
Abstract
Background
Esketamine nasal spray is approved for treatment-resistant depression.
Objective
The objective of this study was to characterize the pharmacokinetics of esketamine and noresketamine in healthy subjects and patients with treatment-resistant depression.
Methods
Esketamine and noresketamine were measured in > 9000 plasma samples collected from 820 individuals who received esketamine by the intranasal, intravenous, and oral routes. An open linear model for esketamine (three compartments) and noresketamine (two compartments) that included a hepato-portal compartment was developed using NONMEM® VII. The effects of covariates on esketamine pharmacokinetics and a model evaluation were performed using conventional methods.
Results
The fraction of a 28-mg intranasal dose absorbed through the nasal cavity (FRn) is 54% (100% of this fraction is completely absorbed); the remaining 46% is swallowed and undergoes intestinal and first-pass metabolism and 18.6% of the swallowed dose reaches the systemic circulation. The absolute bioavailability of 56 and 84 mg of intranasal esketamine is 54 and 51%, respectively. Esketamine volume at steady state and clearance were 752 L and 114 L/h, respectively. Noresketamine volume at steady state and apparent clearance were 185 L and 38 L/h, respectively. Relative to non-Asian subjects, Asian subjects showed a 64.0 and 19.4% decrease in the esketamine elimination rate constant and noresketamine apparent clearance, respectively. Japanese subjects exhibited a 34% increase in FRn vs other races. Hepatic blood flow decreased by 21.9 L/h for each decade in age in subjects aged > 60 years. These changes resulted in esketamine and noresketamine maximum concentration and area under the concentration–time curve after 24 h post-dose values that were up to 36% higher than those observed in other races or in younger adult subjects.
Conclusions
Esketamine and noresketamine pharmacokinetics was successfully characterized in healthy subjects and patients with treatment-resistant depression. The model quantified esketamine absolute nasal and oral bioavailability, its hepatic flow-limited clearance and biotransformation to the major metabolite noresketamine, and the influence of intrinsic and extrinsic factors on esketamine pharmacokinetics.
Clinical trials registration numbers of the studies included in the analysis: ESKETINTRD1001 (NCT01780259), ESKETINTRD1002 (NCT01980303), ESKETINTRD1003 (NCT02129088), ESKETINTRD1008 (NCT02846519), ESKETINTRD1009 (NCT02343289), ESKETINTRD1010 (NCT02568176), ESKETINTRD1012 (NCT02345148), 54135419TRD1015 (NCT02682225), ESKETINTRD2003 (NCT01998958), ESKETINSUI2001 (NCT02133001), ESKETINTRD3001 (NCT02417064), ESKETINTRD3002 (NCT02418585), and ESKETINTRD3005 (NCT02422186).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The well-stirred model was used to characterize the esketamine first-pass metabolism due to drug-metabolizing enzymes in the intestine and liver and enabled the quantification of esketamine absolute nasal bioavailability (56 mg: 54%; 84 mg: 51%) and its hepatic flow-dependent clearance. |
The population pharmacokinetic model successfully described the time course of esketamine and noresketamine plasma concentrations and their variability in healthy subjects and patients with treatment-resistant depression. After intranasal administration, the absolute bioavailability is 63, 54, and 51%, for 28-, 56-, and 84-mg esketamine doses, respectively. Esketamine and noresketamine demonstrated linear elimination, no accumulation with twice-weekly dosing and time-independent pharmacokinetics. |
1 Introduction
Major depressive disorder affects more than 300 million people and is the leading cause of disability worldwide; this common condition is also associated with a 10-year reduction in life expectancy [1, 2]. Nearly all monoamine-based pharmacological drug classes available for the treatment of depression take several weeks to show efficacy [3]. About 30% of patients with major depressive disorder do not respond despite treatment with different antidepressant medications and are considered to have treatment-resistant depression (TRD), which is associated with chronicity, functional disability, and increased morbidity [4]. Therefore, a significant need exists for fast-acting novel treatments based upon relevant pathophysiologic pathways underlying major depressive disorder for the rapid relief of depressive symptoms, especially in patients with TRD.
Esketamine, the S-enantiomer of ketamine, is a N-methyl-d-aspartate receptor antagonist which when administered in the antidepressant dose range, transiently increases the glutamatergic transmission [5,6,7] leading to α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor activation [8]. Esketamine nasal spray (Spravato™) has been approved for use in conjunction with an oral antidepressant in the treatment of TRD in the USA, Europe, and many other countries [9]. Marketing authorization was based on studies in adults with TRD that demonstrated a flexibly dosed nasal esketamine spray plus a newly initiated oral antidepressant produced a statistically significant and clinically meaningful reduction in depressive symptoms, demonstrated by the change in Montgomery–Åsberg Depression Rating Scale total score, as compared to a placebo nasal spray plus a newly initiated oral antidepressant [9, 10]. An effect of esketamine on the reduction of depression symptoms was observed as early as 24 h post-dose [9, 11, 12].
The esketamine pharmacokinetic (PK) profile in plasma following intranasal administration was characterized by fast absorption, moderate absolute bioavailability, high clearance, and a large volume of distribution [13, 14]. After intranasal administration in humans, the expected time to reach peak plasma concentration for esketamine is 20–40 min after the last nasal spray of a treatment session, after which esketamine plasma concentrations declined rapidly, with a mean terminal half-life of 7–12 h [13]. There was no accumulation of esketamine in plasma after repeated administration following twice-weekly intranasal administration or less frequently. Esketamine exposure increased with the dose from 28 mg to 84 mg. The increase in maximum concentration (Cmax) and the area under the concentration–time curve (AUC) values was less than dose proportional between 28 and 56 mg and 28 and 84 mg but was nearly dose proportional between 56 and 84 mg.
Esketamine is extensively metabolized by the cytochrome P450 enzymes (CYP), mainly 3A4 and 2B6. N-demethylation of esketamine to form noresketamine is the major metabolic pathway [15]. Noresketamine is further metabolized by CYP-dependent pathways. After intravenous and oral administration of esketamine, the AUC from time zero to infinity for this metabolite was higher than that of esketamine, particularly after the oral route, but had a mean terminal half-life comparable to that of esketamine [16]. Following intravenous or oral administration of radiolabeled esketamine by human subjects, ≥ 78% of administered radioactivity was recovered in urine [13].
This article describes the population PK analysis conducted to characterize the plasma concentrations of esketamine and noresketamine in healthy subjects and patients with TRD. In addition, the interindividual variability (IIV) and the effects of intrinsic and extrinsic factors on PK parameters of esketamine and noresketamine were also evaluated. The result of the current analyses increased our understanding of the pharmacokinetics of esketamine and noresketamine following nasal administration, by integrating relevant PK information across multiple routes of administrations and dose levels, and by assessing the impact of intrinsic and extrinsic factors on the esketamine and noresketamine pharmacokinetics in healthy subjects and patients with TRD. The population PK model developed proved critical for elucidating the exposure–response relationships and guiding the dose selection.
2 Methods
2.1 Clinical Studies, Sample Collection, and Bioanalytical Method
Rich and sparse plasma concentration data of esketamine and noresketamine obtained from 13 clinical studies, for which the entrance criteria are detailed elsewhere (ESKETINTRD1001 [17], ESKETINTRD1002 [18], ESKETINTRD1003 [19], ESKETINTRD1008 [20], ESKETINTRD1009 [21], ESKETINTRD1010 [22], ESKETINTRD1012 [23], 54135419TRD1015 [24], ESKETINTRD2003 [25], ESKETINSUI2001 [26], ESKETINTRD3001 (TRANSFORM-1) [27], ESKETINTRD3002 (TRANSFORM-2) [28], and ESKETINTRD3005 (TRANSFORM-3) [29]), were pooled for the population PK analysis. The clinical studies were selected based on the available PK data from single and multiple intranasal dosing, as well as intravenous (IV) and oral (PO) administration. Table 1 provides a summary of the study characteristics as well as specific time points the PK samples were collected. The concentrations of esketamine and noresketamine in plasma were measured using a validated liquid chromatography-tandem mass spectroscopy method. The lower limit of quantitation was 0.5 ng/mL for esketamine and noresketamine.
All studies were conducted in accordance with principles for human experimentation as defined in the Declaration of Helsinki and were approved by the human investigational review board of each study center and by the competent authority of each country. Informed consent was obtained from each subject before enrollment in the studies, after being advised of the potential risks and benefits of the study, as well as the investigational nature of the study.
2.2 Software
The plasma concentration–time data of esketamine and noresketamine were used for non-linear mixed-effect modeling using NONMEM® version 7.3.0 (Icon Development Solutions, Ellicott City, MD, USA) [30] in a validated environment, Navigator 9.1.5146, based on Good Automated Manufacturing Practice and in accordance with 21 CFR Part 11 and good clinical practice regulations. The Fortran compiler was Intel® Fortran 64 Compiler Professional, Version 11.1. The first-order conditional estimation method was used. The exploratory and statistical analysis, diagnostic graphics, and post-processing of NONMEM® analysis results were carried out in R version 3.2.3 or higher (Comprehensive R Network, http://cran.r-project.org/) [31].
2.3 Pharmacokinetic Model Development
After IV administration, esketamine disposition in plasma was described by an open, linear, three-compartment model. The model was parameterized in terms of volumes of distribution of the central (Vc), shallow (Vp1) and deep (Vp2) peripheral compartments, and the corresponding inter-compartmental clearances (Q1) and (Q2). In addition, esketamine central compartment was kinetically linked to a hepatic compartment by the effective hepatic flow parameter (Qh) and the hepatic volume of distribution (Vh), which both were estimated from the available data. The hepatic compartment resembles the hepato-portal system and its parameterization is consistent with the well-stirred model [32]. Furthermore, the model assumed that esketamine in the hepatic compartment is metabolized to either noresketamine or other metabolites through a linear process. The biotransformation of esketamine to noresketamine was characterized by a first-order rate constant, kmet, while the elimination of esketamine through other metabolic routes was characterized by the first-order rate constant, kel. Therefore, the product Vh × (kel + kmet) reflects the intrinsic clearance (Clint) of esketamine. Based on the well-stirred model, the hepatic extraction (E) was defined as Clint/(Qh + Clint) and the esketamine plasma clearance was equal to Qh × E. Furthermore, the fraction of esketamine in the hepatic compartment that is converted to noresketamine (Fmet) or is eliminated through other metabolic pathways (Fel) can be derived as follows:
and
Noresketamine disposition was characterized by an open, linear, two-compartment model, which was parameterized in terms of central (Vcn/F) and peripheral (Vpn/F) volumes of distribution, inter-compartmental clearance (Q3/F), and clearance (CLn/F). Noresketamine elimination rate-constant (ken) was derived as a secondary PK parameter as CLn/Vcn. In the absence of noresketamine plasma concentration data after noresketamine IV dosing, the PK parameters of noresketamine were considered “apparent” as the fraction of esketamine that is metabolized to noresketamine cannot be estimated directly from the available data.
To properly characterize esketamine after PO dosing, an oral depot compartment was incorporated into the model and a joint analysis of IV and PO data was conducted. After PO dosing, a fraction of the PO dose is “lost” in the gastrointestinal tract due to intestinal metabolism and the remaining fraction of the dose (Fgut) is released into the oral depot compartment following a zero-order process, characterized by duration, Dpo. Subsequently, esketamine is absorbed from the oral depot compartment into the hepatic compartment, following a first-order process, characterized by the rate constant ka,po. The fraction of the PO dose absorbed that avoids the first-pass metabolism in hepatocytes (Fhep) is equal to 1 − E. In this context, the absolute PO bioavailability (Fpo) is the product of Fgut × Fhep.
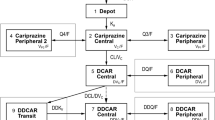
To characterize esketamine after intranasal dosing, a nasal depot compartment, representing the nasal cavity, was incorporated into the model and a joint analysis of IV, PO, and intranasal PK data was conducted. After intranasal administration, a fraction of the esketamine dose (FRn) is absorbed via the vascular plexus into the central compartment, following a first-order process characterized by ka,n. Linking the nasal depot compartment directly to the central compartment allows modeling of the direct and complete absorption of the fraction of the esketamine dose absorbed through the intranasal mucosa, avoiding the intestinal metabolism in the gastrointestinal tract and hepatic first-pass metabolism. The remaining fraction of the intranasal esketamine dose (1 − FRn) is swallowed and enters the systemic circulation via the PO depot compartment following a sequential zero-first-order process characterized by Dsw and ka,sw. Therefore, the absolute nasal depot bioavailability, Fn, is equal to FRn + (1 − FRn) × Fpo. The schematic of the PK model described is displayed in Fig. 1 and the corresponding differential equations used to describe the time course of esketamine and noresketamine after intranasal, IV, and PO dosing were the following:
where \(A_{1}\) at dosing time is equal to the esketamine dose.
\(A_{1}\) at dosing time is equal to the esketamine dose.

Schematic of the population pharmacokinetic model for esketamine and noresketamine. CLn/F apparent clearance of noresketamine, Dpo zero-order absorption duration of the PO solution dose, Dsw zero-order absorption duration of the intranasal dose that is swallowed, Fgut percentage of the PO dose of esketamine that reaches the hepato-portal system after intestinal metabolism, FRn fraction of the intranasal dose absorbed in the nasal cavity, ka,n esketamine first-order nasal absorption rate constant, ka,po esketamine first-order PO solution absorption rate constant, ka,sw esketamine first-order nasal-swallowed absorption rate constant, kel esketamine elimination rate constant, kmet esketamine rate constant of metabolism to noresketamine, PO oral, Q1 esketamine inter-compartmental clearance between the central and the shallow peripheral compartment, Q2 esketamine inter-compartmental clearance between the central and the deep peripheral compartment, Q3/F noresketamine apparent inter-compartmental clearance between the central and the peripheral compartment, Qh hepatic flow, Vc, Vp1, Vp2 esketamine central, shallow, and deep peripheral volumes of distribution, respectively, Vcn/F noresketamine apparent central volume of distribution, Vh hepatic volume of distribution, Vpn/F noresketamine apparent peripheral volume of distribution. Dose effect is the dose-dependent effect in FRn
with A1, A2, A3, A4, A5, A6, A7, and A8 being the corresponding amounts in nasal, oral, esketamine central, esketamine peripheral 1, esketamine peripheral 2, hepato-portal system, noresketamine central, and noresketamine peripheral 1 compartments, respectively. Other abbreviations are already provided in the body text and in Fig. 1.
2.4 Statistical Model
The IIV in the PK model parameters was assumed to follow a log-normal distribution. Residual variability in esketamine and its major metabolite was separately evaluated using an additive error model after natural logarithmic transformation of the observations and model predictions [33].
2.5 Model Assessment
To identify the best pharmaco-statistical model, a series of models were evaluated. For each nested model, the improvement in the fit was assessed by the likelihood ratio test (p = 0.001). In addition, the reduction in IIV and residual variability, the precision and correlation in parameter estimates, examination of diagnostic plots [34], predictive checks, and shrinkage [35] were also employed.
2.6 Covariate Analysis
The effect of demographic factors (sex, age, body weight, race), hepatic function biomarkers (alanine transaminase [ALT], aspartate transaminase [AST], alkaline phosphatase [ALP], gamma glutamyl transpeptidase, lactate dehydrogenase, and total bilirubin (TB)], serum albumin, total protein (TP), estimated glomerular filtration rate, and diagnostic status (healthy subjects vs patients with TRD) on esketamine and noresketamine PK parameters were evaluated. Table 2 shows the summary of the subjects’ characteristics at baseline. The covariate screening was guided by graphical assessment and stepwise linear regression of the relationships between the Bayesian estimates of the random effects and the covariates. Only covariates that were statistically significant (p < 0.001) and had a coefficient of determination r2 > 0.10 with model parameters were considered as potentially clinically relevant and were further tested one by one in NONMEM to evaluate whether they were incorporated in the population PK model process as described elsewhere [36]. The p-values associated with the covariate analysis were selected after considering the sample size available, the number of covariates evaluated, and the number of model parameters where the potential relationship with covariates was evaluated. This adjustment follows the recent guidance that the American Statistical Association [37] and another large coalition of 72 methodologists [38, 39] proposed with respect to p-values. As the p-values can be affected by the sample size, the r2 between the covariate-PK relationship was an additional decision criterion during model assessment because its value indicates the PK variability explained by a covariate of interest. Falk and Miller recommend at least 10% of the total variance and therefore this 10% cut-off for the r2 was considered during the model building [40]. With this methodology, only covariates showing significant and clinically relevant contributions were retained in the population PK model.
To fully understand the impact of the individual covariates on esketamine and noresketamine exposure, exploratory forest plots were created with the post hoc estimates of the individual exposure. The AUC after 24 h post-dose (AUC0–24 h) for esketamine and noresketamine was calculated for each subject. Each exploratory forest plot represents the geometric mean ratio and its 90% confidence interval (CI) for the (AUC0–24 h) of esketamine and noresketamine in a specific subpopulation relative to the value in the reference subpopulation, after adjusting other covariates.
2.7 Model Evaluation
Goodness-of-fit plots, normalized prediction distribution errors (NPDEs) as well as prediction-corrected visual predictive check were used to evaluate the developed population PK model [41].
2.8 Model-Based Simulations
Deterministic simulations were undertaken using the population PK structural model parameters. A full plasma concentration–time profile was simulated to represent the esketamine and noresketamine pharmacokinetics for a subject with given covariate characteristics and the typical value of model parameters. Subsequently, the exposure metrics Cmax and AUC0–24 h for each PK profile were calculated and the magnitude of the covariate effect on esketamine and noresketamine exposure was assessed.
3 Results
In total, 9784 and 9397 plasma concentrations of esketamine and noresketamine, respectively, were collected from 820 subjects, including 256 (31.2%) healthy subjects from phase I studies and 564 (68.8%) subjects with TRD enrolled in phase II and phase III studies who received twice-weekly intranasal administration of esketamine with a dose range of 28–112 mg. Table 2 provides the demographic, clinical, and baseline laboratory characteristics of the subjects included in the population PK analysis. An open, linear, three-compartment disposition model for esketamine and an open, linear, two-compartment disposition model for noresketamine in combination with a separate hepato-portal compartment (“first-pass model”) provided a good description of the time courses of esketamine and noresketamine plasma concentrations and their associated variability after esketamine administration as a nasal spray, IV infusion, or PO solution (Fig. 1).
After intranasal administration of 28 mg of esketamine, 54% of the dose is absorbed through the nasal cavity to plasma (FRn), while the remaining 46% of the esketamine dose is swallowed. The fraction of the dose absorbed through the nasal cavity is reduced by 38% for the subsequent 28-mg doses. Therefore, for the 56-, 84-, and 112-mg doses, a total of 44, 40, and 38% of the esketamine dose is absorbed through the nasal cavity, respectively, while the remaining esketamine dose is swallowed. The fraction of the esketamine dose absorbed through the nasal cavity was complete and fast, as indicated by the mean absorption time (1/ka,n) of 0.341 h. The gastrointestinal absorption of the swallowed dose of esketamine was incomplete and relatively slower than the nasal absorption and had a mean absorption time (Dpo + 1/ka,po + Vh/Qh) of 2.02 h. About 64% of the PO esketamine dose escapes gastrointestinal degradation and reaches the hepato-portal system (Fgut). About 29% of the dose that reaches the hepato-portal system overcomes first-pass metabolism (Fhep = 1 − E) and accesses the systemic central compartment, while the remaining 71% undergoes first-pass metabolism (E) in the liver. This result implies an absolute PO bioavailability (Fpo = Fgut × Fhep = 0.64 × 0.29) of 18.6%. Consequently, after intranasal administration, the absolute bioavailability, taking into account direct absorption through the nasal cavity and PO bioavailability, is 63, 54, 51, and 50% for the 28-, 56-, 84-, and 112-mg esketamine doses, respectively.
Esketamine typical volumes of the central, shallow, and deep peripheral compartment and the corresponding IIV (CV%) were estimated to be 192 L (27.5%), 143 L (49.3%), and 417 L, respectively. The intrinsic clearance and the E were 392 L/h and 0.71, respectively, thus the typical plasma clearance of esketamine was estimated to be 114 L/h. The esketamine terminal half-life was estimated to be 11 h. Approximately 71% of the esketamine in the hepatic compartment is metabolized to noresketamine (Fmet), while the remaining 29% of esketamine in the hepatic compartment is eliminated through other metabolic routes (Fel). The typical PK parameters of noresketamine for the apparent central and peripheral volumes of distribution were estimated to be 70.0 and 115 L, while the apparent inter-compartmental flow and apparent clearance were 26.1 and 38.0 L/h, respectively. The IIV was quantified using an exponential error model for ka,n, ka,po, Vc, Vp1, Vh, Qh, kel, kmet, Vcn/F, and CLn/F and an additive error model in the logit domain for FRn. The shrinkage was lower than 40%, except for Vcn/F (41.7%) and Vh (44.5%).
The covariate analysis showed an effect of being Asian on kel (r2 = 0.168, p < 0.001) and Cln/F (r2 = 0.122, p < 0.001). The simultaneous inclusion of both covariate-PK relationships improved the model fit (∆MVOF: − 78.74, df = 2, p < 0.001). Relative to non-Asian subjects, the Asian subjects showed a 64.0% and a 19.4% decrease in kel and CLn/F, respectively. Sex, body weight, ALT, AST, ALP, gamma glutamyl transpeptidase, TB, albumin, TP, estimated glomerular filtration rate, and disease state (healthy subjects vs patients with TRD) had no discernable impact on the PK parameters of esketamine and noresketamine as the covariate-parameter relationships were found to have r2 values ≤ 10% and p-values > 0.001 in the exploratory analysis.
The effect of Japanese ethnicity and age on esketamine and noresketamine pharmacokinetics was evaluated by performing a sensitivity analysis. After the inclusion of the covariates that met the criteria specified in Sect. 2.6, the strongest relationships between race and age on individual PK model parameters according to r2 and p-values were explored and formally tested one by one in NONMEM®. The sensitivity analysis showed that the Japanese subjects had a 34% increase in FRn (∆MVOF = − 27.20; df = 1; p < 0.001), relative to other races (non-Asian subjects and Asian non-Japanese subjects) whereas Qh, decreased at a rate of 21.9 L/h/10 years from 60 years of age onwards (∆MVOF = − 16.8; df = 1; p < 0.001) and, on average, the Qh was 25% lower in subjects from 77 years of age onwards than in adult subjects younger than 60 years of age. The parameter estimates of the population PK model, including all the covariate-PK relationships, are presented in Table 3. All parameters were estimated with adequate precision, as measured by a relative standard error < 10% for the fixed effects and relative standard error < 25% for the random effects.
Figure 2 depicts the goodness-of-fit plots of the final model for esketamine and noresketamine against the population model prediction and individual model prediction showing a normal random scatter around the identity line and indicates the absence of significant bias. Similarly, the middle and lower panels of Fig. 2 represent the distribution of conditional weighted residuals (CWRES) and the NPDE as a function of the population predictions and time. Both the CWRES and NPDE supported model adequacy. The overall distribution of the NPDE approximately follows a normal distribution with a mean of 0 and a standard deviation of 1. Specifically, the mean and standard deviation of the NPDE for esketamine was 0.017 (95% CI − 0.004 to 0.033) and 0.949 (95% CI 0.935–0.970), respectively, while the mean and standard deviation of the NPDE for noresketamine was 0.040 (95% CI 0.019–0.056) and 0.987 (95% CI 0.972–1.000). These results confirm the model accuracy and precision because the mean and standard deviation of the NPDE for both esketamine and noresketamine were close to 0 and 1, respectively. The results of the prediction-corrected visual predictive check of the phase I, II, and III clinical studies are depicted in Fig. 3. Overall, the population PK model adequately describes the time courses of both esketamine and noresketamine plasma concentrations after intranasal dosing and, therefore, is considered suitable for simulating the time courses of esketamine and noresketamine plasma concentrations and their associated variability. Finally, the exploratory forest plots for esketamine and noresketamine describing the covariate-PK impact on AUC0–24 h are presented in Fig. 4.

Diagnostic plots of the population pharmacokinetic model developed for esketamine (a) and noresketamine (b). Circles (phase I and phase II studies) and triangles (phase III studies) represent the observations/predictions (upper panels), conditional weighted residuals (middle panels), and the normalized prediction distribution errors (lower panels) for esketamine (a) and noresketamine (b); solid black lines represent the identity lines; red dashed lines represent the trend line (locally weighted scatterplot smoothing)

Prediction-corrected visual predictive check of the population pharmacokinetic model for esketamine and noresketamine after intranasal dosing for the 24 h (left panels) and for 6 h (right panels) post-dose. Circles (phase I and phase II studies) and triangles (phase III studies); gray solid lines represent the median, 5th, and 95th percentiles of the simulated data with its 95% confidence interval (light-gray shaded area); purple dashed lines represent the median, 5th, and 95th percentiles of the observed data for esketamine and noresketamine respectively. A total of 100 replicates of the original dataset were used to create these plots

Forest plot for the covariate evaluation for esketamine (a) and noresketamine (b). ALT alanine transaminase, AST aspartate transaminase, AUC area under the concentration–time curve, CI confidence interval, GGT gamma glutamyl transpeptidase, GMR geometric mean ratio, TRD treatment-resistant depression
Model-predicted exposure metrics (Cmax and AUC0–24 h) for 28, 56, and 84 mg are presented in Table 4 stratified by race and age. Esketamine Cmax for a typical Asian non-Japanese subject was similar to a typical Caucasian subject; however, its AUC0–24 h was around 8% higher. With respect to noresketamine, a typical Asian non-Japanese subject had a 1.24- and a 1.36-fold higher Cmax and AUC0–24 h, respectively, when compared with a typical Caucasian subject. Esketamine Cmax and AUC0–24 h were both 1.32-fold higher for a typical Japanese subject when compared with a typical Caucasian subject. With respect to noresketamine, Cmax and AUC0–24 h were 1.22- and 1.48-fold higher in Japanese subjects, when both exposures were compared with a typical Caucasian subject. The impact of age on Qh showed that for a Caucasian subject of 70 years of age, esketamine Cmax and AUC0–24 h were 1.32-fold higher than a Caucasian subject below 60 years of age. Small differences were observed for noresketamine Cmax and AUC0–24 h and both parameters were comparable to those for Caucasian subjects who were below 60 years of age, across all the doses evaluated. Esketamine Cmax for a typical, elderly (aged 70 years) Asian non-Japanese subject was comparable to the Cmax for a Caucasian subject below 60 years of age; however, esketamine AUC0–24 h was 1.16-fold higher for the Asian non-Japanese subject relative to a Caucasian subject below 60 years of age. With regard to noresketamine, Cmax and AUC0–24 h were 1.22- and 1.37-fold higher, respectively, for a typical elderly (aged 70 years) Asian, non-Japanese subject when compared with a Caucasian subject below 60 years of age. Esketamine Cmax and AUC0–24 h were 1.32- and 1.42-fold higher respectively, for a typical elderly (aged 70 years) Japanese subject when compared with a Caucasian subject below 60 years of age. Noresketamine Cmax and AUC0–24 h values were 1.18- and 1.48-fold higher, respectively, for a typical elderly (aged 70 years) Japanese subject when compared with a Caucasian subject below 60 years of age.
4 Discussion
The primary goal of this population PK analysis was to simultaneously characterize the time course of esketamine and its metabolite noresketamine after IV, PO, and nasal dosing. After IV administration, esketamine disposition in plasma was described by an open, linear, three-compartment disposition model in which its central compartment was kinetically linked to a hepatic compartment [32]. The model assumed that esketamine in the hepatic compartment was metabolized to noresketamine through a linear process and once esketamine had been converted to noresketamine, noresketamine exhibited a bi-exponential decay. Similar structural population PK models using three- and two-compartment models have been used to characterize esketamine and noresketamine pharmacokinetics; [42,43,44] however, these models lacked PO data and therefore assumed a series of intermediate transit compartments to account for noresketamine formation time delays, without considering the “well-stirred” liver model [32], which physiologically accounts for the extensive first-pass metabolism in the liver [45].
To properly characterize esketamine after PO dosing and its corresponding first-pass metabolism, an oral depot compartment was incorporated into the model. The major site of first-pass metabolism of most orally administered drugs is often assumed to be the liver; however, the gut wall also plays an important role by directly metabolizing orally administered drugs prior to systemic uptake and thereby reducing drug bioavailability [46]. The absorption profile of esketamine was best described with a first-order absorption rate constant model rather than a transit compartment model [47], similar to the finding reported by Ashraf et al. [44]. Esketamine is mainly metabolized to noresketamine by CYP3A- and CYP2B6-mediated N-demethylation and the presence of CYP3A in the gut wall and CYP3A and CYP2B6 in the liver is well known [48]. The population PK model accounted for this physiological absorption process. About 64% of the PO esketamine dose overcomes gastrointestinal degradation and reaches the hepato-portal system where the first-pass hepatic metabolism takes place before esketamine becomes bioavailable in the systemic circulation. About 29% of the dose that reaches the hepatic compartment accesses directly to the central compartment and the remaining 71% undergoes first-pass metabolism (E). This implies an absolute PO bioavailability of 18.6%, which is consistent with the published estimates of PO bioavailability (14%) obtained from non-compartmental analyses of clinical PK data from subjects who received oral ketamine [13] and falls within the 7–20% range reported by Fanta et al. after simultaneously modeling esketamine and noresketamine in healthy volunteers receiving IV and PO dosing [47].
A nasal depot compartment, representing the nasal cavity, was incorporated into the population PK model and linked to the central disposition compartment of esketamine. Intranasal administration is a non-invasive route for drug delivery, which offers a highly vascularized and permeable surface area enabling drug molecules to be transferred rapidly to the systemic blood circulation without first-pass hepatic and intestinal metabolism [49]. The fraction of dose absorbed through the nasal cavity is reduced by 38% for the subsequent 28-mg doses; this decrement conceivably could be explained by esketamine-induced nasal vasoconstriction, which leads to reduced drug permeability, decreasing the extent of drug absorption [50]. Linking the nasal depot compartment directly to the central compartment allows modeling of the direct and complete absorption of the fraction esketamine dose absorbed through the nasal cavity, avoiding the intestinal metabolism in the gastrointestinal tract and hepatic first-pass metabolism. Consequently, the absolute bioavailability after nasal administration is 63, 54, 51, and 50% for 28-, 56-, 84-, and 112-mg esketamine doses, respectively. These results are comparable to the estimate of the absolute nasal bioavailability, 48%, for a 84-mg dose administered to healthy subjects [13, 14]. In addition, these results were also consistent with the report of Yanagihara et al. [51], which estimated the bioavailability of ketamine administered by a nasal spray to be approximately 45%.
Esketamine Vss was estimated to be 752 L, which is larger compared water in the body (42 L for a 70-kg male individual) and reflective of the large non-specific distribution to peripheral lipophilic tissues. This is similar to the Vss of 709 L calculated by non-compartmental methods [13, 14]. In previous literature of PK analyses, a large variability in the volume of distribution of esketamine has been described from 160 to 550 L [16, 42, 52, 53]. In the current analysis, esketamine Vss was estimated to be larger than this range, probably because of the extensive sampling in the phase I studies included in this analysis, relative to the studies reported in the literature.
The CLint and the E were estimated to be 392 L/h and 0.71, respectively, thus the typical plasma clearance of esketamine was estimated to be 114 L/h, which is consistent with the results of a non-compartmental analysis [13, 14] and the 95.2 L/h reported by Fanta et al. [47], suggesting that it is similar to or exceeds hepatic blood flow in humans [54]. These results are consistent with literature PK studies in which clearance of esketamine had a range of 73–147 L/h, [16, 42, 52, 53, 55]. According to the well-stirred model, a high-extraction drug such as esketamine, whose intrinsic clearance exceeds the hepatic blood flow, behaves such that a decrease in hepatic blood flow causes a decrease in the clearance of the drug when administered systemically [56]. Moreover, a decrease in the drug’s intrinsic clearance would cause only minor changes in its total clearance following systemic administration. Thus, alterations in hepatic blood flow could influence the clearance rate of systemically administered esketamine or the fraction of the esketamine dose that is absorbed directly through the nasal mucosa. In contrast, the pharmacokinetics of intranasally administered esketamine was relatively insensitive to the effects of the potent CYP inducer rifampin and the CYP3A inhibitor clarithromycin [12, 14].
Similar to esketamine, an apparent Vss of 185 L for noresketamine reflects a large non-specific distribution into peripheral tissues. The terminal half-life of noresketamine was calculated to be 7.5 h, shorter than the terminal half-life of esketamine (11 h), but longer than the half-life associated with noresketamine formation from esketamine. Consequently, following a twice-weekly intranasal administration, no accumulation of esketamine and noresketamine in plasma is expected, as the inter-dose interval is greater than the wash-out period (five half-lives).
The dataset used for the population PK analysis included data from study ESKETINTRD1009, in which esketamine was administered via multiple routes, as well as 12 clinical studies where only intranasal esketamine was administered. As a consequence, there is an imbalance between the amount of data obtained following PO and IV administration when compared to intranasal administration, since intranasal was the intended route of administration in the clinic. However, this limitation does not affect the PK parameter estimates since the presence of ESKETINTRD1009 study in the dataset assures there are sufficiently informative data to have an adequate estimation of the model parameters. The identifiability of model parameters is primarily determined by the quality of the PK data collected, and not by an imbalance between the number of available PO and IV data compared to intranasal administration, especially when enough information is included in the dataset as in the present case. Therefore, the low proportion of data after PO and IV administration relative to intranasal administration in the population PK dataset did not impact the adequacy of PK parameter estimation because the dataset contains highly informative PK profiles for the three routes of administration obtained from study ESKETINTRD1009.
The Asian subjects showed a 64.0 and 19.4% decrease in the elimination rate constant of esketamine (kel) and noresketamine (CLn/F), respectively, which resulted on average, in an 8 and 36% increase in esketamine and noresketamine AUC0–24 h, respectively, when compared with the Caucasian population. Esketamine and noresketamine clearance variability can be attributed to variability in the amount of CYP3A- and CYP2B6-mediated N-demethylation enzymes in the liver. Previous pharmacogenomic studies reported a reduced function of the CYP2B6*10 allele in about 38–50% of East Asians but only in about 3% of Caucasians [57,58,59].
Japanese subjects exhibited a 34% increase in FRn relative to other races, which translated into a 1.32-fold higher Cmax and AUC0–24 h when compared with Caucasian subjects. In contrast, noresketamine Cmax and AUC0–24 h was 1.22- and 1.48-fold higher in the Japanese subjects, when both exposures were compared to the Caucasian subjects. These results are in line with phase I studies that indicated the Cmax and AUC of esketamine are approximately 40% higher in Japanese subjects compared with Caucasian subjects [13, 14], while the decline in plasma esketamine concentrations was similar for Japanese and Caucasian subjects. This difference in exposures between Japanese and Caucasian subjects conceivably may be attributable to differences in anatomical and physiological properties of the nasal cavity between these subpopulations. Factors such as membrane permeability, environmental pH, or mucociliary clearance could affect the systemic bioavailability of drugs, which are administered through the intranasal route [60]. However, further studies are needed to test these hypotheses.
A slight decrease in Qh was observed at increasing age. On average, Qh is 25% lower in subjects from 77 years of age onwards than in adult subjects younger than 60 years of age. On average, Cmax and AUC0–24 h for esketamine in the elderly subjects were approximately 21 and 18% higher, respectively, compared with the younger adult subjects. This finding was comparable to the results of a phase I study that compared the pharmacokinetics of 28 mg of intranasal esketamine between healthy elderly subjects (65–81 years of age) and younger subjects (18–55 years of age, inclusive) [13, 14]. Reduced hepatic blood flow changes can affect systemic drug clearance especially for drugs with high extraction ratios such as esketamine (E = 0.71) [56]. Furthermore, it is well known that elderly subjects are more likely to manifest relatively higher drug exposures as a result of a reduction in the hepatic blood flow caused by age. This reduced hepatic blood flow caused by aging leads to a decreased clearance driving an increase in PK exposure in this subpopulation [61].
5 Conclusions
The concept of the well-stirred model was implemented in the population PK model to physiologically account for the extensive first-pass metabolism due to drug-metabolizing enzymes in the intestine and in the liver. The developed model successfully described the time course of esketamine and noresketamine across different dose levels after IV, PO, and intranasal administration in healthy subjects and patients with TRD. In addition, the model enabled the quantification of esketamine absolute nasal and oral bioavailability, its hepatic flow-limited clearance, and its biotransformation to noresketamine, the major circulating metabolite, as well as the influence of intrinsic and extrinsic factors in esketamine pharmacokinetics. In summary, esketamine is a high extraction drug, with relatively low absolute oral bioavailability and a large volume of distribution. It has linear elimination, dose-proportional and time-independent pharmacokinetics, and negligible accumulation after a twice-weekly dosing regimen. Higher exposure was observed in Asian subjects owing to a decrease in esketamine and noresketamine elimination, in Japanese subjects specifically due to an increase in esketamine bioavailability, and in elderly subjects because of a reduction in hepatic blood flow. However, dose adjustments in these subpopulations should be based on the efficacy and tolerability of esketamine. Furthermore, sex, body weight, ALT, AST, ALP, gamma glutamyl transpeptidase, TP, albumin, TB, estimated glomerular filtration rate, and mood disease state had no discernable impact on the PK parameters of esketamine and noresketamine, consequently, no dose adjustment on the basis of these covariates is warranted.
References
WHO. WHO fact sheet on depression 2018. http://www.who.int/news-room/fact-sheets/detail/depression. Accessed 19 Feb 2020.
Walker ER, McGee RE, Druss BG. Mortality in mental disorders and global disease burden implications: a systematic review and meta-analysis. JAMA Psychiatry. 2015;72(4):334–41.
Machado-Vieira R, Salvadore G, Luckenbaugh D, Manji H, Zarate C. Rapid onset of antidepressant action: a new paradigm in the research and treatment of major depression. J Clin Psychiatry. 2008;69:946–58.
Nemeroff CB. Prevalence and management of treatment-resistant depression. J Clin Psychiatry. 2007;68(Suppl. 8):17–25.
Manji H, Drevets W, Charney D. The cellular neurobiology of depression. Nat Med. 2001;7:541–7.
Murrough J, Abdallah C, Mathew S. Targeting glutamate signaling in depression: progress and prospects. Nat Rev Drug Discov. 2017;16:472–86.
Duman R, Aghajanian G, Sanacora G, Krystal J. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med. 2016;22:238–49.
Kaur U, Pathak BK, Singh A, et al. Esketamine: a glimmer of hope in treatment-resistant depression. Eur Arch Psychiatry Clin Neurosci. 2019. https://doi.org/10.1007/s00406-019-01084-z.
Daly EJ, Singh JB, Fedgchin M, et al. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2018;75(2):139–48.
Montgomery S, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–9.
Fedgchin M, Trivedi M, Daly EJ, et al. Efficacy and safety of fixed-dose esketamine nasal spray combined with a new oral antidepressant in treatment-resistant depression: results of a randomized, double-blind, active-controlled study (TRANSFORM-1). Int J Neuropsychopharmacol. 2019;22(10):616–30.
Zanos P, Moaddel R, Morris P, Riggs L, Highland J, Georgiou P, et al. Ketamine and ketamine metabolite pharmacology: insights into therapeutic mechanisms. Pharmacol Rev. 2018;70:621–60.
Spravato: US label. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211243lbl.pdf. Accessed 19 Feb 2020.
Spravato: summary of product characteristics. https://www.ema.europa.eu/en/documents/product-information/spravato-epar-product-information_en.pdf. Accessed 19 Feb 2020.
Yanagihara Y, Kariya S, Ohtani M, et al. Involvement of CYP2B6 in N-demethylation of ketamine in human liver microsomes. Drug Metab Dispos. 2001;29:887–90.
Peltoniemi MA, Saari TI, Hagelberg NM, et al. Rifampicin has a profound effect on the pharmacokinetics of oral S-ketamine and less on intravenous S-ketamine. Basic Clin Pharmacol Toxicol. 2012;111:325–32.
Janssen Research and Development. A study to assess the pharmacokinetics, safety, and tolerability of intranasally administered esketamine in healthy participants [ClinicalTrials.gov Identifier: NCT01780259]. US National Institutes of Health, Clinicaltrials.gov. 2013. https://clinicaltrials.gov/ct2/show/NCT01780259. Accessed 11 Oct 2020.
Janssen Research and Development. A study to assess the pharmacokinetics of intranasally administered esketamine in healthy japanese and caucasian volunteers [ClinicalTrials.gov Identifier: NCT01980303]. US National Institutes of Health, Clinicaltrials.gov. 2014. https://clinicaltrials.gov/ct2/show/NCT01980303. Accessed 11 Oct 2020.
Janssen Research and Development. A pharmacokinetic, safety and tolerability study of esketamine in healthy elderly and adult participants [ClinicalTrials.gov Identifier: NCT02129088]. US National Institutes of Health, Clinicaltrials.gov. 2014. https://clinicaltrials.gov/ct2/show/NCT02129088. Accessed 11 Oct 2020.
Janssen Research and Development. Pharmacokinetic, safety, and tolerability study of intranasally administered esketamine in healthy Han Chinese, Korean, Japanese, and Caucasian participants and the effects of rifampin on the pharmacokinetics of intranasally administered esketamine. [ClinicalTrials.gov Identifier: NCT02846519]. US National Institutes of Health, Clinicaltrials.gov. 2019. https://clinicaltrials.gov/ct2/show/NCT02846519. Accessed 11 Oct 2020.
Janssen Research and Development. A study to evaluate the absolute bioavailability of intranasal and oral esketamine and the effects of clarithromycin on the pharmacokinetics of intranasal esketamine in healthy participants [ClinicalTrials.gov Identifier: NCT02343289]. US National Institutes of Health, Clinicaltrials.gov. 2015. https://clinicaltrials.gov/ct2/show/NCT02343289. Accessed 11 Oct 2020.
Janssen Research and Development. Pharmacokinetic study of intranasal esketamine and its effects on the pharmacokinetics of orally-administered midazolam and bupropion in healthy participants. [ClinicalTrials.gov Identifier: NCT02568176]. US National Institutes of Health, Clinicaltrials.gov. 2017. https://clinicaltrials.gov/ct2/show/NCT02568176. Accessed 11 Oct 2020.
Janssen Research and Development. Pharmacokinetic, safety, and tolerability study of intranasally administered esketamine in elderly and healthy younger adult participants. [ClinicalTrials.gov Identifier: NCT02345148]. US National Institutes of Health, Clinicaltrials.gov. 2017. https://clinicaltrials.gov/ct2/show/NCT02345148. Accessed 11 Oct 2020.
Janssen Research and Development. Crossover study to evaluate the abuse potential of intranasal esketamine compared to racemic intravenous ketamine in nondependent, recreational drug users [ClinicalTrials.gov Identifier: NCT02682225]. US National Institutes of Health, Clinicaltrials.gov. 2017. https://clinicaltrials.gov/ct2/show/NCT02682225. Accessed 11 Oct 2020.
Janssen Research and Development. A study to evaluate the safety and efficacy of intranasal esketamine in treatment-resistant depression (SYNAPSE) [ClinicalTrials.gov Identifier: NCT01998958]. US National Institutes of Health, Clinicaltrials.gov. 2019. https://clinicaltrials.gov/ct2/show/NCT01998958. Accessed 11 Oct 2020.
Janssen Research and Development. A double-blind study to assess the efficacy and safety of intranasal esketamine for the rapid reduction of the symptoms of major depressive disorder, including suicidal ideation, in participants who are assessed to be at imminent risk for suicide [ClinicalTrials.gov Identifier: NCT02133001]. US National Institutes of Health, Clinicaltrials.gov. 2019. https://clinicaltrials.gov/ct2/show/NCT02133001. Accessed 11 Oct 2020.
Janssen Research and Development. A study to evaluate the efficacy, safety, and tolerability of fixed doses of intranasal esketamine plus an oral antidepressant in adult participants with treatment-resistant depression (TRANSFORM-1) [ClinicalTrials.gov Identifier: NCT02417064]. US National Institutes of Health, Clinicaltrials.gov. 2019. https://clinicaltrials.gov/ct2/show/NCT02417064. Accessed 11 Oct 2020.
Janssen Research and Development. A study to evaluate the efficacy, safety, and tolerability of flexible doses of intranasal esketamine plus an oral antidepressant in adult participants with treatment-resistant depression (TRANSFORM-2) [ClinicalTrials.gov Identifier: NCT02418585]. US National Institutes of Health, Clinicaltrials.gov. 2019. https://clinicaltrials.gov/ct2/show/NCT02418585. Accessed 11 Oct 2020.
Janssen Research and Development. A study to evaluate the efficacy, safety, and tolerability of intranasal esketamine plus an oral antidepressant in elderly participants with treatment-resistant depression (TRANSFORM-3) [ClinicalTrials.gov Identifier: NCT02422186]. US National Institutes of Health, Clinicaltrials.gov. 2019. https://clinicaltrials.gov/ct2/show/NCT02422186. Accessed 11 Oct 2020.
Beal SL, Sheiner LB, Boeckmann AJ, Bauer RJ, editors. NONMEM 7.1.0 users guides. Ellicott City: Icon Development Solutions; 1989–2009.
R Development Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2012. http://www.R-project.org/Guidance for Industry. Accessed 11 Oct 2020.
Piotrovskij V, Van Peer A. A model with separate hepato-portal compartment (“first-pass” model): fitting to plasma concentration-time profiles in humans. Pharm Res. 1997;14:230–7.
Girard P. Data transformation and parameter transformations in NONMEM. Available from: http://www.pagemeeting.org/page/page2002/PascalGirardPage2002.pdf. Accessed 19 Feb 2020.
Nguyen TH, Mouksassi MS, Holford N, Al-Huniti N, Freedman I, Hooker AC, et al. Model evaluation of continuous data pharmacometric models: metrics and graphics. CPT Pharmacomet Syst Pharmacol. 2017;6(2):87–109.
Savic RM, Karlsson MO. Importance of shrinkage in empirical Bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11(3):58–69.
Mandema JW, Verotta D, Sheiner LB. Building population pharmacokinetic-pharmacodynamic models. I. Models for covariate effects. J Pharmacokinet Biopharm. 1992;20(5):511–28.
Wasserstein RL, Lazar NA. The ASA’s statement on p-values: context, process, and purpose. Am Stat. 2016;70(2):129–33.
Benjamin DJ, Berger JO, Johnson VE, et al. Redefine statistical significance. Nat Hum Behav. 2018;2:6–10.
Wasserstein RL, Schirm AL, Lazar NA. Moving to a world beyond “p < 0.05”. Am Stat. 2019;73(Suppl):1–19.
Falk FR, Miller NB. A primer for soft modeling. Akron: University of Akron Press; 1992.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13(2):143–51.
Sigtermans M, Dahan A, Mooren R, et al. S (+)-ketamine effect on experimental pain and cardiac output: a population pharmacokinetic-pharmacodynamic modeling study in healthy volunteers. Anesthesiology. 2009;111:892–903.
Jonkman K, Duma A, Olofsen E, et al. Pharmacokinetics and bioavailability of inhaled esketamine in healthy volunteers. Anesthesiology. 2017;127:675–83.
Ashraf MW, Peltoniemi MA, Olkkola KT, et al. Semimechanistic population pharmacokinetic model to predict the drug-drug interaction between S-ketamine and ticlopidine in healthy human volunteers. CPT Pharmacomet Syst Pharmacol. 2018;7:687–97.
Kharasch ED, Labroo R. Metabolism of ketamine stereoisomers by human liver microsomes. Anesthesiology. 1992;77:1201–7.
Rowland M, Tozer TN. Clinical pharmacokinetics: concepts and applications. 3rd ed. Baltimore: Lippincott Williams & Wilkins; 1995.
Fanta S, Kinnunen M, Backman JT, et al. Population pharmacokinetics of S-ketamine and norketamine in healthy volunteers after intravenous and oral dosing. Eur J Clin Pharmacol. 2015;71:441–7.
Paine MF, Hart HL, Ludington SS, et al. The human intestinal cytochrome P450 “pie”. Drug Metab Dispos. 2006;34:880–6.
Türker S, Onur E, Ozer Y. Nasal route and drug delivery systems. Pharm World Sci. 2004;26:137–42.
Pires A, Fortuna A, Alves G, et al. Intranasal drug delivery: how, why and what for? J Pharm Pharm Sci. 2009;12:288–311.
Yanagihara Y, Ohtani M, Kariya S, et al. Plasma concentration profiles of ketamine and norketamine after administration of various ketamine preparations to healthy Japanese volunteers. Biopharm Drug Dispos. 2003;24:37–43.
Persson J, Hasselström J, Maurset A, et al. Pharmacokinetics and non-analgesic effects of S- and R-ketamines in healthy volunteers with normal and reduced metabolic capacity. Eur J Clin Pharmacol. 2002;57:869–75.
Dahan A, Olofsen E, Sigtermans M, et al. Population pharmacokinetic-pharmacodynamic modeling of ketamine-induced pain relief of chronic pain. Eur J Pain. 2011;15:258–67.
Bradley SE, Ingelfinger FJ, Bradley GP, et al. The estimation of hepatic blood flow in man. J Clin Investig. 1945;24:890–7.
Brunette KE, Anderson BJ, Thomas J, et al. Exploring the pharmacokinetics of oral ketamine in children undergoing burns procedures. Paediatr Anaesth. 2011;21:653–62.
Pang KS, Rowland M. Hepatic clearance of drugs. I. Theoretical considerations of a “well-stirred” model and a “parallel tube” model: influence of hepatic blood flow, plasma and blood cell binding, and the hepatocellular enzymatic activity on hepatic drug clearance. J Pharmacokinet Biopharm. 1977;5(6):625–53.
Yasuda SU, Zhang L, Huang SM. The role of ethnicity in variability in response to drugs: focus on clinical pharmacology studies. Clin Pharmacol Ther. 2008;84:417–23.
Hijazi Y, Boulieu R. Contribution of CYP3A4, CYP2B6, and CYP2C9 isoforms to N-demethylation of ketamine in human liver microsomes. Drug Metab Dispos. 2002;30:853–8.
Man M, Farmen M, Dumaual C, et al. Genetic variation in metabolizing enzyme and transporter genes: comprehensive assessment in 3 major East Asian subpopulations with comparison to Caucasians and Africans. J Clin Pharmacol. 2010;50:929–40.
Arora P, Sharma S, Garg S. Permeability issues in nasal drug delivery. Drug Discov Today. 2002;7:967–75.
Woodhouse KW, Wynne HA. Age-related changes in liver size and hepatic blood flow: the influence on drug metabolism in the elderly. Clin Pharmacokinet. 1988;15:287–94.
Acknowledgements
The authors thank the patients, investigators, and their medical, nursing, and laboratory staff who participated in the clinical studies included in the present work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The clinical studies were supported by research funding from Janssen Research and Development, and the analyses presented here were supported by Janssen Research and Development.
Conflict of interest/Competing interests
Stefaan Rossenu, Peter Zannikos, Partha Nandy, Jaskaran Singh, Wayne Drevets, and Juan Jose Perez Ruixo were employees and shareholders of Janssen Pharmaceutical Companies at the time this analysis was conducted. Carlos Pérez-Ruixo was an employee of Janssen Pharmaceutical Companies at the time this analysis was conducted.
Ethics approval
All studies were conducted in accordance with principles for human experimentation as defined in the 1964 Declaration of Helsinki and were approved by the human investigational review board of each study center and by the competent authority of each country.
Consent to participate
Informed consent was obtained from each subject before enrollment in the studies after being advised of the potential risks and benefits of the study, as well as the investigational nature of the study.
Consent for publication
All authors revised the article for important intellectual content and provided final approval of the published version.
Availability of data and material
The datasets generated and/or analyzed during the current study belong to Janssen Research & Development and are not publicly available.
Code availability
Not applicable.
Author contributions
Carlos Perez Ruixo performed the population pharmacokinetic analysis, executed the model-based simulations, and took the lead in writing the manuscript. Stefaan Rossenu, Peter Zannikos, Partha Nandy, Jaskaran Singh, Wayne Drevets, and Juan Jose Perez Ruixo substantially contributed to the design of the clinical studies that were included in the analyses, reviewed early versions of the manuscript, contributed to the scientific content (including data analysis and interpretation of the results), and provided approval of the final version that was submitted for publication.
Rights and permissions
About this article

Cite this article
Perez-Ruixo, C., Rossenu, S., Zannikos, P. et al. Population Pharmacokinetics of Esketamine Nasal Spray and its Metabolite Noresketamine in Healthy Subjects and Patients with Treatment-Resistant Depression. Clin Pharmacokinet 60, 501–516 (2021). https://doi.org/10.1007/s40262-020-00953-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-020-00953-4