
Abstract
Purpose
Transglutaminases (TGs) are multifunctional enzymes exhibiting transglutaminase crosslinking, as well as atypical GTPase/ATPase and kinase activities. Here, we used an integrated comprehensive analysis to assess the genomic, transcriptomic and immunological landscapes of TGs across cancers.
Methods
Gene expression and immune cell infiltration patterns across cancers were obtained from The Cancer Genome Atlas (TCGA) database and Gene Set Enrichment Analysis (GSEA) datasets. Western blotting, immunofluorescence staining, enzyme-linked immunosorbent assays, and orthotopic xenograft models were used to validate our database-derived results.
Results
We found that the overall expression of TGs (designated as the TG score) is significantly upregulated in multiple cancers and related to a worse patient survival. The expression of TG family members can be regulated through multiple mechanisms at the genetic, epigenetic and transcriptional levels. The expression of transcription factors crucial for epithelial-to-mesenchymal transition (EMT) is commonly correlated with the TG score in many cancer types. Importantly, TGM2 expression displays a close connection with chemoresistance to a wide range of chemotherapeutic drugs. We found that TGM2 expression, F13A1 expression and the overall TG score were positively correlated with the infiltration of immune cells in all cancer types tested. Functional and clinical verification revealed that a higher TGM2 expression is linked with a worse patient survival, an increased IC50 value of gemcitabine, and a higher abundance of tumor-infiltrating macrophages in pancreatic cancer. Mechanistically, we found that increased C–C motif chemokine ligand 2 (CCL2) release mediated by TGM2 contributes to macrophage infiltration into the tumor microenvironment.
Conclusions
Our results reveal the relevance and molecular networks of TG genes in human cancers and highlight the importance of TGM2 in pancreatic cancer, which may provide promising directions for immunotherapy and for addressing chemoresistance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Transglutaminases (TGs) are multifunctional, calcium-dependent enzymes. In humans, TGs are classified into nine types: TGM1-7, Factor XIII A (F13A1) and EPB42, depending on their distribution in organs and tissues [1]. All TG genes contain four sequential and structurally distinct domains: a N-terminal β-sandwich, a catalytic core, and two COOH-β-terminal barrel domains. F13A1 and TG1 contain an additional NH2-terminal pro-peptide sequence that can be cleaved to generate the active forms of these enzymes [2,3,4,5,6,7]. The most common function of TGs is to catalyze the formation of isopeptide linkages between the carboxamide moieties of glutamine residues bound to proteins and the ε amino group of lysine residues bound to proteins [8]. TGM2 has protein disulfide isomerase (PDI) [9], atypical GTPase/ATPase [10], and protein kinase [11] activity. In addition, FXIII-A, TGM1 and TGM2 have been reported to play roles in cell signaling and cell–matrix interactions [10].
TG family members perform various physiological functions, well-known examples of which are blood clotting [12,13,14], skin barrier formation [14,15,16] and bone formation [17, 18]. Genetic defects in these enzymes are known to cause severe bleeding [14, 19], skin disorders [20, 21] and several autoimmune diseases [14, 22,23,24]. Recently, increasing evidence has emphasized the contribution of TGs to tumor initiation and development. For instance, TGM1 has been found to promote stemness and chemoresistance in gastric cancer cells by regulating Wnt/β-catenin signaling [25]. TGM2 has been found to be implicated in several biological functions, including but not limited to apoptosis, extracellular matrix formation, cell adhesion and migration. Overexpression of TGM2 has been found to be associated with advanced tumor stages, distant metastases and chemoresistance in cancer [25]. TGM3 has been reported to be dysregulated in various cancers and to functions as a tumor suppressor [26,27,28,29,30,31]. TGM4 has been shown to promote cell–matrix adhesion, invasion and epithelial-mesenchymal transition in prostate cancer [32] and TGM6 may serve as a prognostic biomarker for colon cancer [33]. EPB42 has been shown to predict improved survival outcomes in patients with pancreatic cancer [34]. F13A1 has been found to serve as an effective serological biomarker as part of a screening program for CRC detection [35]. Although the implications of several TGs have been well documented in human cancers, a pancancer landscape of TG family members is lacking.
In this study, we comprehensively analyzed the molecular features of all members of the TG family using a large dataset covering 20 different types of human cancers. For this purpose, we analyzed the potential biological and clinical relevance of TG proteins in cancer progression from the perspectives of their gene expression, underlying cellular pathways, as well as associations with the immune microenvironment and chemoresistance. A high TGM2 expression was found to be a critical cause of chemoresistance to gemcitabine and to increase macrophage infiltration by upregulating CCL2 expression in pancreatic cancer.
2 Materials and methods
2.1 Expression and clinical analyses
Median expression values, corresponding clinicopathological parameters and survival data of TG family members in 20 different tumors and normal tissues were extracted from The Cancer Genome Atlas (TCGA) database and the Genotype Tissue Expression (GTEx) database. The most relevant cancer types including were: bladder urothelial carcinoma (BLCA), breast invasive carcinoma (BRCA), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), colon adenocarcinoma (COAD), esophageal carcinoma (ESCA), glioblastoma multiforme (GBM), head and neck squamous cell carcinoma (HNSC), kidney chromophobe (KICH), kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma (KIRP), liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), pancreatic adenocarcinoma (PAAD), pheochromocytoma and paraganglioma (PCPG), prostate adenocarcinoma (PRAD), rectum adenocarcinoma (READ), stomach adenocarcinoma (STAD), thyroid carcinoma (THCA) and uterine corpus endometrial carcinoma (UCEC). Survival analysis based on TG expression was performed using the Kaplan‒Meier Plotter and the log-rank test. A Cox proportional hazards model was used to determine hazard ratios (HRs).
2.2 Single-sample gene set enrichment analysis (ssGSEA)
ssGSEA was applied to identify TG genes with different enrichment scores between tumor and normal tissues. The ssGSEA score (TG score) of each sample was determined using the R Bioconductor package Gene Set Variation Analysis (GSVA, version 1.22.4) with default parameters. Correlations between TG scores and pathological stages were determined by performing Spearman correlation analysis. Receiver operating characteristic (ROC) curve analysis was used to examine the sensitivity and specificity of risk prediction according to the TG score.
2.3 Mutation and copy number variation analysis
Missense variants, in-frame deletions and in-frame insertions were classified as point mutations. Frameshift mutations, stop-gain mutations, splice acceptor variants and splice donor variants were classified as loss-of-function mutations. Point mutations and loss-of-function mutations were identified using MuTect. Copy number variations (CNVs) were assessed via the Affymetrix genome-wide human SNP array 6.0 platform using GISTIC2.0 (Version 2.0.22).
2.4 DNA methylation analysis
DNA methylation values are presented as beta values for each CpG probe [36]. The methylation value method is considered to be more statistically valid than the beta value method. We converted the beta values to methylation values. An independent samples t test was used to compare the methylation levels of tumor and normal tissues. A two-tailed p value < 0.05 was considered statistically significant.
2.5 Transcription factor and pathway enrichment analyses
Transcription factor data were downloaded from the JASPAR database. We assessed co-expression of TG genes with each transcription factor in 20 different tumors. |R| > 0.3 and p < 0.05 were considered to indicate a statistically significant difference. Pathway enrichment analyses were conducted using GSEA software, and hallmark gene sets were used for analysis.
2.6 Immune cell infiltration and drug sensitivity analyses
The estimated fraction of individual immune cell types in 20 different tumors was calculated using ssGSEA in the R package GSVA. The analysis included 16 adaptive immune cells and 12 innate immune cells, as reported previously [37]. All data for TG family-related drugs were obtained from the Cancer Cell Line Encyclopedia and Genomics of Drug Sensitivity in Cancer database, and an absolute correlation coefficient > 0.3 and a p value < 0.05 were used as the thresholds for significance.
2.7 Immunohistochemistry
Immunohistochemical (IHC) analysis was performed as previously reported [38, 39]. Prepared slides were routinely deparaffinized in xylene and rehydrated in an alcohol gradient, followed by antigen retrieval with citrate buffer (G1202, Servicebio, China). Then, endogenous peroxidase activity was blocked with 0.3% hydrogen peroxide in methanol, and nonspecific binding was blocked with 10% bovine serum albumin (BSA, GC305010, Servicebio, China) for one hour at room temperature. Next, the slides were incubated with primary antibodies at 4°C overnight. The next day, the slides were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:300, Servicebio, GB23303) for one hour at room temperature. The proteins were visualized with diaminobenzidine (DAB, G1212, Servicebio, China), followed by counterstaining with hematoxylin. Scoring was conducted based on the percentage of positively stained cells and the staining intensity. These scores were evaluated in a blinded manner. The antibodies used for immunohistochemistry were specific for TGM2 (1:100, Proteintech, 15100-1-AP) and CD68 (1:100, Servicebio, GB113109).
2.8 Cell culture
Human pancreatic cancer cell lines Panc1 and Patu-8988 were retrived from the Shanghai Cancer Institute, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University. The cells were cultured in DMEM medium (Gibco, Thermo Fisher Scientifific) supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% antibiotic mixture. Human monocyte leukemia cell line THP-1 was purchased from the Chinese Academy of Sciences (China) and cultured in RPMI-1640 medium (Gibco, Thermo Fisher Scientifific) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco) and 1% antibiotic mixture. All cells were incubated at 37°C in a humidified atmosphere with 5% CO2.
2.9 Knockdown assay
A small interfering RNA (siRNA) duplex oligonucleotides targeting TGM2 and lentiviral vectors for knockdown of Tgm2 were purchased from Gene Pharma (Shanghai, China). The siRNA duplex oligonucleotide sequences were si-TGM2-1, 5’-GCCUGAUCCUUCUAGAUGUTT-3’; si-TGM2-2, 5’-GUCGUGACCAACUACAACUTT-3’; and si-TGM2-3, 5’-GGCUGAAGAUCAGCACUAATT-3’. The scrambled negative control siRNA used (designated as siNC) did not have any target site within the human genome. JetPRIME siRNA transfection reagent (Polyplus transfection) was used to transfect siRNA according to the manufacturer’s instructions. Lentiviral particles were transfected into cells in the presence of Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions when the cells were 50% confluent. Then, the lentivirally infected cells were treated with 2 μg/ml puromycin (Gibco, A1113802, USA) for 7 days to select stable knockdown cells. The sequences targeting Tgm2 for knockdown were: sh-Tgm2-1, 5’-CGGCTGTCAGCAAGTGAAGTA-3’; sh-Tgm2-2, 5’-GAGTGGTGACCAACTACAACT-3’.
2.10 Quantitative real-time polymerase chain reaction (qRT‒PCR)
Total RNA was isolated using Trizol reagent (MRC, TR118) and a PrimeScript™ RT Master Mix (Takara, RR036A) was applied for cDNA synthesis according to the manufacturer’s instructions. qRT-PCR was performed using SYBR Premix Ex Taq (Takara, Japan) on a 7500 Real-time PCR system (Applied Biosystems, USA). The primer sequences used for PCR were as follows: TGM2 forward primer 5'-GAGGAGCTGGTCTTAGAGAGG-3' and reverse 5'-CGGTCACGACACTGAAGGTG- 3'; CCL2 forward 5’-CAGCCAGATGCAATCAATGCC-3’ and reverse 5’-TGGAATCCTGAACCCACTTCT-3’; 18 s forward 5'-TGCGAGTACTCAACACCAACA-3' and reverse 5'- GCATATCTTCGGCCCACA-3'. Relative mRNA expression was calculated by the 2(− ΔΔCt) method and normalized to 18 s mRNA levels.
2.11 Western blotting
Cell lysates were prepared using lysis buffer (Beyotime, Shanghai, China), separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to nitrocellulose membranes (Millipore, Danvers, MA). Subsequently, the membranes were washed using Tris-buffered saline with Tween 20 (TBST) and blocked with 5% nonfat milk solution at least 1 h at room temperature. Next, the membranes were incubated with an anti-TGM2 antibody (1:1000, Proteintech, 15100–1-AP) or an anti-β-actin antibody (1:5000, Proteintech, 81115–1-RR) at 4°C overnight, washed with TBST three times and incubated with a HRP-conjugated goat anti-rabbit antibody (1:10,000, Proteintech, 15015). Finally, the bands were detected using an Odyssey imaging system (LI-COR Biosciences, Lincoln, NE, USA).
2.12 Cell viability assay
To investigate the effect of TGM2 knockdown on the response of pancreatic cancer cells to gemcitabine (Selleck, S1149, Texas, USA), cells were inoculated in a 96-well plate (5 × 103 cells/well) and transfected with either TGM2 siRNA or scrambled siRNA for 24 h. Next, the cells were treated with different concentrations of gemcitabine or vehicle control for 48 h. After that, CCK-8 reagent (10 μl/well, CCK-8, Dojindo, Japan) mixed with the serum-free medium (90 μl/well) was added to each well and incubated at 37°C for 1 h. The absorbance was measured using a Power Wave XS microplate reader (BIO-TEK) at a wavelength of 450 nm with a reference at 600 nm. The survival rate (SR) was calculated using the following equation: SR = (OD450Treatment /OD450vehicle control) × 100%.
2.13 THP-1 migration assay
Transwell 24-well plates with filters with an 8 μm diameter pore size (Corning, NY, USA) were used in the migration assay. THP-1 cells (2 × 104 in 200 μl RPMI-1640 medium) were seeded in the upper chamber, and 700 μl of serum-free pancreatic cancer cell supernatant treated with or without an anti-CCL2 antibody (5 μg/ml; R&D Systems) for 30 min was added to the lower chamber before the addition of THP-1 cells. After incubation for 12 h at 37°C in a humidified atmosphere with 5% CO2, THP-1 cells were stained with 2 μM Calcein-AM for 15 minutes, after which the cells on the upper side of the filters were removed. The invaded cells were counted under a microscope at 200× magnification.
2.14 Orthotopic xenograft model
C57BL/6 J adult male mice aged 6 to 8 weeks were used in this study.1 × 106 short hairpin-Control (sh-Ctrl), short hairpin-Tgm2 (sh-Tgm2), overexpression-vector (ov-vector) and ov-Tgm2 (overexpression-Tgm2) KPC1199 cells in 25 μl of DMEM were injected into the body of the pancreas. Next, the mice were randomly divided into groups and treated with PBS, gemcitabine (50 mg/kg), PBS liposomes or clodronate liposomes (70 mg/kg) for three weeks beginning 5 days post-surgery. After 3 weeks of administration, bioluminescence imaging analysis was used to determine the tumor burden, and quantitative analysis was performed using Living Image software, version 4.5.3. The formula for calculating the tumor volume was: volume = 1/2 × (length × width2).
2.15 Statistical analysis
All statistical analyses were conducted using R software (version 3.6.1). The relationships between the expression of TG family members and immune cell fractions were tested by Spearman correlation analysis. One-way ANOVA or Student’s t test was performed to confirm the differences between groups. Values of p < 0.05 were considered statistically significant.
3 Results
3.1 Expression patterns and clinical relevance of TGs in the pancancer landscape
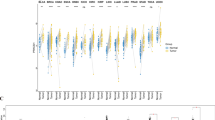
To evaluate the expression patterns of all nine TGs in cancer, we performed a pancancer analysis using the TCGA and GTEx databases. To achieve this, 20 cancer types with available normal counterparts, clinicopathological parameters and survival data were included. Expression heterogeneity across cancers was observed for all 9 TGs. A high expression of EPB42 was found in STAD (Fig. 1A) and of F13A1 in GBM, LIHC and PAAD (Fig. 1B). TGM1 was highly expressed in BLCA, CESC and THCA, and TGM7 was found to be upregulated in UCEC (Fig. 1C and I). Upregulation of TGM2-5 was observed in more than five individual cancer types, together covering 14 cancer types (Fig. 1C–G). GBM, KIRC, LIHC, LUSC, PAAD and UCEC were characterized by coordinated upregulation of more than 3 TGMs. Due to the low detection rates of TGM6 and TGM7 in some cancer types and control tissues, this study did not consider their differential expression between the respective cancer types (Fig. 1H and I). In addition, we found that the TG scores were higher in GBM, PAAD and UCEC (Fig. 1J). These results indicate that TGM2 to TGM5 are vital TGs upregulated in different cancer types and that GBM, KIRC, LIHC, LUSC, PAAD and UCEC are cancers with a frequent upregulation of TGs.

TG expression levels in different cancers in the TCGA. (A–I) Differential expression of TG family genes between TCGA cancer tissues and GTEx normal tissues. (J) TG scores of TCGA cancer tissues and GTEx normal tissues. The green rectangle indicates gene expression levels in normal tissues, and the purple rectangle indicates gene expression levels in cancer tissues. Normalized mRNA expression data are presented as means ± standard deviation (SD). Two-tailed Student’s t test. Ns: nonsignificant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Abbreviations: BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; COAD, colon adenocarcinoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, heck and neck squamous cell carcinoma; KICH, kidney chromophore; KIRC, kidney clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectal cancer; STAD, stomach adenocarcinoma; THCA, thyroid carcinoma and UCEC, uterine corpus endometrial carcinoma
Next, we pooled all patients in the 20 TCGA cancer types and divided them into high and low score groups according to the median value of the TG score. Then, we analyzed the association of the TG score with overall patient survival, which was determined using Kaplan‒Meier Plotter (Fig. 2A). We found that a high TG score correlated with a worse survival [HR = 0.91 (95% CI: 0.84–0.99), p = 0.0353]. We also summarized the number of TG genes with statistically significant effects on OS (Fig. 2B) and found that one or more of the TG genes were associated with survival in KIRC, KIRP, LUAD, LUSC, PAAD, READ, STAD and UCEC. Subsequent heatmap analysis revealed associations between the expression level of each individual TG gene and OS in the 20 cancer types in TCGA in detail (Fig. 2C). TGs predicted OS differentially among different cancer types. We also found that increased expression of TGM2 and TGM4 was associated with increased survival disadvantages, i.e, TGM2 expression predicted a poor prognosis in patients with KIRC, LUSC and PAAD, and TGM4 expression predicted a poor prognosis in patients with KIRC, PAAD and READ (Fig. 2C). Allow expression of EPB42 in LUAD, but a high expression of EPB42 in STAD predicted a worse patient survival. Notably, high expression of each individual TG family member predicted a poor prognosis of patients with KIRC, suggesting that TG upregulation generally contributes to KIRC malignancy.

Associations between TG expression levels and clinical features. (A) Comparison of Kaplan‒Meier survival curves based on high and low TG scores in the overall cohort of cancer patients in TCGA (n = 7744) (optimal cutoff). Hazard ratio (HR) = 0.91 (95% confidence interval (CI): 0.84–0.99, p = 0.0353). (B) In different cancers, the number of TG family genes whose prognostic value in overall survival achieved statistical significance (p < 0.05). BLCA (n = 0); BRCA (n = 0); CESC (n = 0); COAD (n = 0); ESCA (n = 0); GBM (n = 0); HNSC (n = 0); KICH (n = 0); KIRC (n = 5); KIRP (n = 1); LIHC (n = 0); LUAD (n = 1); LUSC (n = 2); PAAD (n = 2); PCPG (n = 0); PRAD (n = 0); READ (n = 1); STAD (n = 2); THCA (n = 0); UCEC (n = 1). (C) TG family gene expression related to overall survival in different cancers in TCGA. The color in the heatmap indicates the hazard ratio (HR). The number indicates the -log10 (p value). Red represents a high risk of death (HR > 1), whereas blue represents a low risk of death (HR < 1). (D) Correlation analysis between the TG score and pathological tumor–node–metastasis (TNM) stage in KIRC, LUSC, PAAD and STAD. (E–G) The associations between the TG score and pathological tumor stage (E), pathological node stage (F), and pathological metastasis stage (G) in KIRC, LUSC, PAAD and STAD. Ns: nonsignificant; *p < 0.05; **p < 0.01
In KIRC, LUSC, PAAD and STAD, we found that more than two TG genes exhibited statistically significant effects on OS. Therefore, we carried out a comprehensive analysis of the relevance of TG scores to clinicopathological parameters in these four cancers through the Wilcoxon test. We found that in KIRC and STAD the TG score was related to the pathologic TNM stage. In KIRC, the TG score was related to the pathological T stage and M stage. In LUSC, the TG score was related to pathological N stage (Fig. 2D–G).
To explore the application of the TG genes in cancer prognosis, ROC analysis was performed to predict the 1-, 3- and 5-year OS of patients stratified by TG gene expression levels in KIRC, LUSC, PAAD and STAD (Fig. S1). We found that the area under the curve (AUC) values for 1-, 3-, and 5-year survival were 0.7, 0.711 and 0.914, respectively, in PAAD (Fig. S1G-I). These results indicate that TG gene expression has a good predictive power for OS in PAAD, which may facilitate efficacy evaluation.
3.2 Mutation, CNV and DNA methylation analyses of TG family members
TG mutations have been found to be related to various diseases [20, 40,41,42]. Here, mutations in TG family genes were also identified in the TCGA cohort (Fig. S2 A). In UCEC, TGs exhibited the highest overall number of mutations. F13A1 had the highest number of mutations in LUAD samples. Next, we summarized the counts of missense variants and loss-of-function mutations of all TG genes in each cancer type (Fig. S2B). We found that UCEC exhibited the highest number of missense variants in TG genes, followed by LUAD, STAD, LUSC and COAD, respectively. UCEC also showed the most loss-of-function mutations in TG genes, followed by LUSC, LUAD, COAD and STAD.
Gene copy number variations are considered critical for modulating cancer occurrence and progression. We analyzed the correlation of TG gene expression and the frequency of CNVs in the corresponding gene in each cancer type (Fig. S2C). We found that a low expression of EPB42 was correlated with copy number deletions in BRCA, UCEC, LUAD and BLCA. A low expression of F13A1 and TGM1 was found to be associated with copy number deletions in KICH and KIRC, respectively. A significant correlation was found between decreased TGM4 expression and copy number deletions in BRCA, LUAD and KIRC. In addition, we found that increased TGM2 expression was associated with its copy number amplification in BRCA and READ, but not in KIRC or THCA. A high TGM3 expression was related to its copy number amplification in BRCA and LUSC, but not in LIHC, PRAD, UCEC or THCA. Upregulation of TGM4 or TGM5 was not associated with CNV in the various cancers tested. These results indicate that CNV constitutes an important way to regulate the expression of TGs in a number of cancers, whereas other regulatory mechanisms remain to be uncovered.
Hypermethylation of the promoters of specific genes may contribute to their downregulation. Therefore, we next analyzed promoter methylation levels of the respective TG genes. We found that the methylation levels of the TG genes were commonly altered in cancer tissues compared to their normal counterparts and that hypomethylation was the most common alteration (Fig. 3A–G). Next, we analyzed correlations between the methylation status and expression levels of TG genes in different cancers. We found that these levels correlated with their methylation levels in most cancers. Specifically, high TGM1 expression was found to be correlated with decreased methylation in THCA and ESCA and high TGM2 expression was correlated with decreased methylation in READ (Fig. 3H). Increased TGM5 expression was found to be associated with decreased methylation in KIRP and UCEC, while a low expression of TGM1 was related to increased methylation in PRAD.

DNA methylation of TGs across cancers. (A–G) DNA methylation of TGs between cancer tissues in TCGA and corresponding normal tissues. The red rectangular boxes indicate tumor tissue, while the gray boxes indicate normal tissue. The methylation levels are presented as the means ± standard deviation (SD). Two-tailed Student’s t test. (H) Bubble map of differential methylation of TGs between normal and tumor tissues in TCGA. The gradient of colors in the bubble map represents the log2-fold change between cancer and normal tissues. The purple nodes indicate high expression in cancer tissues, whereas the green nodes indicate low expression in cancer tissues. The red circles indicate increased methylation, while the blue circles indicate decreased methylation. The size of the nodes represents the statistical significance; the larger the size, the greater the significance. Ns: nonsignificant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
3.3 Transcription, pathway and immune cell infiltration analyses based on TG family members
To further explore the regulatory mechanism of the TG family in cancer, we validated the transcription factors most relevant to the TG family (|R| > 0.3, p < 0.05). We found that KICH exhibited the highest number of transcription factors related to TG family genes, followed by THCA, ESCA and BLCA (Fig. 4B). Twist Family BHLH Transcription Factor 2 (TWIST2), Snail Family Transcriptional Repressor 2 (SNAI2), ETS Transcription Factor ELK3 (ELK3), FOS Like 1, AP-1 Transcription Factor Subunit (FOSL1), Basonuclin 1 (BNC1), Paired Related Homeobox 1 (PRRX1) and Paired Related Homeobox 2 (PRRX2) are master transcription factors of epithelial–mesenchymal transition (EMT) and were found to be positively associated with TG family members in more than 10 cancer types (Fig. 4C). To explore the underlying mechanisms mediated by TGs, we first identified the pathways enriched with proteins that interact with TGs based on the InWeb-IM database. We found that these genes were mainly enriched in the pathways Mitophagy – animal, Shigellosis, Viral carcinogenesis, Proteoglycans in cancer and Cell cycle (Fig. 4A). Next, we analyzed the pathways associated with TG gene expression by GSEA. Four cancer-related pathways, namely, enabling replicative immortality, genome instability and mutation, inducing angiogenesis and reprogramming energy metabolism, were found to be significantly associated with TG genes in nearly all 20 cancer types (Fig. 4D). Furthermore, we found that the TGs were positively associated with more than two cancer-related pathways in most cancers. Specifically, in HNSC, STAD, GBM, LUAD and THCA, TG expression was positively correlated with all four cancer-related pathways. In GBM, HNSC and STAD, TG genes exhibited the strongest association with the angiogenesis-inducing pathway. In BRCA, HNSC and PRAD, TG genes showed the strongest association with energy metabolism reprogramming.

TGs in the pathways and TG transcription factors in cancers in TCGA. (A) Network diagram demonstrating the pathways enriched with proteins interacting with TGs identified based on the InWeb-IM database. The gradient of colors represents the statistical significance; the redder the color, the greater the significance. The size of the nodes represents the correlation strength; the larger the size, the greater the correlation. (B) Histogram showing the counts of transcription factors (p value < 0.05, |R| > 0.3) related to TG family genes in different cancer types. KICH exhibits the most transcription factors related to TG family genes (n = 758), followed by THCA (n = 419), ESCA (n = 345), and BLCA (n = 332). (C) Transcription factors (p value < 0.05, |R| > 0.3) related to TGs in more than ten types of cancer. The relationships between the expression of TG family genes and transcription factors were analyzed by Spearman correlation analysis. (D) Four pathways were most likely to be activated by TG family genes based on the GSEA database. The size of the nodes represents the statistical significance; the larger the size, the greater the significance. The gradient of the colors represents the NES. The red nodes indicate positively correlated pathways, whereas the green nodes indicate negatively correlated pathways. Abbreviations: TWIST2: Twist Family BHLH Transcription Factor 2; SNAI2: Snail Family Transcriptional Repressor 2; PRDM1: PR/SET Domain 1; MAFB: MAF BZIP Transcription Factor B; ELK3: ETS Transcription Factor ELK3; SOX7: SRY-Box Transcription Factor 7; FOSL1: FOS Like 1, AP-1 Transcription Factor Subunit; BNC1: Basonuclin 1; AEBP1: AE Binding Protein 1; ZNF365: Zinc Finger Protein 365; ZNF19: Zinc Finger Protein 19; ZBED2: Zinc Finger BED-Type Containing 2; TSHZ3: Teashirt Zinc Finger Homeobox 3; TFEC: Transcription Factor EC; PRRX2: Paired Related Homeobox 2; PRRX1: Paired Related Homeobox 1; MAF: MAF BZIP Transcription Factor; IKZF1: IKAROS Family Zinc Finger 1; HES2: Hes Family BHLH Transcription Factor 2; FLI1: Fli-1 Proto-Oncogene, ETS Transcription Factor; BNC2: Basonuclin 2
We further evaluated correlations between immune cell infiltration and TG gene expression (Fig. 5A-H). The scores of a total of 28 subpopulations of tumor-infiltrating leukocytes, i.e, 16 adaptive immune cells and 12 innate immune cells, were calculated based on the TCGA database. We found that F13A1 and TGM2 were related to the infiltration of almost all immune cells, such as natural killer T cells, macrophages, central memory CD4 + T cells, CD8 + T cells and myeloid-derived suppressor cells (MDSCs), across different cancers (Fig. 5B and D). The correlation of TGM1, TGM3, TGM4 and TGM5 expression with immune cell infiltration differed among cancers, except in THCA, where all of these TGs were well correlated with the infiltration of immune cells (Fig. 5C, E–G). Furthermore, we found that the TG score was associated with the infiltration of nearly all immune cell types in most cancers, including THCA, PCPG, BRCA and PRAD (Fig. 5H). These results indicate that TG upregulation may affect the formation of the immune microenvironment in cancer.

Relationships between immune cell infiltration and TGs across cancers. Heatmap showing the correlations between mRNA expression of EPB42 (A), F13A1 (B), TGM1 (C), TGM2 (D), TGM3 (E), TGM4 (F), TGM5 (G) and the TG score (H) with immune cell infiltration scores in cancer types in TCGA. After calculating the single-sample gene set enrichment analysis (ssGSEA) score representing immune cells in the TCGA cancer cohort, the correlations between the infiltration of each type of immune cell and the expression of each TG family gene were determined via Spearman correlation analysis. The gradient of colors in the heatmap represents the statistical significance. Red indicates a positive correlation, while blue indicates a negative correlation. “X” means not significant (p > 0.05). Abbreviation: MDSC: myeloid-derived suppressor cell
3.4 TG genes are associated with chemoresistance
Next, we analyzed the correlations between TG gene expression and chemotherapy drug usage based on the Cancer Cell Line Encyclopedia (CCLE) and the Genomics of Drug Sensitivity in Cancer (GDSC) database (Fig. S3 and S4). We found that only TGM2 was related to sensitivity to most chemotherapeutics (|R| > 0.3, p < 0.05). i.e, higher TGM2 expression was commonly correlated with reduced sensitivity to numerous drugs, including the AKT inhibitor GSK690693, the FLT3 inhibitor quizartinib and other drug candidates (Fig. 6A). These results suggest that TGM2 upregulation may be a common inducer of clinical chemoresistance. However, a few exceptions exist, whereby TGM2 is likely to exhibit an opposite association with chemoresistance to a few particular drugs. We found, for instance, that cancer cells with a high TGM2 expression were more sensitive to refametinib and trametinib, two oral MEK inhibitors (Fig. 6A). Interestingly, a common association of high F13A1 expression with increased chemosensitivity was observed, in contrast to the relationships observed for TGM2. These results indicate that TGM2 and F13A1 predict opposite correlations with chemoresistance and suggest that they function differently in the transduction of cancer-related signaling pathways and, as such, may be used in combination for optimal prediction of drug sensitivity.

Higher TGM2 expression contributes to gemcitabine chemoresistance in pancreatic cancer. (A) Scatter plots showing associations between TGM2 expression and drug sensitivity. TGM2 expression positively correlates with sensitivity to GSK690693 (AKT Serine/Threonine Kinase inhibitor), quizartinib (Fms Related Receptor Tyrosine Kinase 3 inhibitor), WZ3105 (neurotrophin receptor kinase inhibitor) and other drug candidates (NPK76-II-72–1, TL-2–105 and XMD13-2). In addition, TGM2 expression negatively correlates with the sensitivity to refametinib and trametinib, two oral MEK inhibitors. (B) Representative H&E staining (upper) and TGM2 immunohistochemical (IHC) staining (lower) patterns showing changes in organizational structure and TGM2 expression from normal pancreas tissue to pancreatic intra-epithelial neoplasia (PanIN) to pancreatic cancer tissue in KrasG12D/ + ; Trp53R172H/ + ; Pdx1-Cre (KPC) mice. Scale bar, 100 μm. (C) IHC analysis of TGM2 expression in a human pancreatic cancer tissue microarray (n = 188). Representative IHC images are shown in the left panel. The percentage of the tissue core displaying low and high staining in pancreatic tumor and normal tissues is shown in the right panel. (D) Survival curves of si-Ctrl and si-TGM2 Panc1 and Patu-8988 cells exposed to gemcitabine or vehicle (n = 6 replicates per group, means ± standard deviation, one of three biological replicates). (E) Orthotopically transplanted xenografts generated with sh-Ctrl or shTgm2 KPC1199 cells treated with gemcitabine or vehicle. Blue dotted circles indicate the tumors (n = 5 replicates per group, means ± standard deviation, 2 biological replicates). (F) Representative images of Ki67 + cells from pancreatic tumor tissues of C57BL/6 J mice treated with gemcitabine or vehicle. Scale bar: 50 μm. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
3.5 Higher TGM2 expression contributes to gemcitabine chemoresistance in pancreatic cancer
Considering the striking association of TGM2 expression with chemotherapeutic sensitivity and patient survival with PAAD, we further validated these relevancies in PAAD using in vitro and in vivo assays. First, we assessed the expression of TGM2 in mouse pancreatic cancer tissues. Using IHC in a genetically engineered LSL-KrasG12D/ + ; LSL-Trp53R172H/ + ; Pdx1-Cre (KPC) mouse model, we found that TGM2 protein expression was elevated in pancreatic intra-epithelial neoplasia (PanIN) and pancreatic cancer tissues compared with normal acinar tissues (Fig. 6B). Next, we performed IHC staining on a tissue microarray containing 188 paired pancreatic cancer and adjacent normal tissues. We found that TGM2 expression was significantly higher in pancreatic cancer tissues than in adjacent tissues (Fig. 6C). To determine whether TGM2 affects the sensitivity of pancreatic cancer cells to gemcitabine, we silenced TGM2 expression using siRNAs in pancreatic cancer cells. Western blotting was used to evaluate the knockdown efficiency of TGM2 in Panc1 and Patu-8988 cells and revealed that TGM2 expression was greatly decreased by both specific siRNAs (Fig. S5A). Using a cell viability assay, we found that TGM2 knockdown improved pancreatic cancer cell sensitivity to gemcitabine (Fig. 6D). TGM2 siRNA-1 and siRNA-2 markedly reduced the IC50 value of gemcitabine in Panc1 and Patu-8988 cells after 48 h (Fig. 6D).
Next, mouse pancreatic cancer KPC1199 cells stably expressing a lentiviral shRNA targeting TGM2 (sh-Tgm2-1) were generated. The knockdown efficiency was validated by Western blotting (Fig. S5B). sh-Tgm2-1 and control KPC1199 cells were subsequently used to establish an orthotopic tumor model by injection into the pancreas. After tumor cell injection, the mice received treatment with gemcitabine (50 mg/kg) or vehicle every 5 days for three weeks. We found that TGM2 knockdown promoted gemcitabine efficacy and resulted in a marked reduction in the tumor burden (Fig. 6E). IHC staining of the proliferation marker Ki67 in the tumor tissues confirmed that Tgm2 knockdown boosted the antitumor effect of gemcitabine (Fig. 6F). Collectively, these results indicate that TGM2 is upregulated in pancreatic cancer and that TGM2 knockdown enhances the sensitivity of pancreatic cancer cells to gemcitabine.
3.6 TGM2 is associated with macrophage infiltration via upregulated CCL2 production in pancreatic cancer
To explore correlation between TGM2 expression and macrophage infiltration, we performed IHC staining of the macrophage markers CD68 and TGM2 in a cohort of samples from 55 pancreatic cancer patients. We found that a high expression of TGM2 was significantly associated with macrophage infiltration (p = 0.0312) (Fig. 7A and B). To further determine the link between TGM2 and macrophage infiltration, we examined the migratory ability of phorbol myristate acetate (PMA, 100 ng/ml)-primed THP-1 (human myeloid leukemia mononuclear) cells upon treatment with conditioned medium (CM) from si-Ctrl and si-TGM2 Panc1 and Patu-8988 cells. Transwell assays revealed that the migration of PMA-primed THP1 cells induced by si-TGM2 CM was significantly reduced compared with that induced by si-Ctrl CM (Fig. 7C).

TGM2 is associated with macrophage infiltration via upregulated CCL2 production in pancreatic cancer. (A) Representative immunohistochemical images of TGM2 (upper) and CD68 (lower) in a pancreatic cancer microarray, n = 55. Scale bar: 100 μm. (B) Heatmap displaying the consistency of TGM2 and CD68 expression in the pancreatic cancer microarray. N = 55, p = 0.0312 (Chi-square test). (C) Effects of conditioned medium from si-Ctrl or si-TGM2 Panc1 and Patu-8988 cells on the migration capacity of PMA-primed THP-1 cells (n = 3 replicates per group, means ± standard deviation, one of three biological replicates). (D) Orthotopic xenografts generated with ov-vector or ov-Tgm2 KPC1199 cells treated with PBS liposomes or clodronate liposomes. The blue dotted circles indicate the tumors (n = 5 replicates per group, means ± standard deviation, 2 biological replicates). (E) Bioluminescence images of ov-vector and ov-Tgm2 tumors treated with PBS liposomes or clodronate liposomes (n = 5 replicates per group, means ± standard deviation, 2 biological replicates). (F) Representative images of F4/80 + cells from pancreatic tumor, liver and lung tissues of C57BL/6 J mice treated with PBS liposomes or clodronate liposomes. Scale bar: 50 μm. Statistical analyses of infiltrated F4/80 + cells are shown in the right panel. (G) Relative mRNA levels of TGM2 and CCL2 in pancreatic cancer cells treated with si-TGM2 or si-Ctrl (n = 3 replicates per group, means ± standard deviation, one of three biological replicates). (H) The concentration of secreted CCL2 in the conditioned medium (CM) from sh-Ctrl and sh-Tgm2 KPC1199 cells (n = 3 replicates per group, means ± standard deviation, one of three biological replicates). (I) Transwell assay of the migration capacity of PMA-primed THP-1 cells stimulated with pancreatic cancer CM in which CCL2 was neutralized by treatment with an anti-antibody, (n = 3 replicates per group, means ± standard deviation, one of three biological replicates). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
Next, we established orthotopic pancreatic cancer models using Tgm2-overexpressing and control KPC1199 cells in C57BL/6 J mice. Clodronate liposomes or PBS liposomes were administered to these mice two weeks before and three weeks after tumor cell implantation. We found that the number of F4/80 + macrophages infiltrating pancreatic tumors was decreased in mice treated with clodronate liposomes, suggesting the effective elimination of macrophages. In addition, we found that tumor tissues with Tgm2 overexpression exhibited more macrophage infiltration than control tissues (Fig. 7F), suggesting a chemotactic effect of Tgm2 overexpression on macrophages. The results indicated that liposomal clodronate treatment largely reversed the increase in tumor growth promoted by Tgm2 overexpression (Fig. 7D and E).
It has been reported that CCL2 plays a significant role in macrophage recruitment into the tumor microenvironment [43]. We, therefore, measured its expression by qRT‒PCR and found that the mRNA level of CCL2 was significantly reduced in si-TGM2 Panc1 and Patu-8988 cells compared to control cells (Fig. 7G). Consistently, we found that CCL2 was upregulated by TGM2 overexpressing in Panc1 and Patu-8988 cells (Sup Fig. 5C). Subsequent ELISA revealed that the CCL2 level was reduced in the conditioned medium (CM) of sh-Tgm2 KPC1199 cells (Fig. 7H). To investigate whether CCL2 is responsible for the macrophage infiltration induced by TGM2 overexpression, an anti-CCL2 antibody (5 μg/ml) was applied in the migration assay. We found that the migration of THP-1 cells induced by ov-TGM2 cell conditioned medium was largely attenuated by the anti-CCL2 antibody (Fig. 7)I. Collectively, these findings indicate that TGM2 may facilitate macrophage infiltration by upregulating CCL2 production in pancreatic cancer.
4 Discussion
Our current data indicate that (1) members of the TG family are tightly regulated by multiple mechanisms at the genetic, epigenetic and transcriptional levels, (2) TGs are likely to be involved in the EMT process and regulation of the immune microenvironment in most cancers, (3) TGM2 is a potential sensitive marker for various chemotherapy drugs and (4) TGM2 is upregulated in pancreatic cancer, reduces sensitivity to gemcitabine and enhances macrophage infiltration by upregulating CCL2 expression.
It has been reported that TGs are differentially expressed in numerous cancers and have considerable impacts on proliferation, apoptosis, stem cell phenotypes, epithelial-mesenchymal transition and chemoresistance [24]. Previously, it has been reported that TGM1 is upregulated in gastric cancer and promotes its proliferation, stemness and chemoresistance via the Wnt signaling pathway [25]. TGM1 was also found to be upregulated in tumor cell aggregates (spheroids) exhibiting a low chemosensitivity in ovarian cancer peritoneal fluid [44]. F13A1 has been identified as a biomarker for a dismal prognosis in various types of cancer [35, 45, 46] and promotes the metastasis of melanoma as well as lung squamous cell carcinoma cells [47, 48]. As a well-characterized transamidase, TGM2 can not only promote protein crosslinking by catalyzing posttranslational modifications, but also function as a G protein-coupled membrane receptor (GTPase), deamidase, adapter/scaffold, protein disulfide isomerase, isopeptidase and kinase [24, 49,50,51,52,53]. It is also responsible for serotonylation, a type of posttranslational modification that plays a role in numerous biological processes, by catalyzing the transamidation reaction and transferring serotonin to substrates [54]. Due to its multiple enzymatic activities, it is involved in diverse physiological processes and the pathophysiology of various diseases, including cell survival, apoptosis, inflammation and fibrosis [55, 56]. Interestingly, it has been found that the diverse subcellular localizations and various biological activities of TGM2 provide it with dual roles in different contexts, leading to either protection from apoptosis or death via apoptosis [57, 58]. We found that the TG score was high in multiple types of cancer and was closely related to a dismal prognosis. Furthermore, we found that the TG score showed a significant relationship to pathological TNM stage and individually to pathological M stage and T stage in KIRC. TG gene expression also exhibited exceptional predictive power for OS, among which TGM2 showed a particularly strong predictive ability. Since the various TGs showed tight correlations with the prognosis of PAAD, these TGs, especially TGM2, may be used as novel markers for the prediction of PAAD patient survival.
CNVs are genetic variations in which the structure of chromosomes is rearranged, resulting in effects such as duplication and deletion of DNA segments. By driving genetic diversity, CNVs can alter the expression of multiple genes and lead to cancer [59]. We found that the occurrence of CNVs was widely and highly consistent with TG expression. DNA methylation is a fundamental epigenetic modification involving the transfer of a methyl group (-CH) to the 5‑carbon on a cytosine ring (5mC) in a CpG dinucleotide via a reaction catalyzed by DNA methyltransferases. It regulates gene transcription and preserves genome stability, thus altering the cell phenotype. DNA methylation also presented a widespread change in the pancancer atlas and was found to be involved in the regulation of TGM1 and TGM5 expression.
Transcription factor binding is another classical mechanism to alter gene expression. We found that several master TFs of EMT, such as TWIST2, SNAI2, BNC2, PRRX1, FOSL1 and ELK3, positively regulate the transcription of TGs, which indicates that TGs may play a role in the EMT process. Consistent with this hypothesis, it has been reported that EMT may be brought about by dysregulation of TGs. In mammary epithelial cells, it has been reported that TGM2 may promote the upregulation of fibronectin, N-cadherin and vimentin, and the downregulation of E-cadherin by altering the expression of Snail1, Zeb1, Zeb2 and Twist1 [60]. TGM2 was also found to be upregulated in hepatocellular carcinoma cells activated by hepatic stellate cells via inflammatory signaling, which further induced the depletion of von Hippel‒Lindau protein and caused a pseudo-hypoxic state that activated EMT [61]. Stable expression of TGM2 was also found to induce a mesenchymal phenotype in ovarian cancer cells, which was mediated by alterations in the level and function of several transcriptional repressors, including Zeb1, via nuclear factor-κB complex activation [62]. TGM3 has been indicated to promote EMT in colorectal cancer cells and hepatocellular carcinoma cells via the AKT signaling pathway [26, 63]. Akt stabilizes the function of Snail and Slug, two classical transcription factors that induce EMT, and thus represses the transcription of E-cadherin [64]. ELK3, a member of the ETS transcription factor family, and FOSL1 are two other basic factors involved in the complex network of EMT [65, 66] and play key roles in positively regulating the expression of TGs. These results indicate that TGs are critical in EMT and the malignant behavior of cancer cells.
Some immune-related transcription factors have also been found to be involved in the regulatory network of TG genes in many types of cancer. MAFB, a member of the Maf family, has been found to regulate the differentiation of macrophages and to promote tumorigenesis [67, 68]. Moreover, as a regulator of the differentiation of B cells as well as T cells, PR domain zinc finger protein 1 (PRDM1) has been found to play a pivotal role in immunosuppression in the tumor environment and to lead to a dismal prognosis [69, 70]. We observed a close relationship between the expression of TGM2 and Friend leukemia virus integration 1 (FLI1), a member of the ETS family. FLI1 has been widely identified as a critical factor that regulates the development and differentiation of lymphocytes. It can regulate the expression of cytokines and chemokines and, thus, manipulate the function of immune cells. Dysregulation of FLI1 is implicated in several autoimmune diseases [71,72,73,74,75]. Furthermore, it has been reported that FLI1 inhibits chromatin accessibility at ETS: RUNX motifs and suppresses effector CD8 + T-cell lineage differentiation, which provides protective immunity in cancer [76]. ZBED2 is a marker of exhausted CD8 + T cells. ZNF365 has been found to be related to inflammatory bowel disease and ankylosing spondylitis [77,78,79]. By binding to LPS, ZNF365 also helps defend against infections with gram-negative bacteria [80]. Thus, TGs may be strongly involved in immune responses in the tumor microenvironment.
According to our findings of antitumor drug resistance, the expression of TGM2 was found to be positively related to the IC50 values of 6 drugs, namely, GSK690693, NPK76-II-72–1, quizartinib, TL-2–105, WZ3105 and XMD13-2, which indicates potential resistance to these drugs. Two drugs, refametinib and trametinib, that were negatively related to the TGM2 level, indicating a good response of patients to these drugs. Refametinib is a selective MEK inhibitor that exhibits a high selectivity for MEK 1/2. Through CCK8 assays and orthotopic mouse models, we found that TGM2 reduces the sensitivity to gemcitabine in pancreatic cancer. Potent activity of refametinib in inhibiting cell proliferation in various cancer cell lines has been shown in vitro, as well as in retarding xenograft growth in models of melanoma, colon and epidermal carcinoma in vivo. Several clinical trials have been conducted to explore its potential clinical application, and clinical benefits have been demonstrated in several advanced solid tumors [81, 82]. The combination of refametinib and sorafenib has shown synergistic antitumor activity in multiple types of cancer, among which patients with RAS mutations derived the best benefit [83,84,85]. Trametinib is also a selective, oral MEK1/MEK2 inhibitor that has been widely tested in several types of cancer and exhibits great benefits [86,87,88,89]Here, we found that both refametinib and trametinib, two selective MEK1/MEK2 inhibitors, may be more effective in patients with high expression of TGM2. Additional studies and related clinical trials are still needed to determine the underlying mechanism.
In addition, we explored relationships between the expression of TGs and immune cell infiltration. We found that a subgroup of TGs is related to the immune cell infiltration landscape, among which F13A1 and TGM2 exhibited an extremely strong relationship. F13A1 is an active transglutaminase that crosslinks fibrin strands and plays a role in stabilizing fibrin clots and blood coagulation [14]. Recent studies have also shown its involvement in basic immunoreactions [90]. In human adipose tissue, differential expression of F13A1 has been found to be related to multiple functions, such as the immune responses, leucocyte and neutrophil activation and the cytokine responses [91]. It has also been identified as a co-expression factor of M2 macrophages and to correlate with clinical outcomes of renal clear cell carcinoma [92]. Consistent with this observation, F13A1-positive macrophages in Hodgkin lymphoma have been found to function in malignant cell proliferation [93]. Consistent with these reports, we found that F13A1 was related to the infiltration of many types of immune cells across cancers, as was TGM2. TGM2 has been widely found to participate in inflammatory signaling in pathological conditions such as celiac disease, cystic fibrosis and sepsis [94,95,96]. TGM2 not only directly influences the malignant behavior of cancer cells [97, 98] but also regulates the immune microenvironment. TGM2 has been reported to regulate the production of interleukin-6 (IL-6) [99], an important cytokine in the immune microenvironment. We found that TGM2 upregulates CCL2 expression and is responsible for the enhancement of macrophage infiltration in pancreatic cancer.
In conclusion, we first assessed the expression and prognostic value of members of the TG family in 20 cancer types and comprehensively characterized genomic alterations, epigenetic modifications, transcriptional regulation, interactive proteins and pathways, the immune tumor microenvironment and chemotherapy responses in human cancers. Moreover, we found that TGM2 enhances resistance to gemcitabine and correlates with macrophage infiltration in pancreatic cancer. Our data provide new insight into the potential value of TG family members as novel biomarkers and therapeutic targets for cancer.
Data availability
All sequencing data were obtained from publicly available database (TCGA https://portal.gdc.cancer.gov and GTEx https://xenabrowser.net) and other data generated or analyzed in this study were included in the article.
References
E.S. Chermnykh, E.V. Alpeeva, E.A. Vorotelyak, Transglutaminase 3: the involvement in epithelial differentiation and cancer. Cells 9, 1996 (2020)
J.E. Folk, P.W. Cole, Identification of a functional cysteine essential for the activity of guinea pig liver transglutaminase. J. Biol. Chem. 241, 3238–3240 (1966)
S.E. Iismaa, S. Holman, M.A. Wouters, L. Lorand, R.M. Graham, A. Husain, Evolutionary specialization of a tryptophan indole group for transition-state stabilization by eukaryotic transglutaminases. Proc. Natl. Acad. Sci. U. S. A. 100, 12636–12641 (2003)
K.N. Lee, S.A. Arnold, P.J. Birckbichler, M.K. Patterson Jr., B.M. Fraij, Y. Takeuchi, H.A. Carter, Site-directed mutagenesis of human tissue transglutaminase: Cys-277 is essential for transglutaminase activity but not for GTPase activity. Biochim. Biophys. Acta 1202, 1–6 (1993)
R. Micanovic, R. Procyk, W. Lin, G.R. Matsueda, Role of histidine 373 in the catalytic activity of coagulation factor XIII. J. Biol. Chem 269, 9190–9194 (1994)
S.N. Murthy, S. Iismaa, G. Begg, D.M. Freymann, R.M. Graham, L. Lorand, Conserved tryptophan in the core domain of transglutaminase is essential for catalytic activity. Proc. Natl. Acad. Sci. U. S. A. 99, 2738–2742 (2002)
L.C. Pedersen, V.C. Yee, P.D. Bishop, I. Le Trong, D.C. Teller, R.E. Stenkamp, Transglutaminase factor XIII uses proteinase-like catalytic triad to crosslink macromolecules. Protein Sci. 3, 1131–1135 (1994)
S.Y. Kim, T.M. Jeitner, P.M. Steinert, Transglutaminases in disease. Neurochem. Int. 40, 85–103 (2002)
G. Hasegawa, M. Suwa, Y. Ichikawa, T. Ohtsuka, S. Kumagai, M. Kikuchi, Y. Sato, Y. Saito, A novel function of tissue-type transglutaminase: protein disulphide isomerase. Biochem. J. 373, 793–803 (2003)
S.E. Iismaa, B.M. Mearns, L. Lorand, R.M. Graham, Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol. Rev. 89, 991–1023 (2009)
S. Mishra, L.J. Murphy, Tissue transglutaminase has intrinsic kinase activity: identification of transglutaminase 2 as an insulin-like growth factor-binding protein-3 kinase. J. Biol. Chem. 279, 23863–23868 (2004)
L. Muszbek, R. Adány, M. Kávai, Z. Boda, S. Lopaciuk, Monocytes of patients congenitally deficient in plasma factor XIII lack factor XIII subunit a antigen and transglutaminase activity. Thromb. Haemost. 59, 231–235 (1988)
L. Lorand, N. Barnes, J.A. Bruner-Lorand, M. Hawkins, M. Michalska, Inhibition of protein cross-linking in Ca2+-enriched human erythrocytes and activated platelets. Biochemistry 26, 308–313 (1987)
L. Lorand, Factor XIII: structure, activation, and interactions with fibrinogen and fibrin. Ann. N. Y. Acad. Sci. 936, 291–311 (2001)
H.C. Kim, Z. Nemes, W.W. Idler, C.C. Hyde, P.M. Steinert, B. Ahvazi, Crystallization and preliminary X-ray analysis of human transglutaminase 3 from zymogen to active form. J. Struct. Biol. 135, 73–77 (2001)
Z. Nemes, L.N. Marekov, L. Fésüs, P.M. Steinert, A novel function for transglutaminase 1: attachment of long-chain omega-hydroxyceramides to involucrin by ester bond formation. Proc. Natl. Acad. Sci. U. S. A. 96, 8402–8407 (1999)
D. Aeschlimann, O. Kaupp, M. Paulsson, Transglutaminase-catalyzed matrix cross-linking in differentiating cartilage: identification of osteonectin as a major glutaminyl substrate. J. Cell. Biol. 129, 881–892 (1995)
M. Wozniak, A. Fausto, C.P. Carron, D.M. Meyer, K.A. Hruska, Mechanically strained cells of the osteoblast lineage organize their extracellular matrix through unique sites of alphavbeta3-integrin expression. J. Bone Miner. Res. 15, 1731–1745 (2000)
H. Mikkola, V.C. Yee, M. Syrjälä, R. Seitz, R. Egbring, P. Petrini, R. Ljung, J. Ingerslev, D.C. Teller, L. Peltonen, A. Palotie, Four novel mutations in deficiency of coagulation factor XIII: consequences to expression and structure of the A-subunit. Blood 87, 141–151 (1996)
M. Huber, I. Rettler, K. Bernasconi, E. Frenk, S.P. Lavrijsen, M. Ponec, A. Bon, S. Lautenschlager, D.F. Schorderet, D. Hohl, Mutations of keratinocyte transglutaminase in lamellar ichthyosis. Science 267, 525–528 (1995)
Z. Nemes, M. Demény, L.N. Marekov, L. Fésüs, P.M. Steinert, Cholesterol 3-sulfate interferes with cornified envelope assembly by diverting transglutaminase 1 activity from the formation of cross-links and esters to the hydrolysis of glutamine. J. Biol. Chem. 275, 2636–2646 (2000)
M. Sárdy, S. Kárpáti, B. Merkl, M. Paulsson, N. Smyth, Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J. Exp. Med. 195, 747–757 (2002)
W. Dieterich, T. Ehnis, M. Bauer, P. Donner, U. Volta, E.O. Riecken, D. Schuppan, Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 3, 797–801 (1997)
L. Lorand, R.M. Graham, Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 4, 140–156 (2003)
H. Huang, Z. Chen, X. Ni, Tissue transglutaminase-1 promotes stemness and chemoresistance in gastric cancer cells by regulating Wnt/β-catenin signaling. Exp. Biol. Med. (Maywood) 242, 194–202 (2017)
J.W. Hu, Z.F. Yang, J. Li, B. Hu, C.B. Luo, K. Zhu, Z. Dai, J.B. Cai, H. Zhan, Z.Q. Hu, J. Hu, Y. Cao, S.J. Qiu, J. Zhou, J. Fan, X.W. Huang, TGM3 promotes epithelial-mesenchymal transition and hepatocellular carcinogenesis and predicts poor prognosis for patients after curative resection. Dig. Liver Dis. 52, 668–676 (2020)
E. Méndez, W. Fan, P. Choi, S.N. Agoff, M. Whipple, D.G. Farwell, N.D. Futran, E.A. Weymuller Jr., L.P. Zhao, C. Chen, Tumor-specific genetic expression profile of metastatic oral squamous cell carcinoma. Head Neck 29, 803–814 (2007)
A. Negishi, M. Masuda, M. Ono, K. Honda, M. Shitashige, R. Satow, T. Sakuma, H. Kuwabara, Y. Nakanishi, Y. Kanai, K. Omura, S. Hirohashi, T. Yamada, Quantitative proteomics using formalin-fixed paraffin-embedded tissues of oral squamous cell carcinoma. Cancer Sci. 100, 1605–1611 (2009)
J. Liu, Y. Zhou, J. Wan, Z. Liu, Expression of TGM3 protein and its significance in laryngeal carcinoma. Lin Chung Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 26, 101–103 (2012)
B.S. Chen, M.R. Wang, X. Xu, Y. Cai, Z.X. Xu, Y.L. Han, M. Wu, Transglutaminase-3, an esophageal cancer-related gene. Int. J. Cancer 88, 862–865 (2000)
A. Smirnov, L. Anemona, M. Montanaro, A. Mauriello, M. Annicchiarico-Petruzzelli, E. Campione, G. Melino, E. Candi, Transglutaminase 3 is expressed in basal cell carcinoma of the skin. Eur. J. Dermatol. 29, 477–483 (2019)
Z. Cao, Y. Wang, Z.Y. Liu, Z.S. Zhang, S.C. Ren, Y.W. Yu, M. Qiao, B.B. Zhai, Y.H. Sun, Overexpression of transglutaminase 4 and prostate cancer progression: a potential predictor of less favourable outcomes. Asian J. Androl. 15, 742–746 (2013)
Y. Zhou, Y. Zang, Y. Yang, J. Xiang, Z. Chen, Candidate genes involved in metastasis of colon cancer identified by integrated analysis. Cancer Med. 8, 2338–2347 (2019)
M. Meng, S. Liu, C. Wang, X. Gu, E. Linghu, X. Xue, Mass spectrum analysis of membrane proteins reveals that CASK, CD36 and EPB42 are differentially expressed in pancreatic adenocarcinoma. Oncol. Lett. 20, 376 (2020)
J. Peltier, J.P. Roperch, S. Audebert, J.P. Borg, L. Camoin, Activation peptide of the coagulation factor XIII (AP-F13A1) as a new biomarker for the screening of colorectal cancer. Clin. Proteomics 15, 15 (2018)
Cancer Genome Atlas Research Network. Electronic address: andrew_aguirre@dfci.harvard.edu; Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32, 185–203.e13 (2017)
P. Charoentong, F. Finotello, M. Angelova, C. Mayer, M. Efremova, D. Rieder, H. Hackl, Z. Trajanoski, Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 18, 248–262 (2017)
S.H. Jiang, J. Li, F.Y. Dong, J.Y. Yang, D.J. Liu, X.M. Yang, Y.H. Wang, M.W. Yang, X.L. Fu, X.X. Zhang, Q. Li, X.F. Pang, Y.M. Huo, J. Li, J.F. Zhang, H.Y. Lee, S.J. Lee, W.X. Qin, J.R. Gu, Y.W. Sun, Z.G. Zhang, Increased serotonin signaling contributes to the Warburg effect in pancreatic tumor cells under metabolic stress and promotes growth of pancreatic tumors in mice. Gastroenterology 153, 277-291.e19 (2017)
Q. Li, X.X. Zhang, L.P. Hu, B. Ni, D.X. Li, X. Wang, S.H. Jiang, H. Li, M.W. Yang, Y.S. Jiang, C.J. Xu, X.L. Zhang, Y.L. Zhang, P.Q. Huang, Q. Yang, Y. Zhou, J.R. Gu, G.G. Xiao, Y.W. Sun, J. Li, Z.G. Zhang, Coadaptation fostered by the SLIT2-ROBO1 axis facilitates liver metastasis of pancreatic ductal adenocarcinoma. Nat. Commun. 14, 861 (2023)
M. Pigors, D. Kiritsi, C. Cobzaru, A. Schwieger-Briel, J. Suárez, F. Faletra, H. Aho, L. Mäkelä, J.S. Kern, L. Bruckner-Tuderman, C. Has, TGM5 mutations impact epidermal differentiation in acral peeling skin syndrome. J. Invest. Dermatol. 132, 2422–2429 (2012)
K. Chen, Y. Lu, F. Peng, H.L. Yu, J.Y. Wu, Y. Tan, Y.X. Zhao, TGM6 variants in Parkinson’s disease: clinical findings and functional evidence. J. Integr. Neurosci. 19, 51–64 (2020)
A. Schulze-Krebs, F. Canneva, J. Stemick, A.C. Plank, J. Harrer, G.P. Bates, D. Aeschlimann, J.S. Steffan, S. von Hörsten, Transglutaminase 6 is colocalized and interacts with mutant huntingtin in Huntington disease rodent animal models. Int. J. Mol. Sci. 22, 8914 (2021)
N.V. Serbina, E.G. Pamer, Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7, 311–317 (2006)
Y. Ding, V. Labitzky, K. Legler, M. Qi, U. Schumacher, B. Schmalfeldt, C. Stürken, L. Oliveira-Ferrer, Molecular characteristics and tumorigenicity of ascites-derived tumor cells: mitochondrial oxidative phosphorylation as a novel therapy target in ovarian cancer. Mol. Oncol. 15, 3578–3595 (2021)
Y. Luo, B. Li, J. Li, Y. Zhang, M. Deng, C. Hu, W. Yan, Z. Zhou, G. Zhang, Coagulation factor XIII Subunit A Is a biomarker for curative effects and prognosis in malignant solid tumors, especially non-small cell lung cancer. Front. Oncol. 11, 719085 (2021)
Y. Sawai, Y. Yamanaka, S. Nomura, Clinical significance of factor XIII activity and monocyte-derived microparticles in cancer patients. Vasc. Health Risk Manag. 16, 103–110 (2020)
A. Porrello, P.L. Leslie, E.B. Harrison, B.K. Gorentla, S. Kattula, S.K. Ghosh, S.H. Azam, A. Holtzhausen, Y.L. Chao, M.C. Hayward, T.A. Waugh, S. Bae, V. Godfrey, S.H. Randell, C. Oderup, L. Makowski, J. Weiss, M.D. Wilkerson, D.N. Hayes, H.S. Earp, A.S. Baldwin, A.S. Wolberg, C.V. Pecot, Factor XIIIA-expressing inflammatory monocytes promote lung squamous cancer through fibrin cross-linking. Nat. Commun. 9, 1988 (2018)
J.S. Palumbo, K.A. Barney, E.A. Blevins, M.A. Shaw, A. Mishra, M.J. Flick, K.W. Kombrinck, K.E. Talmage, M. Souri, A. Ichinose, J.L. Degen, Factor XIII transglutaminase supports hematogenous tumor cell metastasis through a mechanism dependent on natural killer cell function. J. Thromb. Haemost. 6, 812–819 (2008)
H. Nakaoka, D.M. Perez, K.J. Baek, T. Das, A. Husain, K. Misono, M.J. Im, R.M. Graham, Gh: a GTP-binding protein with transglutaminase activity and receptor signaling function. Science 264, 1593–1596 (1994)
G.E. Begg, L. Carrington, P.H. Stokes, J.M. Matthews, M.A. Wouters, A. Husain, L. Lorand, S.E. Iismaa, R.M. Graham, Mechanism of allosteric regulation of transglutaminase 2 by GTP. Proc. Natl. Acad. Sci. U. S. A. 103, 19683–19688 (2006)
D. Aeschlimann, M. Paulsson, K. Mann, Identification of Gln726 in nidogen as the amine acceptor in transglutaminase-catalyzed cross-linking of laminin-nidogen complexes. J. Biol. Chem. 267, 11316–11321 (1992)
M.T. Kaartinen, A. Pirhonen, A. Linnala-Kankkunen, P.H. Mäenpää, Transglutaminase-catalyzed cross-linking of osteopontin is inhibited by osteocalcin. J. Biol. Chem. 272, 22736–22741 (1997)
C.S. Greenberg, P.J. Birckbichler, R.H. Rice, Transglutaminases: multifunctional cross-linking enzymes that stabilize tissues. FASEB J. 5, 3071–3077 (1991)
S.H. Jiang, Y.H. Wang, L.P. Hu, X. Wang, J. Li, X.L. Zhang, Z.G. Zhang, The physiology, pathology and potential therapeutic application of serotonylation. J. Cell Sci. 134, 257337 (2021)
D. Telci, M. Griffin, Tissue transglutaminase (TG2)–a wound response enzyme. Front. Biosci. 11, 867–882 (2006)
S. Oliverio, A. Amendola, C. Rodolfo, A. Spinedi, M. Piacentini, Inhibition of “tissue” transglutaminase increases cell survival by preventing apoptosis. J. Biol. Chem. 274, 34123–34128 (1999)
A. Chhabra, A. Verma, K. Mehta, Tissue transglutaminase promotes or suppresses tumors depending on cell context. Anticancer Res. 29, 1909–1919 (2009)
M.A. Antonyak, J.M. Jansen, A.M. Miller, T.K. Ly, M. Endo, R.A. Cerione, Two isoforms of tissue transglutaminase mediate opposing cellular fates. Proc. Natl. Acad. Sci. U. S. A. 103, 18609–18614 (2006)
J. Sebat, B. Lakshmi, J. Troge, J. Alexander, J. Young, P. Lundin, S. Månér, H. Massa, M. Walker, M. Chi, N. Navin, R. Lucito, J. Healy, J. Hicks, K. Ye, A. Reiner, T.C. Gilliam, B. Trask, N. Patterson, A. Zetterberg, M. Wigler, Large-scale copy number polymorphism in the human genome. Science 305, 525–528 (2004)
A. Kumar, J. Xu, S. Brady, H. Gao, D. Yu, J. Reuben, K. Mehta, Tissue transglutaminase promotes drug resistance and invasion by inducing mesenchymal transition in mammary epithelial cells. PLoS ONE 5, e13390 (2010)
H. Ma, L. Xie, L. Zhang, X. Yin, H. Jiang, X. Xie, R. Chen, H. Lu, Z. Ren, Activated hepatic stellate cells promote epithelial-to-mesenchymal transition in hepatocellular carcinoma through transglutaminase 2-induced pseudohypoxia. Commun. Biol. 1, 168 (2018)
M. Shao, L. Cao, C. Shen, M. Satpathy, B. Chelladurai, R.M. Bigsby, H. Nakshatri, D. Matei, Epithelial-to-mesenchymal transition and ovarian tumor progression induced by tissue transglutaminase. Cancer Res. 69, 9192–9201 (2009)
Y. Feng, D. Ji, Y. Huang, B. Ji, Y. Zhang, J. Li, W. Peng, C. Zhang, D. Zhang, Y. Sun, Z. Xu, TGM3 functions as a tumor suppressor by repressing epithelial-to-mesenchymal transition and the PI3K/AKT signaling pathway in colorectal cancer. Oncol. Rep. 43, 864–876 (2020)
S.J. Grille, A. Bellacosa, J. Upson, A.J. Klein-Szanto, F. van Roy, W. Lee-Kwon, M. Donowitz, P.N. Tsichlis, L. Larue, The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res. 63, 2172–2178 (2003)
Q. Zhao, Y. Ren, H. Xie, L. Yu, J. Lu, W. Jiang, W. Xiao, Z. Zhu, R. Wan, B. Li, ELK3 mediated by ZEB1 facilitates the growth and metastasis of pancreatic carcinoma by activating the Wnt/β-Catenin pathway. Front. Cell Dev. Biol. 9, 700192 (2021)
T.Z. Li, S.M. Kim, W. Hur, J.E. Choi, J.H. Kim, S.W. Hong, E.B. Lee, J.H. Lee, S.K. Yoon, Elk-3 contributes to the progression of liver fibrosis by regulating the epithelial-mesenchymal transition. Gut Liver 11, 102–111 (2017)
Y. Chen, T. Wang, M. Huang, Q. Liu, C. Hu, B. Wang, D. Han, C. Chen, J. Zhang, Z. Li, C. Liu, W. Lei, Y. Chang, M. Wu, D. Xiang, Y. Chen, R. Wang, W. Huang, Z. Lei, X. Chu, MAFB promotes cancer stemness and tumorigenesis in osteosarcoma through a Sox9-mediated positive feedback loop. Can. Res. 80, 2472 (2020)
T. Nemoto, Y. Shibata, S. Inoue, A. Igarashi, Y. Tokairin, K. Yamauchi, T. Kimura, M. Sato, K. Sato, H. Nakano, S. Abe, M. Nishiwaki, M. Kobayashi, S. Yang, Y. Minegishi, K. Furuyama, H. Machida, I. Kubota, MafB silencing in macrophages does not influence the initiation and growth of lung cancer induced by urethane. EXCLI J. 16, 914–920 (2017)
S. Mzoughi, Y.X. Tan, D. Low, E. Guccione, The role of PRDMs in cancer: one family, two sides. Curr. Opin. Genet. Dev. 36, 83–91 (2016)
Y.H. Zhu, J.H. Zheng, Q.Y. Jia, Z.H. Duan, H.F. Yao, J. Yang, Y.W. Sun, S.H. Jiang, D. J. Liu, Y. M. Huo, Immunosuppression, immune escape, and immunotherapy in pancreatic cancer: focused on the tumor microenvironment. Cell Oncol (Dordr) 46, 17–48 (2022)
E.M. Richard, T. Thiyagarajan, M.A. Bunni, F. Basher, P.O. Roddy, L.J. Siskind, P.J. Nietert, T.K. Nowling, Reducing FLI1 levels in the MRL/lpr lupus mouse model impacts T cell function by modulating glycosphingolipid metabolism. PLoS ONE 8, e75175 (2013)
K.P. Sundararaj, T. Thiyagarajan, I. Molano, F. Basher, T.W. Powers, R.R. Drake, T.K. Nowling, FLI1 levels impact CXCR3 expression and renal infiltration of T cells and renal glycosphingolipid metabolism in the MRL/lpr lupus mouse strain. J. Immunol. 195, 5551–5560 (2015)
K. Kim, S.Y. Bang, H.S. Lee, S.K. Cho, C.B. Choi, Y.K. Sung, T.H. Kim, J.B. Jun, D.H. Yoo, Y.M. Kang, S.K. Kim, C.H. Suh, S.C. Shim, S.S. Lee, J. Lee, W.T. Chung, J.Y. Choe, H.D. Shin, J.Y. Lee, B.G. Han, S.K. Nath, S. Eyre, J. Bowes, D.A. Pappas, J.M. Kremer, M.A. Gonzalez-Gay, L. Rodriguez-Rodriguez, L. Ärlestig, Y. Okada, D. Diogo, K.P. Liao, E.W. Karlson, S. Raychaudhuri, S. Rantapää-Dahlqvist, J. Martin, L. Klareskog, L. Padyukov, P.K. Gregersen, J. Worthington, J.D. Greenberg, R.M. Plenge, S.C. Bae, High-density genotyping of immune loci in Koreans and Europeans identifies eight new rheumatoid arthritis risk loci. Ann. Rheum. Dis. 74, e13 (2015)
S. Bradshaw, W.J. Zheng, L.C. Tsoi, G. Gilkeson, X.K. Zhang, A role for Fli-1 in B cell proliferation: implications for SLE pathogenesis. Clin. Immunol. 129, 19–30 (2008)
Y.S. He, X.K. Yang, Y.Q. Hu, K. Xiang, H.F. Pan, Emerging role of Fli1 in autoimmune diseases. Int. Immunopharmacol. 90, 107127 (2021)
Z. Chen, E. Arai, O. Khan, Z. Zhang, S.F. Ngiow, Y. He, H. Huang, S. Manne, Z. Cao, A.E. Baxter, Z. Cai, E. Freilich, M.A. Ali, J.R. Giles, J.E. Wu, A.R. Greenplate, M.A. Hakeem, Q. Chen, M. Kurachi, K. Nzingha, V. Ekshyyan, D. Mathew, Z. Wen, N.A. Speck, A. Battle, S.L. Berger, E.J. Wherry, J. Shi, In vivo CD8(+) T cell CRISPR screening reveals control by Fli1 in infection and cancer. Cell 184, 1262-1280.e22 (2021)
D.Y. van Haaften-Visser, M. Harakalova, E. Mocholi, J.M. van Montfrans, A. Elkadri, E. Rieter, K. Fiedler, P.M. van Hasselt, E.M.M. Triffaux, M.M. van Haelst, I.J. Nijman, W.P. Kloosterman, E.E.S. Nieuwenhuis, A.M. Muise, E. Cuppen, R.H.J. Houwen, P.J. Coffer, Ankyrin repeat and zinc-finger domain-containing 1 mutations are associated with infantile-onset inflammatory bowel disease. J. Biol. Chem. 292, 7904–7920 (2017)
O. Küçükşahin, A. Ateş, N. Türkçapar, M. Törüner, M. Turgay, T. Duman, A. Şahin, M.T. Yıldızgören, A.K. Okoh, E. Külahçıoğlu, Ş Erten, G. Kınıklı, S. Assadpour, N. Düzgün, Association between single nucleotide polymorphisms in prospective genes and susceptibility to ankylosing spondylitis and inflammatory bowel disease in a single centre in Turkey. Turk. J. Gastroenterol. 27, 317–324 (2016)
Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–78 (2007)
X. Du, Y. Zhou, L. Song, X. Wang, S. Zhang, Zinc finger protein 365 is a new maternal LPS-binding protein that defends zebrafish embryos against gram-negative bacterial infections. FASEB J. 32, 979–994 (2018)
T. Bekaii-Saab, M.A. Phelps, X. Li, M. Saji, L. Goff, J.S. Kauh, B.H. O’Neil, S. Balsom, C. Balint, R. Liersemann, V.V. Vasko, M. Bloomston, W. Marsh, L.A. Doyle, G. Ellison, M. Grever, M.D. Ringel, M.A. Villalona-Calero, Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J. Clin. Oncol. 29, 2357–2363 (2011)
C.D. Weekes, D.D. Von Hoff, A.A. Adjei, D.P. Leffingwell, S.G. Eckhardt, L. Gore, K.D. Lewis, G.J. Weiss, R.K. Ramanathan, G.K. Dy, W.W. Ma, B. Sheedy, C. Iverson, J.N. Miner, Z. Shen, L.T. Yeh, R.L. Dubowy, M. Jeffers, P. Rajagopalan, N.J. Clendeninn, Multicenter phase I trial of the mitogen-activated protein kinase 1/2 inhibitor BAY 86–9766 in patients with advanced cancer. Clin. Cancer Res. 19, 1232–1243 (2013)
H.Y. Lim, J. Heo, H.J. Choi, C.Y. Lin, J.H. Yoon, C. Hsu, K.M. Rau, R.T. Poon, W. Yeo, J.W. Park, M.H. Tay, W.S. Hsieh, C. Kappeler, P. Rajagopalan, H. Krissel, M. Jeffers, C.J. Yen, W.Y. Tak, A phase II study of the efficacy and safety of the combination therapy of the MEK inhibitor refametinib (BAY 86–9766) plus sorafenib for Asian patients with unresectable hepatocellular carcinoma. Clin. Cancer Res. 20, 5976–5985 (2014)
R. Schmieder, F. Puehler, R. Neuhaus, M. Kissel, A.A. Adjei, J.N. Miner, D. Mumberg, K. Ziegelbauer, A. Scholz, Allosteric MEK1/2 inhibitor refametinib (BAY 86–9766) in combination with sorafenib exhibits antitumor activity in preclinical murine and rat models of hepatocellular carcinoma. Neoplasia 15, 1161–1171 (2013)
A.A. Adjei, D.A. Richards, A. El-Khoueiry, F. Braiteh, C.H. Becerra, J.J. Stephenson Jr., A.F. Hezel, M. Sherman, L. Garbo, D.P. Leffingwell, C. Iverson, J.N. Miner, Z. Shen, L.T. Yeh, S. Gunawan, D.M. Wilson, K.J. Manhard, P. Rajagopalan, H. Krissel, N.J. Clendeninn, A phase I study of the safety, pharmacokinetics, and pharmacodynamics of combination therapy with Refametinib plus Sorafenib in patients with advanced cancer. Clin. Cancer Res. 22, 2368–2376 (2016)
J.R. Infante, L.A. Fecher, G.S. Falchook, S. Nallapareddy, M.S. Gordon, C. Becerra, D.J. DeMarini, D.S. Cox, Y. Xu, S.R. Morris, V.G. Peddareddigari, N.T. Le, L. Hart, J.C. Bendell, G. Eckhardt, R. Kurzrock, K. Flaherty, H.A. Burris 3rd., W.A. Messersmith, Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 13, 773–781 (2012)
G. Kan, Z. Wang, C. Sheng, G. Chen, C. Yao, Y. Mao, S. Chen, Dual inhibition of DKC1 and MEK1/2 synergistically restrains the growth of colorectal cancer cells. Adv. Sci. (Weinh) 8, 2004344 (2021)
S.A. Blankenstein, M.W. Rohaan, W.M.C. Klop, B. van der Hiel, B.A. van de Wiel, M.J. Lahaye, S. Adriaansz, K. Sikorska, H. van Tinteren, A. Sari, L.G. Grijpink-Ongering, W.J. van Houdt, M. Wouters, C.U. Blank, S. Wilgenhof, J.V. van Thienen, A.C.J. van Akkooi, J. Haanen, Neoadjuvant Cytoreductive treatment with BRAF/MEK inhibition of prior unresectable regionally advanced melanoma to allow complete surgical resection, REDUCTOR: A prospective, single-arm, open-label phase II trial. Ann. Surg. 274, 383–389 (2021)
X. Zhou, A. Zhu, X. Gu, G. Xie, Inhibition of MEK suppresses hepatocellular carcinoma growth through independent MYC and BIM regulation. Cell Oncol. (Dordr) 42, 369–380 (2019)
L. Paragh, D. Törőcsik, Factor XIII Subunit A in the skin: applications in diagnosis and treatment. Biomed. Res. Int. 2017, 3571861 (2017)
M.T. Kaartinen, M. Arora, S. Heinonen, A. Rissanen, J. Kaprio, K.H. Pietiläinen, Transglutaminases and obesity in humans: Association of F13A1 to adipocyte hypertrophy and adipose tissue immune response. Int. J. Mol. Sci. 21, 8289 (2020)
Y. Wang, K. Yan, J. Lin, J. Li, J. Bi, Macrophage M2 co-expression factors correlate with the immune microenvironment and predict outcome of renal clear cell carcinoma. Front. Genet. 12, 615655 (2021)
R. Adány, Z. Nemes, L. Muszbek, Characterization of factor XIII containing-macrophages in lymph nodes with Hodgkin’s disease. Br. J. Cancer 55, 421–426 (1987)
L. Maiuri, A. Luciani, I. Giardino, V. Raia, V.R. Villella, M. D’Apolito, M. Pettoello-Mantovani, S. Guido, C. Ciacci, M. Cimmino, O.N. Cexus, M. Londei, S. Quaratino, Tissue transglutaminase activation modulates inflammation in cystic fibrosis via PPARgamma down-regulation. J. Immunol. 180, 7697–7705 (2008)
C. Klöck, T.R. Diraimondo, C. Khosla, Role of transglutaminase 2 in celiac disease pathogenesis. Semin. Immunopathol. 34, 513–522 (2012)
T. Su, X.Y. Qin, Y. Furutani, Transglutaminase 2 as a marker for inflammation and therapeutic target in sepsis. Int J Mol Sci 22, 1897 (2021)
L. Cao, M. Shao, J. Schilder, T. Guise, K.S. Mohammad, D. Matei, Tissue transglutaminase links TGF-β, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene 31, 2521–2534 (2012)
J. Fu, Q.Y. Yang, K. Sai, F.R. Chen, J.C. Pang, H.K. Ng, A.L. Kwan, Z.P. Chen, TGM2 inhibition attenuates ID1 expression in CD44-high glioma-initiating cells. Neuro Oncol. 15, 1353–1365 (2013)
K. Oh, H.G. Moon, D.S. Lee, Y.B. Yoo, Tissue transglutaminase-interleukin-6 axis facilitates peritoneal tumor spreading and metastasis of human ovarian cancer cells. Lab. Anim. Res. 31, 188–197 (2015)
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 82173153, 81902370, 81972582 and 92168111) and the Natural Science Foundation of Shanghai (19ZR1452500).
Author information
Authors and Affiliations
Contributions
Conception and design: Q. Yang, XM Yang, FY Dong, ZG Zhang; Development of methodology: S. Zhang, Q. Yang; Acquisition of data: S. Zhang, H. Li, Q. Yang, HF Yao, T. Su, W. Hao, SH Jiang; Analysis and interpretation of data: S. Zhang, Q. Yang, XM Yang, T. Su, H. Li, FY Dong, SH Jiang; Writing, review and/or revision of the manuscript: S. Zhang, Q. Yang, XM Yang, FY. Dong, ZG Zhang; Administrative, technical or material support: Q. Yang, FY Dong, ZG Zhang, XM Yang. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethical approval
Studies using human PDAC samples were approved by the Research Ethics Committee of Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University (No. RA-2019–116). The animal experiments according to the protocols were approved by the Shanghai Jiao Tong University Animal Care Commission and all mice received humane care in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this paper.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Shan Zhang, Hong-Fei Yao, Hui Li and Tong Su share co-first authorship.
Supplementary Information
Below is the link to the electronic supplementary material.

Supplementary Figure 1
The prognostic value of the TG score in KIRC, LUSC, PAAD, and STAD. Time-dependent receiver operating characteristic (ROC) curves of the prognostic TG score model in TGCA-KIRC (A–C), TGCA-LUSC (D–F), TGCA-PAAD (G–I), and TGCA-STAD (J–L). The X-axis shows the false-positive rates, and the Y-axis shows the true-positive rates (PNG 233 kb)

Supplementary Figure 2
Mutation and CNV of TG family genes across cancers. (A) Point mutation frequency in TG family genes across TCGA cancer types. (B) The frequencies of missense variants and loss-of-function mutations in TG family genes in 20 TCGA cancer types. The highest frequency of mutations in TG family genes was observed in UCEC. (C) The frequency of copy number variations in TG family genes in different TCGA cancers. The gradient of colors in the bubble map represents the log2-fold change between cancer and normal tissues. The purple nodes indicate high expression in cancer, while the green nodes indicate low expression in cancer. The red circles represent copy number amplification, whereas the blue circles represent deletion (PNG 197 kb)

Supplementary Figure 3
The relationships between TG expression and drug sensitivity based on the Cancer Cell Line Encyclopedia (CCLE) database. The gradient of colors in the bubble map represents the R-value. Purple indicates a positive correlation with sensitivity to the drug candidates, while green indicates a negative correlation. The size of the nodes represents the statistical significance; the larger the size is, the greater the significance (PNG 119 kb)

Supplementary Figure 4
The relationship between TG expression and drug sensitivity based on the Genomics of Drug Sensitivity in Cancer (GDSC) database. The gradient of colors in the bubble map represents the R-value. Purple indicates a positive correlation with sensitivity to the drug candidates, while green indicates a negative correlation. The size of the nodes represents the statistical significance; the larger the size is, the greater the significance (PNG 585 kb)

Supplementary Figure 5
(A) Western blotting analysis of the knockdown efficiency of TGM2 in panc1 and patu-8988 cells. (B) Western blotting analysis of the knockdown and overexpression efficiency of Tgm2 in KPC1199 cells. (C) Analysis of CCL2 expression in TGM2 overexpression and control pancreatic cancer cells (n=3 replicates per group, means ± standard deviations, one of three biological replicates). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 (PNG 922 kb)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Zhang, S., Yao, HF., Li, H. et al. Transglutaminases are oncogenic biomarkers in human cancers and therapeutic targeting of TGM2 blocks chemoresistance and macrophage infiltration in pancreatic cancer. Cell Oncol. 46, 1473–1492 (2023). https://doi.org/10.1007/s13402-023-00824-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-023-00824-7