
Abstract
The colonization of humans with commensals is critical for our well-being. This tightly regulated symbiotic relationship depends on the flora and an intact mucosal immune system. A disturbance of either compound can cause intestinal inflammation. This review summarizes extrinsic and intrinsic factors contributing to intestinal dysbiosis and inflammatory bowel disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As the largest epithelial surface colonized with trillions of microbes the human intestine is permanently challenged by controversial tasks. At the same time, it has to tolerate symbionts, control facultative pathogens and fight obligate pathogens in order to maintain intestinal homeostasis. A disturbance of intestinal homeostasis results in intestinal dysbiosis which is a hallmark of inflammatory bowel diseases (IBD) including Crohns’s disease (CD) and ulcerative colitis (UC) characterized by episodic, relapsing or continuous inflammatory processes affecting distinct layers and sections of the intestinal wall (Kaser et al. 2010).
Twin studies have shown a concordance rate among monozygotic twins of about 6% for UC and of about 58% for CD indicating pathogenic roles of both intrinsic and extrinsic factors (Tysk et al. 1988). On the one hand, genome-wide association studies (GWAS) have identified single-nucleotide polymorphisms (SNPs) in genes such as nucleotide-binding oligomerization domain containing 2 (NOD2) and interleukin 23 receptor (IL-23R) representing innate and adaptive immunity, respectively. On the other hand, molecular analysis of the gut microbiota has revealed a higher bacterial diversity in samples from healthy individuals compared to samples from Crohn’s patients. In addition, monocygotic healthy twins display a more similar microbiota than monocygotic twins who are discordant for CD (Dicksved et al. 2008). Thus, IBD seems to result from inflammatory immune responses triggered by intestinal dysbiosis both of which are favoured by environmental factors and a genetic susceptibility of the host (Fig. 1).

Extrinsic and intrinsic factors trigger intestinal dysbiosis and inflammation. A crosstalk between environmental factors (e.g. stress, smoking, antibiotic treatment and mode of delivery), compounds of the gut flora with protective or colitogenic effects, genetic susceptibility and cellular as well as humoral compounds of the immune system determine the extent of intestinal dysbiosis and intestinal inflammation both of which enhance each other. Escherichia coli (E. coli), Faecalibacterium prausnitzii (F. prausnitzii), Saccharomyces boulardii (S. boulardii), small-chain-fatty-acids (SCFA), segmented filamentous bacteria (SFB), adenosine triphosphate (ATP), adherent-invasive Escherichia coli (AIEC), nucleotide-oligomerisation domain 2 (NOD2), adipose triglyceride lipase 16 (ATGL16), interleukin 23 receptor (IL-23R), chemokine receptor (CCR), NACHT, LRR and PYD domains-containing protein 3 (NLRP3), Toll-like receptor 4 (TLR4), caspase-recruitment domain 9 (CARD9), immunity-related GTPase family M (IRGM), x-box-binding protein 1 (XBP1), inducible co-stimulator ligand (ICOSL), peroxisome proliferator-activated receptor γ (PPAR-γ), pathogen recognition receptor (PRR), dendritic cells (DCs), non-steroidal anti-inflammatory drugs (NSAID)
The importance of the enteric flora for the development of IBD has hence been acknowledged. In this review we will discuss the role of the enteric flora in (1) maintaining the intestinal barrier, (2) interacting with distinct intestinal epithelial cells (IECs), (3) becoming recognized by intestinal antigen uptake pathways, (4) inducing tolerogenic immune responses, (5) mediating resistance towards pathogens (6) initiating destructive inflammatory immune responses and (7) being manipulated for the treatment of IBD.
Enteric Flora Maintains Intestinal Homeostasis
Comprising more than 400 m2 the intestinal tract is colonized by the highest bacterial burden with approximately 100 trillion individual microorganisms (Macdonald and Monteleone 2005). The bacterial concentration increases along the intestinal tract with 104 cells/g of luminal contents in stomach and duodenum up to a density of 1011 to 1014 cells/g in the large intestine (Whitman et al. 1998). In total, the number of intestinal bacteria is approximately ten times the number of cells constituting the human body.
Based on 16S rRNA sequencing, it was possible to identify up to 40,000 bacterial species in the intestine including non-culturable bacteria (Frank and Pace 2008). Phylotype census showed that 99% of intestinal bacteria are constituted by four phyla including Proteobacteria, actinobacteria and the two major phyla cytophaga-flavobacterium-bacteroides and Firmicutes (Manson et al. 2008). While Bacteroidetes species show a large variety between subjects, a high number of Firmicutes species belong to butyrate-producing clostridial clusters (Eckburg et al. 2005). The collective genome, also referred to as microbiome, contains 100-fold more genes than the entire human genome (Tsai and Coyle 2009). Accordingly, a tightly regulated relationship has evolved between the human intestine and its microbiota. If this relationship is of gain for both partners it is referred to as symbiosis whereas a commensal relationship is characterized by a benefit for one and neither gain or loss for the other partner and a parasitic relationship is beneficial for one but harmful for the other partner. The human intestine aims for a symbiotic relationship which plays a fundamental role in human homeostasis including growth, metabolism and immunity. Experiments with germ-free (GF), gnotobiotic, conventional (CV) and conventionalized animals have opened new ways to analyse this interaction.
It has been demonstrated that 1–20% of the indispensable amino acids lysine and threonine in human plasma are produced by the gut flora and intestinal bacteria are an important source of essential vitamins, such as vitamin K and group B vitamins as well as minerals like iron and copper (Metges 2000; Reddy et al. 1965). Furthermore, GF animals need 30% more calories than CV animals to keep a stable weight approving an important role of bacterial enzymes especially in the digestion of complex polysaccharides (Wostmann et al. 1983). This finding explains a dramatic enlargement of the cecum in GF mice which is mainly caused by an accumulation of undegraded mucus (Gustafsson et al. 1970). The comparison of GF and CV rats has also revealed differences in intestinal motility. GF rats have a delayed gastric emptying and a prolonged intestinal transit caused by a slower spread of migrating motor complexes (Husebye et al. 1994). There is also accumulating evidence for immunomodulatory functions of microbial metabolites including short-chain fatty acids (SCFA) such as butyrate and products from the citric cycle such as succinate. Upon binding to the G-protein coupled receptor GPR109A on IECs, butyrate dampens immune responses by suppressing NFκB activation via neddylation of cullin-1 in IECs (Collier-Hyams et al. 2005). GPR43 is another receptor of SCFA. Similar to GF mice, GPR43 deficient animals suffer from exacerbated inflammatory responses in models of asthma, arthritis and colitis indicating a protective role of SCFA (Maslowski et al. 2009). Succinate binds to GPR91 on dendritic cells (DCs) and induces production of proinflammatory cytokines improving antigen-specific T cell responses (Rubic et al. 2008). Hence, antigen-specific T cell responses are impaired in GPR91 deficient mice.
The Mucosal Barrier Prevents the Translocation of the Flora
The intestinal epithelium consists of four different types of polarized IECs: absorptive enterocytes, mucus producing goblet cells, enteroendocrine cells and Paneth cells (Table 1). The renewal of IECs starts from multipotent stem cells in mucosal invaginations, called crypts. It has been estimated that the human intestine produces 50 × 106 cells/day which equates to approximately 250 g of cells (Croft and Cotton 1973). Accordingly, the self-renewal of the intestinal epithelium takes about 5 days. While enterocytes, goblet cells and enteroendocrine cells migrate upward from the crypt to the villus tip, Paneth cells migrate downward to the base of the crypt. Moreover, Paneth cells predominantly exist in the terminal ileum and have a prolonged lifetime of about 20 days (Bjerknes and Cheng 1981a, b). The development of IECs depends on the presence of the enteric flora. GF mice show a delayed expression of lysozyme in Paneth cells and the induction of fucosyltransferase as well as fucosylated glycoconjugates are decreased in IECs from GF animals (Falk et al. 1998). The analysis of intestinal angiogenesis has shown an impaired development of intestinal capillary networks and Paneth cells in the absence of microbiota (Stappenbeck et al. 2002). The integrity of the intestinal tract is based on an intact epithelial layer which is often endangered by invading pathogens (Sakaguchi et al. 2002). Consequently, the intestine is challenged by fighting pathogenic and concurrently cooperating with symbiotic microbes in order to maintain homeostasis. To fulfil this complex task distinct strategies have evolved.
Globlet Cells and the Mucus Layer
Goblet cells produce large amounts of highly glycosylated proteins forming a two layered mucus that extends up to 150 μm (Johansson et al. 2008). While the inner layer contains antimicrobial peptides (AMPs) and is attached to the epithelium, the outer membrane is detachable, colonized by bacteria and its volume is enlarged by proteolytic cleavage of Muc2 mucin. Notably, Muc2 deficient animals suffer from intestinal inflammation and tumorigenesis underlining the protective function of the mucin layer which reduces microbial adherence to IECs. In addition, goblet cells secret the protease-resistant intestinal trefoil factor (ITF) which is supposed to attenuate intestinal injuries by protecting IECs from apoptosis and promoting epithelial cell migration (Dignass et al. 1994; Taupin et al. 2000). Accordingly, ITF-deficient mice are characterized by impaired wound healing and accelerated colitis after challenge with dextran sodium sulfate (DSS) (Mashimo et al. 1996). Moreover, defects in globlet cell differentiation have been associated with Crohn’s disease (CD) (Gersemann et al. 2009).
Paneth Cells and Autophagy
Paneth cells are critical for the maintainance of the intestinal barrier by producing zinc and AMPs such as lysozyme, RegIIIγ, cathelicidin and defensins. The production of AMPs depends on the activation of myeloid-differentiation factor 88 (MyD88)-signalling pathways in Paneth cells (Vaishnava et al. 2008). In line, genetic ablation of Paneth cells which express high levels of Toll-like receptors (TLRs) results in an enhanced translocation of intestinal bacteria causing systemic infections. Moreover, Paneth cell function depends on autophagy-related pathways. Autophagy displays a highly conserved catabolic process which is triggered by cell stress signals such as hypoxia, nutrient deprivation or growth factor depletion. The autophagosome is specialized for the recycling of amino acids by degrading defective proteins (Levine and Kroemer 2008). In addition, autophagosomal enzymes enhance immunity by destroying intracellular pathogens such as Mycobacterium tuberculosis and Streptococcus pyogenes (Gutierrez et al. 2004; Nakagawa et al. 2004). In this context, successful antigen presentation via major histocompatibility complex (MHC) class II molecules and concomitant induction of CD4+ T cell responses depend on detection of intracellular pathogens by intracellular receptors such as NOD2 and consecutive activation of the autophagosome (Cooney et al. 2010). Interestingly, not only NOD2 mutations but also mutations in the autophagy-related gene ATGL16 are significantly associated with CD and thereby provide strong evidence for a role of autophagy in the pathogenesis of IBD (Cadwell et al. 2008).
Sensing of the Microbiota by Epithelial Cells
IECs are equipped with pathogen recognition receptors (PRRs) such as TLRs, NLRs, c-type lectin receptors and retinoid acid inducible gene-I like receptors detecting pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs). TLRs consist of highly conserved transmembrane receptors expressed on the cell membrane and on endosomal compartments (Takeda et al. 2003). TLRs 1, 2, 4, 5, 6 form homo- and heterodimers on the cell membrane and bind bacterial cell wall compounds such as lipoteichoic acid (TLR-2/6), lipopolysaccharide (LPS; TLR-4) and flagellin (TLR-5) whereas homodimers of TLRs 3, 7, 8, 9 are expressed on endosomal membranes and sense nucleic acids such as single-stranded ribonucleic acid (ss-RNA; TLR-3), double-stranded RNA (TLR-7,8) and CpG-rich deoxyribonucleic acid (double-stranded deoxyribonucleic acid; TLR-9). NLRs are cytoplasmic receptors including the NALP-, NAIP- and NOD-families which detect microbial compounds such as bacterial DNA as well as DAMPs like uric acid (Meylan et al. 2006). NOD2 binds peptidoglycan and subsequently induces secretion of proinflammatory cytokines via activation of the NFκB pathway. About 15% of all CD patients carry homozygous or compound heterozygous mutations in the NOD2 gene causing an impaired NOD2 function and a reduced antimicrobial response as possible pathomechanism (Clayton et al. 2005; Hugot et al. 2001). Since symbiotic and pathogenic microbes express overlapping PAMPs, PRRs can hardly distinguish between pathogens and symbionts. In this context, compartmentalization and strength of PRR expression may help to avoid excessive inflammatory responses to the commensals. IECs express high levels of Toll-inhibitor-protein (Tollip) but relative low levels of TLR-2 resulting in an unresponsiveness to corresponding ligands such as peptidoglycan or lipoteichoic acid (Melmed et al. 2003). In addition, results from IEC lines demonstrate a decreased expression of TLR-4 and its co-receptor CD14/MD2 (Abreu et al. 2001). By contrast, TLR-5 is highly but exclusively expressed on the basolateral membrane of IECs (Gewirtz et al. 2001). This expression profile suggests that only invasive pathogens induce inflammation by contacting the basolateral membrane whereas the interaction between symbiotic microbes and IECs induces a minor inflammatory response as it is restricted to the apical membrane. Nevertheless, MyD88, TLR-2 as well as TLR-4 deficient animals are highly susceptible towards DSS colitis and DSS colitis in antibiotic-treated mice can be ameliorated by oral application of TLR-2 or TLR-4 ligands (Rakoff-Nahoum et al. 2004). In addition to TLRs, IECs express the carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6) which mediates adherence of adherent-invasive Escherichia coli (AIEC) on the apical membrane. Interestingly, CEACAM6 is upregulated on the inflamed epithelium of IBD patients indicating a pathomechanism which enhances intestinal inflammation (Barnich et al. 2007). These results point to the theory that symbiotic microbes induce a continuous production of AMPs by IECs during steady state which are essential to maintain intestinal homeostasis whereas changes in PRR expression or the presence of invasive enteropathogens favour intestinal inflammation.
Presentation of Luminal Antigens by Epithelial Cells
In addition to antigen recognition and antigen uptake, IECs are able to process and present antigen. IECs express molecules resembling the antigen presenting machinery including MHC class I and II molecules, HLA-DM, the Invariant chain, Lamp-1 as well as CD63 and CD68 molecules (Lin et al. 2005; Mayer et al. 1991). During steady state IECs induce CD4+ regulatory T cells (Tregs) expressing high levels of IL-10 and forkhead-box-protein 3 (Foxp3) whereas inflammatory conditions instruct IECs to prime interferon (IFN)-γ producing CD4+ T cells (Dotan et al. 2007; Westendorf et al. 2006).
Sensing of the Microflora by Intestinal Lymph Follicles
The gut-associated lymphoid tissue (GALT) contains about 70% of all immunocytes and thereby represents the largest secondary lymphoid organ (Corr et al. 2008). While intraepithelial lymphocytes (IELs) and lamina propria (lp) lymphocytes including lpDCs are scattered cell populations which constitute the effector site of the GALT, mesenteric lymph nodes (mLN), isolated lymphoid follicles (iLF) and intestinal lymph aggregates (iLA) such as Peyer’s patches (PP) represent the inductive site of the GALT (Niess 2008) (Table 1). In opposite to mLN residing at the mesenterium and receiving intestinal antigens through afferent lymph vessels, iLF and iLA develop in the intestinal wall and obtain antigens through the follicle-associated epithelium (FAE). Interestingly, GF and CV mice differ in the GALT architecture. The number of αβ TCR-positive IELs increases after colonization of GF mice and GF mice possess underdeveloped PP, fewer CD4+ T cells and decreased numbers of IgA-producing plasma cells in the lamina propria (Macdonald and Monteleone 2005). Notably, development of iLF depends on the recognition of peptidoglycan from Gram-negative bacteria by the NOD1 receptor (Bouskra et al. 2008).
The co-operation between FAE and adjacent lymphoid tissue has been extensively studied in PP located at the antimesenteric side of the small intestine. Here, the FAE consists of absorptive enterocytes and specialized epithelial cells without microvilli but with microfolds on the luminal surface. Therefore, these cells were named microfold cells or M-cells (Owen and Jones 1974). M-cell progenitors have been detected in the early proliferative zone of the crypts indicating a common origin of M-cells and enterocytes (Gebert and Pabst 1999). In vitro experiments using adenocarcinoma cells (Caco-2) have suggested a relationship between M-like cell development and PP lymphocytes (Kerneis et al. 1997). However, other studies showed that M-like cells develop from Caco-2 cells even in the absence of lymphocytes (Blanco and DiRita 2006). Interestingly, M-cells do hardly secrete glycoproteins and thus have only a thin mucus layer which allows a close contact to luminal antigens of the intestinal tract. In addition, the basolateral membrane of M-cells forms a pocket-like invagination that is charged with DCs, macrophages, T and B lymphocytes. Based on these features, M-cells are specialized for trans-epithelial transport of particles, macromolecules and microorganisms, also referred to as transcytosis directed from the intestinal lumen to underlying immune cells. On the one hand, transcytosis allows a rapid induction of immune responses against intestinal pathogens but on the other hand, pathogens have evolved strategies to benefit from this entry site. For example, Salmonella typhimurium equipped with the Salmonella pathogenicity island 1 (SPI1) invades M-cells, destroys the FAE and causes large ulcera which pave the way to lymphoid follicles of PP (Jones et al. 1994). In turn, chemokine receptor 6 (CCR6+) DCs become activated, migrate to the subepithelial cell dome of PP and induce antigen-specific T cell responses. The impact of this response has been shown by infection of CCR6-deficient animals with S. typhimurium. CCR6-deficient animals showed an enhanced susceptibility to S. typhimurium due to a decreased migratory capacity of CCR6-deficient DCs in PP (Salazar-Gonzalez et al. 2006). Interestingly, a SNP in the human CCR6 gene is associated with CD (Barrett et al. 2008).
Recognition of Intestinal Microbes by Myeloid Cells
Intestinal myeloid cells are a heterogenous cell population that can be divided in CD11c+B220+ plasmacytoid DCs (pDCs) and CD11c+B220− conventional DCs (cDCs) (Shortman and Liu 2002), macrophages and monocytes. cDCs can be further divided into CD4+, CD8+ and CD4–CD8− DCs. In the lamina propria of the small and large intestine CD4−CD8− cDCs are the predominant population. In addition, intestinal myeloid cells can be categorized according to the expression of CD103, the ligand of E-cadherin and CX3CR1, the receptor for fraktalkine/CX3CL1. Moreover, recent work has identified monocyte-derived DCs (Mo-DCs) which are characterized by the expression of E-cadherin and drive intestinal inflammation by promoting Th17 responses (Siddiqui et al. 2010). Recently, Steinman and colleagues could show that microbial stimuli such as LPS drive differentiation of Mo-DCs marked by the c-type lectin receptor DC-SIGN/CD209 (Cheong et al. 2010).
CD103−CD11b+CD14+CX3CR1+ mononuclear phagocytes develop from Ly6Chigh monocytes in the presence of granulocyte-macrophage colony-stimulating factor (Varol et al. 2009). CX3CR1+ myeloid cells do hardly migrate and seem to control local immune responses at the mucosal surface (Schulz et al. 2009). Further analysis revealed that lp CX3CR1+CD11c+ cells express F4/80 and CD68 (Niess and Adler 2010). Because CX3CR1 is expressed by pDCs, cDCs, monocytes and macrophages, CX3CR1 cannot be considered as a classic lineage marker for DCs (Geissmann et al. 2010; Niess and Adler 2010). However, CX3CR1+ mononuclear phagocytes stay in direct contact with intestinal microbes by forming transepithelial dendrites and expressing tight-junction proteins such as claudin and occludin which preserve integrity of the epithelial layer (Niess et al. 2005; Rescigno et al. 2001). In this way, CX3CR1+ mononuclear phagocytes bypass IECs and gain access to luminal antigens. CX3CR1 deficient mice lack this ability and show an enhanced susceptibility to entero-invasive pathogens such as Salmonella species (Niess et al. 2005). Moreover, a reduction of intestinal microbes by antibiotic treatment as well as genetic ablation of the TLR adaptor molecule MyD88 in IECs decrease the number of transepithelial dendrites of CX3CR1+ mononuclear phagocytes (Chieppa et al. 2006; Niess et al. 2005). In line, colonization of the intestine guides the accumulation of CX3CR1+ mononuclear phagocytes in the small and large intestinal lamina propria (Niess and Adler 2010). CX3CR1+CD11c+F4/80−CD68− mononuclear cells induce differentiation of Th1 and Th17 cells (Atarashi et al. 2008; Denning et al. 2007). In humans, a unique CX3CR1+CCR9+ mononuclear cell that expresses DC (CD205, CD209) and macrophage markers (CD14, CD33, CD68) contributes to the pathogenesis of CD by inducing Th1 differentiation (Kamada et al. 2008).
In contrast to CX3CR1+ mononuclear phagocytes, CD103+CX3CR1− lpDCs derive from macrophage-DC precursors (depending on fms-like tyrosine kinase 3) ligand signalling (Bogunovic et al. 2009; Varol et al. 2009). CD103+ lpDCs have an enhanced migratory potential which enables this subset to carry antigen to mLNs (Schulz et al. 2009). This process is critically influenced by expression of CCR7 on lpDCs and the presence of the corresponding ligands chemokine ligand (CCL) 21 and CCL19 (Jang et al. 2006). Homed to mLNs, antigen presenting lpDCs prime gut tropic T cells by secreting retinoic acid (RA) which induces expression of CCR9 and α4β7 in T cells (Iwata et al. 2004; Johansson-Lindbom et al. 2003).
In addition, recent work has identified an innate lymphoid cell which is characterized by expression of Thy1, stem cell antigen 1, retinoic acid receptor-related orphan receptor γ t (RORγt) and IL-23R and produces IL-17 and IFN-γ upon stimulation with IL-23 (Buonocore et al. 2010). Accordingly, depletion of Thy1+ cells diminished acute and chronic intestinal inflammation.
The Enteric Flora is Tolerized by Intestinal Regulatory T Cells
Tregs are antigen specific T cells which prevent autoimmunity and preserve tolerance towards harmless non-self antigens. The latter function is of importance for the intestine since it stays in permanent contact with large amounts of foreign but essential antigen. Hence, the GALT has evolved a unique pathway to induce Tregs. Depending on the presence of transforming growth factor (TGF)-β and RA, CD103+ mesenteric lymph node DCs are capable to induce CD4+ Foxp3+ Tregs. (Coombes et al. 2007; Sun et al. 2007). Notably, TGF-β but not RA is sufficient to drive Treg development which becomes enhanced by a synergism of both cytokines (Chen et al. 2003). Bioactive RA is synthesized by CD103+ DCs through oxidation of the vitamin A derivate retinal. Thus, RA acts like a natural adjuvant improving oral tolerance. Functionally, RA operates even indirectly by inhibiting the release of proinflammatory cytokines such as IL-4, IFN-γ and IL-21 from CD44high T cells (Hill et al. 2008). The source of TGF-β has so far not been clearly defined. On the one hand, CD103+ DCs are assumed to produce TGF-β and on the other hand, in vitro experiments indicate that TGF-β secreted from IECs primes tolerogenic CD103+ DCs (Iliev et al. 2009).
Treg cells produce the effector cytokines TGF-β and IL-10. Treg cell-specific ablation of IL-10 results in spontaneous colitis and expression of TGF-β1 by Treg cells is required to inhibit the development of colitogenic Th1 cells in a T cell transfer model (Li et al. 2007; Rubtsov et al. 2008). Moreover, genetic ablation of Foxp3+ Treg cells causes lethal multi-organ autoimmunity, similar to the phenotype of scurfy mice and the human X-linked syndrome (IPEX) (Bennett et al. 2001; Lahl et al. 2007). In line, Treg cells with decreased or without Foxp3 expression preferentially convert into Th2 cells that cause inflammation of the intestinal tract (Lahl et al. 2009; Wan and Flavell 2007). Therefore, functional Treg cells are indispensable for the maintainance of intestinal homeostasis. Regarding microbial factors that support Treg cell function, it has been observed that polysaccharide A (PSA) from Bacteroides fragilis inhibits Th17 cells and enhances IL-10 producing T cells conferring protection against Helicobacter hepaticus-induced colitis (Mazmanian et al. 2008). Moreover, distinctly phosphorylated products from the uracil, guanine and adenine metabolism influence the function of Treg cells. For example, adenosine inhibits proinflammatory T cells and induces Tregs upon binding to the A2A receptor on T cells (Huang et al. 1997; Zarek et al. 2008). Consequently, CD25+ T cells from A2A receptor deficient animals are not able to suppress colitis induced by CD45RBhigh T cell transfer (Naganuma et al. 2006). Further, an increased frequency of the Firmicutes strain Faecalibacterium prausnitzii is associated with reduced recurrence rates in CD patients and increased frequencies of IL-10 producing T cells (Sokol et al. 2008).
Resistance to Pathogens is Mediated by the Enteric Flora
Accumulating evidence reveals that host tolerance controls not only host immunity but indirectly mediates host defense carried out by the enteric flora. For example, Shigella flexneri-induced enteritis in GF mice can be attenuated by prior colonization with specific commensal microbes and S. typhimurium—the most common cause of infectious diarrhea in humans world-wide—causes a more severe gastroenteritis in GF mice compared to CV mice (Maier and Hentges 1972; Nardi et al. 1989). Moreover, it has been demonstrated that S. typhimurium infection elicits a protective soluble (s)IgA response. However, sIgA deficiency does not impair pathogen clearance whereas mice with a low complex enteric flora produce normal sIgA levels but are incapable to clear the pathogen (Endt et al. 2010). In this context, several defense mechanisms of the enteric flora have been described. One defense mechanism is mediated by the secretion of antibacterial proteins. E. coli, for example, produces antibacterial proteins such as colicins and microcins (Destoumieux-Garzon et al. 2002; Riley and Wertz 2002). Colicins are proteins with 25–80 kDa whereas microcins are much smaller with less than 10 kDa. Both groups of antibacterial proteins fight Gram-negative enteropathogens by damaging bacterial membranes or destabilizing membrane potentials, respectively. E. coli itself owns resistance towards its toxins. The fact that colonization of gnotobiotic rats with a strain of Peptostreptococcus confers protection against pathogenic bacteria such as Clostridium (C.) perfringens and C. difficile indicates the production of further, so far unknown toxins by this microbe (Ramare et al. 1993). Moreover, a strain of Ruminococcus gnavus has been isolated from human feces which secrets ruminococcin A, an antibacterial toxin that mediates resistance towards various pathogenic clostridia (Dabard et al. 2001). In addition, a protease from S. boulardii digests C. difficile toxin A and thereby inhibits intestinal ileal secretion and histologic damage in C. difficile colitis (Castagliuolo et al. 1996).
Apart from direct antibacterial effects, commensal microbes do also enhance host defense mechanisms. The analysis of Toxoplasma gondii infection has shown an impaired immune response in the absence of commensal microbes triggering innate and adaptive immune responses via TLRs and DCs (Benson et al. 2009). Accordingly, MyD88 deficient animals are even less protected towards gut injury (Rakoff-Nahoum et al. 2004). Furthermore, it has been observed that TLR9 deficient animals are more susceptible to oral infection with the microsporidium Encephalitozoon cuniculi (Hall et al. 2008). This effect was accompanied by decreased levels of IFN-γ in IELs but increased frequencies of Tregs in the epithelium, PP and lp as well as increased systemic levels of IL-4 and IL-10 in E. cuniculi-infected TLR9 deficient mice. DNA extracted from gut flora inhibited induction of Tregs in vitro and oral application of CpG-rich DNA restored immune responses towards E. cuniculi in antibiotic-treated animals indicating that commensal microbes enhance immune responses towards enteropathogens via TLR9 signalling. Lactobacilli have been shown to induce secretion of intestinal mucins from IECs. In vitro experiments suggest that lactobacilli inhibit adhesion of enteropathogenic E. coli by upregulating expression and secretion of MUC2 and MUC3 from IECs (Mack et al. 1999). Moreover, the secretion of PSA from Bacteroides fragilis protects animals against Helicobacter hepaticus infection by dampening pro-inflammatory intestinal T cell responses (Mazmanian et al. 2008). Together, these results demonstrate that only a symbiotic relationship between the enteric flora and the host organism provides a substantial defense towards enteropathogens.
Intestinal Th17 Responses: a Double-Edged Sword
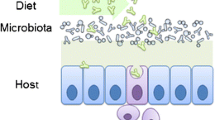
The fact that enteric flora from IBD patients significantly differs from the one of healthy people indicates that intestinal dysbiosis triggers harmful T cell responses in IBD. Apart from Treg cells, intestinal CD4+ T cells can be subdivided into Th1, Th2 and Th17 cells. IL-17 producing Th17 cells are frequent cells in the lamina propria with highest frequency in the terminal ileum and ceacum. In the absence of enteric flora, Th17 cells are reduced in the lamina propria (Ivanov et al. 2008; Niess and Adler 2010). A member of the non-pathogenic Clostridium coccoides species, the segmented filamentous bacterium (SFB), specifically attachs to the FAE of PP, recruits lymphocytes and induces differentiation of Th17 cells during steady state as shown in Fig. 2 (Gaboriau-Routhiau et al. 2009; Ivanov et al. 2009). Th17 cells are defined by transcription factors such as RORγt, interferon regulatory factor 4 (IRF4) and signal transducer of activated T cells 3 (STAT3) as well as the aryl hydrocarbon receptor. (Brustle et al. 2007; Harris et al. 2007; Ivanov et al. 2006; Veldhoen et al. 2008). The differentiation of naive T cells into Th17 cells occurs in the presence of IL-6 or IL-21 and TGF-β whereas IL-23 seems to be essential for maintainance and expansion of Th17 cells (Korn et al. 2007; Veldhoen et al. 2006; Zhou et al. 2007). However, recent work has shown that even a combination of IL-1 with IL-6 or IL-23 generates Th17 cells from naive T cells (Ghoreschi et al. 2010). At the same time, this cytokine pattern antagonizes Foxp3+ Treg cells (Bettelli et al. 2006). Moreover, it has been shown that adenosine triphosphate (ATP) promotes inflammatory intestinal Th17 responses (Atarashi et al. 2008). Th17 responses decrease upon degradation of intestinal flora by antibiotic treatment indicating that bacterial ATP production plays a critical role. Correspondingly, mice with genetic ablation of the CD39/ENTPD1 gene encoding an ATP-hydrolase suffer from exacerbated DSS colitis and the analysis of SNPs in a human case–control cohort revealed significant associations between SNPs in the human CD39 gene and an enhanced susceptibility to CD (Friedman et al. 2009). In a similar manner, the NOD2 ligand muramyldipeptide (MDP) induces IL-17 production in human memory T cells by enhancing the release of IL-23 and IL-1 from monocyte-derived DCs primed by TLR agonists (van Beelen et al. 2007). However, TLR-primed Mo-DCs from patients with double-dose NOD2 mutations in the leucine-rich repeat domain fail to produce IL-23 upon stimulation with MDP. Moreover, the absence of IL-23 increases susceptibility to Citrobacter rodentium-driven colitis due to an impaired Th17 response (Mangan et al. 2006). In terms of fungal infections, the c-type lectin receptor Dectin-1 binds Candida albicans and subsequently induces IL-23 via Syk-CARD9 signalling (LeibundGut-Landmann et al. 2007). CARD9 deficient animals show a decreased Th17 response towards C. albicans and rapidly succumb to the fungus (Gross et al. 2006). IL-23 is constituted by IL-12p40 and p19, whereas IL-12 consists of p40 and p35 (Oppmann et al. 2000). Mice deficient for p19 or p35 show different phenotypes in a colitis model. (Uhlig et al. 2006). While the absence of p19 protects mice from intestinal pathology but not from wasting disease, p35 deficient animals show an increased intestinal inflammation but decreased weight loss. On the other hand, IL-23 is necessary to maintain chronic intestinal inflammation by inducing IL-17 and IL-6 producing T cells and IL-23 promotes extraintestinal disorders such as experimental allergic encephalomyelitis, joint inflammation and psoriasis (Chan et al. 2006; Cua et al. 2003; Murphy et al. 2003; Yen et al. 2006). Moreover, a large GWAS identified a highly significant association between the human IL-23R subunit IL-23R located on chromosome 1p31 and CD (Duerr et al. 2006). In line, IRF4-deficient animals are protected from chemical-induced colitis by a decreased production of IL-6 in intestinal T cells and the blockade of IL-6 signalling ameliorates experimental colitis by induction of apoptosis in lamina propria T cells (Atreya et al. 2000; Mudter et al. 2008). Except IL-6, Th17 cells produce the effector cytokines IL-17A, IL-17F, IL-22 and IL-26 (Andoh et al. 2008). IL-26 has been detected in intestinal Th17 cells (Dambacher et al. 2009). Stimulation of IEC lines with IL-26 results in cell proliferation and enhanced expression of proinflammatory cytokines such as IL-8 and IL-22. Transfer of RORγ-null, IL-17A−/−, IL-17F−/− as well as IL-22−/− T cells into RAG1 deficient mice results in a more severe colitis compared to transfer of wildtype T cells. Moreover, colitis induced by transfer of IL-17F deficient T cells becomes attenuated by concomitant neutralization of IL-17A indicating redundant functions of IL-17A and IL-17F in IBD (Yang et al. 2008; Zenewicz et al. 2008). Correspondingly, GWAS have identified risk loci for UC in 7q22 and 22q13 (IL17REL) (McGovern et al. 2010). Taken together, these data demonstrate an important role of Th17 responses in host defense but concomitantly reveal that defects in genes regulating corresponding cytokine pathways predispose to develop harmful uncontrolled Th17 responses triggered by intestinal microbes. Thus, selected IBD patients might benefit from therapeutics which target Th17 associated pathways such IL-23 signalling or antagonize intestinal microbes triggering Th17 responses.

Gut flora and fauna shape intestinal t cell responses. Francisella prausnitzii (FP), strains of Bifidobacteria (BB) such as Bifidobacterium S17 and parasites such as Encephalitozoon cuniculi (EC) promote regulatory T cell (Treg) responses. Hence, worms such as Trichuris muris induce Th2 responses and segmented filamentous bacteria (SFB) as well as intestinal adenosine triphosphate (ATP) favour Th17 responses whereas Th1 responses become enhanced by Citrobacter rodentium (CR)
Enteric Flora Drives Inflammatory Th1 Responses
In the presence of IL-12, type I and type II IFNs naive T cells differentiate into Th1 cells. This process is characterized by activation of STAT1 and STAT4 inducing T-box transcription factor expressed in T cells (T-bet) and IFN-γ, both hallmarks of Th1 cells (Murphy et al. 1999). Elevated levels of IFN-γ were detected in sera of CD patients and increased frequencies of IFN-γ and T-bet expressing CD4+ T cells were found in intestinal biopsies of CD patients (Fuss et al. 1996). Moreover, lamina propria CD4+ T cells isolated from CD patients express elevated levels of IL-12Rβ1, IL-12Rβ2 and IL-18R representing typical features of Th1 cells (Okazawa et al. 2002). Accordingly, application of anti-IL-12- antibodies to mice with 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis ameliorated disease and transfer of T-bet deficient CD45RBhigh T cells into RAG-deficient mice did not cause colitis indicating a pivotal role of Th1 cells in IBD (Neurath et al. 1995; Neurath et al. 2002). Based on these results, it has been hypothesized that CD patients benefit from therapies antagonizing Th1 responses. However, neither application of anti-IL-12 antibodies nor treatments with fontolizumab, an anti-IFN-γ antibody, could produce convincing results (Kaser et al. 2010). Interestingly, RAG2−/−T-bet−/− mice develop colitis despite the absence of adaptive immunity and wildtype mice develop colitis upon ingestion of intestinal flora from RAG2−/−T-bet−/− mice indicating that intestinal inflammation driven by innate immunity selects a bacterial flora with proinflammatory potential (Garrett et al. 2007). The influence of the microbiota on intestinal Th1 responses has been analysed in IL-2 and IL-10 deficient mice, respectively. Both genotypes spontaneously develop colitis caused by enhanced Th1 responses. However, only IL-10 deficient but not IL-2 deficient animals showed an ameliorated disease with decreased Th1 responses in the absence of TLR-MyD88 signalling (Rakoff-Nahoum et al. 2006). Furthermore, IL-10−/−/TLR9−/− animals showed an accelerated colitis progression compared to IL-10−/−/TLR4−/− mice indicating that bacterial DNA triggers Th1 responses via TLR-9 signalling (Gonzalez-Navajas et al. 2010).
Tumor necrosis factor (TNF)-α is another proinflammatory cytokine which is associated with Th1 responses and higher expressed by intestinal cells from IBD patients compared to healthy controls (MacDonald et al. 1990). Neutralization of TNF-α or genetic deletion of TNF-α reduced Th1 responses and intestinal inflammation in animal models (Kontoyiannis et al. 2002; Neurath et al. 1997). In line, intestinal inflammation is attenuated in Tnfr2−/−TnfΔARE mice, even absent in Tnfr1−/−TnfΔARE mutants and limited to superficial layers in RAG−/−TnfΔARE mice (Kontoyiannis et al. 1999). Accordingly, anti-TNF-α antibody treatment was tested in humans and has established a very efficacious therapy for human IBD (Baumgart and Sandborn 2007). However, the exact molecular mode of action of anti-TNF-α antibody therapy is only partially understood. Regarding the role of microbes, it has been demonstrated that intestinal flora drives TNF-α production by lp CD4+ T cells (Niess et al. 2008). Moreover, E. coli strains isolated from feces of CD patients showed an enhanced mucosal adherence and invasiveness compared to E. coli strains isolated from healthy controls (Darfeuille-Michaud et al. 2004). Based on these features, AIEC enter the lamina propria and become phagocytosed by macrophages which subsequently produce large amounts of TNF-α (Barnich et al. 2007). Citrobacter rodentium infection elicits a Th1 response in mice which causes intestinal lesions similar to those in murine IBD models (Higgins et al. 1999).
Taken together, these results show that the enteric flora can induce colitogenic Th1 responses. However, the fact that IBD patients only benefit from anti-TNF-α, but not from anti-IL-12 or anti-IFN-γ therapies indicates specific changes in the human genome and probably even in the microbiome that particularly enhance TNF-α production. In this way, NOD2 polymorphisms may favour bacterial dysbiosis triggering TNF-α production in IBD.
The Commensal Microflora Shape Intestinal Th2 Responses
Th2 cells are characterized by the transcription factor GATA-3 and become induced by antigen-presenting cells (APCs) secreting cytokines such as IL-4, IL-5 and IL-13 (Zheng and Flavell 1997). Not only classical APCs like macrophages and DCs but also basophils can promote differentiation of Th2 cells (Sokol et al. 2009). On the one hand, Th2 responses are essential to fight helminth and parasite infections but on the other hand, uncontrolled Th2 responses cause hyperinflammatory responses including excessive IgE production and mast cell activation which cause atopic diseases such as asthma. Interestingly, Th2 responses are even associated with IBD. IL-13 is produced in an oxazalone colitis model by nonclassical natural killer (NK) T cells that are restricted to the MHC molecule CD1d but do not respond to the invariant NKT ligand α-galactosylceramide (Fuss et al. 2004). In line, activation of IL-13R α2 induces fibrogenic factors in TNBS colitis and IL-13R α2 is upregulated in IECs from UC patients (Fichtner-Feigl et al. 2008; Mandal and Levine 2010). Neutralization of IL-13 as well as depletion of NKT cells prohibits colitis in animal models (Heller et al. 2002). Moreover, GWAS could detect highly significant associations between SNPs in the ORMDL3 gene located on chromosome 17 and CD as well as childhood asthma indicating common pathomechanisms (Barrett et al. 2008; Moffatt et al. 2007). Regarding intestinal microflora and macrofauna, it has been shown that protective immunity against the worm Trichuris muris relies on Th2 responses driven by thymic stromal lymphopoietin (TSLP) from IECs. Being released, TSLP binds to its receptor on lpDCs and triggers release of Th2 cytokines via IKKβ activation (Taylor et al. 2009; Zaph et al. 2007). TSLP-receptor deficient animals as well as DCs from IkbkbVillin-Cre mice produce increased levels of Th1 associated cytokines such as IFN-γ and TNF-α which again decrease upon neutralization of IL-12/23p40. Surprisingly, antibiotic treatment of mice infected with Trichuris muris leads to a reduction of the microflora and the macrofauna accompanied by an increased Th2 response (Hayes et al. 2010). In this context, the infection of mice with the intestinal helminth Heligmosomoides polygyrus has shown a selective de novo induction of Treg cells but not Th2 cells through stimulation of the TGF-β receptor with a parasite-secreted protein (Grainger et al. 2010). Thus, one can speculate that not only the microflora but also the macrofauna contributes to intestinal homeostasis. Accordingly, a small prospective study tested the safety and efficacy of live eggs from the porcine whipworm, Trichuris suis in IBD patients (Summers et al. 2003). The treatment could improve the Crohn’s Disease Activity Index (CDAI) and the IBD Quality of Life Index without having side effects. Based on these results large randomized, double-blind studies have been planned.
Manipulating the Enteric Flora for the Benefit of the Host
The perception that not only host immunity controls intestinal microflora but vice versa intestinal microbes influence host immune responses resulted in the development of therapies manipulating the intestinal flora in order to dampen harmful immune responses in IBD by dissolving bacterial dysbiosis (Tables 2, 3).
In this context, the terms probiotics, prebiotics and synbiotics need to be mentioned. Probiotics have been defined as “a live microbial feed supplement which beneficially affects the host animal by improving its intestinal microbial balance” (Fuller 1991). The term prebiotic has been introduced to describe “a non-digestible food ingredient that beneficially affects the host by selectively stimulating the growth and/or activity of one or a limited number of bacteria in the colon” (Gibson and Roberfroid 1995). Thus, the therapeutic effect of prebiotics relies on the presence of probiotics. Finally, products which contain both pro- and prebiotics that act synergistically are referred to as synbiotics (Roberfroid 1998).
Lactobacillus (L.) species were one of the first bacteria whose probiotic potential has been evaluated in IBD. Animal studies showed that intracolonic application of L. reuteri R2LC to rats with acetic acid-induced colitis significantly ameliorated the disease and intragastric administration of L. reuteri R2LC to rats with methotrexate-induced enterocolitis resulted in decreased weight loss and reduced intestinal permeability (Fabia et al. 1993; Mao et al. 1996). Furthermore, Lactobacillus species attenuated colitis in IL-10 deficient mice (Madsen et al. 1999). Accordingly, IL-10 and TGF-β producing Treg cells became induced by co-culturing murine splenocytes with L. paracasei NCC2461 or by co-culturing human DC-SIGN+ Mo-DCs with L. reuteri or L. casei (Smits et al. 2005; von der Weid et al. 2001). Notably, anti-DC-SIGN antibodies blocked the induction of Tregs indicating a tolerogenic role of the c-type lectin receptor. Based on these promising results, Lactobacillus species were tested in IBD patients. However, except one open-label pediatric study, no randomized placebo-controlled study could so far show a significant benefit for treatments with Lactobacillus in CD (Gupta et al. 2000; Rolfe et al. 2006). Two randomized placebo-controlled trials tested the effect of Lactobacillus johnsonii, LA1 on the recurrence of CD after lleo-cecal resection and in both trials application of LA1 could not decrease recurrence rates (Marteau et al. 2006; Van Gossum et al. 2007). Regarding UC, a randomized-controlled trial could show that the onset of pouchitis after ileal pouch-anal anastomosis became delayed by a daily intake of L. rhamnosusGG (Gosselink et al. 2004). Promising data were also obtained from bifidobacteria. Here, a randomized-controlled trial could show that bifidobacteria-fermented milk supplement significantly reduced relapses and maintained remission in patients with UC (Ishikawa et al. 2003). In line, Cui et al. (2004) observed fewer relapses in UC patients treated with bifidobacteria during remission. However, it needs to be considered that bifidobacteria strains differ in their anti-inflammatory potential which is particular strong in Bifidobacterium bifidum S17 (Preising et al. 2010).
VSL#3 is a mixture of eight bacteria including L. acidophilus, L. casei, L. plantarum, L. bulgaricus, Bifidobacterium (B.) longum, B. breve, B. infantis and Streptococcus thermophilus. The safety and efficacy of VSL#3 has been evaluated in an uncontrolled study that included 34 UC patients with mild or moderate disease activity. Here, no clinical or biochemical adverse effects occured (Bibiloni et al. 2005). A double-blind, placebo-controlled study tested the probiotic potential of VSL#3 in 147 patients with mild or moderate UC. Randomized into two groups, the patients received either 3.6 × 1012 colony forming units (CFU) of VSL#3 or placebo twice daily for 12 weeks. After this time, significantly more patients treated with VSL#3 achieved remission and reported a decreased Ulcerative Colitis Disease Activity Index (Sood et al. 2009). Similar results were also obtained from a double-blind, placebo-controlled trial with UC patients treated about 8 weeks with VSL#3 (3,600 billion CFU/day) or placebo in addition to their immunosuppressive drugs (Tursi et al. 2010). Another prospective double-blind, placebo-controlled study analysed the efficacy of VSL#3 in children with newly diagnosed active UC. After 1 year, significant higher remission rates and significant less relapses were observed in children treated with VSL#3 (Miele et al. 2009). Regarding its biochemical mechanism, in vitro experiments suggest that compounds of VSL#3 downregulate secretion of TNF-induced IFN-γ-induced protein 10 (Hoermannsperger et al. 2009).
Similar results were obtained from the non-pathogenic strain E. coli Nissle 1917. Compared to a standard therapy with mesalazine, UC patients treated with E. coli Nissle 1917 achieved and maintained equal remission rates (Kruis et al. 2004; Kruis et al. 1997). In addition, application of E. coli Nissle 1917 enemas increased remission rates of left-sided UC in a dose dependent manner (Matthes et al. 2010). One study tested E. coli Nissle 1917 in patients with active CD treated with steroids. However, no beneficial effect of the probiotic was observed (Malchow 1997). Functionally, E. coli Nissle 1917 is able to inhibit adhesion and invasion of AIEC and in vitro experiments showed that E. coli Nissle 1917 induces production of human β-defensin-2 in IECs (Boudeau et al. 2003; Wehkamp et al. 2004). Moreover, colonization with E. coli Nissle 1917 decreased levels of TNF-α in a model of LPS-induced sepsis (Arribas et al. 2009). Thus, intestinal microflora as well as adaptive immunity are indispensable for the safety of E. coli Nissle 1917 (Gronbach et al. 2010).
The analysis of mucosa-associated microbiota in CD patients at the time of surgical resection and 6 months later revealed a significant association between reduced counts of the Firmicutes strain Faecalibacterium (F.) prausnitzii and a higher risk of ileal recurrence of CD (Sokol et al. 2008). Correspondingly, oral administration of F. prausnitzii or its supernatant to mice ameliorated TNBS-colitis. In vitro experiments suggest that these effects are mediated by a yet-to-be-defined secreted product(s) which inhibits NFκB activity, reduces levels of IL-12 and IFN-γ and increases levels of IL-10.
Saccharomyces (S.) boulardii is a non-pathogenic yeast which interferes with multiple pro-inflammatory pathways such as NFκB and mitogen-activated protein kinases and thereby inhibits production of inflammatory cytokines (Sougioultzis et al. 2006). In addition, a protease from S. boulardii cleaves C. difficile toxin A and its receptor, respectively, indicating a probiotic potential of this microbe (Castagliuolo et al. 1996). Accordingly, a clinical trial has compared the efficacy of S. boulardii and mesalazine versus mesalazine alone in 32 CD patients. Notably, clinical relapses were less frequent in patients who received mesalazine plus S. boulardii (Guslandi et al. 2000). Results from a pilot trial with S. boulardii suggest that it also improves remission rates in UC patients treated with mesalazine (Guslandi et al. 2003).
As mentioned above, even the macrofauna represented by helminths plays in important role in gut homeostasis as it induces Tregs and balances Th1, Th2 and Th17 responses. Accordingly, Summers and colleagues performed a randomized, double-blind, placebo-controlled study wherein 54 patients with active UC received 2,500 Trichuris suis ova or placebo orally at 2-week intervals for 12 weeks (Summers et al. 2005b). Notably, 43.3% of the ova-treated patients but only 16.7% of the placebo group reached the primary efficacy variable which was defined as improvement of the Disease Activity Index to ≥4. Analogue to this study, the effect of Trichuris suis ova was analysed in an open-label study which included 29 patients with active CD (Summers et al. 2005a). Here, 23 patients responded (decrease in CDAI >100 or CDAI <150) and 21 patients remitted (CDAI <150). In both studies, no adverse side effects were observed.
Apart from native microbes, the probiotic therapy has been extended to genetically manipulated bacteria expressing cytokines and/or delivering drugs and vaccines. For example, intragastric application of an IL-10 secreting Lactococcus (L.) lactis significantly decreased murine DSS-colitis and could even prevent colitis in IL-10 deficient mice (Steidler et al. 2000). Correspondingly, Foligne and colleagues could show that genetically engineered.
L. lactis secreting the Y. pseudotuberculosis protein LcrV prevented TNBS- and DSS-colitis in mice (Foligne et al. 2007). Moreover, intranasal administration of tetanus toxin expressing L. plantarum elicited antigen specific humoral and cellular immune responses that protected mice from the toxin (Grangette et al. 2001). Nevertheless, it still needs pre-clinical trials to guarantee the safety of this new therapeutical approach.
Conclusions and Future Perspectives
The analysis of the human microbiome has revealed significant differences between the microbiota of IBD patients and healthy controls. Additional studies could show that not only genetic factors but also environmental factors influence each individual’s microbiota. Therefore, strategies have been developed to treat IBD by manipulating the intestinal flora. Especially, probiotic bacteria seem to be capable of restoring intestinal symbiosis by unfolding antiinflammatory effects. Selective colonization of GF mice with genetically manipulated bacteria will help to identify molecular mechanisms mediating antiinflammatory effects. These results are important to improve our knowledge about the interaction between the host and its microbiota in order to develop more specific therapies targeting intestinal dysbiosis in IBD.
Abbreviations
- IBD:
-
Inflammatory bowel disease
- CD:
-
Crohn’s disease
- UC:
-
Ulcerative colitis
- GWAS:
-
Genome-wide association studies
- SNPs:
-
Single-nucleotide polymorphisms
- NOD:
-
Nucleotide-binding oligomerization domain
- IL-23R:
-
Interleukin 23 receptor
- GF:
-
Germ-free
- CV:
-
Conventional
- SCFA:
-
Short-chain fatty acids
- IECs:
-
Intestinal epithelial cells
- ITF:
-
Intestinal trefoil factor
- DSS:
-
Dextran sodium sulfate
- AMPs:
-
Antimicrobial peptides
- MyD88:
-
Myeloid-differentiation factor 88
- TLRs:
-
Toll-like receptors
- AIEC:
-
Adherent-invasive Escherichia coli
- MHC:
-
Major histocompatibility complex
- PRRs:
-
Pathogen recognition receptors
- PAMPs:
-
Pathogen-associated molecular patterns
- DAMPs:
-
Danger-associated molecular patterns
- LPS:
-
Lipopolysaccharide
- ss-RNA:
-
Single-stranded ribonucleic acid
- CEACAM6:
-
Carcinoembryonic antigen-related cell adhesion molecule 6
- Foxp:
-
Forkhead-box-protein
- IFN-γ:
-
Interferon γ
- GALT:
-
Gut-associated lymphoid tissue
- IELs:
-
Intraepithelial lymphocytes
- mLN:
-
Mesenteric lymph nodes
- iLF:
-
Isolated lymphoid follicles
- iLA:
-
Intestinal lymph aggregates
- PP:
-
Peyer’s patches
- FAE:
-
Follicle-associated epithelium
- M-cell:
-
Microfold cell
- DC:
-
Dendritic cell
- pDCs:
-
Plasmacytoid DCs
- cDCs:
-
Conventional DCs
- PSA:
-
Polysaccharide A
- RA:
-
Retinoic acid
- CCR:
-
Chemokine receptor
- CLL:
-
Chemokine ligand
- TGF-β:
-
Transforming growth factor β
- ATP:
-
Adenosine triphosphate
- STAT:
-
Signal transducer of activated T cells
- IRF:
-
Interferon regulatory factor
- MDP:
-
Muramyldipeptide
- T-bet:
-
T-box transcription factor expressed in T cells
- TNF:
-
Tumor necrosis factor
- TSLP:
-
Thymic stromal lymphopoietin
- CDAI:
-
Crohn’s Disease Activity Index
References
Abreu MT, Vora P, Faure E et al (2001) Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J Immunol 167:1609–1616
Andoh A, Yagi Y, Shioya M et al (2008) Mucosal cytokine network in inflammatory bowel disease. World J Gastroenterol 14:5154–5161
Arribas B, Rodriguez-Cabezas ME, Camuesco D et al (2009) A probiotic strain of Escherichia coli, Nissle 1917, given orally exerts local and systemic anti-inflammatory effects in lipopolysaccharide-induced sepsis in mice. Br J Pharmacol 157:1024–1033
Atarashi K, Nishimura J, Shima T et al (2008) ATP drives lamina propria T(H)17 cell differentiation. Nature 455:808–812
Atreya R, Mudter J, Finotto S et al (2000) Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med 6:583–588
Barnich N, Carvalho FA, Glasser AL et al (2007) CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J Clin Invest 117:1566–1574
Barrett JC, Hansoul S, Nicolae DL et al (2008) Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet 40:955–962
Baumgart DC, Sandborn WJ (2007) Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet 369:1641–1657
Bennett CL, Christie J, Ramsdell F et al (2001) The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 27:20–21
Benson A, Pifer R, Behrendt CL et al (2009) Gut commensal bacteria direct a protective immune response against Toxoplasma gondii. Cell Host Microbe 6:187–196
Bettelli E, Carrier Y, Gao W et al (2006) Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441:235–238
Bibiloni R, Fedorak RN, Tannock GW et al (2005) VSL#3 probiotic-mixture induces remission in patients with active ulcerative colitis. Am J Gastroenterol 100:1539–1546
Bjerknes M, Cheng H (1981a) The stem-cell zone of the small intestinal epithelium. I. Evidence from Paneth cells in the adult mouse. Am J Anat 160:51–63
Bjerknes M, Cheng H (1981b) The stem-cell zone of the small intestinal epithelium. III. Evidence from columnar, enteroendocrine, and mucous cells in the adult mouse. Am J Anat 160:77–91
Blanco LP, DiRita VJ (2006) Bacterial-associated cholera toxin and GM1 binding are required for transcytosis of classical biotype Vibrio cholerae through an in vitro M cell model system. Cell Microbiol 8:982–998
Bogunovic M, Ginhoux F, Helft J et al (2009) Origin of the lamina propria dendritic cell network. Immunity 31:513–525
Boudeau J, Glasser AL, Julien S et al (2003) Inhibitory effect of probiotic Escherichia coli strain Nissle 1917 on adhesion to and invasion of intestinal epithelial cells by adherent-invasive E. coli strains isolated from patients with Crohn’s disease. Aliment Pharmacol Ther 18:45–56
Bouskra D, Brezillon C, Berard M et al (2008) Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456:507–510
Brustle A, Heink S, Huber M et al (2007) The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol 8:958–966
Buonocore S, Ahern PP, Uhlig HH et al (2010) Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464:1371–1375
Cadwell K, Liu JY, Brown SL et al (2008) A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456:259–263
Castagliuolo I, LaMont JT, Nikulasson ST et al (1996) Saccharomyces boulardii protease inhibits Clostridium difficile toxin A effects in the rat ileum. Infect Immun 64:5225–5232
Chan JR, Blumenschein W, Murphy E et al (2006) IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med 203:2577–2587
Chen W, Jin W, Hardegen N et al (2003) Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 198:1875–1886
Cheong C, Matos I, Choi JH et al (2010) Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell 143:416–429
Chieppa M, Rescigno M, Huang AY et al (2006) Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J Exp Med 203:2841–2852
Clayton DG, Walker NM, Smyth DJ et al (2005) Population structure, differential bias and genomic control in a large-scale, case-control association study. Nat Genet 37:1243–1246
Collier-Hyams LS, Sloane V, Batten BC et al (2005) Cutting edge: bacterial modulation of epithelial signaling via changes in neddylation of cullin-1. J Immunol 175:4194–4198
Coombes JL, Siddiqui KR, Arancibia-Carcamo CV et al (2007) A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med 204:1757–1764
Cooney R, Baker J, Brain O et al (2010) NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 16:90–97
Corr SC, Gahan CC, Hill C (2008) M-cells: origin, morphology and role in mucosal immunity and microbial pathogenesis. FEMS Immunol Med Microbiol 52:2–12
Croft DN, Cotton PB (1973) Gastro-intestinal cell loss in man. Its measurement and significance. Digestion 8:144–160
Cua DJ, Sherlock J, Chen Y et al (2003) Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421:744–748
Cui HH, Chen CL, Wang JD et al (2004) Effects of probiotic on intestinal mucosa of patients with ulcerative colitis. World J Gastroenterol 10:1521–1525
Dabard J, Bridonneau C, Phillipe C et al (2001) Ruminococcin A, a new lantibiotic produced by a Ruminococcus gnavus strain isolated from human feces. Appl Environ Microbiol 67:4111–4118
Dambacher J, Beigel F, Zitzmann K et al (2009) The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut 58:1207–1217
Darfeuille-Michaud A, Boudeau J, Bulois P et al (2004) High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 127:412–421
Denning TL, Wang YC, Patel SR et al (2007) Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol 8:1086–1094
Destoumieux-Garzon D, Peduzzi J, Rebuffat S (2002) Focus on modified microcins: structural features and mechanisms of action. Biochimie 84:511–519
Dicksved J, Halfvarson J, Rosenquist M et al (2008) Molecular analysis of the gut microbiota of identical twins with Crohn’s disease. ISME J 2:716–727
Dignass A, Lynch-Devaney K, Kindon H et al (1994) Trefoil peptides promote epithelial migration through a transforming growth factor beta-independent pathway. J Clin Invest 94:376–383
Dotan I, Allez M, Nakazawa A et al (2007) Intestinal epithelial cells from inflammatory bowel disease patients preferentially stimulate CD4+ T cells to proliferate and secrete interferon-gamma. Am J Physiol Gastrointest Liver Physiol 292:G1630–G1640
Duerr RH, Taylor KD, Brant SR et al (2006) A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314:1461–1463
Eckburg PB, Bik EM, Bernstein CN et al (2005) Diversity of the human intestinal microbial flora. Science 308:1635–1638
Endt K, Stecher B, Chaffron S et al (2010) The microbiota mediates pathogen clearance from the gut lumen after non-typhoidal Salmonella diarrhea. PLoS Pathog 6:e1001097
Fabia R, Ar’Rajab A, Johansson ML et al (1993) The effect of exogenous administration of Lactobacillus reuteri R2LC and oat fiber on acetic acid-induced colitis in the rat. Scand J Gastroenterol 28:155–162
Falk PG, Hooper LV, Midtvedt T et al (1998) Creating and maintaining the gastrointestinal ecosystem: what we know and need to know from gnotobiology. Microbiol Mol Biol Rev 62:1157–1170
Fichtner-Feigl S, Young CA, Kitani A et al (2008) IL-13 signaling via IL-13R alpha2 induces major downstream fibrogenic factors mediating fibrosis in chronic TNBS colitis. Gastroenterology 135:2003–2013, 2013 e1–7
Foligne B, Dessein R, Marceau M et al (2007) Prevention and treatment of colitis with Lactococcus lactis secreting the immunomodulatory Yersinia LcrV protein. Gastroenterology 133:862–874
Frank DN, Pace NR (2008) Gastrointestinal microbiology enters the metagenomics era. Curr Opin Gastroenterol 24:4–10
Friedman DJ, Künzli BM, A-Rahim YI et al (2009) From the cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc Natl Acad Sci USA 106:16788–16793
Fuller R (1991) Probiotics in human medicine. Gut 32:439–442
Fuss IJ, Neurath M, Boirivant M et al (1996) Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol 157:1261–1270
Fuss IJ, Heller F, Boirivant M et al (2004) Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest 113:1490–1497
Gaboriau-Routhiau V, Rakotobe S, Lecuyer E et al (2009) The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 31:677–689
Garrett WS, Lord GM, Punit S et al (2007) Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131:33–45
Gebert A, Pabst R (1999) M cells at locations outside the gut. Semin Immunol 11:165–170
Geissmann F, Manz MG, Jung S et al (2010) Development of monocytes, macrophages, and dendritic cells. Science 327:656–661
Gersemann M, Becker S, Kubler I et al (2009) Differences in goblet cell differentiation between Crohn’s disease and ulcerative colitis. Differentiation 77:84–94
Gewirtz AT, Navas TA, Lyons S et al (2001) Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol 167:1882–1885
Ghoreschi K, Laurence A, Yang XP et al (2010) Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 467:967–971
Gibson GR, Roberfroid MB (1995) Dietary modulation of the human colonic microbiota: introducing the concept of prebiotics. J Nutr 125:1401–1412
Gonzalez-Navajas JM, Fine S, Law J et al (2010) TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental colitis in mice. J Clin Invest 120:570–581
Gosselink MP, Schouten WR, van Lieshout LM et al (2004) Delay of the first onset of pouchitis by oral intake of the probiotic strain Lactobacillus rhamnosus GG. Dis Colon Rectum 47:876–884
Grainger JR, Smith KA, Hewitson JP et al (2010) Helminth secretions induce de novo T cell Foxp3 expression and regulatory function through the TGF-beta pathway. J Exp Med 207:2331–2341
Grangette C, Muller-Alouf H, Goudercourt D et al (2001) Mucosal immune responses and protection against tetanus toxin after intranasal immunization with recombinant Lactobacillus plantarum. Infect Immun 69:1547–1553
Gronbach K, Eberle U, Muller M et al (2010) Safety of probiotic Escherichia coli strain Nissle 1917 depends on intestinal microbiota and adaptive immunity of the host. Infect Immun 78:3036–3046
Gross O, Gewies A, Finger K et al (2006) Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 442:651–656
Gupta P, Andrew H, Kirschner BS et al (2000) Is Lactobacillus GG helpful in children with Crohn’s disease? Results of a preliminary, open-label study. J Pediatr Gastroenterol Nutr 31:453–457
Guslandi M, Mezzi G, Sorghi M et al (2000) Saccharomyces boulardii in maintenance treatment of Crohn’s disease. Dig Dis Sci 45:1462–1464
Guslandi M, Giollo P, Testoni PA (2003) A pilot trial of Saccharomyces boulardii in ulcerative colitis. Eur J Gastroenterol Hepatol 15:697–698
Gustafsson BE, Midtvedt T, Strandberg K (1970) Effects of microbial contamination on the cecum enlargement of germfree rats. Scand J Gastroenterol 5:309–314
Gutierrez MG, Master SS, Singh SB et al (2004) Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119:753–766
Hall JA, Bouladoux N, Sun CM et al (2008) Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity 29:637–649
Harris TJ, Grosso JF, Yen HR et al (2007) Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol 179:4313–4317
Hayes KS, Bancroft AJ, Goldrick M et al (2010) Exploitation of the intestinal microflora by the parasitic nematode Trichuris muris. Science 328:1391–1394
Heller F, Fuss IJ, Nieuwenhuis EE et al (2002) Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity 17:629–638
Higgins LM, Frankel G, Douce G et al (1999) Citrobacter rodentium infection in mice elicits a mucosal Th1 cytokine response and lesions similar to those in murine inflammatory bowel disease. Infect Immun 67:3031–3039
Hill JA, Hall JA, Sun CM et al (2008) Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi cells. Immunity 29:758–770
Hoermannsperger G, Clavel T, Hoffmann M et al (2009) Post-translational inhibition of IP-10 secretion in IEC by probiotic bacteria: impact on chronic inflammation. PLoS One 4:e4365
Huang S, Apasov S, Koshiba M et al (1997) Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood 90:1600–1610
Hugot JP, Chamaillard M, Zouali H et al (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411:599–603
Husebye E, Hellstrom PM, Midtvedt T (1994) Intestinal microflora stimulates myoelectric activity of rat small intestine by promoting cyclic initiation and aboral propagation of migrating myoelectric complex. Dig Dis Sci 39:946–956
Iliev ID, Spadoni I, Mileti E et al (2009) Human intestinal epithelial cells promote the differentiation of tolerogenic dendritic cells. Gut 58:1481–1489
Ishikawa H, Akedo I, Umesaki Y et al (2003) Randomized controlled trial of the effect of bifidobacteria-fermented milk on ulcerative colitis. J Am Coll Nutr 22:56–63
Ivanov II, McKenzie BS, Zhou L et al (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17 + T helper cells. Cell 126:1121–1133
Ivanov II, de Frutos RL, Manel N et al (2008) Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4:337–349
Ivanov II, Atarashi K, Manel N et al (2009) Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139:485–498
Iwata M, Hirakiyama A, Eshima Y et al (2004) Retinoic acid imprints gut-homing specificity on T cells. Immunity 21:527–538
Jang MH, Sougawa N, Tanaka T et al (2006) CCR7 is critically important for migration of dendritic cells in intestinal lamina propria to mesenteric lymph nodes. J Immunol 176:803–810
Johansson ME, Phillipson M, Petersson J et al (2008) The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA 105:15064–15069
Johansson-Lindbom B, Svensson M, Wurbel MA et al (2003) Selective generation of gut tropic T cells in gut-associated lymphoid tissue (GALT): requirement for GALT dendritic cells and adjuvant. J Exp Med 198:963–969
Jones BD, Ghori N, Falkow S (1994) Salmonella typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer’s patches. J Exp Med 180:15–23
Kamada N, Hisamatsu T, Okamoto S et al (2008) Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest 118:2269–2280
Kaser A, Zeissig S, Blumberg RS (2010) Inflammatory bowel disease. Annu Rev Immunol 28:573–621
Kerneis S, Bogdanova A, Kraehenbuhl JP et al (1997) Conversion by Peyer’s patch lymphocytes of human enterocytes into M cells that transport bacteria. Science 277:949–952
Kim SW, Kim HM, Yang KM et al (2010) Bifidobacterium lactis inhibits NF-kappaB in intestinal epithelial cells and prevents acute colitis and colitis-associated colon cancer in mice. Inflamm Bowel Dis 16:1514–1525
Kontoyiannis D, Pasparakis M, Pizarro TT et al (1999) Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity 10:387–398
Kontoyiannis D, Boulougouris G, Manoloukos M et al (2002) Genetic dissection of the cellular pathways and signaling mechanisms in modeled tumor necrosis factor-induced Crohn’s-like inflammatory bowel disease. J Exp Med 196:1563–1574
Korn T, Bettelli E, Gao W et al (2007) IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 448:484–487
Kruis W, Schutz E, Fric P et al (1997) Double-blind comparison of an oral Escherichia coli preparation and mesalazine in maintaining remission of ulcerative colitis. Aliment Pharmacol Ther 11:853–858
Kruis W, Fric P, Pokrotnieks J et al (2004) Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut 53:1617–1623
Lahl K, Loddenkemper C, Drouin C et al (2007) Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med 204:57–63
Lahl K, Mayer CT, Bopp T et al (2009) Nonfunctional regulatory T cells and defective control of Th2 cytokine production in natural scurfy mutant mice. J Immunol 183:5662–5672
Lee SK, Kim YW, Chi SG et al (2009) The effect of Saccharomyces boulardii on human colon cells and inflammation in rats with trinitrobenzene sulfonic acid-induced colitis. Dig Dis Sci 54:255–263
LeibundGut-Landmann S, Gross O, Robinson MJ et al (2007) Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol 8:630–638
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132:27–42
Li MO, Wan YY, Flavell RA (2007) T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 26:579–591
Lin XP, Almqvist N, Telemo E (2005) Human small intestinal epithelial cells constitutively express the key elements for antigen processing and the production of exosomes. Blood Cells Mol Dis 35:122–128
Macdonald TT, Monteleone G (2005) Immunity, inflammation, and allergy in the gut. Science 307:1920–1925
MacDonald TT, Hutchings P, Choy MY et al (1990) Tumour necrosis factor-alpha and interferon-gamma production measured at the single cell level in normal and inflamed human intestine. Clin Exp Immunol 81:301–305
Mack DR, Michail S, Wei S et al (1999) Probiotics inhibit enteropathogenic E. coli adherence in vitro by inducing intestinal mucin gene expression. Am J Physiol 276(4 Pt 1):G941–G950
Madsen KL, Doyle JS, Jewell LD et al (1999) Lactobacillus species prevents colitis in interleukin 10 gene-deficient mice. Gastroenterology 116:1107–1114
Maier BR, Hentges DJ (1972) Experimental Shigella infections in laboratory animals. I. Antagonism by human normal flora components in gnotobiotic mice. Infect Immun 6:168–173
Malchow HA (1997) Crohn’s disease and Escherichia coli. A new approach in therapy to maintain remission of colonic Crohn’s disease? J Clin Gastroenterol 25:653–658
Mandal D, Levine AD (2010) Elevated IL-13Ralpha2 in intestinal epithelial cells from ulcerative colitis or colorectal cancer initiates MAPK pathway. Inflamm Bowel Dis 16:753–764
Mangan PR, Harrington LE, O’Quinn DB et al (2006) Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441:231–234
Manson JM, Rauch M, Gilmore MS (2008) The commensal microbiology of the gastrointestinal tract. Adv Exp Med Biol 635:15–28
Mao Y, Nobaek S, Kasravi B et al (1996) The effects of Lactobacillus strains and oat fiber on methotrexate-induced enterocolitis in rats. Gastroenterology 111:334–344
Marteau P, Lemann M, Seksik P et al (2006) Ineffectiveness of Lactobacillus johnsonii LA1 for prophylaxis of postoperative recurrence in Crohn’s disease: a randomised, double blind, placebo controlled GETAID trial. Gut 55:842–847
Mashimo H, Wu DC, Podolsky DK et al (1996) Impaired defense of intestinal mucosa in mice lacking intestinal trefoil factor. Science 274:262–265
Maslowski KM, Vieira AT, Ng A et al (2009) Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461:1282–1286
Matthes H, Krummenerl T, Giensch M et al (2010) Clinical trial: probiotic treatment of acute distal ulcerative colitis with rectally administered Escherichia coli Nissle 1917 (EcN). BMC Complement Altern Med 10:13
Mayer L, Eisenhardt D, Salomon P et al (1991) Expression of class II molecules on intestinal epithelial cells in humans. Differences between normal and inflammatory bowel disease. Gastroenterology 100:3–12
Mazmanian SK, Round JL, Kasper DL (2008) A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453:620–625
McGovern DP, Gardet A, Torkvist L et al (2010) Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet 42:332–337
Melmed G, Thomas LS, Lee N et al (2003) Human intestinal epithelial cells are broadly unresponsive to Toll-like receptor 2-dependent bacterial ligands: implications for host-microbial interactions in the gut. J Immunol 170:1406–1415
Metges CC (2000) Contribution of microbial amino acids to amino acid homeostasis of the host. J Nutr 130:1857S–1864S
Meylan E, Tschopp J, Karin M (2006) Intracellular pattern recognition receptors in the host response. Nature 442:39–44
Miele E, Pascarella F, Giannetti E et al (2009) Effect of a probiotic preparation (VSL#3) on induction and maintenance of remission in children with ulcerative colitis. Am J Gastroenterol 104:437–443
Moffatt MF, Kabesch M, Liang L et al (2007) Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 448:470–473
Mudter J, Amoussina L, Schenk M et al (2008) The transcription factor IFN regulatory factor-4 controls experimental colitis in mice via T cell-derived IL-6. J Clin Invest 118:2415–2426
Murphy KM, Ouyang W, Szabo SJ et al (1999) T helper differentiation proceeds through Stat1-dependent, Stat4-dependent and Stat4-independent phases. Curr Top Microbiol Immunol 238:13–26
Murphy CA, Langrish CL, Chen Y et al (2003) Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med 198:1951–1957
Naganuma M, Wiznerowicz EB, Lappas CM et al (2006) Cutting edge: Critical role for A2A adenosine receptors in the T cell-mediated regulation of colitis. J Immunol 177:2765–2769
Nakagawa I, Amano A, Mizushima N et al (2004) Autophagy defends cells against invading group A Streptococcus. Science 306:1037–1040
Nardi RM, Silva ME, Vieira EC et al (1989) Intragastric infection of germfree and conventional mice with Salmonella typhimurium. Braz J Med Biol Res 22:1389–1392
Neurath MF, Fuss I, Kelsall BL et al (1995) Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med 182:1281–1290
Neurath MF, Fuss I, Pasparakis M et al (1997) Predominant pathogenic role of tumor necrosis factor in experimental colitis in mice. Eur J Immunol 27:1743–1750
Neurath MF, Weigmann B, Finotto S et al (2002) The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J Exp Med 195:1129–1143
Niess JH (2008) Role of mucosal dendritic cells in inflammatory bowel disease. World J Gastroenterol 14:5138–5148
Niess JH, Adler G (2010) Enteric flora expands gut lamina propria CX3CR1+ dendritic cells supporting inflammatory immune responses under normal and inflammatory conditions. J Immunol 184:2026–2037
Niess JH, Brand S, Gu X et al (2005) CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 307:254–258
Niess JH, Leithauser F, Adler G et al (2008) Commensal gut flora drives the expansion of proinflammatory CD4 T cells in the colonic lamina propria under normal and inflammatory conditions. J Immunol 180:559–568
Okazawa A, Kanai T, Watanabe M et al (2002) Th1-mediated intestinal inflammation in Crohn’s disease may be induced by activation of lamina propria lymphocytes through synergistic stimulation of interleukin-12 and interleukin-18 without T cell receptor engagement. Am J Gastroenterol 97:3108–3117
Oppmann B, Lesley R, Blom B et al (2000) Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13:715–725
Owen RL, Jones AL (1974) Epithelial cell specialization within human Peyer’s patches: an ultrastructural study of intestinal lymphoid follicles. Gastroenterology 66:189–203
Preising J, Philippe D, Gleinser M et al (2010) Selection of bifidobacteria based on adhesion and anti-inflammatory capacity in vitro for amelioration of murine colitis. Appl Environ Microbiol 76:3048–3051
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F et al (2004) Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118:229–241
Rakoff-Nahoum S, Hao L, Medzhitov R (2006) Role of toll-like receptors in spontaneous commensal-dependent colitis. Immunity 25:319–329
Ramare F, Nicoli J, Dabard J et al (1993) Trypsin-dependent production of an antibacterial substance by a human Peptostreptococcus strain in gnotobiotic rats and in vitro. Appl Environ Microbiol 59:2876–2883
Reddy BS, Wostmann BS, Pleasants JR (1965) Iron, copper, and manganese in germfree and conventional rats. J Nutr 86:159–168
Rescigno M, Urbano M, Valzasina B et al (2001) Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol 2:361–367
Riley MA, Wertz JE (2002) Bacteriocin diversity: ecological and evolutionary perspectives. Biochimie 84:357–364
Roberfroid MB (1998) Prebiotics and synbiotics: concepts and nutritional properties. Br J Nutr 80:S197–S202
Rolfe VE, Fortun PJ, Hawkey CJ et al (2006) Probiotics for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev (4):CD004826
Rubic T, Lametschwandtner G, Jost S et al (2008) Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol 9:1261–1269
Rubtsov YP, Rasmussen JP, Chi EY et al (2008) Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28:546–558
Sakaguchi T, Kohler H, Gu X et al (2002) Shigella flexneri regulates tight junction-associated proteins in human intestinal epithelial cells. Cell Microbiol 4:367–381
Salazar-Gonzalez RM, Niess JH, Zammit DJ et al (2006) CCR6-mediated dendritic cell activation of pathogen-specific T cells in Peyer’s patches. Immunity 24:623–632
Schulz O, Jaensson E, Persson EK et al (2009) Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J Exp Med 206:3101–3114
Shortman K, Liu YJ (2002) Mouse and human dendritic cell subtypes. Nat Rev Immunol 2:151–161
Siddiqui KR, Laffont S, Powrie F (2010) E-cadherin marks a subset of inflammatory dendritic cells that promote T cell-mediated colitis. Immunity 32:557–567
Smits HH, Engering A, van der Kleij D et al (2005) Selective probiotic bacteria induce IL-10-producing regulatory T cells in vitro by modulating dendritic cell function through dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin. J Allergy Clin Immunol 115:1260–1267
Sokol H, Pigneur B, Watterlot L et al (2008) Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA 105:16731–16736
Sokol CL, Chu NQ, Yu S et al (2009) Basophils function as antigen-presenting cells for an allergen-induced T helper type 2 response. Nat Immunol 10:713–720
Sood A, Midha V, Makharia GK et al (2009) The probiotic preparation, VSL#3 induces remission in patients with mild-to-moderately active ulcerative colitis. Clin Gastroenterol Hepatol 7:1202–1209, 1209 e1
Sougioultzis S, Simeonidis S, Bhaskar KR et al (2006) Saccharomyces boulardii produces a soluble anti-inflammatory factor that inhibits NF-kappaB-mediated IL-8 gene expression. Biochem Biophys Res Commun 343:69–76
Stappenbeck TS, Hooper LV, Gordon JI (2002) Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci USA 99:15451–15455
Steidler L, Hans W, Schotte L et al (2000) Treatment of murine colitis by Lactococcus lactis secreting interleukin-10. Science 289:1352–1355
Summers RW, Elliott DE, Qadir K et al (2003) Trichuris suis seems to be safe and possibly effective in the treatment of inflammatory bowel disease. Am J Gastroenterol 98:2034–2041
Summers RW, Elliott DE, Urban JF Jr et al (2005a) Trichuris suis therapy in Crohn’s disease. Gut 54:87–90
Summers RW, Elliott DE, Urban JF Jr et al (2005b) Trichuris suis therapy for active ulcerative colitis: a randomized controlled trial. Gastroenterology 128:825–832
Sun CM, Hall JA, Blank RB et al (2007) Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med 204:1775–1785
Takeda K, Kaisho T, Akira S (2003) Toll-like receptors. Annu Rev Immunol 21:335–376
Taupin DR, Kinoshita K, Podolsky DK (2000) Intestinal trefoil factor confers colonic epithelial resistance to apoptosis. Proc Natl Acad Sci USA 97:799–804
Taylor BC, Zaph C, Troy AE et al (2009) TSLP regulates intestinal immunity and inflammation in mouse models of helminth infection and colitis. J Exp Med 206:655–667
Tsai F, Coyle WJ (2009) The microbiome and obesity: is obesity linked to our gut flora? Curr Gastroenterol Rep 11:307–313
Tursi A, Brandimarte G, Papa A et al (2010) Treatment of relapsing mild-to-moderate ulcerative colitis with the probiotic VSL#3 as adjunctive to a standard pharmaceutical treatment: a double-blind, randomized, placebo-controlled study. Am J Gastroenterol 105:2218–2227
Tysk C, Lindberg E, Jarnerot G et al (1988) Ulcerative colitis and Crohn’s disease in an unselected population of monozygotic and dizygotic twins. A study of heritability and the influence of smoking. Gut 29:990–996
Uhlig HH, McKenzie BS, Hue S et al (2006) Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 25:309–318
Vaishnava S, Behrendt CL, Ismail AS et al (2008) Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci USA 105:20858–20863
van Beelen AJ, Zelinkova Z, Taanman-Kueter EW et al (2007) Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity 27:660–669
Van Gossum A, Dewit O, Louis E et al (2007) Multicenter randomized-controlled clinical trial of probiotics (Lactobacillus johnsonii, LA1) on early endoscopic recurrence of Crohn’s disease after lleo-caecal resection. Inflamm Bowel Dis 13:135–142
Varol C, Vallon-Eberhard A, Elinav E et al (2009) Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity 31:502–512
Veldhoen M, Hocking RJ, Atkins CJ et al (2006) TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24:179–189
Veldhoen M, Hirota K, Westendorf AM et al (2008) The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453:106–109
von der Weid T, Bulliard C, Schiffrin EJ (2001) Induction by a lactic acid bacterium of a population of CD4(+) T cells with low proliferative capacity that produce transforming growth factor beta and interleukin-10. Clin Diagn Lab Immunol 8:695–701
Wan YY, Flavell RA (2007) Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature 445:766–770
Wehkamp J, Harder J, Wehkamp K et al (2004) NF-kappaB- and AP-1-mediated induction of human beta defensin-2 in intestinal epithelial cells by Escherichia coli Nissle 1917: a novel effect of a probiotic bacterium. Infect Immun 72:5750–5758
Westendorf AM, Bruder D, Hansen W et al (2006) Intestinal epithelial antigen induces CD4 + T cells with regulatory phenotype in a transgenic autoimmune mouse model. Ann N Y Acad Sci 1072:401–406
Whitman WB, Coleman DC, Wiebe WJ (1998) Prokaryotes: the unseen majority. Proc Natl Acad Sci USA 95:6578–6583
Wostmann BS, Larkin C, Moriarty A et al (1983) Dietary intake, energy metabolism, and excretory losses of adult male germfree Wistar rats. Lab Anim Sci 33:46–50
Yang XO, Chang SH, Park H et al (2008) Regulation of inflammatory responses by IL-17F. J Exp Med 205:1063–1075
Yen D, Cheung J, Scheerens H et al (2006) IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest 116:1310–1316
Zaph C, Troy AE, Taylor BC et al (2007) Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature 446:552–556
Zarek PE, Huang CT, Lutz ER et al (2008) A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 111:251–259
Zenewicz LA, Yancopoulos GD, Valenzuela DM et al (2008) Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 29:947–957
Zheng W, Flavell RA (1997) The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89:587–596
Zhou L, Ivanov II, Spolski R et al (2007) IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol 8:967–974
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft, No. Ni575/6-2 and Ni575/7-1.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Stephani, J., Radulovic, K. & Niess, J.H. Gut Microbiota, Probiotics and Inflammatory Bowel Disease. Arch. Immunol. Ther. Exp. 59, 161–177 (2011). https://doi.org/10.1007/s00005-011-0122-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-011-0122-5