
Abstract
Thymic stromal lymphopoietin (TSLP) primes dendritic cells to promote a Th2 inflammatory response. Its action is mediated by a heterodimeric receptor which consists of the interleukin-7 receptor α chain and the TSLP receptor chain (TSLPR). TSLPR resembles the common γ chain subunit utilized by many type 1 cytokine receptors. Normal epithelial cells, keratinocytes, and stromal cells constitutively express TSLP. Dendritic cells that are activated by TSLP promote the development of CD4+ T cells into pro-inflammatory Th2 cells. TSLP thus plays a potentially important role in the pathogenesis of allergic inflammation in asthma and atopic dermatitis. TSLP also has direct effects on other types of cells in the bronchial mucosa. It is over-expressed in the bronchial mucosa in chronic obstructive pulmonary disease (COPD), which is traditionally described as a Th1-related disease, as well as severe asthma, which is traditionally described as a Th2-related disease. In this review we will discuss TSLP expression, function, and available and potential mechanisms in both allergic inflammation and COPD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Asthma and atopic dermatitis (AD) are allergic inflammatory disorders characterized by the infiltration and accumulation of memory-like T-helper (Th)2 cells and eosinophils (Kay 2001). The inflammatory profile in these disorders is predominantly allergen-driven Th2-coordinated inflammation with the release of interleukin (IL)-4, IL-5, IL-9, and IL-13. Up-regulation of the cytokines IL-4 (Leung et al. 2005; Matsunaga et al. 2006; Quaedvlieg et al. 2006; Shahid et al. 2002), IL-5 (Kim et al. 2003), IL-9 (Shimbara et al. 2000), and IL-13 (Bodey et al. 1999; Huang et al. 1995) can be detected in exhaled breath condensate, sputum, bronchoalveolar lavage (BAL) fluid, and endobronchial biopsies of asthmatics. These cytokines play a key role in allergic inflammation and remodeling in asthma, such as inducing IgE production and vascular cell adhesion molecule-1 expression, maturation of eosinophils, mast cell development, mucus overproduction, and airway hyperresponsiveness (Corry 2002; Kay 2003). The Th2 cytokine release pattern is also of pathophysiological importance in AD, at least in the initial stages of AD (Incorvaia et al. 2008; Numerof and Asadullah 2006).
As antigen-presenting cells, dendritic cells (DCs) play a critical role in directing T cell responses to an antigen, priming T cells to develop into Th1, Th2, and Th17 memory/effector cells. DCs activated by thymic stromal lymphopoietin (TSLP) in vitro promote the development of Th2 memory/effector cells (Liu et al. 2007). The involvement of TSLP in the Th2 inflammatory pathway is suggested by the fact that the expression of TSLP is increased in the bronchial mucosa of asthmatics and its expression is correlated with Th2-attracting chemokine expression and disease severity (Kato and Schleimer 2007; Ying et al. 2005). The role of TSLP in chronic obstructive pulmonary disease (COPD) is less clear, although its expression at the mRNA and protein level is elevated in COPD (Ying et al. 2008). This suggests that TSLP not only plays a key role in allergic inflammation, but may also play a role in non-allergic airway inflammation. In this review we will discuss the factors and mechanisms involved in the expression of TSLP, the function and molecular mechanisms underlying TSLP’s role in allergic inflammation, and the potential role and mechanism of TSLP in COPD.
TSLP/TSLP Signaling Pathway
TSLP, an IL-7-like cytokine, was first isolated from the supernatant of a murine thymic stromal cell line and initially recognized as a pre-B-cell growth factor (Friend et al. 1994; Ray et al. 1996). Despite exhibiting poor homology (amino-acid correspondence is 43%), human and murine TSLP exert similar biological functions (Liu et al. 2007; Quentmeier et al. 2001). In humans, TSLP is expressed predominantly by epithelial cells (mostly of the lung, skin, and gut) and keratinocytes (Allakhverdi et al. 2007; Liu et al. 2007). Lung fibroblasts, DCs, mast cells, and smooth muscle cells all have the potential to produce TSLP (Liu et al. 2007; Soumelis et al. 2002). TSLP is also produced by allergen-activated basophils (Sokol et al. 2008), but not by most other hematopoietic cells, including B cells, T cells, natural killer cells, granulocytes, and macrophages (Liu et al. 2007; Zhang et al. 2007).
The TSLP receptor is a heterodimeric receptor consisting of the IL-7 receptor α chain (IL-7Rα) and a specific subunit, the TSLP receptor chain (TSLPR). The IL-7R, on the other hand, is a heterodimer of IL-7Rα and the common γ chain (γc). Human and murine TSLPR share 39% amino-acid identity, while human TSLPR and human γc share 24%. Co-expression of human TSLPR and IL-7Rα is found in activated mast cells, CD4+ T cells, CD8+ T cells, DCs, and monocytes (Allakhverdi et al. 2007; Park et al. 2000; Reche et al. 2001). The TSLPR, like its murine counterpart (see below), has been found to induce phosphorylation of signal transducer and activator of transcription 5 (STAT5) in the absence of Janus family tyrosine kinase 3 (JAK3) activation (Quentmeier et al. 2001; Reche et al. 2001). Human TSLP receptor is also able to utilize STAT3 (Reche et al. 2001).
In contrast to humans, murine TSLPR (mTSLPR) is expressed in many tissues, including liver, brain, heart, kidney, testis, bone marrow, spleen, and thymus, and on a variety of cell types, including T cells, B cells, DCs, and monocytes (Pandey et al. 2000; Park et al. 2000; Tonozuka et al. 2001). mTSLPR is similar to the γc subunit, with a 47% similarity at the protein level (Pandey et al. 2000; Park et al. 2000; Tonozuka et al. 2001). TEC family kinases might be involved in TSLP-mediated STAT5 activation, while JAKs might be involved in mIL-7 signaling (Isaksen et al. 1999). A member of the TEC family kinases is thought to play a role in TSLP-mediated STAT5 activation, such as SOCS-1 (suppressors of cytokine signaling-1, an inhibitor of JAK and TEC kinases) which is able to inhibit TSLP-mediated signaling (Isaksen et al. 1999, 2002). Furthermore mTSLP receptor signaling requires STAT5, while mIL-7 receptor is also able to utilize STAT1 and STAT3 as well as STAT5 in its signaling (Isaksen et al. 1999; Lin et al. 1995; van der Plas et al. 1996).
Induction of TSLP Expression
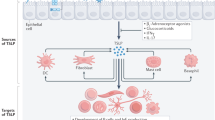
TSLP expression can be triggered and enhanced by a variety of stimuli, so that a diverse array of signaling pathways may be involved in the induction of its expression (Fig. 1).

The regulation of TSLP expression in epithelial cells. Viral dsRNA, viral protein, and bacterial peptidoglycans induce the expression of TSLP mRNA and protein via TLRs and the NF-κB pathways. IL-1β, lysophosphatidic acid, and TNF-α also use the NF-κB pathway following activation of their corresponding receptors to induce the expression of TSLP mRNA and protein. IL-4, however, utilizes the JAK-STAT6 pathway to induce expression of both TSLP mRNA and protein. TGF-β, IFN-β, and IL-13 are able to induce the expression of TSLP mRNA, although the pathway for this has yet to be elucidated and the increased expression of mRNA has not been accompanied by increased protein expression. It is also postulated that cigarette smoke and DEPs cause an increase in intracellular oxidative stress, which leads to NF-κB activation and increased TSLP expression. In mice, vitamin D3, via its nuclear receptor VDR, has been shown to increase the expression of TSLP
-
1.
Infection: viral infection in synergy with cytokine production is likely to be a primary driver for TSLP production. Rhinovirus and respiratory syncytial virus (RSV) can up-regulate the expression of TSLP in human bronchial epithelial cells (HBECs) (Tu et al. 2007). TSLP expression evoked by rhinoviral double-stranded ribonucleic acid (dsRNA) and RSV proteins acting on Toll-like receptors (TLR) is synergistically enhanced by IL-4, suggesting that nuclear factor-κB (NF-κB) and STATs cooperate in the transcriptional regulation of the TSLP gene (Allakhverdi et al. 2007; Kato et al. 2007). dsRNA synthesized by rhinoviruses is a ligand for TLR3. The induction of TSLP by dsRNA is dependent on TLR3, NF-κB, and IRF-3, while enhancement of dsRNA-dependent TSLP expression by IL-4 is STAT6 dependent (Kato et al. 2007)
-
2.
Cytokines and other mediators: pro-inflammatory cytokines (IL-1β and tumor necrosis factor (TNF)-α), bacterial peptidoglycan, lysophosphatidic acid (LPA), and other TLR ligands (such as LTA, i.e. lipoteichoic acid, and poly I:C, i.e. polyinosinic:polycytidylic acid) are also able to induce the production of TSLP by HBECs (Allakhverdi et al. 2007; Lee and Ziegler 2007; Medoff et al. 2009). Additionally, the cytokines IL-4, IL-13, interferon (IFN)-β, and transforming growth factor (TGF)-β are able to up-regulate the expression of TSLP mRNA by HBECs, but have not been shown to be able to induce the production of TSLP at the protein level (Allakhverdi et al. 2007; Kato et al. 2007). Mutations in NF-κB binding sites and 9-cis-retinoic acid (a NF-κB inhibitor) have both been shown to inhibit the expression of TSLP stimulated by IL-1β in HBECs, indicating that pro-inflammatory cytokine induction of TSLP is regulated by NF-κB in HBECs (Lee and Ziegler 2007; Lee et al. 2008). The induction of TSLP by LPA in HBECs is also mediated by NF-κB and this activation of NF-κB is dependent on CARMA3 (Medoff et al. 2009). This is further evidence to support the regulation of TSLP by NF-κB in HBECs. IL-1β and TNF-α can also induce the production of TSLP by human airway smooth muscle cells (HASMCs) independently and synergistically. Unlike HBECs, the expression of TSLP by IL-1β and TNF-α is mediated via the ERK1/2 and p38 MAPK signaling pathways (Zhang et al. 2007). Mast cells were found to produce, store, and release TSLP following FcεRI aggregation. This was augmented in the presence of IL-4. TSLP released from mast cells was rapidly degraded not only by mast cell-derived proteases (tryptase, chymase, or both), but also by other undefined mediators (Okayama et al. 2009). In human skin keratinocytes, stimulation with IL-1α or TNF-α in combination with Th2 cytokines (IL-4 or IL-13) is able to induce the production of TSLP in a synergistic manner (Okayama et al. 2009). Poly I:C, which mimics dsRNA and signals via TLR3, also induces the production of TSLP by human airway epithelial cells (Allakhverdi et al. 2007) and human keratinocytes. This induction of TSLP by dsRNA was enhanced by IL-4, IL-13 TNF-α, IFN-α, or IFN-β, while it was suppressed by IFN-γ, TGF-β, or IL-17 (Kinoshita et al. 2009)
-
3.
Smoking and air pollution: the effects of cigarette smoking and pollution on TSLP production illustrate the importance of TSLP as a key cytokine at the interface between the environment and epithelium. Exposure of HBEC to diesel exhaust particles (DEPs) has been shown to up-regulate the expression of TSLP at both the mRNA and protein level. TSLP induction by DEPs was found to be mediated by an oxidant-induced pathway and was inhibited by increased free-radical scavenging by N-acetyl cysteine(Bleck et al. 2008). The action of DEPs is most likely mediated by NF-κB as it is both involved in the regulation of TSLP production (Kato et al. 2007; Lee and Ziegler 2007; Lee et al. 2008; Medoff et al. 2009) and is known to be activated by oxidative stress in HBECs (Rahman 2000). Further indirect confirmation that oxidative stress may induce the release of TSLP can be found in our data showing that TSLP mRNA is elevated in the bronchial mucosa and the protein is elevated in the BAL fluid of patients with COPD (Ying et al. 2008). In a BALB/c ovalbumin mouse model of asthma, cigarette smoke extract has been shown to be able to induce the expression of TSLP mRNA and protein in mouse lung. TLSP release was also inhibited by N-acetyl cysteine, highlighting the importance of the activation of TSLP via an oxidant-induced pathway (Nakamura et al. 2008)
The use of animal models has implicated several other cellular and nuclear pathways that may play an important role in the regulation of TSLP in humans. Keratinocytes from retinoid X receptor α and β knockout mice (RXRαβep−/− mice) express high levels of TSLP and exhibit a skin and systemic syndrome similar to AD. 1α,25(OH)2D3 (calcitriol), the physiologically active ligand of the vitamin D receptor (VDR), is able to stimulate murine keratinocytes to induce the expression of TSLP through binding of its nuclear receptor, VDR. Since the nuclear receptors RXRαβ and unliganded VDR bind to form a heterodimer which acts as a co-repressor, binding of calcitriol to VDR or knockout of RXRαβ will result in removal of this inhibition and TSLP expression by murine keratinocytes (Li et al. 2006). The peptidyl-prolyl isomerase PIN1, a regulator of phosphorylation signaling, catalytically regulates the conformation of substrates after their phosphorylation and so regulates protein function (Lu and Zhou 2007). In rats, PIN1 blockade reduces TSLP expression. PIN1 is thus likely to act as a potential modulator of TSLP expression at the post-transcriptional level (Esnault et al. 2008).
The Role of TSLP in DCs and T Cell Subtypes
TSLP strongly induces the expression of major histocompatibility complex class I and II as well as co-stimulatory molecules, such as CD40, CD80, and CD86, on DCs. It also prolongs DC survival and enhances DC-T cell conjugate formation, resulting in a strong proliferation of CD4+ and CD8+ T cells (Akamatsu et al. 2008; Gilliet et al. 2003; Soumelis et al. 2002; Watanabe et al. 2004). Unlike DCs, which express TSLPR, unstimulated CD4+ and CD8+ T cells do not express TSLPR and are not sensitive to TSLP. T cell receptor stimulation results in cell-surface expression of the TSLP receptor, allowing TSLP to act directly on activated CD4+ and CD8+ T cells, promoting proliferation (Akamatsu et al. 2008; Gilliet et al. 2003; Rochman et al. 2007).
In atopic dermatitis, TSLP expression by keratinocytes is associated with the activation and migration of Langerhans cells (LCs) from the epidermis to the dermis (Soumelis et al. 2002). TSLP enhances epidermal LC survival and migration and stimulates their production of thymus and activation-regulated chemokine (TARC) (Ebner et al. 2007). TSLP-activated DCs (TSLP-DCs) produce the chemokines IL-8, eotaxin-2, TARC, and macrophage-derived chemokine (MDC) as well as the OX40 ligand (OX40L) (Isaksen et al. 2002; Ito et al. 2005; Soumelis et al. 2002). TSLP-DCs do not, however, produce the Th1-polarizing cytokine IL-12 or the proinflammatory cytokines TNF-α, IL-1β, and IL-6 (Soumelis et al. 2002). TSLP-DCs also do not produce the Th2-polarizing cytokine IL-4 or the anti-inflammatory cytokine IL-10 (Soumelis et al. 2002). OX40L, a member of the TNF superfamily, has been implicated as the key cytokine by which TSLP-DCs polarize naïve CD4+ T cells to a pro-inflammatory Th2 response. Inhibition of OX40L by neutralizing antibodies results in strong inhibition of IL-4, IL-13, and TNF-α production while promoting the production of IFN-γ and IL-10 by CD4+ T cells primed by TSLP-DCs. This effect is not seen in CD4+ T cells primed by DCs stimulated by other ligands, such as TLR ligands and IFNs. The induction of Th2 response priming by TSLP-DCs can be inhibited and switched towards a Th1 response by the presence of exogenous IL-12, Mycobacterium bovis bacillus Calmette-Guérin (which down-regulates OX40L and stimulates IL-12 production), or simultaneous blockade of both OX40L (released by TSLP-DCs) and IL-4 (released by Th2 T cells) by neutralizing antibodies (Ito et al. 2005; Yokoi et al. 2008). On the other hand, TSLP-DCs induce the proliferation of naïve CD8+ T cells and induce CD8+ T cells to differentiate into cytotoxic CD8+ T cells that produce IL-5, IL-13, TNF-α, and IFN-γ (Gilliet et al. 2003).
T-regulatory (Treg) cells are involved in the regulation of the peripheral immune response and likely play a central role in determining the incidence and severity of several immune pathologies, including autoimmune disease, infectious disease, allergy, and asthma (Hawrylowicz and O’Garra 2005). The Treg subsets which have been identified are CD8+ Treg cells, natural killer (NK) cells, and CD4+ Treg cells (van der Plas et al. 1996). Naturally occurring Treg cells are CD4+CD25+Foxp3+ cells which originate in the thymus and are also possibly induced in the periphery (Hawrylowicz and O’Garra 2005). The epithelial cells of Hassall’s corpuscles in the thymus express high levels of TSLP, which prime the DCs in the Hassall’s corpuscles. These TSLP-DCs induce the proliferation and differentiation of CD4+CD8−CD25− thymocytes into CD4+CD25+Foxp3+ T regs, which have suppressive functions similar to those of naturally occurring Tregs. This induction is dependent upon IL-2 and CD28 signaling (Watanabe et al. 2005).
The Role of TSLP in Other Cell Types
Mast cells play an important role in allergic inflammation, particularly in asthma, acting via multiple mechanisms. Mast cells in patients with asthma produce Th2-type cytokines, induce IgE synthesis in B cells, up-regulate the production of a variety of cytokines/chemokines by epithelial cells and fibroblasts, and induce the recruitment of basophils, T cells, and eosinophils to sites of allergic inflammation as well as promote their own intraepithelial accumulation (Bradding et al. 2006; Hart 2001). Mast cells not only express TSLP mRNA (Ying et al. 2008), but are also a target cell for TSLP, expressing both TSLPR and IL-7Rα (Allakhverdi et al. 2007). Stimulation of human mast cells in vitro with IL-1β and TNF-α in the presence of TSLP strongly augments the production of the pro-inflammatory cytokines and chemokines while blocking endogenous TSLP released by activated primary human epithelial cells completely inhibits IL-1β and TNF-α induced production of IL-13 by mast cells (Allakhverdi et al. 2007).
In the mouse, TSLP promotes an early stage of B-cell and T cell development (Levin et al. 1999; Sims et al. 2000). In contrast, in humans TSLP does not have direct effects on B cells, NK cells, or neutrophils (Liu et al. 2007).
TSLP Triggers AD and Asthma
Allergic diseases such as asthma and AD are characterized by their Th2 inflammatory profile with Th2 cytokine and chemokine release along with infiltration of their corresponding leukocyte targets. TSLP released by both bronchial epithelial cells and keratinocytes primes DCs to induce the expansion of naïve CD4+ T cells into pro-inflammatory CD4+ Th2 cells. This in turn drives and maintains the inflammation seen in these diseases (Bogiatzi et al. 2007; Ebner et al. 2007; Soumelis et al. 2002). The pro-inflammatory cytokines released by TSLP-DC-primed T cells are characteristic of asthma and AD which are possibly mediated by IL-4, IL-13, and TNF-α, but not by IL-10 or IFN-γ (Ebner et al. 2007; Soumelis et al. 2002).
The effects of TSLP in vitro on DCs and T cells are mirrored by the in vivo findings in disease. In AD, the production of TSLP is restricted to keratinocytes of the apical layers and seems to be associated with the migration of activated LCs from the epidermis to the dermis (Bogiatzi et al. 2007; Soumelis et al. 2002), although the mechanisms of the migration are unclear. In asthma we have found that TSLP expression is increased in asthmatic airways and correlates with the expression of the Th2-attracting chemokines TARC/CCL17 and MDC/CCL22 in endobronchial biopsies and with disease severity. We found that epithelial cells, endothelial cells, neutrophils, macrophages, and mast cells expressed TSLP (Ying et al. 2005). Further evidence of the key role that TSLP plays in asthma and AD is available from animal “models” of these diseases. For example, transgenic mice that have inducible TSLP over-expression in keratinocytes develop an AD-like phenotype following TSLP induction characterized by eczematous skin lesions containing inflammatory cell infiltrates, a dramatic increase in circulating Th2 cells, and elevated serum IgE (Yoo et al. 2005). Transgenic mice that over-expressed TSLP in the lungs had an augmented Th2 inflammatory response (infiltration of leukocytes and release of both Th2 cytokines and IgE) and remodeling (goblet cell hyperplasia and subepithelial fibrosis) when challenged with ovalbumin. Conversely, TSLP receptor-deficient mice (TSLPR knockout) showed an attenuated Th2 inflammatory response and remodeling when challenged with ovalbumin (Zhou et al. 2005). Murine models have also shown that the Th2 inflammatory response induced by TSLP is T cell and eosinophil dependent (Jessup et al. 2008). Furthermore, the response to inhaled antigen can be restored in TSLPR knock-out mice by transfer of CD4+ T cells from wild-type mice into the knock-out mice (Al-Shami et al. 2005). Attenuation of the Th2 inflammatory response and remodeling in this ovalbumin murine “model” of asthma can also be achieved by administration of anti-TSLPR antibodies prior to ovalbumin sensitization (Shi et al. 2008), suggesting that this blockade might reduce various functions of TSLPR+ cells involved in the pathogenesis of asthma. The potential role of TSLP in asthma is summarized in Fig. 2.

The expression and function of TSLP in asthma and atopic dermatitis (AD). The expression of TSLP in asthma and AD is allergen driven. Allergen activates mast cells via IgE and FcεRI. This causes the mast cell to release TSLP, various inflammatory cytokines, and other mediators such as proteases. These inflammatory cytokines are likely to be the signal that causes epithelial cells/keratinocytes to produce TSLP. It is not known if allergen can directly act on epithelial cells/keratinocytes to induce the production of TSLP. The TSLP release primes DCs to secrete Th2-attracting chemokines as well as OX40 ligand. TSLP also promotes a subtype of DCs, Langerhans cells, to migrate from the site of induction to the lymph nodes. OX40 ligand is one of the signals required from DCs to prime naïve T cells to differentiate into CD4+ Th2-type T cells and enhance their proliferation and expansion. TSLP is also able to act on activated T cells to enhance proliferation and expansion. Th2 cytokines released by T cells feedback positively to the epithelial cells and stimulate TSLP production. TNF-α also stimulates airway smooth muscle to release TSLP, therefore creating another positive-feedback loop
The Role of TSLP in COPD
COPD is a disease characterized by inflammation of the central and peripheral airways which, as with asthma, has been circumstantially implicated in contributing to airway obstruction but, in contrast to asthma, is poorly responsive to anti-inflammatory therapy such as corticosteroid. A second key mechanism in COPD is destruction of the lung parenchyma, reducing elastic support for the small airways, which causes them to collapse, and eliminating alveoli, thus impairing gas transfer (Di Stefano et al. 2004). Cigarette smoking is a major risk factor for the development of COPD, with over 90% of COPD patients in the developed world being cigarette smokers (Pauwels et al. 2001; Siafakas et al. 1995). The cellular inflammation in COPD is characterized by increased numbers of neutrophils, macrophages, and T lymphocytes, with a preponderance of CD8+ (cytotoxic) over CD4+ (helper) cells in the bronchial mucosa and increased macrophages and neutrophils in BAL fluid and induced sputum (Di Stefano et al. 1998; Keatings et al. 1996; Pesci et al. 1998; Saetta et al. 1998; Sarir et al. 2008). The key inflammatory mediators in COPD are thought to be the cytokine TNF-α and the chemokine IL-8 (Reimold 2002; Sarir et al. 2008; Weathington et al. 2006). IFN-γ and IL-13 have also been implicated in causing emphysema, at least in animal “models” (Elias et al. 2006).
There is considerable evidence that increased oxidative stress plays an important role in the pathogenesis of COPD. Cigarette smoke, which is rich in oxidative substances, increases the oxidative burden in the lungs, compounding the innate oxidative stress created in inflammatory processes through the generation of reactive oxygen and other species by inflammatory, immune, and epithelial cells in the airways, leading to further inflammation and tissue destruction (Elias et al. 2006; Rahman and MacNee 1996; Repine et al. 1997). Furthermore NF-κB, which regulates the release of many cytokines and chemokines, is responsive to the oxidative stress (Rahman 2000). We postulate that the activation of NF-κB by oxidative stress from cigarette smoke contributes to elevated TSLP production in the bronchial mucosa in COPD (Kato et al. 2007; Lee and Ziegler 2007; Lee et al. 2008; Medoff et al. 2009; Rahman 2000; Ying et al. 2008). We discovered elevated bronchial mucosal expression of mRNA encoding TSLP and the chemokines TARC/CCL17, MDC/CCL22, and IFN-inducible protein 10/CXCL10, with corresponding elevation of these proteins in the BAL fluid in COPD (Ying et al. 2008). We also found a similar pattern of expression at the protein level (although there was low level of TSLP) in smokers without COPD (Ying et al. 2008). These data are consistent with the hypothesis that this expression is most likely due at least partly to the effects of cigarette smoke on TSLP expression and the corresponding effects of TSLP on DC release of chemokines in COPD. The fact that not all smokers develop COPD suggests that there are likely additional factors which drive the development of COPD in particular individuals. Oxidative stress is most likely not the only mechanism by which TSLP expression in elevated in COPD (Fig. 3). The cytokines TNF-α and IL-1β, which are associated with COPD, may also play a key role in the elevation of TSLP expression (Sarir et al. 2008). Each of these cytokines is able to induce the expression of TSLP by HBECs and HASMCs (Allakhverdi et al. 2007; Lee and Ziegler 2007; Zhang et al. 2007). Another mechanism that may play a role in the induction of TSLP in COPD is chronic infection. There is increasing evidence for persisting bacterial or viral infection in stable COPD (Miyata et al. 2008; Sethi and Murphy 2008). Again, bacterial peptidoglycan and TLR ligands (LTA, dsRNA, RSV proteins) have been shown to be able to induce HBECs to produce TSLP (Allakhverdi et al. 2007; Kato et al. 2007; Tu et al. 2007) (Fig. 1). As we found a significant elevation of TSLP protein in the BAL fluid of COPD patients but not smokers without COPD, it is possible that these cytokines (TNF-α and IL-1β), persistent viral/bacterial infection, and oxidative stress all act synergistically to enhance the production and expression of TSLP in COPD (Ying et al. 2008).

The expression and function of TSLP in COPD. The expression of TSLP in COPD is most likely multi-factorial, with oxidative stress from cigarette smoke and pollution, bacterial, and viral infections as well as inflammatory mediators released from macrophages stimulated directly by cigarette smoke and pollution. Cigarette smoke and pollution also directly stimulate epithelial cells to produce IL-1β and TNF-α, which could act in an autocrine fashion stimulating the release of TSLP. The exact function of TSLP in COPD is yet to be determined. TSLP could possibly prime DCs to induce the secretion of Th2-attracting chemokines and induce cytotoxic CD8+ Th2-type T cell proliferation and expansion
TSLP is able to promote the expansion of both CD4+ and CD8+ T cells either directly following activation or indirectly via DCs. In vitro and in vivo studies have suggested that TSLP primes the T cell response towards a Th2-type inflammatory response (Akamatsu et al. 2008; Gilliet et al. 2003; Rochman et al. 2007; Watanabe et al. 2004). While TSLP may contribute to expansion of CD8+ T cells, of which there is a predominance in COPD (Di Stefano et al. 2004; Saetta et al. 1998; Sarir et al. 2008), it is as yet not clear why, despite elevated expression of TSLP and Th2 chemokines in COPD, there does not appear to be a Th2 cellular response and corresponding cytokine response. It is alternatively possible that TSLP is induced nonspecifically in certain inflammatory processes in which it does not play a key functional role.
Conclusion
TSLP is a cytokine that is highly expressed by skin keratinocytes in AD and bronchial epithelial cells in asthma and COPD. A variety of stimuli can induce its expression, including bacterial and viral infection, oxidant stress, and the local cytokine milieu. Activation of NF-κB plays a key role in the expression of TSLP in these diseases. However, there are most likely other signaling pathways involved in TSLP expression that have yet to be discovered. These may interact in various ways to regulate the expression of TSLP mRNA and protein in different circumstances. The precise mechanism which results in the production of TSLP by structural cells following exposure to allergen has yet to be determined in humans. The activation of mast cells by allergen and the release of its inflammatory products (TNF-α, IL-4, and IL-13) (Bradding et al. 2006; Hart 2001) is one possible mechanism. A murine model of allergic rhinitis supports this theory, as mast cell-deficient or Fc receptor γ chain-deficient mice show attenuated TSLP expression in epithelial cells in the response to allergen (Miyata et al. 2008). The evidence is clear that TSLP not only initiates, but also enhances and propagates the Th2 inflammatory response in allergic inflammation. Although it is clear that the signaling pathway of TSLPR involves STAT5 activation, the mechanism by which this occurs is unknown, as JAK activation is not apparently initiated by the TSLPR. The most intriguing finding is that TSLP is elevated in COPD. The mechanism by which this occurs and the roles which TSLP plays in COPD need further investigation.
Abbreviations
- AD:
-
Atopic dermatitis
- BAL:
-
Bronchoalveolar lavage
- CCL:
-
Chemokine (C–C motif) ligand
- COPD:
-
Chronic obstructive pulmonary disease
- DC:
-
Dendritic cell
- DEP:
-
Diesel exhaust particle
- dsRNA:
-
Double-stranded ribonucleic acid
- HASMC:
-
Human airway smooth muscle cell
- HBEC:
-
Human bronchial epithelial cell
- IL:
-
Interleukin
- IL-7Rα:
-
IL-7 receptor α chain
- JAK:
-
Janus family tyrosine kinases
- LC:
-
Langerhans cell
- LPA:
-
Lysophosphatidic acid
- LTA:
-
Lipoteichoic acid
- MDC:
-
Macrophage-derived chemokine
- NF-κB:
-
Nuclear factor-κB
- NK:
-
Natural killer
- OX40L:
-
OX40 ligand
- poly I:C:
-
Polyinosinic:polycytidylic acid
- RSV:
-
Respiratory syncytial virus
- STAT:
-
Signal transducer and activator of transcription
- TARC:
-
Thymus activation-regulated chemokine
- Th:
-
T-helper
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- Treg:
-
T-regulatory
- TSLP:
-
Thymic stromal lymphopoietin
- TSLPR:
-
TSLP receptor chain
- TSLP-DC:
-
TSLP-activated dendritic cell
- TSLP-LC:
-
TSLP-activated Langerhans cell
- γc :
-
Common γ chain
- VDR:
-
Vitamin D receptor
References
Akamatsu T, Watanabe N, Kido M et al (2008) Human TSLP directly enhances expansion of CD8+ T cells. Clin Exp Immunol 154:98–106
Allakhverdi Z, Comeau MR, Jessup HK et al (2007) Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med 204:253–258
Al-Shami A, Spolski R, Kelly J et al (2005) A role for TSLP in the development of inflammation in an asthma model. J Exp Med 202:829–839
Bleck B, Tse DB, Curotto de Lafaille MA et al (2008) Diesel exhaust particle-exposed human bronchial epithelial cells induce dendritic cell maturation and polarization via thymic stromal lymphopoietin. J Clin Immunol 28:147–156
Bodey KJ, Semper AE, Redington AE et al (1999) Cytokine profiles of BAL T cells and T-cell clones obtained from human asthmatic airways after local allergen challenge. Allergy 54:1083–1093
Bogiatzi SI, Fernandez I, Bichet JC et al (2007) Cutting edge: proinflammatory and Th2 cytokines synergize to induce thymic stromal lymphopoietin production by human skin keratinocytes. J Immunol 178:3373–3377
Bradding P, Walls AF, Holgate ST (2006) The role of the mast cell in the pathophysiology of asthma. J Allergy Clin Immunol 117:1277–1284
Corry DB (2002) Emerging immune targets for the therapy of allergic asthma. Nat Rev Drug Discov 1:55–64
Di Stefano A, Capelli A, Lusuardi M et al (1998) Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med 158:1277–1285
Di Stefano A, Caramori G, Ricciardolo FL et al (2004) Cellular and molecular mechanisms in chronic obstructive pulmonary disease: an overview. Clin Exp Allergy 34:1156–1167
Ebner S, Nguyen VA, Forstner M et al (2007) Thymic stromal lymphopoietin converts human epidermal Langerhans cells into antigen-presenting cells that induce proallergic T cells. J Allergy Clin Immunol 119:982–990
Elias JA, Kang MJ, Crothers K et al (2006) State of the art mechanistic heterogeneity in chronic obstructive pulmonary disease: insights from transgenic mice. Proc Am Thorac Soc 3:494–498
Esnault S, Rosenthal LA, Shen ZJ et al (2008) Thymic stromal lymphopoietin expression in allergic pulmonary inflammation is Pin1-dependent. J Allergy Clin Immunol 121:1289–1290
Friend SL, Hosier S, Nelson A et al (1994) A thymic stromal cell line supports in vitro development of surface IgM+ B cells and produces a novel growth factor affecting B and T lineage cells. Exp Hematol 22:321–328
Gilliet M, Soumelis V, Watanabe N et al (2003) Human dendritic cells activated by TSLP and CD40L induce proallergic cytotoxic T cells. J Exp Med 197:1059–1063
Hart PH (2001) Regulation of the inflammatory response in asthma by mast cell products. Immunol Cell Biol 79:149–153
Hawrylowicz CM, O’Garra A (2005) Potential role of interleukin-10-secreting regulatory T cells in allergy and asthma. Nat Rev Immunol 5:271–283
Huang SK, Xiao HQ, Kleine-Tebbe J et al (1995) IL-13 expression at the sites of allergen challenge in patients with asthma. J Immunol 155:2688–2694
Incorvaia C, Frati F, Verna N et al (2008) Allergy and the skin. Clin Exp Immunol 153(suppl 1):27–29
Isaksen DE, Baumann H, Trobridge PA et al (1999) Requirement for stat5 in thymic stromal lymphopoietin-mediated signal transduction. J Immunol 163:5971–5977
Isaksen DE, Baumann H, Zhou B et al (2002) Uncoupling of proliferation and Stat5 activation in thymic stromal lymphopoietin-mediated signal transduction. J Immunol 168:3288–3294
Ito T, Wang YH, Duramad O et al (2005) TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med 202:1213–1223
Jessup HK, Brewer AW, Omori M et al (2008) Intradermal administration of thymic stromal lymphopoietin induces a T cell- and eosinophil-dependent systemic Th2 inflammatory response. J Immunol 181:4311–4319
Kato A, Schleimer RP (2007) Beyond inflammation: airway epithelial cells are at the interface of innate and adaptive immunity. Curr Opin Immunol 19:711–720
Kato A, Favoreto S Jr, Avila PC et al (2007) TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J Immunol 179:1080–1087
Kay AB (2001) Allergy and allergic diseases: second of two parts. N Engl J Med 344:109–113
Kay AB (2003) Immunomodulation in asthma: mechanisms and possible pitfalls. Curr Opin Pharmacol 3:220–226
Keatings VM, Collins PD, Scott DM et al (1996) Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 153:530–534
Kim CK, Kim SW, Park CS et al (2003) Bronchoalveolar lavage cytokine profiles in acute asthma and acute bronchiolitis. J Allergy Clin Immunol 112:64–71
Kinoshita H, Takai T, Le TA et al (2009) Cytokine milieu modulates release of thymic stromal lymphopoietin from human keratinocytes stimulated with double-stranded RNA. J Allergy Clin Immunol 123:179–186
Lee HC, Ziegler SF (2007) Inducible expression of the proallergic cytokine thymic stromal lymphopoietin in airway epithelial cells is controlled by NFkappaB. Proc Natl Acad Sci USA 104:914–919
Lee HC, Headley MB, Iseki M et al (2008) Cutting edge: Inhibition of NF-kappaB-mediated TSLP expression by retinoid X receptor. J Immunol 181:5189–5193
Leung TF, Wong GW, Ko FW et al (2005) Analysis of growth factors and inflammatory cytokines in exhaled breath condensate from asthmatic children. Int Arch Allergy Immunol 137:66–72
Levin SD, Koelling RM, Friend SL et al (1999) Thymic stromal lymphopoietin: a cytokine that promotes the development of IgM+ B cells in vitro and signals via a novel mechanism. J Immunol 162:677–683
Li M, Hener P, Zhang Z et al (2006) Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc Natl Acad Sci USA 103:11736–11741
Lin JX, Migone TS, Tsang M et al (1995) The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity 2:331–339
Liu YJ, Soumelis V, Watanabe N et al (2007) TSLP: an epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu Rev Immunol 25:193–219
Lu KP, Zhou XZ (2007) The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol 8:904–916
Matsunaga K, Yanagisawa S, Ichikawa T et al (2006) Airway cytokine expression measured by means of protein array in exhaled breath condensate: correlation with physiologic properties in asthmatic patients. J Allergy Clin Immunol 118:84–90
Medoff BD, Landry AL, Wittbold KA et al (2009) CARMA3 mediates lysophosphatidic acid-stimulated cytokine secretion by bronchial epithelial cells. Am J Respir Cell Mol Biol 40:286–294
Miyata M, Hatsushika K, Ando T et al (2008) Mast cell regulation of epithelial TSLP expression plays an important role in the development of allergic rhinitis. Eur J Immunol 38:1487–1492
Nakamura Y, Miyata M, Ohba T et al (2008) Cigarette smoke extract induces thymic stromal lymphopoietin expression, leading to T(H)2-type immune responses and airway inflammation. J Allergy Clin Immunol 122:1208–1214
Numerof RP, Asadullah K (2006) Cytokine and anti-cytokine therapies for psoriasis and atopic dermatitis. BioDrugs 20:93–103
Okayama Y, Okumura S, Sagara H et al (2009) FcepsilonRI-mediated thymic stromal lymphopoietin production by IL-4-primed human mast cells. Eur Respir J 34:425–435
Pandey A, Ozaki K, Baumann H et al (2000) Cloning of a receptor subunit required for signaling by thymic stromal lymphopoietin. Nat Immunol 1:59–64
Park LS, Martin U, Garka K et al (2000) Cloning of the murine thymic stromal lymphopoietin (TSLP) receptor: formation of a functional heteromeric complex requires interleukin 7 receptor. J Exp Med 192:659–670
Pauwels RA, Buist AS, Calverley PM et al (2001) Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) workshop summary. Am J Respir Crit Care Med 163:1256–1276
Pesci A, Balbi B, Majori M et al (1998) Inflammatory cells and mediators in bronchial lavage of patients with chronic obstructive pulmonary disease. Eur Respir J 12:380–386
Quaedvlieg V, Henket M, Sele J et al (2006) Cytokine production from sputum cells in eosinophilic versus non-eosinophilic asthmatics. Clin Exp Immunol 143:161–166
Quentmeier H, Drexler HG, Fleckenstein D et al (2001) Cloning of human thymic stromal lymphopoietin (TSLP) and signaling mechanisms leading to proliferation. Leukemia 15:1286–1292
Rahman I (2000) Regulation of nuclear factor-kappa B, activator protein-1, and glutathione levels by tumor necrosis factor-alpha and dexamethasone in alveolar epithelial cells. Biochem Pharmacol 60:1041–1049
Rahman I, MacNee W (1996) Oxidant/antioxidant imbalance in smokers and chronic obstructive pulmonary disease. Thorax 51:348–350
Ray RJ, Furlonger C, Williams DE et al (1996) Characterization of thymic stromal-derived lymphopoietin (TSLP) in murine B cell development in vitro. Eur J Immunol 26:10–16
Reche PA, Soumelis V, Gorman DM et al (2001) Human thymic stromal lymphopoietin preferentially stimulates myeloid cells. J Immunol 167:336–343
Reimold AM (2002) TNFalpha as therapeutic target: new drugs, more applications. Curr Drug Targets Inflamm Allergy 1:377–392
Repine JE, Bast A, Lankhorst I (1997) Oxidative stress in chronic obstructive pulmonary disease: oxidative stress study group. Am J Respir Crit Care Med 156:341–357
Rochman I, Watanabe N, Arima K et al (2007) Cutting edge: direct action of thymic stromal lymphopoietin on activated human CD4+ T cells. J Immunol 178:6720–6724
Saetta M, Di Stefano A, Turato G et al (1998) CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 157:822–826
Sarir H, Henricks PA, van Houwelingen AH et al (2008) Cells, mediators and toll-like receptors in COPD. Eur J Pharmacol 585:346–353
Sethi S, Murphy TF (2008) Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med 359:2355–2365
Shahid SK, Kharitonov SA, Wilson NM et al (2002) Increased interleukin-4 and decreased interferon-gamma in exhaled breath condensate of children with asthma. Am J Respir Crit Care Med 165:1290–1293
Shi L, Leu SW, Xu F et al (2008) Local blockade of TSLP receptor alleviated allergic disease by regulating airway dendritic cells. Clin Immunol 129:202–210
Shimbara A, Christodoulopoulos P, Soussi-Gounni A et al (2000) IL-9 and its receptor in allergic and nonallergic lung disease: increased expression in asthma. J Allergy Clin Immunol 105:108–115
Siafakas NM, Vermeire P, Pride NB et al (1995) Optimal assessment and management of chronic obstructive pulmonary disease (COPD) The European Respiratory Society Task Force. Eur Respir J 8:1398–1420
Sims JE, Williams DE, Morrissey PJ et al (2000) Molecular cloning and biological characterization of a novel murine lymphoid growth factor. J Exp Med 192:671–680
Sokol CL, Barton GM, Farr AG et al (2008) A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat Immunol 9:310–318
Soumelis V, Reche PA, Kanzler H et al (2002) Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol 3:673–680
Tonozuka Y, Fujio K, Sugiyama T et al (2001) Molecular cloning of a human novel type I cytokine receptor related to delta1/TSLPR. Cytogenet Cell Genet 93:23–25
Tu HY, Chen X, Li J (2007) Signal transduction in respiratory syncytial virus infection-induced thymic stromal lymphopoietin expression in human epithelial cells. Nan Fang Yi Ke Da Xue Xue Bao 27:1581–1583
van der Plas DC, Smiers F, Pouwels K et al (1996) Interleukin-7 signaling in human B cell precursor acute lymphoblastic leukemia cells and murine BAF3 cells involves activation of STAT1 and STAT5 mediated via the interleukin-7 receptor alpha chain. Leukemia 10:1317–1325
Watanabe N, Hanabuchi S, Soumelis V et al (2004) Human thymic stromal lymphopoietin promotes dendritic cell-mediated CD4+ T cell homeostatic expansion. Nat Immunol 5:426–434
Watanabe N, Wang YH, Lee HK et al (2005) Hassall’s corpuscles instruct dendritic cells to induce CD4+ CD25+ regulatory T cells in human thymus. Nature 436:1181–1185
Weathington NM, van Houwelingen AH, Noerager BD et al (2006) A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med 12:317–323
Ying S, O’Connor B, Ratoff J et al (2005) Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J Immunol 174:8183–8190
Ying S, O’Connor B, Ratoff J et al (2008) Expression and cellular provenance of thymic stromal lymphopoietin and chemokines in patients with severe asthma and chronic obstructive pulmonary disease. J Immunol 181:2790–2798
Yokoi T, Amakawa R, Tanijiri T et al (2008) Mycobacterium bovis Bacillus Calmette-Guerin suppresses inflammatory Th2 responses by inducing functional alteration of TSLP-activated dendritic cells. Int Immunol 20:1321–1329
Yoo J, Omori M, Gyarmati D et al (2005) Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J Exp Med 202:541–549
Zhang K, Shan L, Rahman MS et al (2007) Constitutive and inducible thymic stromal lymphopoietin expression in human airway smooth muscle cells: role in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol 293:L375–L382
Zhou B, Comeau MR, De Smedt T et al (2005) Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol 6:1047–1053
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Fang, C., Siew, L.Q.C., Corrigan, C.J. et al. The Role of Thymic Stromal Lymphopoietin in Allergic Inflammation and Chronic Obstructive Pulmonary Disease. Arch. Immunol. Ther. Exp. 58, 81–90 (2010). https://doi.org/10.1007/s00005-010-0064-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-010-0064-3