
Abstract
The influence of iron on immune function has been long appreciated. However, the molecular basis for this interaction is less well understood. Recently, there have been several important advances that have shed light on the mechanisms that regulate mammalian iron metabolism. The new insights provide a conceptual framework for understanding and manipulating the cross-talk between iron homeostasis and the immune system. This article will review what is currently known about how disturbances of iron metabolism can affect immunity and how activation of the immune system can lead to alterations in iron balance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Iron is essential for almost all living organisms and takes part in a number of important biological processes. Its ability to switch between multiple oxidation states makes it an important co-factor in electron transfer and oxidation–reduction reactions, and also allows it to interact reversibly with other atoms, especially oxygen, sulfur and nitrogen (Beard 2001). However, free iron can be cytotoxic when present at high concentrations because it can catalyze the formation of oxidative radicals that damage proteins, lipids and nucleic acids. Thus, both iron deficiency and iron excess can have adverse effects on a variety of cell, tissue and organ functions, but it has been difficult, in general, to link such functional abnormalities to alterations in specific iron-dependent biochemical reactions. Given the problems associated with having too little or too much iron, it should come as no surprise that mammals have evolved mechanisms for precise regulation of extra- and intracellular iron levels. States of iron deficiency or overload can occur when these mechanisms go awry or when the amount of iron entering the system falls outside physiologic limits. Such disorders of iron metabolism are seen either as a primary abnormality or secondary to other disease states. Iron deficiency resulting from inadequate dietary intake is the most widely prevalent of these disorders, particularly in the developing world (Oppenheimer 2001). Iron overload is less frequently seen, but affects significant numbers of people in the form of hereditary hemochromatosis or in association with hemolytic anemias, repeated blood transfusions, dietary excess, certain infections and alcoholic liver disease (Beaumont and Delaby 2009; Lee and Beutler 2009).
There is a well-described, but complex and rather poorly understood, interaction between iron status and immune function, particularly with respect to the incidence and course of infectious disease (Schaible and Kaufmann 2004). Iron deficiency has been reported to be associated with increased susceptibility to infection in both humans and experimental animals, but the results of many of these studies have been conflicting and hard to interpret because of variations in baseline iron status, the severity of the deficiency and the possibility of co-existing nutritional problems (Oppenheimer 2001). The situation is further complicated by observations showing that iron supplementation in humans, particularly in the tropics, can increase the risk of infections such as malaria and tuberculosis (Murray et al. 1978; Oppenheimer 2001; Sazawal et al. 2006). Iron overload caused by dietary excess, abnormal hemolysis or inherited disorders is also associated with heightened susceptibility to such infections (Gangaidzo et al. 2001; Magnus et al. 1999; Moyo et al. 1997; Wanachiwanawin 2000). While some of these associations may reflect the effects of iron on pathogen growth, they may also be the outcome of iron’s influence on the host response to the pathogen. The latter possibility has not received much attention until relatively recently, and there is a need for additional work to characterize the various ways in which iron can alter the immune response. Abnormalities of iron-dependent biochemical reactions undoubtedly contribute to the effects of iron on the immune system but, as alluded to earlier, connecting such changes in biochemistry to alterations in immune cell function has not been possible except in a few instances. The situation is even more difficult when trying to make links to immune dysfunction at the level of a clinical disorder. Accordingly, this review will focus largely on how changes in systemic iron homeostasis affect immune responses (and vice versa) and, for the most part, will not deal with the more thorny issue of trying to explain immunological abnormalities on the basis of alterations in specific biochemical reactions. That will have to be left as a challenge for the future.
Molecular Control of Iron Homeostasis
The last few years have seen a number of dramatic advances in our understanding of the mechanisms that regulate normal iron balance (Andrews 2008). In particular, the identification, cloning and characterization of the molecules that mediate the movement of iron into and out of cells, and elucidation of the ways in which these molecules are influenced by iron status, have led to important insights into how iron absorption, recycling and distribution are modulated in response to changing requirements. This information will provide the conceptual framework for discussing the interactions between iron and immunity.
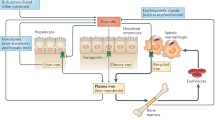
Iron metabolism is tightly regulated so that the amounts of the metal entering the circulation from its two major sources—macrophages that recycle iron from red blood cells (RBCs) and duodenal epithelial cells that absorb iron from the diet—are kept in balance with systemic requirements (Andrews and Schmidt 2007). A simplified scheme of what is involved in normal iron homeostasis is shown in Fig. 1. Iron released from phagocytosed RBCs is transported from the phagosomal lumen into the cytosol by the divalent metal transporter natural resistance-associated macrophage protein 2 (Nramp2), with the related protein Nramp1 also contributing to this process (Biggs et al. 2001; Soe-Lin et al. 2008, 2009). Nramp2 functions similarly at the apical surface of duodenal enterocytes to absorb iron from the intestinal lumen. Iron is effluxed into the plasma from both macrophages and duodenal enterocytes by the transporter ferroportin (FPN), which is expressed on the plasma membrane of the former cell type and on the basolateral surface of the latter. Iron circulates complexed to transferrin and is used by cells following internalization by the type I transferrin receptor (TfR1). The levels of FPN, and therefore the amount of iron released into circulation, are regulated in response to needs. A key factor in this regulation is the peptide hepcidin, which is secreted by hepatocytes. Hepcidin binds to FPN and induces its Janus kinase 2 (Jak2)-dependent phosphorylation, internalization and lysosomal degradation, thereby decreasing cellular iron efflux from macrophages and duodenal enterocytes (De Domenico et al. 2007, 2009; Nemeth et al. 2004b). Conditions that increase the demand for iron (iron deficiency, hypoxia, anemia) lead to decreased expression of hepcidin, resulting in higher levels of FPN and a corresponding increase in circulating iron. Conversely, elevated iron levels, as well as inflammatory cytokines, lead to increased hepcidin production and consequent down-regulation of FPN, with intracellular retention of iron and decreased circulating levels of the element.

Regulation of systemic iron homeostasis. Hepcidin controls entry of iron into the circulation by modulating expression of FPN on phagocytes that recycle iron from aged RBCs and on duodenal enterocytes that absorb dietary iron from the intestinal lumen. Hepcidin expression in the liver is regulated by systemic iron levels and requirements, and by inflammatory signals. Green arrows indicate activating responses, red lines with circles designate inhibitory effects, and black arrows indicate movement of iron
The mechanisms that regulate hepcidin expression have been revealed by recent studies (Fig. 2). Circulating iron-transferrin complexes are sensed by HFE, a non-classical class I MHC protein expressed on the surface of hepatocytes and other cells (Feder et al. 1996). When iron-transferrin levels are high, HFE is displaced from its association with TfR1 and binds to the hepatocyte-specific TfR2. The latter interaction activates signals that induce the transcriptional up-regulation of hepcidin (Goswami and Andrews 2006; Schmidt et al. 2008). Recent findings indicate that HFE-dependent signals function together with signals induced by the binding of bone morphogenetic proteins (BMPs) to the BMP receptor and the co-receptor hemojuvelin (HJV; Andriopoulos et al. 2009; Babitt et al. 2006; Meynard et al. 2009). These signals lead to activation of the similar to mothers against decapentaplegic 4 transcription factor, which acts directly on the hepcidin promoter (Wang et al. 2005). Pro-inflammatory cytokines, including interleukin (IL)-6, tumor necrosis factor (TNF)-α and IL-1, constitute another important stimulus for the transcriptional up-regulation of hepcidin (Lee et al. 2004, 2005; Nemeth et al. 2004a). The effect of IL-6 on hepcidin expression is mediated by the signal transducer and activator of transcription 3 (STAT3) transcription factor and is a major factor in the pathogenesis of the anemia associated with chronic inflammation, an issue that will be discussed in more detail later in this article (Nemeth et al. 2004a; Verga Falzappa et al. 2007; Wrighting and Andrews 2006). Mechanisms that actively inhibit transcription of the hepcidin gene are brought into play under conditions of increased iron demand, including up-regulation of the transcription factor hypoxia-inducible factor (HIF)-1α by hypoxia and iron deficiency (Peyssonnaux et al. 2007), increased levels of growth differentiation factor 15 in certain hemolytic anemias (Tanno et al. 2007), and cleavage of membrane HJV by the serine protease matriptase 2 in response to iron deficiency (Silvestri et al. 2008).

Regulation of hepcidin expression in hepatocytes. Hepcidin expression is regulated mainly at the level of transcription and is modulated by activating (green arrows) or inhibitory (red lines with circles) signals induced by systemic iron status, conditions that alter iron requirements, and inflammation
Intracellular free iron concentrations depend on circulating levels, but are also controlled by regulated changes in expression of TfR1 and the cytoplasmic iron storage protein ferritin. The expression of these proteins is controlled post-transcriptionally via iron response elements (IREs) that are found in the 3′ untranslated region of the TfR1 mRNA and in the 5′ untranslated region of the ferritin mRNA. Under conditions of low cytosolic iron, trans-acting regulatory proteins (IRE-binding proteins, IRPs) bind to the IREs and promote mRNA stability in the case of TfR1, and inhibit mRNA translation in the case of ferritin (Muckenthaler et al. 2008). These changes lead to increased TfR1 expression and decreased ferritin expression, both of which facilitate an increase in cytosolic iron levels. The IRE-IRP system thus ensures that the expression levels of proteins involved in cellular iron uptake and storage are coordinately regulated to maintain normal intracellular iron concentrations.
Mutations in the HFE gene are associated with the most common inherited disorder of iron metabolism, type I hemochromatosis (Pietrangelo 2006). In individuals with this disease, as well as in Hfe knock-out mice, the absence of functional HFE impairs iron sensing, leading to abnormally low hepcidin levels, elevated FPN expression, increased iron release from macrophages and enterocytes and, ultimately, an iron overload state. Other forms of hemochromatosis (types II and III) are also associated with inappropriately low levels of hepcidin and are linked to mutations in the gene for hepcidin itself, or in those genes such as TfR2 and HJV that are involved in inducing hepcidin expression. Type IV hemochromatosis is caused by mutations in the FPN gene and has a variable clinical phenotype that includes accumulation of iron within phagocytic cells.
Iron and Innate Immunity
The innate immune response is the first line of defense against infectious and noxious challenges. It is activated rapidly, within minutes to hours, and typically involves cells at cutaneous and mucosal surfaces. These cells use germ-line encoded pattern recognition receptors to sense the presence of microbial molecules or products of tissue damage, and mount a stereotypic response that includes the activation of anti-microbial mechanisms and the secretion of chemotactic molecules that recruit other cells of the immune system to the site of infection or damage (Kawai and Akira 2010). The effects of iron on innate anti-microbial defenses have been studied for some time using tissue culture and animal models (Wang and Cherayil 2009). The data from these studies have not always been concordant. Iron chelators such as desferrioxamine inhibit the activity of phagocyte oxidase and reduce reactive oxygen intermediate-dependent killing of bacterial pathogens, both in vitro and in vivo (Collins et al. 2002). On the other hand, iron chelation increases, and iron inhibits, expression of another important anti-microbial molecule, inducible nitric oxide synthase (iNOS), effects that are likely to be mediated by iron-dependent changes in transcription of the iNOS gene (Dlaska and Weiss 1999; Melillo et al. 1997; Weiss et al. 1994). Iron has also been shown to influence the activation of NF-κB, a transcription factor that is required for the expression of a number of genes involved in innate immunity and inflammation (Vallabhapurapu and Karin 2009). Elevated intracellular iron promotes the activation of NF-κB, in part by increasing the production of reactive oxygen intermediates, while reduced intracellular iron inhibits the phosphorylation of the RelA sub-unit of NF-κB that is required for its activity (Bubici et al. 2006; Chen et al. 2007; Seldon et al. 2007). Another iron-regulated transcription factor that has been shown to play an important role in innate immune responses, particularly the expression of inflammatory cytokines and anti-microbial peptides by macrophages, is HIF-1α (Nizet and Johnson 2009). Under basal conditions, HIF-1α is prolyl hydroxylated by the action of an iron- and oxygen-dependent prolyl hydroxylase and thereby targeted for degradation by the proteasome (Kaelin and Ratcliffe 2008). When intracellular iron concentrations are low, prolyl hydroxylation of HIF-1α is reduced and the protein is spared from degradation. Thus, in contrast to the effects on NF-κB, low intracellular iron promotes HIF-1α-dependent gene expression by preventing the degradation of the transcription factor.
How is one to make sense of all these conflicting observations? One interesting idea is that a deviation of intracellular iron levels away from the norm could act as a “danger” signal. In this context, it would be logical that both an abnormal increase and a decrease in intracellular iron would activate inflammatory and anti-microbial mechanisms via NF-κB and HIF-1, respectively. In keeping with this idea, it has been shown recently that siderophores released by pathogenic bacteria can activate HIF-1-dependent responses, demonstrating that lowering of intracellular iron can function as an indicator of infection (Hartmann et al. 2008).
The discovery of FPN’s role in cellular iron efflux sparked considerable interest in the potential involvement of this protein in immune function, particularly since it is found on macrophages and since its expression and/or function can be altered in anemias, iron deficiency states and hemochromatosis (Andrews and Schmidt 2007; Pietrangelo 2006). Several groups have reported that increased expression of FPN on macrophages inhibited the intracellular growth of pathogens such as Salmonella typhimurium, Mycobacterium tuberculosis, Chlamydia psittaci, Chlamydia trachomatis and Legionella pneumophila by lowering cellular iron levels (Chlosta et al. 2006; Olakanmi et al. 2007; Paradkar et al. 2008). Conversely, hepcidin-induced down-regulation of FPN, or expression of a dominant negative mutant of FPN that blocked iron efflux, had the opposite effect (Chlosta et al. 2006; Paradkar et al. 2008). Taken together, these studies strongly support the notion that changes in FPN expression or function can have a significant impact on intracellular pathogen growth by altering the amount of iron available for microbial acquisition. In this context, the transcriptional up-regulation of FPN that occurs following infection with S. typhimurium or M. tuberculosis or following exposure to interferon (IFN)-γ (Nairz et al. 2007; Van Zandt et al. 2008) could be viewed as an innate anti-microbial defense mechanism based on iron deprivation. This increase in FPN expression could be particularly important in counteracting the post-translational down-regulation of FPN mediated by the increased circulating levels of hepcidin associated with infection (Andrews and Schmidt 2007), and would complement other iron sequestration mechanisms mediated by proteins such as Nramp1, lactoferrin and siderocalin, all of which have well-described roles in innate immunity (Goetz et al. 2002; Marquis and Gros 2008; Wang and Cherayil 2009; Ward et al. 2005).
In contrast to the results of the tissue culture experiments described above, the dominant effect of altered macrophage FPN expression in vivo may be on the inflammatory response rather than on intracellular pathogen growth. Wang et al. (2008) found that Hfe knock-out mice, a model of type I hemochromatosis in which macrophage FPN levels are elevated, had significantly attenuated Salmonella-induced intestinal inflammation compared to wild-type controls, whereas their tissue pathogen burden was increased. Macrophages isolated from the knock-out animals produced lower amounts of TNF-α and IL-6 than wild-type cells when infected with Salmonella or treated with lipopolysaccharide (LPS) in vitro. This abnormality, which resembles that seen in humans with hereditary hemochromatosis (Gordeuk et al. 1992), was associated with impaired activation of specific signaling pathways downstream of Toll-like receptor 4 (TLR4), the receptor for LPS, and could be corrected by treating the cells with hepcidin or ferrous sulfate, consistent with the idea that the low intra-macrophage iron concentrations characteristic of Hfe deficiency contributed to the impaired cytokine response (Wang et al. 2009). The biochemical mechanism responsible for the effects of low intra-macrophage iron levels on TLR4 signaling awaits clarification. It should be noted that the effects of FPN on inflammatory responses have not been consistently seen by all investigators. Nairz et al. (2009) found that Hfe knock-out mice were more resistant to Salmonella infection than wild-type animals and did not observe any abnormalities of inflammatory cytokine expression in the former. The explanation for the discordant results in the two studies is currently unclear. Very recently published work has revealed yet another aspect of hepcidin’s effects on macrophage inflammatory responses. De Domenico et al. (2010) found that the hepcidin-FPN interaction, in addition to activating Jak2-dependent phosphorylation of FPN, also activated Jak2-mediated phosphorylation of STAT3 and the consequent transcriptional modulation of a large number of macrophage genes. Strikingly, the hepcidin-induced changes in gene expression resulted in the suppression of TNF-α and IL-6 production, and correspondingly, hepcidin-deficient mice had higher serum levels of these cytokines following exposure to LPS. One consequence of this anti-inflammatory effect was that mice administered hepcidin were protected from LPS-induced lethality. Thus it appears that hepcidin may influence the inflammatory response in multiple ways, including alteration of intra-macrophage iron levels and by inducing Jak2/STAT3-dependent changes in gene expression. It should also be noted in this context that macrophages themselves have been shown to express hepcidin, as have neutrophils (Koening et al. 2009; Liu et al. 2005; Peyssonnaux et al. 2006). This local production of hepcidin at sites of infection, which occurs in a TLR-dependent fashion, could add to the effects of the liver-derived peptide.
Iron and Adaptive Immunity
Adaptive immunity develops over the course of days to weeks following exposure to a foreign substance and depends on the activation, proliferation and differentiation of antigen-specific B and T lymphocytes, which are involved in antibody- and cell-mediated responses, respectively (Bonilla and Oettgen 2010). The clonotypic receptors that these cells use to recognize and respond to antigen are derived from a highly ordered and regulated process of somatic gene rearrangement that occurs during development in the bone marrow (in the case of B cells) or the thymus (in the case of T cells). Lymphocyte development in the mouse is dependent on the ability to acquire iron via TfR1. Recombination activating gene 2-deficient blastocyst complementation experiments demonstrated that the absence of TfR1 resulted in the complete arrest of T cell differentiation at the triple negative (CD3−CD4−CD8−) stage, while B cell development was less severely affected and allowed the emergence of some IgM+ cells (Ned et al. 2003). It is likely that TfR1-dependent iron acquisition is required to support the extensive cell proliferation that is involved in lymphocyte development, but why the T cell lineage is particularly sensitive to iron deprivation is not clear. Interestingly, patients with congenital atransferrinemia do not appear to have any abnormalities in the generation of B and T lymphocytes, suggesting that transferrin-independent pathways of iron acquisition may be sufficient to support this process in humans (Goya et al. 1972; Hamill et al. 1991). Nevertheless, the in vitro polyclonal proliferation of human B and T lymphocytes can be inhibited by antibodies to TfR1, indicating a requirement for iron uptake during cell division (Kemp et al. 1989; Neckers et al. 1984). In keeping with these observations, several studies in animals and humans have shown that nutritional iron deficiency is associated with impaired phytohemagglutinin-induced lymphocyte proliferation and delayed-type hypersensitivity responses with relative preservation of humoral immunity (e.g., Kuvibidila et al. 1981; Macdougall et al. 1975; Srikantia et al. 1976; also reviewed in Oppenheimer 2001). There is also some evidence to suggest that iron deficiency in humans can alter the cytokine expression profile of activated lymphocytes, leading to a higher proportion of cells expressing IFN-γ and a lower proportion expressing IL-4 (Jason et al. 2001).
The data on the effects of iron overload on lymphocyte function are not very informative. Some studies on patients with HFE-associated hemochromatosis indicate a decrease in circulating lymphocytes, with CD8+ cells being particularly affected (Barton et al. 2005; Macedo et al. 2010), but these abnormalities have not been observed consistently. Experimental iron overload in rodents is not associated with altered lymphocyte numbers, but has been suggested to affect proliferative and cytokine responses (Melo et al. 1997; Mencacci et al. 1997; Wu et al. 1990). This is an area that would benefit from further research, especially in the context of the more recent developments in the iron metabolism field.
Effects of Inflammation on Iron Homeostasis
Just as iron status influences immune function, immune responses can also lead to alterations in iron metabolism. Chronic inflammatory conditions in both humans and experimental animals result in the development of low serum iron levels and a mild to moderate, normocytic, normochromic anemia known as the anemia of chronic disease (Andrews 2008; Ganz and Nemeth 2009). The anemia is typically refractory to oral iron supplementation and can be an important factor in the quality of life of affected individuals. The pathophysiology of this condition has become clearer following the discovery of the central role played by hepcidin in iron homeostasis. Pro-inflammatory cytokines such as IL-6, TNF-α and IL-1 have been shown to increase expression of hepcidin, which in turn leads to down-regulation of FPN on enterocytes and macrophages and consequent intracellular sequestration of iron. The resultant hypoferremia may have adaptive value by depriving extracellular pathogens of an essential nutrient, but it also leads to impaired erythropoiesis and the development of anemia. Hepcidin levels have been found to be elevated in inflammatory bowel disease, rheumatoid arthritis and lupus, at least in some studies (Demirag et al. 2009; Semrin et al. 2006; Zhang et al. 2008). Obesity, which is increasingly being recognized as a chronic inflammatory state, also has been associated recently with elevated hepcidin levels and iron deficiency (del Giudice et al. 2009; McClung and Karl 2009). Adipocyte-derived inflammatory mediators, as well as adipokines such as leptin, are likely to contribute to the induction of hepcidin in the liver, but interestingly, adipocytes themselves have been shown to be a source of hepcidin in obese individuals (Bekri et al. 2006; Chung et al. 2007; Lago et al. 2007).
Recent experiments in mice have raised the possibility that the interaction between iron metabolism and inflammation can be manipulated for therapeutic purposes. Studies with Hfe knock-out mice indicate that low circulating hepcidin levels are associated with attenuation of inflammatory responses in vitro and in vivo (Wang et al. 2008). On the other hand, addition of hepcidin to macrophages results in enhanced production of inflammatory cytokines (Wang et al. 2009). Thus, hepcidin could be considered to have pro-inflammatory effects in addition to its well-known role in iron homeostasis. Furthermore, since chronic inflammatory conditions such as inflammatory bowel disease are often associated with up-regulation of hepcidin expression (Andrews 2008; Demirag et al. 2009; Ganz and Nemeth 2009; Semrin et al. 2006; Zhang et al. 2008), the observations of Wang et al. (2009) suggest that hepcidin may contribute to a vicious cycle that perpetuates the inflammatory state and, therefore, that blocking hepcidin expression or function could help to reduce inflammation. There is experimental support for this idea since it has been shown recently that pharmacologic inhibition of hepcidin expression attenuated intestinal inflammation in mouse models of both infectious and noninfectious colitis (Wang et al. 2009). This approach has the added potential benefit of helping to correct the anemia of chronic disease that results from the elevated hepcidin levels (Andrews 2008; Ganz and Nemeth 2009). Further studies are required to determine whether this novel anti-inflammatory strategy is sufficiently robust for clinical application. It is also important to note that manipulating hepcidin expression and iron homeostasis may have adverse effects. Although it is formally possible that inhibiting hepcidin expression may promote an iron overload state, this is unlikely in practice since inflammatory conditions are associated with elevated hepcidin levels and the goal of treatment would be to normalize these levels. A more important consideration is whether lowering intramacrophage iron by inhibiting hepcidin expression may compromise anti-microbial defenses, especially since iron is an important factor in microbicidal mechanisms such as phagocyte oxidase and iNOS (Flannagan et al. 2009). This is an issue that will need to be addressed before anti-inflammatory strategies based on hepcidin inhibition can be applied.
Conclusion
It is clear from the foregoing discussion that both iron deficiency and iron excess can influence the functioning of the innate and adaptive arms of the immune system. Iron can also have direct effects on the growth and virulence of microbial pathogens. Indeed, an important component of innate anti-microbial defense is based on depriving pathogens of this nutrient. Changes in iron status can thus affect the immune response in multiple ways, particularly in the context of infection, an idea that is worth remembering when considering the value of iron supplementation in areas of the world where infections such as malaria and tuberculosis are highly prevalent. Conversely, chronic immune activation can lead to alterations in iron homeostasis that may impair erythropoiesis and contribute to immunopathology.
With the recent advances in our understanding of how iron metabolism is regulated at the molecular level, we are gaining significant new insights into the bidirectional interaction between iron and immunity. Additional information on how systemic and cellular iron homeostasis is regulated at the molecular level will undoubtedly emerge over the next few years. The challenge for immunologists will be to integrate this information with what is known about the functioning of the immune system under both normal and pathological circumstances. One area that would benefit from this type of integration is the impact of iron on lymphocyte biology and adaptive immunity, particularly with respect to T effector cell function. At the same time, the ever-increasing numbers of cytokines, T cell subsets and immunomodulators that are being discovered by immunologists deserve attention from those in the metabolism field as potential regulators of iron homeostasis. Such immunological factors could help to explain the derangements of iron balance that are associated with chronic infectious and inflammatory diseases. Returning to an issue raised earlier in this article, it is also important to mention that the link between the abnormalities of immune function associated with disorders of iron homeostasis and the actual biochemical reactions that are affected by changes in cellular iron levels remains a black box in most cases. Some light is starting to appear in the darkness, however. The transcription factor HIF-1α, which has well-known functions in the immune response, is a good example of successful tracing of the effects of iron down to the biochemical level (Kaelin and Ratcliffe 2008). Iron-dependent modulation of NF-κB activation (Bubici et al. 2006; Chen et al. 2007; Seldon et al. 2007) and TLR4 signaling (Wang et al. 2009), as well as the recent identification of an iron-dependent ubiquitin ligase (Salahudeen et al. 2009; Vashisht et al. 2009) and the role of iron in regulating the tyrosine phosphatase SHP-1 (Gomez et al. 2010), represent starting points for a similar biochemical dissection of the effects of iron on immunologically relevant processes and molecules. There is much that remains to be learned from studies at the interphase between iron metabolism and immunology. Knowledge gleaned from such studies will be both fundamentally interesting and practically relevant to a broad spectrum of clinical problems.
Abbreviations
- BMP:
-
Bone morphogenetic protein
- FPN:
-
Ferroportin
- HIF:
-
Hypoxia-inducible factor
- HJV:
-
Hemojuvelin
- IFN:
-
Interferon
- IL:
-
Interleukin
- iNOS:
-
Inducible nitric oxide synthase
- IRE:
-
Iron response element
- IRP:
-
IRE-binding protein
- Jak2:
-
Janus kinase 2
- LPS:
-
Lipopolysaccharide
- Nramp:
-
Natural resistance-associated macrophage protein
- RBC:
-
Red blood cell
- TfR:
-
Transferrin receptor
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- STAT3:
-
Signal transducer and activator of transcription 3
References
Andrews NC (2008) Forging a field: the golden age of iron biology. Blood 112:219–230
Andrews NC, Schmidt PJ (2007) Iron homeostasis. Annu Rev Physiol 69:69–85
Andriopoulos B, Corradini E, Xia Y et al (2009) BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet 41:482–487
Babitt JL, Huang FW, Wrighting DM et al (2006) Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet 38:531–539
Barton JC, Wiener HW, Acton RT et al (2005) Total blood lymphocyte counts in hemochromatosis probands with HFE C282Y homozygosity: relationship to severity of iron overload and HLA-A and -B alleles and haplotypes. BMC Blood Disord 5:5
Beard JL (2001) Iron biology in immune function, muscle metabolism and neuronal functioning. J Nutr 131:568S–580S
Beaumont C, Delaby C (2009) Recycling iron in normal and pathological states. Semin Hematol 46:328–338
Bekri S, Gual P, Anty R et al (2006) Increased adipose tissue expression of hepcidin in severe obesity is independent from diabetes and NASH. Gastroenterology 131:788–796
Biggs TE, Baker ST, Botham MS et al (2001) Nramp1 modulates iron homeostasis in vivo and in vitro: evidence for a role in cellular iron release involving de-acidification of intracellular vesicles. Eur J Immunol 31:2060–2070
Bonilla FA, Oettgen HC (2010) Adaptive immunity. J Allergy Clin Immunol 125(suppl 2):S33–S40
Bubici C, Papa S, Dean K et al (2006) Mutual cross-talk between reactive oxygen species and NF-kappaB: molecular basis and biological significance. Oncogene 25:6731–6748
Chen L, Xiong S, She H et al (2007) Iron causes interactions of TAK1, p21ras and phosphatidylinositol-3-kinase in caveolae to activate I-kappaB kinase in hepatic macrophages. J Biol Chem 282:5582–5588
Chlosta S, Fishman DS, Harrington L et al (2006) The iron efflux protein ferroportin regulates the intracellular growth of Salmonella enterica. Infect Immun 74:3065–3067
Chung B, Matak P, McKie AT et al (2007) Leptin increases the expression of the iron regulatory hormone hepcidin in HuH7 human hepatoma cells. J Nutr 137:2366–2370
Collins HL, Kaufmann SH, Schaible UE (2002) Iron chelation via deferoxamine exacerbates experimental salmonellosis via inhibition of the nicotinamide adenine dinucleotide phosphate oxidase-dependent respiratory burst. J Immunol 168:3458–3463
De Domenico I, Ward DM, Langelier C et al (2007) The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol Biol Cell 18:2569–2578
De Domenico I, Lo E, Ward DM et al (2009) Hepcidin-induced internalization of ferroportin requires binding and cooperative interaction with Jak2. Proc Natl Acad Sci USA 106:3800–3805
De Domenico I, Zhang TY, Koening CL et al (2010) Hepcidin mediates transcriptional changes that modulate acute cytokine-induced inflammatory responses in mice. J Clin Invest 120:2395–2405
del Giudice EM, Santoro N, Amato A et al (2009) Hepcidin in obese children as a potential mediator of the association between obesity and iron deficiency. J Clin Endocrinol Metab 94:5102–5107
Demirag MD, Haznedaroglu S, Sancak B et al (2009) Circulating hepcidin in the crossroads of anemia and inflammation associated with rheumatoid arthritis. Intern Med 48:421–426
Dlaska M, Weiss G (1999) Central role of transcription factor NF-IL6 for cytokine and iron-mediated regulation of murine inducible nitric oxide synthase expression. J Immunol 162:6171–6177
Feder JN, Gnirke A, Thomas W et al (1996) A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 13:399–408
Flannagan RS, Cosio G, Grinstein S (2009) Anti-microbial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol 7:355–366
Gangaidzo IT, Moyo VM, Mvundura E et al (2001) Association of pulmonary tuberculosis with increased dietary iron. J Infect Dis 184:936–939
Ganz T, Nemeth E (2009) Iron sequestration and anemia of inflammation. Semin Hematol 46:387–393
Goetz DH, Holmes MA, Borregaard N et al (2002) The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell 10:1033–1043
Gomez MA, Alisaraie L, Shio MT et al (2010) Protein tyrosine phosphatases are regulated by mononuclear iron dicitrate. J Biol Chem 285:24620–24628
Gordeuk VR, Ballou S, Lozanski G et al (1992) Decreased concentrations of tumor necrosis factor-alpha in supernatants of monocytes from homozygotes for hereditary hemochromatosis. Blood 79:1855–1860
Goswami T, Andrews NC (2006) Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalin iron sensing. J Biol Chem 281:28494–28498
Goya N, Miyazaki S, Kodate S et al (1972) A family of congenital atransferrinemia. Blood 40:239–245
Hamill RL, Woods JC, Cook BA (1991) Congenital atransferrinemia. A case report and review of the literature. Am J Clin Pathol 96:215–218
Hartmann H, Eltzschig HK, Wurz H et al (2008) Hypoxia-independent activation of HIF-1 by enterobacteriaceae and their siderophores. Gastroenterology 134:756–767
Jason J, Archibald LK, Nwanyanwu OC et al (2001) The effects of iron deficiency on lymphocyte cytokine production and activation: preservation of hepatic iron but not at all cost. Clin Exp Immunol 126:466–473
Kaelin WG Jr, Ratcliffe PJ (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30:393–402
Kawai T, Akira S (2010) The role of pattern recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11:373–384
Kemp JD, Thorson JA, Gomez F et al (1989) Inhibition of lymphocyte activation with anti-transferrin receptor Mabs: a comparison of three reagents and further studies of their range of effects and mechanism of action. Cell Immunol 122:218–230
Koening C, Miller JC, Nelson JM et al (2009) Toll-like receptors mediate induction of hepcidin in mice infected with Borrelia burgdorferi. Blood 114:1913–1918
Kuvibidila SR, Baliga BS, Suskind RM (1981) Effects of iron deficiency anemia on delayed cutaneous hypersensitivity in mice. Am J Clin Nutr 34:2635–2640
Lago F, Dieguez C, Gomez-Reino J et al (2007) The emerging role of adipokines as mediators of inflammation and immune responses. Cytokine Growth Factor Rev 18:313–325
Lee PL, Beutler E (2009) Regulation of hepcidin and iron-overload disease. Annu Rev Pathol 4:489–515
Lee P, Peng H, Gelbart T et al (2004) The IL-6- and lipopolysaccharide-induced transcription of hepcidin in HFE-, transferrin receptor 2-, and beta 2-microglobulin-deficient hepatocytes. Proc Natl Acad Sci USA 101:9263–9265
Lee P, Peng H, Gelbart T et al (2005) Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc Natl Acad Sci USA 102:1906–1910
Liu XB, Nguyen NB, Marquess KD et al (2005) Regulation of hepcidin and ferroportin expression by lipopolysaccharide in splenic macrophages. Blood Cells Mol Dis 35:47–56
Macdougall LG, Anderson R, McNab GM et al (1975) The immune response in iron-deficient children: impaired cellular defense mechanisms with altered humoral components. J Pediatr 86:833–843
Macedo MF, Porto G, Costa M et al (2010) Low numbers of CD8+T lymphocytes in hereditary hemochromatosis are explained by a decrease of the most mature CD8+ effector memory T cells. Clin Exp Immunol 159:363–371
Magnus SA, Hambleton IR, Moosdeen F et al (1999) Recurrent infections in homozygous sickle cell disease. Arch Dis Child 80:537–541
Marquis JF, Gros P (2008) Genetic analysis of resistance to infections in mice: A/J meets C57BL6/J. Curr Top Microbiol Immunol 321:27–57
McClung JP, Karl JP (2009) Iron deficiency and obesity: the contribution of inflammation and diminished iron absorption. Nutr Rev 67:100–104
Melillo G, Taylor LS, Brooks A et al (1997) Functional requirement of the hypoxia-responsive element in the activation of the inducible nitric oxide synthase promoter by the iron chelator desferrioxamine. J Biol Chem 272:12236–12243
Melo RA, Garcia AB, Viana SR et al (1997) Lymphocyte subsets in experimental hemochromatosis. Acta Haematol 98:72–75
Mencacci A, Cenci E, Boelaert JR et al (1997) Iron overload alters innate and T helper cell responses to Candida albicans in mice. J Infect Dis 175:1467–1476
Meynard D, Kautz L, Darnaud V et al (2009) Lack of BMP6 induces massive iron overload. Nat Genet 41:478–481
Moyo VM, Gangaidzo IT, Gordeuk VR et al (1997) Tuberculosis and iron overload in Africa: a review. Cent Afr J Med 43:334–339
Muckenthaler MU, Galy B, Hentze MW (2008) Systemic iron homeostasis and the IRE/IRP regulatory network. Annu Rev Nutr 28:197–213
Murray MJ, Murray AB, Murray MB et al (1978) The adverse effect of iron repletion on the course of certain infections. Br Med J 2:1113–1115
Nairz M, Theurl I, Ludwiczek S et al (2007) The co-ordinated regulation of iron homeostasis in murine macrophages limits the availability of iron for intracellular Salmonella typhimurium. Cell Microbiol 9:2126–2140
Nairz M, Theurl I, Schroll A et al (2009) Absence of functional Hfe protects mice from invasive Salmonella enterica serovar Typhimurium infection via induction of lipocalin-2. Blood 114:3642–3651
Neckers LM, Yenokida G, James SP (1984) The role of the transferrin receptor in human B lymphocyte activation. J Immunol 133:2437–2441
Ned RM, Swat W, Andrews NC (2003) Transferrin receptor 1 is differentially required in lymphocyte development. Blood 102:3711–3718
Nemeth E, Rivera S, Gabayan V et al (2004a) IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 113:1271–1276
Nemeth E, Tuttle MS, Powelson J et al (2004b) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306:2090–2093
Nizet V, Johnson RS (2009) Interdependence of hypoxic and innate immune responses. Nat Rev Immunol 9:609–617
Olakanmi O, Schlesinger LS, Britigan BE (2007) Hereditary hemochromatosis results in decreased iron acquisition and growth by Mycobacterium tuberculosis within human macrophages. J Leukoc Biol 81:195–204
Oppenheimer SJ (2001) Iron and its relation to immunity and infectious disease. J Nutr 131:616S–635S
Paradkar PN, De Domenico I, Durchfort N et al (2008) Iron depletion limits intracellular bacterial growth in macrophages. Blood 112:866–874
Peyssonnaux C, Zinkernagel AS, Datta V et al (2006) TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 107:3727–3732
Peyssonnaux C, Zinkernagel AS, Schuepbach RA et al (2007) Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest 117:1926–1932
Pietrangelo A (2006) Hereditary hemochromatosis. Annu Rev Nutr 26:251–270
Salahudeen AA, Thompson JW, Ruiz JC et al (2009) An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 326:722–726
Sazawal S, Black RE, Ramsan M et al (2006) Effects of routine prophylactic supplementation with iron and folic acid on admission to hospital and mortality in preschool children in a high malaria transmission setting: community-based, randomised, placebo-controlled trial. Lancet 367:133–143
Schaible UE, Kaufmann SH (2004) Iron and microbial infection. Nat Rev Microbiol 2:946–953
Schmidt PJ, Toran PT, Giannetti AM et al (2008) The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab 7:205–214
Seldon MP, Silva G, Pejanovic N et al (2007) Heme oxygenase-1 inhibits the expression of adhesion molecules associated with endothelial cell activation via inhibition of NF-kappaB RelA phosphorylation at serine 276. J Immunol 179:7840–7851
Semrin G, Fishman DS, Bousvaros A et al (2006) Impaired intestinal iron absorption in Crohn’s disease correlates with disease activity and markers of inflammation. Inflamm Bowel Dis 12:1101–1106
Silvestri L, Pagani A, Nai A et al (2008) The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab 8:502–511
Soe-Lin S, Sheftel AD, Wasyluk B et al (2008) Nramp1 equips macrophages for efficient iron recycling. Exp Hematol 36:929–937
Soe-Lin S, Apte SS, Andriopoulos B Jr et al (2009) Nramp1 promotes efficient macrophage recycling of iron following erythrophagocytosis in vivo. Proc Natl Acad Sci USA 106:5960–5965
Srikantia SG, Prasad JS, Bhaskaram C et al (1976) Anemia and the immune response. Lancet 1:1307–1309
Tanno T, Bhanu NV, Oneal PA et al (2007) High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med 13:1096–1101
Vallabhapurapu S, Karin M (2009) Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol 27:693–733
Van Zandt KE, Sow FB, Florence WC et al (2008) The iron export protein ferroportin 1 is differentially expressed in mouse macrophage populations and is present in the mycobacterial-containing phagosome. J Leukoc Biol 84:689–700
Vashisht AA, Zumbrennen KB, Huang X et al (2009) Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 326:718–721
Verga Falzappa MV, Vujic Spasic M, Kessler R et al (2007) STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 109:353–358
Wanachiwanawin W (2000) Infections in E-beta thalassemia. J Pediatr Hematol Oncol 22:581–587
Wang L, Cherayil BJ (2009) Ironing out the wrinkles in host defense: interactions between iron homeostasis and innate immunity. J Innate Immun 1:455–464
Wang RH, Li C, Xu X et al (2005) A role of SMAD4 in iron metabolism through the regulation of hepcidin expression. Cell Metab 2:399–409
Wang L, Johnson EE, Shi HN et al (2008) Attenuated inflammatory responses in hemachromatosis reveal a role for iron in the regulation of macrophage cytokine translation. J Immunol 181:2723–2731
Wang L, Harrington L, Trebicka E et al (2009) Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. J Clin Invest 119:3322–3328
Ward PP, Paz E, Conneely OM (2005) Multifunctional roles of lactoferrin: a critical overview. Cell Mol Life Sci 62:2540–2548
Weiss G, Werner-Felmayer G, Werner ER et al (1994) Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J Exp Med 180:969–976
Wrighting DM, Andrews NC (2006) IL-6 induces hepcidin expression through STAT3. Blood 108:3204–3209
Wu WH, Meydani M, Meydani SN et al (1990) Effect of dietary iron overload on lipid peroxidation, prostaglandin synthesis and lymphocyte proliferation in young and old rats. J Nutr 120:280–289
Zhang X, Jin M, Wu H et al (2008) Biomarkers of lupus nephritis determined by serial urine proteomics. Kidney Int 74:799–807
Acknowledgments
Work in the author’s laboratory is supported by grants from the National Institutes of Health (R56 AI089700), the Broad Medical Research Program (IBD-0253) and the Crohn’s and Colitis Foundation of America.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Cherayil, B.J. Iron and Immunity: Immunological Consequences of Iron Deficiency and Overload. Arch. Immunol. Ther. Exp. 58, 407–415 (2010). https://doi.org/10.1007/s00005-010-0095-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-010-0095-9