipathic molecules or complexes enhance interaction of the antigen with the cell membrane and promote receptor-mediated endocytosis (Marciani 2003).
The only adjuvants widely employed in human vaccines are aluminum hydroxide and aluminum phosphate. While these have been very useful in augmenting TH2 type humoral responses to bacterial toxoids and other antigens, they have limitations (Allison 1999). The principal limitation of aluminum salts is their inability to elicit cell-mediated immunity, including the CTL response (Gupta et al. 1995), which is important in resistance against some microbial agents and tumors.
The use of particulate adjuvants (e.g. emulsions, microparticles, iscoms, liposomes) as alternatives to immunostimulatory adjuvants has been evaluated by many groups. Immunostimulatory adjuvants may also be included in delivery systems to enhance the level of response, or to focus the response. In addition, formulating potent immunostimulatory adjuvants into delivery systems may limit adverse events by restricting the systemic circulation of the adjuvant (Vajdy et al. 2004).
Liposomes and liposome-like emulsions are good candidates for artificial adjuvants. Martinon and his collaborators (Martinon et al. 1993) have demonstrated that priming virus-specific CTLs in mice immunized with liposome-encapsulated RNA corresponding to the influenza nucleoprotein elicited effective immune protection. Our studies have also confirmed that nanoemulsions are good adjuvants for mucosal vaccines (Bielinska et al. 2008; Myc et al. 2003).
GM-CSF has been another adjuvant employed in some clinical trials. Among other effects, it has been thought that GM-CSF would promote the recruitment and maturation of DCs, and thus enhance the immune response to a given vaccination (Zarei et al. 2009). With this expectation, GM-CSF has been employed as an adjuvant either as the product of transfected whole tumor cells or as a recombinant protein. In fact, early results with autologous peptide vaccines administered with GM-CSF were promising and reportedly immunogenic (Disis et al. 1996). In retrospect, one of the shortfalls of this work and of subsequent clinical trials has been the use of surrogate endpoints in assessing immunostimulation rather than tumor regression or survival (Eggermont 2009). Additionally, the ability of GM-CSF to expand MDSCs which in turn activate Tregs has been noted, this immunosuppressive action being attributed to higher doses of GM-CSF (Parmiani et al. 2007). Recently, these dangers have been realized, bringing GM-CSF under much scrutiny and criticism for its apparent worse overall survival rates versus controls (Eggermont 2009).
Another adjuvant strategy involves the co-administration of α-GalCer with ovalbumin (OVA) to induce full maturation of DCs. This generates functional Ag-specific TH1 CD4+ T cells and CTLs that are resistant to OVA-expressing tumors (Fujii et al. 2004).
In closing, the search for new vectors for direct transfer of TAA proteins or genes to APCs and redirection of the response from tumor tolerance to tumor cytotoxicity is still open and investigated worldwide (Schlom et al. 1999).
The Combination of Vaccinations with Other Conventional Therapies
Because of the widespread use of chemotherapy for the treatment of most malignancies, it is rational to design combinatorial approaches using vaccines alongside standard chemotherapeutic agents. Like radiotherapy, the use of various types of chemotherapy in combination with vaccines has resulted in enhanced anti-tumor immune responses. In general, anti-cancer drugs can kill tumor cells, leading to DC antigen “presentation” to T cells (Tesniere et al. 2010), alternatively they can modulate the phenotype of the tumor cells making them more susceptible to immune-mediated killing. For example, it has been shown that treatment of human colon carcinoma cell lines with 5-fluorouracil or cisplatin enhances their lytic sensitivity to antigen-specific CD8+ cytotoxic T lymphocytes, being associated with an increased expression of intercellular adhesion molecule 1 and Fas (Bergmann-Leitner and Abrams 2001). Although it has been demonstrated that the use of chemotherapeutic agents can enhance anti-tumor responses when used in combination with cancer vaccine modalities, further studies are necessary to investigate optimum schedules and dosing for various chemotherapeutic agents in combination with cancer vaccines (Palena and Schlom 2010).
Conclusions
In this article we have attempted to review some of the most problematic issues facing the development of cancer vaccines. It is critical to be reminded that with the prospect of immunosuppression an ill-designed cancer vaccine can be more harmful than a no-benefit therapy. We have noted that immunoediting and immunodominance are the premier setbacks in peptide-based vaccines. Augmenting these concerns is the Treg mediated immune suppression observed with both peptide and adjuvant approaches. Further complicating these intricacies is the immunocompromised status of most cancer patients, especially those that are the subjects of enrollment in experimental clinical trials. It is also unlikely that a single approach will be broadly applicable to all or even multiple cancer types as each possesses a unique and variable array of targetable antigens. Ultimately, in order to recruit the immune “network” to fight against cancer it appears necessary not only to manipulate the activity of a vast number of principal components (i.e. cells, cytokines, chemokines, expression of antigens on immunocompetent cells, etc.) but also to finely tune their concentrations in time and space. In this labyrinthine milieu it is at least doubtful that any reliable anti-cancer vaccine strategy will emerge in the near future.
References
Allison AC (1999) Squalene and squalane emulsions as adjuvants. Methods 19:87–93
American Cancer Society (2009) Cancer, Facts & Figures 2009. http://www.cancer.org/downloads/STT/500809web.pdf
Banat GA, Christ O, Cochlovius B et al (2001) Tumour-induced suppression of immune response and its correction. Cancer Immunol Immunother 49:573–586
Banchereau J, Palucka AK (2005) Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol 5:296–306
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392:245–252
Banchereau J, Briere F, Caux C et al (2000) Immunobiology of dendritic cells. Annu Rev Immunol 18:767–811
Baratelli F, Lin Y et al (2005) Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J Immunol 175:1483–1490
Barber MA, Zhang T, Gagne BA et al (2007) NK cells negatively regulate antigen presentation and tumor-specific CTLs in a syngeneic lymphoma model. J Immunol 178:6140–6147
Bellone M, Iezzi G, Imro MA et al (1999) Cancer immunotherapy: synthetic and natural peptides in the balance. Immunol Today 20:457–462
Bennett SR, Carbone FR, Toy T et al (1998) B cells directly tolerize CD8(+) T cells. J Exp Med 188:1977–1983
Bergmann-Leitner ES, Abrams SI (2001) Treatment of human colon carcinoma cell lines with anti-neoplastic agents enhances their lytic sensitivity to antigen-specific CD8+ cytotoxic T lymphocytes. Cancer Immunol Immunother 50:445–455
Bettelli E, Korn T, Oukka M et al (2008) Induction and effector functions of T(H)17 cells. Nature 453:1051–1057
Bielinska AU, Janczak KW, Landers JJ et al (2008) Nasal immunization with a recombinant HIV gp120 and nanoemulsion adjuvant produces Th1 polarized responses and neutralizing antibodies to primary HIV type 1 isolates. AIDS Res Hum Retrovir 24:271–281
Blansfield JA, Beck KE, Tran K et al (2005) Cytotoxic T-lymphocyte-associated antigen-4 blockage can induce autoimmune hypophysitis in patients with metastatic melanoma and renal cancer. J Immunother 28:593–598
Borowski AB, Boesteanu AC, Mueller YM et al (2007) Memory CD8+ T cells require CD28 costimulation. J Immunol 179:6494–6503
Carbone FR, Kurts C, Bennett SR et al (1998) Cross-presentation: a general mechanism for CTL immunity and tolerance. Immunol Today 19:368–373
Casares N, Lasarte JJ, de Cerio AL et al (2001) Immunization with a tumor-associated CTL epitope plus a tumor-related or unrelated Th1 helper peptide elicits protective CTL immunity. Eur J Immunol 31:1780–1789
Cazet A, Julien S, Bobowski M et al (2010) Consequences of the expression of sialylated antigens in breast cancer. Carbohydr Res 345:1377–1383
Chang CC, Ciubotariu R, Manavalan JS et al (2002) Tolerization of dendritic cells by T(S) cells: the crucial role of inhibitory receptors ILT3 and ILT4. Nat Immunol 3:237–243
Cox JC, Coulter AR (1997) Adjuvants—a classification and review of their modes of action. Vaccine 15:248–256
Darzynkiewicz Z (2006) Will cancer immunotherapy fail? Scientist 20:14 (letter to editor)
DeNardo DG, Andreu P, Coussens LM (2010) Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev 29:309–316
Disis ML, Bernhard H, Shiota FM et al (1996) Granulocyte–macrophage colony-stimulating factor: an effective adjuvant for protein and peptide-based vaccines. Blood 88:202–210
Durántez M, López-Vázquez AB, de Cerio AL et al (2009) Induction of multiepitopic and long-lasting immune responses against tumour antigens by immunization with peptides, DNA and recombinant adenoviruses expressing minigenes. Scand J Immunol 69:80–89
Eggermont AM (2009) Immunostimulation versus immunosuppression after multiple vaccinations: the woes of therapeutic vaccine development. Clin Cancer Res 15:6745–6747
Ernstoff MS, Crocenzi TS, Seigne JD et al (2007) Developing a rational tumor vaccine therapy for renal cell carcinoma: immune yin and yang. Clin Cancer Res 13(2 Pt 2):733s–740s
Fay JW, Palucka AK, Paczesny S et al (2006) Long-term outcomes in patients with metastatic melanoma vaccinated with melanoma peptide-pulsed CD34(+) progenitor-derived dendritic cells. Cancer Immunol Immunother 55:1209–1218
Finn OJ (2003) Cancer vaccines: between the idea and the reality. Nat Rev Immunol 3:630–641
Fong L, Engleman EG (2000) Dendritic cells in cancer immunotherapy. Annu Rev Immunol 18:245–273
Fujii S, Liu K, Smith C et al (2004) The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med 199:1607–1618
Gallichan WS, Rosenthal KL (1996) Long-lived cytotoxic T lymphocyte memory in mucosal tissues after mucosal but not systemic immunization. J Exp Med 184:1879–1890
Ghiringhelli F, Ménard C, Terme M et al (2005) CD4+ CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med 202:1075–1085
Gorelik L, Flavell RA (2001) Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med 7:1118–1122
Gupta RK, Rost BE, Relyveld E et al (1995) Adjuvant properties of aluminium and calcium compounds. In: Powell MF, Newman MJ (eds) Vaccine design: the subunit and vaccine approach. Plenum, New York, pp 229–248
Gurunathan S, Sacks DL, Brown DR et al (1997) Vaccination with DNA encoding the immunodominant LACK parasite antigen confers protective immunity to mice infected with Leishmania major. J Exp Med 186:1137–1147
Gurunathan S, Klinman DM, Seder RA (2000) DNA vaccines: immunology, application, and optimization. Annu Rev Immunol 18:927–974
Han TH, Jin P, Ren J et al (2009) Evaluation of 3 clinical dendritic cell maturation protocols containing lipopolysaccharide and interferon-gamma. J Immunother 32:399–407
Hayakawa Y, Screpanti V, Yagita H et al (2004) NK cell TRAIL eliminates immature dendritic cells in vivo and limits dendritic cell vaccination efficacy. J Immunol 172:123–129
Huang B, Zhao J, Li H et al (2005) Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res 65:5009–5014
Jäger E, Jäger D, Knuth A (2002) Clinical cancer vaccine trials. Curr Opin Immunol 14:178–182
Janssen EM, Lemmens EE, Wolfe T et al (2003) CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 421:852–856
Julien S, Picco G, Sewell R et al (2009) Sialyl-Tn vaccine induces antibody-mediated tumour protection in a relevant murine model. Br J Cancer 100:1746–1754
Kaczmarek R (2010) Alterations of Lewis histo-blood group antigen expression in cancer cells. Postepy Hig Med Dosw 64:87–99
Kalams SA, Walker BD (1998) The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J Exp Med 188:2199–2204
Khazaie K, Bonertz A, Beckhove P (2009) Current developments with peptide-based human tumor vaccines. Curr Opin Oncol 21:524–530
Ko BK, Kawano K, Murray JL et al (2003) Clinical studies of vaccines targeting breast cancer. Clin Cancer Res 9:3222–3234
Kolar P, Knieke K, Hegel JK et al (2009) CTLA-4 (CD152) controls homeostasis and suppressive capacity of regulatory T cells in mice. Arthritis Rheum 60:123–132
Kraman M, Bambrough PJ, Arnold JN et al (2010) Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 330:827–830
Langermann S (1996) New approaches to mucosal immunization. Semin Gastrointest Dis 7:12–18
Levine MM, Dougan G (1998) Optimism over vaccines administered via mucosal surfaces. Lancet 351:1375–1376
Levitsky HI, Lazenby A, Hayashi RJ et al (1994) In vivo priming of two distinct antitumor effector populations: the role of MHC class I expression. J Exp Med 179:1215–1224
Marciani DJ (2003) Vaccine adjuvants: role and mechanisms of action in vaccine immunogenicity. Drug Discov Today 8:934–943
Martinon F, Krishnan S, Lenzen G et al (1993) Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur J Immunol 23:1719–1722
Martin-Orozco N, Muranski P, Chung Y et al (2009) T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity 31:787–798
McGilvray RW, Eagle RA, Watson NF et al (2009) NKG2D ligand expression in human colorectal cancer reveals associations with prognosis and evidence for immunoediting. Clin Cancer Res 15:6993–7002
McMahan RH, McWilliams JA, Jordan KR et al (2006) Relating TCR-peptide-MHC affinity to immunogenicity for the design of tumor vaccines. J Clin Invest 116:2543–2551
Miyahara Y, Odunsi K, Chen W et al (2008) Generation and regulation of human CD4+ IL-17-producing T cells in ovarian cancer. Proc Natl Acad Sci USA 105:15505–15510
Motamedi M, Arab S, Moazzeni SM et al (2009) Improvement of a dendritic cell-based therapeutic cancer vaccine with components of Toxoplasma gondii. Clin Vaccine Immunol 16:1393–1398
Muller D, Pederson K, Murray R et al (1991) A single amino acid substitution in an MHC class I molecule allows heteroclitic recognition by lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes. J Immunol 147:1392–1397
Myc A, Kukowska-Latallo JF, Bielinska AU et al (2003) Development of immune response that protects mice from viral pneumonitis after a single intranasal immunization with influenza A virus and nanoemulsion. Vaccine 21:3801–3814
Nakanishi Y, Lu B, Gerard C et al (2009) CD8(+) T lymphocyte mobilization to virus-infected tissue requires CD4(+) T-cell help. Nature 462:510–513
Nestle FO, Alijagic S, Gilliet M et al (1998) Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med 4:328–332 [See comment]
Novellino L, Castelli C, Parmiani G (2005) A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol Immunother 54:187–207
Numasaki M, Watanabe M, Suzuki T et al (2005) IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J Immunol 175:6177–6189
Ochsenbein AF (2002) Principles of tumor immunosurveillance and implications for immunotherapy. Cancer Gene Ther 9:1043–1055
Ostrand-Rosenberg S (2008) Immune surveillance: a balance between protumor and antitumor immunity. Curr Opin Genet Dev 18:11–18
Ostrand-Rosenberg S (2010) Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother 59:1593–1600
Palena C, Schlom J (2010) Vaccines against human carcinomas: strategies to improve antitumor immune responses. J Biomed Biotechnol 2010:380697
Pardoll DM (1998) Cancer vaccines. Nat Med 4(5 suppl):525–531
Parmiani G, Castelli C, Dalerba P et al (2002) Cancer immunotherapy with peptide-based vaccines: what have we achieved? Where are we going? J Natl Cancer Inst 94:805–818
Parmiani G, Castelli C, Pilla L et al (2007) Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann Oncol 18:226–232
Paul S, Acres B, Limacher JM et al (2007) Cancer vaccines: challenges and outlook in the field. IDrugs 10:324–328
Qin Z, Blankenstein T (2000) CD4+ T cell-mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity 12:677–686
Rabinovich GA, Gabrilovich D, Sotomayor EM (2007) Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol 25:267–296
Renkvist N, Castelli C, Robbins PF et al (2001) A listing of human tumor antigens recognized by T cells. Cancer Immunol Immunother 50:3–15
Rosenberg SA (2001) Progress in human tumour immunology and immunotherapy. Nature 411:380–384
Rosenberg SA, Yang JC, Schwartzentruber DJ et al (1998) Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med 4:321–327 [See comment]
Schlom J, Tsang KY, Kantor JA et al (1999) Strategies in the development of recombinant vaccines for colon cancer. Semin Oncol 26:672–682
Schuler-Thurner B, Dieckmann D, Keikavoussi P et al (2000) Mage-3 and influenza-matrix peptide-specific cytotoxic T cells are inducible in terminal stage HLA-A2.1 + melanoma patients by mature monocyte-derived dendritic cells. J Immunol 165:3492–3496
Schuler-Thurner B, Schultz ES, Berger TG et al (2002) Rapid induction of tumor-specific type 1 T helper cells in metastatic melanoma patients by vaccination with mature, cryopreserved, peptide-loaded monocyte-derived dendritic cells. J Exp Med 195:1279–1288
Sfanos KS, Bruno TC, Maris CH et al (2008) Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin Cancer Res 14:3254–3261
Shadidi M, Sorensen D, Dybwad A et al (2008) Mucosal vaccination with phage-displayed tumour antigens identified through proteomics-based strategy inhibits the growth and metastasis of 4T1 breast adenocarcinoma. Int J Oncol 32:241–247
Somasundaram R, Jacob L, Swoboda R et al (2002) Inhibition of cytolytic T lymphocyte proliferation by autologous CD4+/CD25+ regulatory T cells in a colorectal carcinoma patient is mediated by transforming growth factor-beta. Cancer Res 62:5267–5272
Speiser DE, Liénard D, Pittet MJ et al (2002) In vivo activation of melanoma-specific CD8(+) T cells by endogenous tumor antigen and peptide vaccines. A comparison to virus-specific T cells. Eur J Immunol 32:731–741
Srivastava MK, Sinha P, Clements VK et al (2010) Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res 70:68–77
Steinman RM, Hawiger D, Nussenzweig MC (2003) Tolerogenic dendritic cells. Annu Rev Immunol 21:685–711
Stills HF Jr (2005) Adjuvants and antibody production: dispelling the myths associated with Freund’s complete and other adjuvants. ILAR J 46:280–293
Tesniere A, Schlemmer F, Boige V et al (2010) Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 29:482–491
Toes RE, Ossendorp F, Offringa R et al (1999) CD4 T cells and their role in antitumor immune responses. J Exp Med 189:753–756
Tüting T, Gambotto A, DeLeo A et al (1999) Induction of tumor antigen-specific immunity using plasmid DNA immunization in mice. Cancer Gene Ther 6:73–80
Vajdy M, Srivastava I, Polo J et al (2004) Mucosal adjuvants and delivery systems for protein-, DNA- and RNA-based vaccines. Immunol Cell Biol 82:617–627
van der Burg SH, Bijker MS, Welters MJ et al (2006) Improved peptide vaccine strategies, creating synthetic artificial infections to maximize immune efficacy. Adv Drug Deliv Rev 58:916–930
Vogel FR, Powell MF (1995) A compendium of vaccine adjuvants and excipients. Pharm Biotechnol 6:141–228
Waldmann TA (2003) Immunotherapy: past, present and future. Nat Med 9:269–277
Yamazaki S, Iyoda T, Tarbell K et al (2003) Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J Exp Med 198:235–247
Zaks TZ, Rosenberg SA (1998) Immunization with a peptide epitope (p369–377) from HER-2/neu leads to peptide-specific cytotoxic T lymphocytes that fail to recognize HER-2/neu + tumors. Cancer Res 58:4902–4908
Zarei S, Schwenter F, Luy P et al (2009) Role of GM-CSF signaling in cell-based tumor immunization. Blood 113:6658–6668
Zhang Z, Sun P, Liu J et al (2008) Suppression of FUT1/FUT4 expression by siRNA inhibits tumor growth. Biochim Biophys Acta 1783:287–296
Zhang JP, Yan J, Xu J et al (2009) Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J Hepatol 50:980–989
Zorn E, Hercend T (1999) A MAGE-6-encoded peptide is recognized by expanded lymphocytes infiltrating a spontaneously regressing human primary melanoma lesion. Eur J Immunol 29:602–607
Acknowledgments
We gratefully acknowledge Dr. James R. Baker, Jr. for review and critical remarks of the manuscript. We also would like to acknowledge financial support from the University of Michigan Comprehensive Cancer Center Cancer Research Committee.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Myc, L.A., Gamian, A. & Myc, A. Cancer Vaccines. Any Future?. Arch. Immunol. Ther. Exp. 59, 249–259 (2011). https://doi.org/10.1007/s00005-011-0129-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-011-0129-y