
Abstract
Mitochondria play an essential role in maintaining energy homeostasis and cellular survival. In the brain, higher ATP production is required by mature neurons for communication. Most of the mitochondrial proteins transcribe in the nucleus and import in mitochondria through different pathways of the mitochondrial protein import machinery. This machinery plays a crucial role in determining mitochondrial morphology and functions through mitochondrial biogenesis. Failure of this machinery and any alterations during mitochondrial biogenesis underlies neurodegeneration resulting in Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), and Parkinson’s disease (PD) etc. Current knowledge has revealed the different pathways of mitochondrial protein import machinery such as translocase of the outer mitochondrial membrane complex, the presequence pathway, carrier pathway, β-barrel pathway, and mitochondrial import and assembly machinery etc. In this review, we have discussed the recent studies regarding protein import machinery, beyond the well-known effects of increased oxidative stress and bioenergetics dysfunctions. We have elucidated in detail how these types of machinery help to import and locate the precursor proteins to their specific location inside the mitochondria and play a major role in mitochondrial biogenesis. We further discuss their involvement in mitochondrial dysfunctioning and the induction of toxic aggregates in neurodegenerative diseases like AD and PD. The review supports the importance of import machinery in neuronal functions and its association with toxic aggregated proteins in mitochondrial impairment, suggesting a critical role in fostering and maintaining neurodegeneration and therapeutic response.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mitochondria are double-membrane organelles that are widely distributed in cells. These are commonly regarded as either “bioenergetics center” or “apoptosis regulator” and play both functional and detrimental roles respectively [1, 2]. Healthy mitochondria maintain cellular homeostasis, energy production, and cell survival etc. The formation of the new functional mitochondria, i.e., mitochondrial biogenesis, is one of the crucial processes of the cell. New mitochondria generate from the pre-existing ones via mitochondrial fission-fusion processes and get involved in the mitochondrial DNA (mtDNA) replication, transcription, translation, and also in the loading of membrane phospholipids and proteins [3,4,5,6]. Additionally, mitochondria play a crucial role in the synthesis of essential metabolites, heme and lipid metabolism, calcium homeostasis, and regulation of the intrinsic pathway of apoptosis [3,4,5, 7, 8].
Maintenance of newly forming mitochondria takes place by both nuclear and mitochondrial genome, which comprises approximately 1500 mitochondrial proteins [9,10,11]. The mitochondrial genome encodes 13 essential proteins belonging to the mitochondrial respiratory chain and requires for the proper functioning of mitochondria [12]. The majority of mitochondrial proteins transcribes on the nuclear genome. Furthermore, they translate in the cytosolic ribosome and import into mitochondria through different pathways of protein import machinery of mitochondria [4, 13]. Mitochondrial protein import machinery is complex of multimeric proteins known as translocases [14]. Translocase of outer membrane complex, i.e., TOM complex, provides general gate entry through which all cytosolic translated mitochondrial proteins enter and localize at their specific location [13, 14]. Furthermore, these localized proteins ensure the central role of mitochondria in bioenergetics, calcium homeostasis, metabolism, and cellular health [13, 15]. For successful mitochondrial biogenesis, the nuclear genome and mitochondrial genome work in a coordinated manner [5]. Any disruption in this co-ordination can impair mitochondrial biogenesis that leads to mitochondrial dysfunction and oxidative stress [16]. Furthermore, the mitochondrial network can also be compromised and disturbed due to imbalanced mitochondrial dynamics (mitochondrial fission-fusion balance) [17]. Altogether, mitochondrial dysfunction results in the form of neurodegeneration and causes incurable neonatal developmental disorders and adult-onset neurodegenerative diseases (Table 1) [5, 18, 33, 34]. Neurodegenerative diseases are the incurable diseases, which lead to slow, and progressive memory loss, behavioral impairment, and uncontrolled motor functions caused by pathogenic abnormal assembly of proteins in the nervous system [35, 36]. The brain cells are susceptible to mitochondrial dysfunction as they require the high number of healthy mitochondria near synapse to provide calcium and adenosine triphosphate (ATP) as an energy source that regulates neuronal communication and survival [37]. Therefore, healthy mitochondria require healthy protein import machinery for appropriate mitochondrial biogenesis and strictly essential for cell viability [38, 39] (Table 1). However, several studies reported the role of the pathogenic protein aggregates such as α-synuclein, amyloid-β, and tau in the impairment of these import machineries that lead to mitochondrial pathophysiology and neurodegeneration [19, 23, 40,41,42]. Mitochondrial defects and dysfunctions have been reported in the association with pathogenesis of several neurodegenerative disorders [43,44,45,46]. Therefore, targeting mitochondrial dysfunction is a promising therapeutic approach for these debilitating diseases [47,48,49,50,51,52,53].
Overall, this review is mainly focused on our emerging understanding of the underlying role of protein import machinery-mediated mitochondrial biogenesis in the context of neuronal vulnerability and neurodegeneration. We discuss the regulation and interplay between different import pathways responsible for the defective mitochondrial network. We also discuss the influence of the defective import on the functioning of mitochondrial biogenesis associated with key toxic protein aggregates and their implications in the pathogenesis of various neurodegenerative diseases like Alzheimer’s disease (AD) and Parkinson’s disease (PD).
Mitochondrial Protein Import Machinery
The field of mitochondrial protein import machinery linked with mitochondrial biogenesis has experienced rapid expansion in recent years [13, 34, 54,55,56]. Several studies have explored the direct connection between mitochondrial dysfunction and neurodegeneration [54, 57, 58]. However, mitochondrial import machinery linked studies with mitochondrial dysfunction and neurodegeneration have been accomplished in the last 20 years [38,39,40, 59,60,61]. Initially, the pre-sequence pathway and the carrier pathway were only known, but with the time, three important import pathways such as sorting and assembly machinery, mitochondrial intermembrane space import and assembly machinery, and mitochondrial import complex have been explored. The integral networks of these different types of protein import machinery regulate mitochondrial function, biogenesis, architecture, and dynamics as the main regulatory center [39]. Here, we have discussed the respective area detail.
Mitochondrial Biogenesis and Dynamics
Mitochondria are known for its formation from pre-existing mitochondria, where it inherits in daughter cells in a cell cycle-dependent manner [62,63,64]. There are many essential processes, which work in a regulated and coordinated manner to complete healthy mitochondrial biogenesis [3]. These processes include transcription and translation of nuclear DNA-encoded mitochondrial proteins, post-translational modification of mitochondrial proteome, correct integration of mitochondrial targeting sequence (MTS) on mitochondrial pre-proteins followed by accurate import, folding, and assembly inside the mitochondria [3, 60]. Altogether, these processes maintain mitochondrial proteostasis and its turnover, which make functional mitochondria. Furthermore, calcium signaling, ATP production, oxidative phosphorylation (OXPHOS), intracellular signaling, regulation of cytochrome-c, and apoptosis etc. are the main functions of mitochondria [60, 65].
Generally, the human mitochondrial genome is built of 16,569 base pairs, where it encodes mRNA essential for 13 OXPHOS proteins, 2 ribosomal ribonucleic acid (rRNA), and 22 transfer ribonucleic acid (tRNA) synthesis [66]. Newly encoded mtDNA proteins localize itself in the mitochondria and play an essential role in the electron transport chain and adenosine triphosphate (ATP) production. Also, un-folding or re-folding of imported mitochondrial proteins is essentially required for the normal functioning of mtDNA proteins [41]. Nuclear DNA (nDNA) and mtDNA act in a tight regulation to also promote assembly of mtDNA encoded factors [41]. Therefore, coordinated communication between the nucleus and mitochondria is required for successful mitochondrial biogenesis. The fusion-fission processes of mitochondrial dynamics are associated with precursor protein import and mtDNA-related protein translation. Altogether, these processes make successful mitochondrial biogenesis and regulate mitochondrial mass and number [67, 68].
Generally, mitochondrial dynamics is the tightly regulated continuous process of fusion and fission events [67]. Fusion is regulated by optic atrophy protein 1 (OPA1), mitofusins (MFN) 1&2, and fission by dynamin-related protein 1 (DRP1) and mitochondrial fission 1 protein (Fis-1). Mitochondrial dynamics regulates the mitochondrial morphology and maintains the healthy mitochondrial pool in neurons [47]. Under the overfed condition, mitochondria follow the fission (fragmentation) process to create new mitochondria. While under the starvation condition, mitochondrial fusion follows mitochondrial elongation to avoid mitochondrial degradation and increases ATP production [67, 69]. These fission-fusion processes are required as a quality control mechanism [67]. Following the fission process, defective mitochondria are removed by mitophagy process [67, 70], while healthy mitochondria are selected based on the number of mtDNA copies, optimum matrix metabolites, and the component of the mitochondrial membrane [67]. During a high level of stress, fission enhances prolong mitochondrial fragmentation that leads to cellular apoptosis [70, 71]. In contrast, the impaired fusion process allows uneven mitochondrial elongation [70]. These impairments accumulate defective mitochondria, and affect mitochondrial functions as well as morphology [70].
The translocase of the outer membrane (TOM) complex, a general entry gate on the mitochondrial outer membrane, allows the import of all mitochondrial precursor proteins [62, 72]. The higher level of TOM complex imports a high level of mitochondrial pre-proteins across the membrane. Therefore, during fission, TOM complex level (Tom6) elevates in the mitotic (M) phase of the cell cycle to induce mitochondrial biogenesis and transfer their copy in next-generation cells via regulated mitochondrial dynamics [62, 73].
Moreover, large-sized developing and mature neurons are highly sensitive for their parental organelle mitochondria, as they need high ATP production on their synapses and during cell to cell communication/neurotransmission [74]. For accurate mitochondrial biogenesis in neurons, nuclear genes and mitochondrial genes must work in a coordinated manner, while the import of nuclear-encoded mitochondrial pre-proteins must be in a regulated manner. Anterograde signaling is associated with nuclear encoding mitochondrial transcription factor, translation, and mitochondrial biogenesis [41], while retrograde signaling is triggered by mitochondrial stress such as reactive oxygen species (ROS) generation, mtDNA defects, and mitochondrial membrane potential (MMP) loss. This stress further activates the retrograde signaling by which mitochondria send signals to trigger cell death or support cell adaptation by the nucleus [41]. Several studies have reported defects in different pathways of mitochondrial import machinery leading to mitochondrial stress [34, 75, 76]. Defect in TOM, translocase of the inner membrane (TIM), mitochondrial intermembrane space assembly (MIA) pathway results in failure of respiratory chain complex I-V function, and ATP production, due to which ROS generation occurs [75]. Any mutation in gene-encoding subunits of import machinery can be responsible for the pathogenesis of mitochondrial diseases. This can also lead to neurodegeneration, muscular dystrophy, and congenital defects etc. [14]. Additionally, this can affect the import and correct folding of pre-proteins that lead to the formation of defective mitochondria. It can further lead to apoptosis and possibly play an important role in the pathogenesis of neurodegenerative diseases (Table 1) [5, 73].
Role of Protein Import Machinery in Maintaining Mitochondrial Architecture and Biogenesis
Double membrane mitochondria consist of an outer and inner membrane (cristae), separating intermembrane space and matrix [77]. Five different mitochondrial import machineries are comprising of the most prominent protein complexes residing on the mitochondrial membrane. Most of the mitochondrial functional proteins characterized by specific mitochondrial targeting signal and cellular metabolites are efficiently transferred across the membrane via these types of machinery [39]. Generally, the cytosolic precursor proteins are imported firstly via the TOM complex, which is a general entry gate, located on the outer membrane [56]. Then, pre-proteins containing specific targeting signals follow the specific pathway of machinery respective to their destination [56, 78]. The pre-sequence pathway involves the import of the cleavable amino-terminus (N-terminal) pre-sequence which acts as a targeting signal for the inner membrane and most of the matrix precursor proteins. Following the TOM complex, precursor proteins import via further pre-sequence TIM23 complex towards their destination [39, 60]. Precursor proteins carrying pre-sequence are firstly identified by TOM20 and TOM22 receptors (members of TOM complex). These receptors import proteins via the TOM40 channel across the outer membrane. Furthermore, TIM50, a receptor of TIM23 complex recognizes and binds with precursor protein, which further transfers to TIM23, a channel-forming protein, closely associated with TIM17. The import of matrix proteins is further driven by ATP-dependent mitochondrial heat-shock protein 70 (mtHsp70) and the pre-sequence translocase-associated motor (PAM). The mtHSP70 and TIM44 associate on the TIM23 channel. Mitochondrial processing peptidase (MPP) and other peptidases cleave off the pre-sequence of precursor proteins in the matrix [39, 60]. Furthermore, chaperones heat shock proteins such as HSP60 and HSP10 promote the matrix proteins folding. Many of the precursor proteins carrying hydrophobic sorting signal laterally import in the inner membrane after removing the sorting signal by inner-membrane peptidase (IMP) and further release into IMS. The TIM23 complex interacts with complex III and IV of the electron transport chain for lateral sorting of pre-proteins and encourages their energy-driven import [39].
It is very clear that the TOM complex imports both cleavable and non-cleavable sequences containing precursor proteins. Other than pre-sequence pathway, all four contain a non-cleavable sequence in the form of an internal targeting signal. The carrier pathway is the second import pathway, where hydrophobic multispanning metabolic carrier proteins of the inner membrane firstly bind to cytosolic chaperone HSP70 then both bind with the TOM70 receptor of TOM complex [39]. Next, pre-proteins import across the outer membrane in a loop form, after interacting with TOM40. The N-terminal of TOM40 round-ups with the small Tim chaperones from the intermembrane space, which further plays a role to deliver pre-proteins onto the carrier translocase of the inner membrane 22 (TIM22). Furthermore, TIM54 intermembrane space domain interacts with small Tim chaperones, which results in a change in MMP. Due to which, pre-proteins destine at their location in the inner membrane [39, 79].
Sorting assembly machinery 50 kDa subunit (SAM50) and related core proteins of sorting and assembly machinery (SAM) in mitochondrial outer membrane make the third import pathway, i.e., the β-barrel pathway. This pathway regulates the importing of nuclear-encoded outer membrane mitochondrial proteins, which are known as β-barrel precursor protein via TOM, SAM, and small Tim chaperones [39]. TOM22 receptor of the TOM complex and Sam37 membrane protein of SAM machinery together create a transient large complex to import outer membrane precursor proteins. Next, Sam35 and Sam50 fold pre-protein in the β-barrel form, which further imports in the lipid layer of the outer membrane [39].
The mitochondrial inter-membrane space pre-proteins contain cysteine motifs, which import via the fourth import pathway, i.e., the mitochondrial inter-membrane space import and assembly (MIA) machinery, which follows redox-regulated import pathway [39, 80]. This pathway comprises major proteins such as Mia40 and Erv1 (essential for respiration and vegetative growth) in yeast or Gfer (growth factor erv1-like) or ALR (augmenter of liver regeneration) in mammals and helper of Tim of 13 kDa (Hot13) [80,81,82]. This pathway is chiefly involved in the oxidative folding of proteins through the insertion of disulfide bonds. These disulfide bonds are necessary for proper folding, stability, and activity of the essential mitochondrial proteins [83]. Some of the specific substrates or precursor proteins are retained in the reduced and unfolding form in the cytosol, from where these are recognized and import by the TOM40 channel. Furthermore, Mia40 acts as a receptor to bind with reduced pre-proteins via hydrophobic interaction and acquires intermolecular bonds. After binding, Mia40 shows its oxidoreductase activity and oxidizes pre-proteins by forming disulfide bonds in the inter-membrane space [39]. Mia40 itself re-oxidizes via FAD dependent ALR sulfhydrl oxidase activity with the help of Hot13, a zinc binding protein. Electron coming from the earlier stage of pre-protein to Mia40 to ALR transfers to the cytochrome-c (Cytc) or to the dioxygen molecule (O2), an essential part of complex IV of the electron transport chain. Overall, this electron-based pathway is known as a mitochondrial disulfide relay system (MDRS) [39, 84]. Therefore, besides import, this pathway also contributes in respiratory chain and cellular survival. Additionally, some matrix and inner membrane proteins are also imported via the MIA pathway along with IMS proteins [39].
The fifth pathway is the mitochondrial import (MIM) complex, which promotes the import of α-helical transmembrane segments containing outer membrane proteins. N-terminal signal-anchor sequence contains by these outer membrane precursor proteins recognizes by Tom70 and transfers them to MIM specific insertase to destine proteins efficiently [39, 85]. The MIM imported small outer membrane proteins such as Tom5 and Tom6 further participate and make a complex with newly inserted Tom40 via the SAM pathway. Altogether, SAM and MIM pathways together encourage the steps of TOM subunits early assembly. Therefore, the MIM pathway is also a versatile and important protein translocase complex [86].
Taken together, these studies suggest the role of different types of protein import machinery in protein importing, folding, and localization within mitochondria. These destined proteins play a multifunctional role in respiration, ATP generation, mitochondrial morphology, biogenesis, apoptosis, and cellular homeostasis.
Interplay Between Different Pathways of Protein Import Machinery in Maintaining Mitochondrial Architecture and Biogenesis
The interplay between different pathways of protein import machinery is a complex biological process and different translocases play an important role in its regulation. The versatility and abundance of different translocases are reported by identification of the absolute copy number of their components [34, 87]. The TOM complex is identified as the most abundant translocase, which makes supercomplex with distinct protein machineries such as TOM-SAM, and TOM-TIM23-pre-proteins super-complexes [88, 89]. The TOM-SAM super-complex is required to import β-barrel pre-proteins [88]. However, the separate pool or one large dynamic pool of the TOM complex is still in debate for the freely interchangeable complexes. Apart from these super-complexes, the TOM complex might also comprise of simple TOM complex, where only the TOM40 channel may help in the import of intermembrane space precursors, which are recognized by Mia40 receptors [34].
The coenzyme Q (CoQ) or ubiquinone is a key molecule for respiratory chain activity, which is located in the inner membrane towards the matrix side [90]. CoQ transfers electrons from complex I and II to complex III and also acts as a cofactor for several enzymes [90]. The subunits of the CoQ complex encode in the nucleus and import into mitochondria via the mutual function of TOM and TIM23 complexes [34]. Therefore, these import types of machinery are also functionally involved in the CoQ biosynthetic pathway and related respiratory functions [34]. On the other hand, the respiratory chain-dependent MMP is also required to import the precursors through TOM-TIM23 super-complexes [34]. Therefore, due to any disruption in respiratory activities, it is also essentially required to maintain the MMP-dependent import machineries [34].
The components of different import types of machinery can collaborate to generate a new pathway [34]. For example Om45, a single-spanning protein of the outer membrane, lies towards IMS [34, 91]. Om45 precursors import via TOM and TIM23 complexes towards IMS, but escaping IMS, Om45 transport topological in opposite action via MIM complex to locate into outer membrane [34, 91, 92].
The outer membrane TOM and SAM machineries are one of the central components of ER-mitochondrial organizing network (ERMIONE) [34]. These machineries are also associated with another central component which is abundant on cristae junctions of the inner membrane, i.e., mitochondrial contact site and cristae organizing system (MICOS) complex [34]. The Mic60, the largest subunit of the MICOS complex, plays a crucial role in the outer and inner membrane contact site formation. All these components are functionally required for the mitochondrial architecture, dynamics, cellular respiration, transport of ion and metabolites, and lipid and protein biogenesis [34, 93]. Additionally, MICOS complex cooperates in the close interaction of TOM complex with downstream positioned SAM and MIA machineries, to promote the import of β-barrel proteins into the outer membrane and cysteine-rich proteins into IMS [34]. The ERMOINE-mediated TOM-SAM-MICOS network also interacts with other machineries, which makes a large and complex mitochondrial functional network [34]. The MICOS and TOM complex also helps in the accumulation of PTEN-induced kinase-1 (PINK1) on the mitochondrial outer membrane during damaged mitochondria, which induces mitophagy [34].
Altogether, the interplay between the different import machineries shows their potential involvement in mitochondrial major functions and in maintaining architecture via ER linked contact sites. However, more information still needs to be explored with the other import machineries, cytosolic proteins, and mitochondrial outer membrane [34].
Regulation of Mitochondrial Protein Import
The constitutively active mitochondrial protein import machinery regulates mitochondrial composition, biogenesis, turnover, and overall homeostasis, which are required for organelle fitness and cellular survival. However, mitochondrial protein import itself is regulated at the different stages associated with cell signaling pathway, metabolism, stress, and pathological conditions [13].
Cytosolic precursor proteins essentially require different types of protein import machinery to be imported into mitochondria [94]. The import of these proteins is regulated by targeting signals and unfolded state. To import into mitochondria, the targeting signals containing precursor proteins first bind with mitochondrial receptors and import in an unfolded state [94]. Any hindrance or modifications or masking in targeting signals and unstable unfolded state or stable folded state of proteins lead to the failure of mitochondrial import. The folded state of precursor proteins can itself mask the targeting signals and impair the import [13]. Depending on metabolic conditions, feedback inhibition directly regulates the import of crucial mitochondrial matrix enzyme 5-aminolevulinate synthase, which is essential for an earlier stage in heme biosynthesis. The metabolite heme binds to enzyme precursor protein and impairs its recognition by TOM receptors [13].
Kinases play a major role in the regulation of mitochondrial import, dynamics, and turnover [13, 95]. Critical protein kinases such as c-Jun N-terminal kinase (JNK), protein kinase A (PKA), casein kinase (CK), and AMP-dependent protein kinase (AMPK) also translocate on the mitochondrial outer membrane [95]. These kinases further phosphorylate their specific protein substrates or precursor proteins on the outer membrane [13, 95]. This phosphorylation gets involved in the crucial function of mitochondrial protein import, dynamics, respiration chain activity, and apoptosis [95]. The cytochrome P450 monooxygenase isoenzymes contain dual mitochondria and ER targeting sequences. To import this enzyme into mitochondria, PKA phosphorylates its mitochondria targeting sequence, which increases the affinity of this enzyme to import via TOM complex, TIM23, and HSP60 while impairing its transport towards ER [13]. Next, the subunits of the TOM complex also act as a substrate for different kinases [95]. CK1 regulates the import of TOM22 in the TOM complex [13]. MIM1/2 and TOM22/70 are the substrates of CK2, which import and assemble into mitochondria following phosphorylation [94, 95]. This phosphorylation is needed for the import of other TOM proteins. Any disruption in CK2 can impair mitochondrial biogenesis and impair the pre-sequence and carrier pathway [94]. Next, in the case of glucose abundance, PKA phosphorylates TOM22, TOM40, and inhibits their biogenesis, while phosphorylates TOM70 to inhibit its activity [94, 95]. Due to elevated adenosine 3′,5′-cyclic monophosphate (cAMP) level, PKA also phosphorylates the Hsp70 chaperone binding pocket of TOM70, which inhibits binding of Hsp70 to TOM70 and related carrier import [94, 95].
The chaperones, ATP levels, and electron transport chain activity also regulate the protein import as a sensitive indicator of mitochondrial energetic stress and fitness [94]. First, the TIM23 and TIM22 complexe-mediated pathways are dependent on MMP and are intimately associated with mitochondrial ATP generating energetic state. Precursor proteins importing towards the inner membrane are dependent only on MMP. To make this import successful, mitochondrial genome-required protein 2 (Mgr2) and Tim21 (subunits of TIM23 complex) bind with supercomplex of cytochrome c oxidase (complex IV) and bc1-complex (complex III) of the electron transport chain. This association triggers the MMP-dependent import of pre-proteins [94]. Second, along with MMP, matrix pre-proteins require ATP as well to drive mtHSP70 chaperone, to import towards the matrix. The mtHSP70 helps in the import of pre-proteins by unfolding their cytosolic side [94]. Third, the TIM23 complex interacts with the TOM complex to import matrix protein from the outer membrane to the inner membrane. Altogether, any disruption in respiratory chain activity, ATP production, and mtHSP70 chaperone activity due to high occupancy by misfolded proteins directly affect the activity of the protein import pathway [94].
Sometimes, new coming precursor proteins get trapped across the TOM complex and clogged the import channel. Therefore, in addition to chaperones, surveillance of protein machinery by ubiquitin-like (UBX)-domain-containing protein 2 (Ubx2) protein associated with the TOM complex is also reported [96,97,98]. In this case, Ubx2 closely interacts with the TOM complex and recruits Cdc48-mediated ubiquitylation, which targets misfolded proteins and clears clogged channels [96]. Ubx2 is a well-known populated protein involved in both endoplasmic-reticulum associated degradation (ERAD) pathway and mitochondrial outer membrane translocated protein associated degradation (MitoTAD) pathway [96, 98]. The failure of the Ubx2-mediated MitoTAD pathway can cause blockage in protein import capacity, which leads to proteotoxic stress in mitochondria [96].
To maintain nuclear and mitochondrial communication, retrograde (mitochondria to nuclear) and anterograde signaling (nucleus to mitochondria) actively work in the cell [41, 99, 100]. In the case of retrograde signaling, increased nuclear gene expression takes place due to the numerous mitochondrial stress and known as a mitochondrial stress response (MSR) [41, 67]. MSR induces in the response of mitochondrial disruption such as the depletion of mtDNA and OXPHOS content, energy metabolism, hypoxia, loss of MMP, ATP decline, and defects in calcium homeostasis etc. [41]. The mitochondrial stress triggers the series of calcium-dependent retrograde signaling, i.e., JNK/mitogen-activated protein kinases (JNK/MAPK), calcium/calmodulin-dependent protein kinases (CAMKs), and calcium/calcineurin-mediated cAMP response element-binding protein (CREB) [101, 102]. These signalings further activate peroxisome proliferator-activated receptor-γ coactivator (PGC-1α)-mediated mitochondrial biogenesis and OXPHOS function related mitochondrial factors in the nucleus [103, 104]. Apart from other factors responsible for MSRs, mitochondrial unfolded protein response (UPRmt) activates during misfolding and accumulation of mitochondrial proteins [105]. For the proper folding and correct functioning of the proteins, co-translational and post-translational protein folding, proteins maturation, and degradation processes are tightly regulated [105]. However, dysregulation in mitochondrial protein homeostasis, i.e., proteostasis triggers UPRmt via activating transcription factor associated with stress-1 (ATFS-1) in yeast and ATF4, ATF5 and C/EBP homologous protein (CHOP)-CCAAT/enhancer-binding proteins (C/EBPβ) system in mammals [67, 76, 102]. On the other hand, the nucleus works in an anterograde signaling manner to induce mitochondrial protein biogenesis, promote the functioning of the mitochondrial network by translating mitochondrial proteins into cytosol, and import across the mitochondrial membrane [41].
The PTEN-induced putative kinase 1 (PINK1)-Parkin (PINK-PRKN) pathway is a well-known quality control system, where it couples TOM machinery-mediated defects in protein import to the mitophagy under severe mitochondrial stress [106]. Generally, PINK1 imports into mitochondria through TOM40-mediated channel in MMP-dependent manner and cleaves via PINK1/PGAM5-associated rhomboid-like protease (PARL) and MPP proteolytic process [11, 107]. This process suppresses the PINK1 levels constitutively [11]. But, whenever mitochondrial MMP collapses (Fig. 2), PINK1 associates with TOM22, TOM20, TOM40, and TOM70 via TOM complex and accumulates on the mitochondrial outer membrane [11]. It causes blockage in protein import and PINK1 acts as a flag to trigger the Parkin E3 ubiquitin-protein ligase protein (PRKN/PARK2) recruitment [107]. The auto-phosphorylation of PINK1 by its kinase activity is required in PARKIN recruitment [108]. Initially, recruited PARKIN on the outer membrane associates with TOM70 and TOM22, where it eliminates all outer membrane proteins via ubiquitylation and leads to the elimination of impaired mitochondria (i.e., mitophagy) [19, 109,110,111].
Overall, these are the distinct regulatory pathways, which regulate the protein import. However, the interaction between different import machinery and regulation under pathological and physiological conditions is still a major challenge to define the integrated mitochondrial network and related fitness [13].
Intersections Between Different Protein Import Machineries and Neurodegeneration
Here, we have discussed the interlinked mechanism of the β-Barrel pathway, pre-sequence pathway, and redox-regulated and carrier pathways of protein import machinery with the accumulated toxic proteins such as amyloid-β (Aβ), tau, α-synuclein (α-syn) and their association with the pathogenesis of neurodegenerative disorders [19, 112]. The aggregation of these toxic proteins result in defective mitochondrial import, respiratory chain failure, disruption in mtDNA maintenance, and ROS generation and finally neuronal demise [19, 112]. Besides protein toxicity, the related important novel findings such as a mutation or any defect in nuclear-encoded mitochondrial factors, a mutation in translocation machinery proteins, a mutation in mitochondria targeting signals, and mitochondrial chaperones are also required to understand the central mechanism of mitochondrial machinery dysfunction-mediated neurodegeneration [39, 113]. Furthermore, in this part of the review, we have focused on the defects in protein import machinery due to toxic aggregates, causing different neurodegenerative disorders.
Protein Import Machinery Dysfunction in AD
AD is one of the most prevalent worldwide late-onset neurodegenerative diseases (Fig. 1) [114, 115]. AD pathogenesis involved loss of neurons and impaired synaptic communication in the cortex, limbic system, hippocampus, and basal forebrain that leads to cognitive deficits [116]. The process of adult neurogenesis is also compromised in AD patients and animal models [117,118,119,120] due to altered axonal pathology, spine morphology, and synaptic plasticity etc. [116, 121, 122]. The adult hippocampal neurogenesis contributes to the storage and processing of new information and plays a crucial role in learning and memory function [116]. The Aβ neurite plaques and hyperphosphorylated Tau neurofibrillary tangles (NFT) are the most common pathological hallmark in these brain regions in AD, which are located intracellular and extracellular, respectively [115, 123]. The Aβ alters neurogenesis in the hippocampus and cortex which is responsible for altered cAMP/CREB signaling cascade and promotes impairment of learning and memory in AD [124].

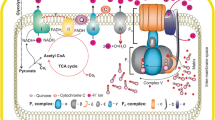
Schematic representation of the import of pathogenic proteins in AD. The possible takeover of mitochondrial proteins by pathogenic proteins as they possess mitochondrial targeting signals such as APP, Aβ, and Tom40 mutant. (a) Aβ imports via Tom40 and blocks (red dotted line) the function of inner membrane complex and matrix enzymes. (b) APP blocks Tom40-Tim23 pathway, leading to no import of OXPHOS proteins causing oxidative stress. (c) Aβ import changes the MMP. (d) Aβ somehow recognizes by Tom22 receptor, from where it imports via Tom40 and shows Aβ-mediated neurotoxicity. (e) APOE*4 allele mutates Tom40 gene, due to which it forms mutated and dysfunctional Tom40 protein in TOM complex. It causes blockage of imported new proteins. (f) Aβ directly inhibits the PreP peptidase, due to which cleaving of signal sequence affected and immature new proteins located in mitochondria leading to mitochondrial dysfunctioning in AD
The energy-driven mitochondria also play an essential role in adult neurogenesis and maturation of newborn neurons [124]. In recent studies, mitochondrial defects and dysfunctions are well reported in causing neurodegeneration and related AD pathophysiology, which aggravates AD symptoms [125,126,127]. A study also reported that the Aβ downregulates the PGC-1α-mediated mitochondrial biogenesis leading to reduced mitochondrial mass [124]. Aβ also inhibits mitochondrial import of nuclear-encoded proteins by blocking mitochondrial import machinery, which affects mitochondrial biogenesis in AD [68]. Aβ and tau imbalances mitochondria dynamics by increasing Drp1 levels [68] and decreasing mitofusins and OPA-1 [124]. Besides tau-mediated mitophagy [68], Aβ also elevates the levels of the mature form of microtubule-associated protein 1A/1B-light chain 3 (LC3), i.e., LC3-II and sequestosome 1 (p62/SQSTM1), which triggers excess mitophagy [124]. Therefore, these mitochondria-related impairments are the intracellular earlier steps, which affect neurogenesis and further lead to the weakening of synaptic communication and neurodegeneration in AD [68, 124].
Aβ is a natural metabolic product from the amyloid precursor protein (APP), and its distribution is reported in the endoplasmic reticulum (ER), mitochondria, Golgi apparatus, endosome, lysosome, and nucleus etc. [128]. Aβ is derived from the catalytic action of β- and γ-secretase enzyme on the APP, where Aβ1–42 is known the most toxic aggregates due to high hydrophobic nature [115]. Generally, APP keeps signal peptide for targeting both ER and mitochondria. In normal conditions, the APP targets ER and moves towards the plasma membrane through the secretory pathway [129]. Although due to APP high proportion, APP interacts with mitochondrial translocation channels [129]. Furthermore, its acidic domain interacts with active γ-secretase complex through mitochondria-associated endoplasmic reticulum membranes (MAM) [20, 130]. This results in the formation of Aβ from APP cleavage in MAM, instead of inside the mitochondria [115]. The newly formed Aβ is imported into the mitochondria via the TOM complex, where mitochondrial pre-sequence protease (PreP) degrades Aβ to control its mitochondrial toxicity and imbalance to keep it in precise concentration in the cell [115]. PreP is a mitochondrial matrix pre-sequence peptidase that degrades mitochondrial targeting signal sequence (MTS) or pre-sequence possessing N-terminal from mitochondrial matrix pre-proteins [131, 132].
The Aβ peptides accumulate on and inside the mitochondria and mediates toxicity, which is well reported in the AD patients [133]. The toxic aggregation of Aβ protein directly blocks the mitochondrial import channels and later blocks the importing of nuclear-encoded mitochondrial proteins in AD [20, 115, 129]. The import of toxic Aβ occurs by the TOM40 channel and further, it targets the inner membrane complex and matrix enzymes causing mitochondrial dysfunctioning in AD [19] (Fig. 1), which leads to cognitive deficits and memory loss [134]. A study reported that Aβ physically interacts with mitochondria and impairs cellular respiration by altering the complex I and IV enzyme activity and disturbs the fission-fusion process [135]. Additionally, alteration leads to increased ROS production and distorted mitochondrial morphology etc., resulting in mitochondrial dysfunctioning [115, 131].
A study reported that Aβ toxic peptides affect PreP-mediated pre-sequence processing machinery, i.e., late step of mitochondrial import reaction (Fig. 1) [136]. Generally, PreP/Cym provides them mature form, after degrading signal sequence. However, Aβ is reported to block this PreP/Cym activity, and therefore, pre-sequence containing matrix precursor proteins become abnormally matured due to interfering with the activity of MPP and contributes to AD pathology by dysfunctioning mitochondria [131, 136]. While in another study, the co-aggregation of Aβ peptides and precursor proteins were reported, where co-aggregation of Aβ42 showed a stronger inhibitory effect against mitochondrial protein import and significantly reduced the import of precursor proteins [136].
A study on human AD pathology reported that the truncated form of non-glycosylated APP makes stable complex through MitN-CytoC orientation with both outer membrane TOM40 and inner membrane TIM23 and inhibits import and localization of mitochondrial cytochrome C oxidase IV and Vb proteins via two import pathways–mediated state (Fig. 1) [20]. Therefore, the lower import of these proteins in mitochondria increases oxidative stress by increasing the hydrogen peroxide (H2O2) level [20]. Due to this mitochondrial accumulation cholinergic, gamma-aminobutyric acid (GABAergic), dopaminergic, and glutaminergic neurons in different brain regions such as the amygdala, hippocampus, and cortex are affected in AD [20].
The gene variants of mitochondrial import pathways themselves transport from the cytoplasm and import inside mitochondria through their import pathways. Rather they could have associated with mitochondrial dysfunction via a defect in their protein structure and related faulty import [41]. However, in a study (Fig. 1), the outer membrane TOMM40 gene is reported as the possible late-onset Alzheimer’s disease (LOAD) risk factor, where the mutated TOMM40 gene becomes responsible for the abnormal TOM40 protein [19, 137]. Generally, the TOMM40 gene remains in upstream and close proximity to apolipoprotein E and C (APOE and APOC) gene, located on the 19q chromosome [19, 137]. These genes are present in the tight cluster as TOMM40-APOE-APOC1-APOC4-APOC2 in the strong linkage disequilibrium (LD) pattern [19]. However, genome-wide association studies (GWAS) reported that the LD locus of APOE has the strongest association signal, which is assigned to APOE*4 allele haplotype, known as the strong pathogenic factor in LOAD [138]. Therefore, other genes present in the LD pattern are also associated with strong significant signals contributing to disease risk [19]. Therefore, TOMM40 single nucleotide polymorphisms (SNPs)-related genetic variations are also associated with LOAD [19, 139]. These variations contain polyT repeats in variable-length within intron 6 at a single locus of TOMM40, known as “TOMM40–523”, which varies in length by race and individuality [19, 139]. As a result, the mutated TOMM40-related cognate TOM40 protein (general import pore) causes APP translocation arrest and cause AD-mediated mitochondrial dysfunctioning [137]. Due to blockage of the channel, the entry of other mitochondrial proteins also hinders, which causes ROS production, decreased COX activity, and mitochondrial defects [140]. In one study, in yeast mitochondria (Fig. 1), cytosolic Aβ was specifically recognized by the outer membrane TOM22 (Tomm22 in the human) [141]. Based on this study, it is further reported that Aβ transfers to another subunit of TOM complex, TOM40, and imports into the mitochondria through the TOM channel. This recognition is essential for the Aβ accumulation in mitochondria in AD [141].
Generally, mitochondrial matrix proteins follow a common inner membrane import route, where they require appropriate MMP [142, 143]. Aβ does not follow this common route as the C-terminal of Aβ is hydrophobic and can bind to import receptors directly or import via vesicle transport [142]. Therefore, Aβ import can depend on its length and hydrophobicity etc. [142]. This study is related to the uptake of Aβ inside the mitochondria that cause Aβ accumulation-mediated mitochondrial dysfunction in AD patients [142]. One of the studies on the AD brain reported that (Fig. 1) Aβ enters through TOM general entry gate into cristae and becomes responsible for lower MMP [133].
In a study, PC12 cells were transfected with GFP protein (mtGFP) tagged N′-terminal mitochondrial targeting sequence followed by sub-lethal exposure of Aβ (10 μmol/L) for 48 h [144]. As a result, GFP tagged mitochondrial precursor proteins were not able to import across mitochondria. Therefore, their levels were significantly higher in the cytoplasm, representing Aβ-mediated neurotoxicity. This study also reported the defective import of TOM20 and mtHSP70/mortalin (nuclear-encoded mitochondrial proteins) inside the mitochondria [144]. Generally, mortalin belongs to the mitochondrial chaperone mtHSP70 protein family, which helps in mitochondrial protein import, protein folding, and cell survival [145]. Therefore, incomplete import of mortalin and TOM40 itself leads to the reduced MMP, increased ROS generation, and altered mitochondrial morphology, contributing to AD [144, 146]. In a study, Aβ-mediated TOM complex interaction also affects OXPHOS complex activities. Reduction in TOM20 and TOM70 subunits also showed deficits in OXPHOS complex I, III, IV, and V activities [147].
In a study, it is reported that any defect in protein import machinery can lead to precursor protein accumulation in the cytosol [148]. The over-accumulation creates stress in the cell that trigger apoptosis. This over-accumulation can also trigger protein aggregation and mistargeting of proteins, which further stimulate unfolded protein response (UPR), a mechanism of cell survival [148,149,150]. In a study, reduced expression of TOM40 leads to the accumulation of protein aggregates in the cytosol and mistargeting of the proteins [148]. Defect in TOM40 also reduced the proteasome activity, ATP production, and increased ROS generation. This defect results in the failure of autophagy and leading to protein aggregation and neurodegeneration [148].
Altogether, these integrated facts in the context of neuronal cells suggest that defective protein import causing impaired mitochondria formation and their dysfunction. Therefore, defective mitochondria have not any replacement and result in neurodegeneration and worse the AD pathogenesis.
Protein Import Machinery Dysfunction in PD
PD is the second well -nown adult-onset neurodegenerative disease, where α-synuclein (α-syn) aggregates form Lewy bodies and affect dopamine neurons of the substantia nigra pars compacta (SNpc) (Fig. 2). It is characterized by the loss of dopaminergic neurons degeneration along with reduced tyrosine hydroxylase expression in the SNpc of PD patients and animal models [43, 53, 151, 152]. α-syn is a known causative agent, which directly inhibits mitochondrial enzyme activity causing oxidative stress and leading to mitochondrial dysfunction in PD [153]. Generally, α-syn is known for its function in suppression of apoptosis, regulation of dopamine biosynthesis, chaperone activity, and neuronal differentiation etc. [154]. But any truncation in the carboxyl-terminus (C-terminal) of α-syn leads to aggregation, which is responsible for PD pathology [154].

Schematic representation of the import of pathogenic proteins in PD. The possible seizing of mitochondrial proteins by pathogenic proteins as they possess mitochondrial targeting signals such as PINK1, DJ-1, and α-syn. (a) α-syn imports via Tom40 channel and blocks inner membrane complex function and inhibits complex I activity, leading to redox misbalancing. (b) Mutated mortalin import via Tom40-Tim23-Tim44 channel and folded by HSP60, leading to immature mitochondrial biogenesis. (c) DJ-1 binds with mortalin and affects their functionality, causes oxidative stress. (d) In case of change in MMP in healthy mitochondria, PINK1 exports outside mitochondria, where it recruits Park2. Park2 recognizes via Tom70 and Tom22, leading to mitophagy. (e) Generally, wild-type PINK1 imports via Tom40-Tim23 channel and cleaved by MPP in the matrix. But in the case of mutant PINK1, it imports via the same channel but does not cleaved properly, leading to reduced neuroprotection
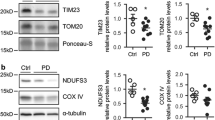
It has been reported that likewise AD, the causative agent of PD, i.e., truncated α-syn, interferes in mitochondrial protein import across the membrane where it triggers mitochondrial impairment and causes α-synucleinopathy [155]. A study on post-mortem brain samples in human PD subjects and α-syn transgenic mice have found the significantly lower levels of TOM40, while no significant change in TOM20 levels [11]. Therefore, TOM40 has been reported as the main target in PD. Oxidative damage and deletions in mitochondrial DNA, reduced energy production, and reduced complex I protein levels are associated with reduced TOM40 in α-syn-transgenic mice [11]. Interestingly, after lentivirus-mediated TOM40 overexpression, the reduced activities were ameliorated in transgenic mice [11]. In another study, cytosolic aggregated α-syn interacts via TOM40 general pore to localize in mitochondria (Fig. 2) [156]. After importing inside mitochondria, α-syn interferes with complex I activity of the respiratory chain and induces ROS production and cytochrome c release [156]. One study on cytosolic modified α-syn specific interaction with TOM20 showed defects in mitochondrial protein import, which causes reduced OXPHOS and increased ROS production [157]. However, no interaction with TOM40, TIM23, and TOM22 was observed [11, 157]. Overexpression of TOM20 also ameliorated all deleterious effects, showing potential therapeutic benefits against α-synucleinopathy and neurodegeneration [11, 23, 157]. Another study further showed the mimicking nature of α-syn with mitochondrial targeting sequence (MTS), through which interaction of α-syn and TOM20 occurs [158]. This interaction prevents the binding of TOM20 with co-receptor TOM22 and disrupts the protein import [157]. This interaction blocks the import of Ndufs3, a nuclear-encoded mitochondrial protein as well as induces aberrant oligomerization and aggregation of α-syn, in the rotenone-induced mouse PD models [158].
These studies also indicate the strong relationship between α-syn overexpression and reduced MIM expression, where decreased OXPHOS activity, imbalanced mitochondrial dynamics, and mtDNA damage etc. are also reported [159]. However, the effectiveness of physical exercise is previously reported for reducing mitochondrial dysfunctions and motor deficits etc. [159]. It is also suggested that the levels of TH induce due to exercise on a treadmill, which further reduces dopaminergic neuronal loss and alleviates PD-like symptoms [159]. Interestingly, treadmill exercise also induces the antioxidant enzyme levels, which further reduces the levels of α-syn protein [159]. On the other hand, several studies have also reported the association of physical exercise with MIM, where TOM20, TOM22, TOM40, and TIM23 expression increases in skeletal muscle due to 7 days of electrical stimulation to PD mouse models [159,160,161]. Altogether, these studies suggested the association of reduced α-syn and induced MIM by physical exercise, which increases protein import and improves mitochondrial function. This also further reduces the dopaminergic neuronal cell death and may improve the partial motor deficits in MPTP-induced PD mouse models [159].
It has been observed that PD linked defects in protein machinery including the TOM complex also interfere in cell quality control mechanism and survival [106]. Several studies have also reported the role of PINK-PARKIN pathway-mediated protein import in PD pathology [106, 162,163,164,165]. Mutation in PINK-PARKIN affects the mitophagy and causes mitochondrial pathology, which later becomes the reason for the dopaminergic neuronal loss, causing autosomal recessive PD [19]. In one study on familial PD, the mutation in C-terminal negative charged motif of PINK1 or deletion of TOM7 causes defects in import arrest [166]. Later, PINK1 fails to locate and accumulates on the outer membrane and imports inside depolarized mitochondria. Furthermore, OMA1 protease degrades PINK1, instead of PARL and affects PARKIN-mediated mitophagy, leading to PD [166]. Interestingly, on another side, subunits of the TOM complex act as a molecular switch in the PINK-PARKIN pathway regulated mitophagy process [110]. During PD causing loss of function mutation in the Parkin gene, PARKIN loses the interaction with TOM70 and TOM40 subunits, while overproduction of TOM22 and TOM40 reverses the mitophagy regulated by the PINK-PARKIN pathway followed by MMP loss. This dysregulation of mitophagy leads to the accumulation of defective mitochondria [110, 167]. Therefore, the loss of coupling between import machinery and mitophagy contributes to the pathogenesis of PD [11, 110].
One of the studies on the PINK1 reported that PINK1 interacts with TOM40-TIM23 multi-subunit complex and gets cleaved by proteolytic activity [168], while mutant PINK1 following the same route of import as wild type but cleavage of PINK1 impairs PINK1 cleavage site that affects on functional and structural level (Fig. 2). Due to which, full-length protein PINK1 does not accumulate on the OMM during stress in mitochondria. This leads to loss of kinase activity of PINK1 that results in neurodegeneration and PD pathogenesis [169, 170].
Unlike mitophagy, which eliminates defective mitochondria, mitochondrial unfolded protein response (UPRmt) rescues and recovers most of the organelle, which can be salvaged and show a protective role [171]. Efficient mitochondrial protein import machinery regulates those upstream regulatory proteins, which further regulate both mitophagy and UPRmt [171]. Protein import acts as a post-translational process and regulates the function of individual organelle as well as the complete organelle pool. In the case of UPRmt, like PINK1, ATFS-1 keeps the MTS signal and import into mitochondria and degrades via Lon protease in healthy mitochondria. While in the case of mitochondrial stress, ATFS-1 increases its levels throughout the cytosol in proportion to mitochondrial stress levels [171,172,173]. Avoiding degradation in the cytosol, ATFS-1 with its targeting nuclear localization sequence (NLS) migrates into the nucleus and induces transcription of more than 400 nuclear genes [171,172,173]. These ATFS-1-induced nuclear-encoded proteins are involved in detoxification, protein import, homeostasis, components of mitochondrial fission, and glycolysis process etc. [171]. These proteins must import into mitochondria to recover mitochondria via protein import machinery. One study reported that rotenone-induced PD-like symptoms including impair complex I activity and paraquat-induced superoxide radicals trigger the UPRmt response [171], while another study on transgenic nematodes revealed the dysregulation of UPRmt by co-expression of α-syn and ATFS-1, causing dopaminergic neurotoxicity and neurodegeneration as characteristics of PD [172].
Due to the crucial role of protein import machinery, these types of machinery remain under surveillance of chaperones [60, 174]. The essential mitochondrial chaperone and only ATPase or core subunit of PAM motor, mortalin mtHSP70, helps to import mitochondrial proteins and can extend the life span of normal human cell and nematodes [174, 175]. Generally, mortalin interacts with N-terminal domain of TIM44 (a member of TIM23 complex) (Fig. 1) and acts as the main platform to connect the inner membrane translocation channel and matrix import motor [174,175,176], whereby energy is driven through ATPase activity of mortalin that lead to MTS containing pre-proteins import in the matrix through TIM44 [174, 175, 177]. Later, mortalin interacts with HSP60 chaperone that helps in protein folding and assembly [145]. Any loss in the mortalin function can cause a fault in protein quality control and impair mitochondrial function and dynamics in PD [178]. Cellular redox sensor DJ-1 protein interacts with mortalin (Fig. 1) and manages oxidative stress and mitochondrial homeostasis, which is critically involved in neurodegeneration [178, 179]. But due to mutation in mortalin, mitochondrial morphology and functions diminish and modification occurs in response to oxidative stress that leads to PD [178].
Besides the role of chaperones in organelle proteostasis, chaperones are also involved in providing degradation tag and eliminating selective proteins for autophagy [180, 181]. Generally, leucine-rich repeat kinase 2 (LRRK2) is degraded by chaperone-mediated autophagy (CMA), while mutated LRRK2 is not able to degrade via this pathway and cause PD [181]. The autosomal dominant mutation, G2019S, is the pathogenic mutant form of LRRK2 which causes familial PD [167, 181]. In many studies, LRRK2 involvement has been reported in the mitochondrial fission/fusion process and has been associated with mitochondrial outer membrane proteins [167]. Generally, LRRK2 recruits Drp1 on the outer membrane and stimulates mitochondrial fission and acts as an essential regulator of mitochondrial dynamics [182]. However, two mutant forms of LRRK2 (G2019S and R1441C) increase their interaction with Drp1. As a result, Drp1 hyperphosphorylation leads to excessive fragmentation, oxidative stress, mitochondrial dysfunctioning, and neuronal anomalies [42, 183]. Besides Drp1, the association of LRRK2 with ubiquitous mitochondrial creatine kinase (uMtCK) is also reported [184]. The uMtCK is involved in mitochondrial membrane permeability and plays an important role in energy transduction [184]. LRRK2 mutant directly inhibits the processing of immature form of uMtCK precursor protein and block its import in the IMS inside mitochondria by retaining it on the mitochondrial outer membrane [184]. This results in the lack of uMtCK and energy channeling inhibition in the mitochondria, which promotes the voltage-dependent anion channel (VDAC) and (adenine nucleotide translocator (ANT) interaction. This VDAC/ANT interaction allows the opening permeabilization transition pore, cytochrome c release that leads to the neuronal intrinsic pathway of apoptosis [184].
The importance of the specific localization of mitochondrial protein machinery is as crucial as their function for cell survival [185]. However, their localization takes place in mitochondrial outer membrane, inner membrane, and intermembrane space [186]. Therefore, the healthy composition of the mitochondrial membrane is important for successful localization of mitochondrial protein import machineries. Phospholipids are the major building block for these membranes, where cardiolipin (CL) is the most unique enriched phospholipid in mitochondrial inner membrane [185, 187, 188]. The function of CL is involved in protein import, mitochondrial membrane morphology, biogenesis and dynamics, electron transport chain and ATP production, mitophagy, and mitochondrial specific role in apoptosis etc. [185, 187]. CL shows its involvement in the biogenesis of TOM complexes and SAM machinery [185]. In the case of protein import of precursor protein, when matrix associated precursor protein import via TOM complex and inter-membrane space, then it interacts with TIM23 machinery [189]. Furthermore, membrane-bound subunit TIM23 and polypeptide substrate receptor TIM50 interaction occurs which is dependent on MMP [189]. This interaction is modulated and stabilized by CL in such a manner, where the TIM50 domain binds with a specific site of TIM23. Furthermore, precursor proteins import into the matrix, which is driven via ATPase dependent PAM complex [189]. Altogether, CL plays a major role in TIM23 dependent import [189]. Therefore, any disruption in CL metabolism or its deficiency can also interfere in this transmembrane import that can lead to mitochondrial dysfunction [185, 189]. Next, the role of CL has also been established in mitophagy, where CL gets externalize from the inner membrane to the outer membrane [190]. CL on the outer membrane is identified by the basic amino acid cluster of microtubule-associated protein 1A/1B-light chain 3 (LC3), which also leads to mitophagy. In one study, it has been reported that the mutation in the basic amino acid of LC3 leads to the failure of LC3 recruitment on mitophagosome [190]. Another study reported that the depletion of CL can interfere or inhibit the mitophagy in rotenone or 6-hydroxydopamine-induced parkinsonian models [190]. However, extensive studies on the effects of CL with protein machinery in case of PD still need more understanding. Altogether, these studies on protein import machinery and linked essential components play a crucial role in understanding pathophysiology of PD.
Conclusion and Future Perspectives
Mitochondria have emerged as a potential target not only in chronic diseases like cancer, diabetes, obesity, and ischemia-reperfusion injury but also in several neurodegenerative diseases [191]. Neurons are highly sensitive for mitochondria as they highly require ATP for synaptic communication and brain functions [192]. Therefore, healthy mitochondrial biogenesis and proper functioning are essential for neuronal homeostasis, which is dependent on nuclear and mitochondrial genome co-ordination [193]. Protein interactions through this co-ordination are further regulated by different pathways of mitochondrial import machineries [193]. These different types of import machinery play a crucial role in the regulation of coordination of nuclear and mitochondrial genome, mitochondrial biogenesis, dynamics, and neuronal cell communication etc. [41]. However, any error in the import of precursor proteins or members of protein machineries itself undoubtedly affects new mitochondrial formation and functions. This leads to apoptosis and neurodegeneration, which are associated with the pathogenesis of neurodegenerative diseases [5, 40].
The TOM, TIM23, TIM22, SAM50, MIA-ALR complexes, and TOM-SAM-mediated complex are involved in the general entry gate, pre-sequence, carrier, SAM, MIA pathways, and MIM complex respectively, as the essential different pathways of mitochondrial import machineries [39, 194]. These pathways import and locate the precursor proteins at their specific location in the outer membrane, IMS, inner membrane, and matrix in the mitochondria [39, 194]. To an extent, several studies have reported the interplay between different import machineries [39, 88, 89, 194]. Although, there is still a need for deeper studies related to their interaction with each other, whether TOM-mediated supercomplexes act as a single complex or freely interchangeable separate way and regulate respiratory functions [34]. The import machineries and electron transport chain derived MMP also work in a coordinated manner to import several nuclear-encoded respiratory proteins such as subunits of CoQ complexes etc. Any disruption directly affects protein import and respiratory functions [34]. Import machineries like TOM and SAM act as a central component of ER-mitochondrial (ERMIONE) network and help in maintaining mitochondrial architecture and network with other organelles [34]. These import machineries are also involved in the contact site of the outer and inner membrane and are associated with another central component of mitochondrial architecture, i.e., MICOS complex [34].
Due to the crucial role of import machineries, they are strictly regulated by several physiological and pathological conditions [13]. They are regulated by targeting sequence and folding state of precursor proteins, metabolic conditions, several kinases, chaperones, respiration activity (ATP level and MMP), surveillance proteins, mitochondrial stress response (UPRmt), cell signaling, stress, and mitophagy etc. [13]. Altogether, these are essentially required for mitochondrial import, folding, biogenesis, architecture, dynamics, cellular respiration, turnover, and overall mitochondrial fitness and cellular survival [13].
Any disruption or impairment in components of import machineries or their regulatory mechanism can lead to the neurodegeneration and cause several chronic neurodegenerative diseases [40, 59]. For an instance, an error in the TOM complex during the M phase of the cell cycle can lead to the failure of mitochondrial biogenesis and dynamics [62]. Another instance of the error in the MPP (mitochondrial processing peptidase)-mediated proteolytic maturation of the newly imported matrix proteins can lead to the defective protein [195]. Additionally, any genomic mutation encoding mitochondrial proteins or aggregated protein like α-syn, Aβ toxicity are the basis of the failure of mitochondrial protein import, respiratory failure, ROS generation, mitochondrial dysfunctioning, and apoptosis, leading to neurodegeneration in AD and PD [59, 196, 197]. In this review, we have discussed the alterations in protein import being linked to mitochondrial bioenergetics, mitophagy, morphology, dynamics, biogenesis, and apoptosis etc. in AD and PD.
Since mitochondrial protein import occurs via MMP generation in the inner membrane and this process is the sensor for the health status of mitochondria and cells, therefore, protein import has become a new therapeutic strategy against neurodegenerative diseases [68]. Recent findings suggested the role of the PINK1/PARKIN pathway as a sensor of import defects, and give us a hint to understand this pathway based mitochondrial regeneration in the case of PD. Still, much work lies ahead to understand the role of protein import and PINK1/PARKIN pathway in the context of healthy and unhealthy mitochondria [106]. Herein, we have also summarized the basic current understanding of the import pathways and their role in mitochondrial biogenesis and dynamics. Furthermore, we have also highlighted the import defects, the underlying general mechanism of mitochondrial pathophysiology in the case of AD and PD. However, the complete mechanism of the import of precursor proteins is still uncovered and needs more understanding in the future.
The next challenge is to better understand the molecular interaction between different pathways of import machineries, which might be a therapeutic target against AD and PD in future studies. To achieve this, induced pluripotent stem cells (iPSCs)–derived PD model approach to assess PD neurons is reported useful for understanding mitochondrial phenotyping [198].
Besides this, the connection between lipids especially CL and membrane insertion of proteins is also an emerging field [141]. The essential role of CL is well reported in maintaining the mitochondrial membrane architecture and morphology. However, it needs further studies about connectivity and cross-talk between several factors involved in CL biosynthesis, cristae formation and remodeling, the establishment of protein import machinery and mitochondrial dynamics [187]. More studies are needed to understand how CL biosynthesis impairment can lead to the failure of import machinery establishment and causes neurodegenerative diseases etc. Effects of therapeutic drugs such as melatonin have been reported to preserve CL integrity while their knowledge is still rudimentary regarding protein machinery-mediated disease progression [187]. Altogether, under stress conditions, the regulation of protein import machineries and their downstream effect on cellular homeostasis and proteostasis are exciting topics to cover the pathophysiology of mitochondrial biogenesis in neurodegeneration in future studies.
The mitochondrial protein import machinery is highly conserved from lower to higher eukaryotes, i.e., yeast to humans. However, their regulatory mechanism may differ as per cell types in different organisms. Till date, most of the regulatory studies of protein import are studied in a limited number of organisms. Therefore, it is important to discuss the conserved or non-conserved regulatory studies in specific species in future studies [13]. Therefore, extensive and variable studies to discover more key proteins and regulatory approaches associated with the network of import machineries may offer promising therapeutic strategies targeting mitochondrial protein import and related proteostasis in neurodegenerative diseases.
References
Martinez J, Marmisolle I, Tarallo D, Quijano C (2020) Mitochondrial bioenergetics and dynamics in secretion processes. Front Endocrinol (Lausanne) 11:319. https://doi.org/10.3389/fendo.2020.00319
Li MX, Dewson G (2015) Mitochondria and apoptosis: emerging concepts. F1000Prime Rep 7:42. https://doi.org/10.12703/P7-42
Ploumi C, Daskalaki I, Tavernarakis N (2017) Mitochondrial biogenesis and clearance: a balancing act. FEBS J 284(2):183–195. https://doi.org/10.1111/febs.13820
Osellame LD, Blacker TS, Duchen MR (2012) Cellular and molecular mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab 26(6):711–723. https://doi.org/10.1016/j.beem.2012.05.003
Franco-Iborra S, Vila M, Perier C (2018) Mitochondrial quality control in neurodegenerative diseases: focus on Parkinson's disease and Huntington's disease. Front Neurosci 12:342. https://doi.org/10.3389/fnins.2018.00342
Seo AY, Joseph AM, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C (2010) New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci 123(Pt 15):2533–2542. https://doi.org/10.1242/jcs.070490
Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G (2008) Viral control of mitochondrial apoptosis. PLoS Pathog 4(5):e1000018. https://doi.org/10.1371/journal.ppat.1000018
Jan R, Chaudhry GE (2019) Understanding apoptosis and apoptotic pathways targeted cancer therapeutics. Adv Pharm Bull 9(2):205–218. https://doi.org/10.15171/apb.2019.024
Stotland A, Gottlieb RA (2015) Mitochondrial quality control: easy come, easy go. Biochim Biophys Acta 1853(10 Pt B):2802–2811. https://doi.org/10.1016/j.bbamcr.2014.12.041
Battersby BJ, Richter U (2013) Why translation counts for mitochondria - retrograde signalling links mitochondrial protein synthesis to mitochondrial biogenesis and cell proliferation. J Cell Sci 126(Pt 19):4331–4338. https://doi.org/10.1242/jcs.131888
Heinemeyer T, Stemmet M, Bardien S, Neethling A (2019) Underappreciated roles of the translocase of the outer and inner mitochondrial membrane protein complexes in human disease. DNA Cell Biol 38(1):23–40. https://doi.org/10.1089/dna.2018.4292
Barchiesi A, Vascotto C (2019) Transcription, processing, and decay of mitochondrial RNA in health and disease. Int J Mol Sci 20(9):2221. https://doi.org/10.3390/ijms20092221
Harbauer AB, Zahedi RP, Sickmann A, Pfanner N, Meisinger C (2014) The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab 19(3):357–372. https://doi.org/10.1016/j.cmet.2014.01.010
Anderson AJ, Jackson TD, Stroud DA, Stojanovski D (2019) Mitochondria-hubs for regulating cellular biochemistry: emerging concepts and networks. Open Biol 9(8):190126. https://doi.org/10.1098/rsob.190126
Jadiya P, Tomar D (2020) Mitochondrial protein quality control mechanisms. Genes (Basel) 11(5):563. https://doi.org/10.3390/genes11050563
Pichaud N, Berube R, Cote G, Belzile C, Dufresne F, Morrow G, Tanguay RM, Rand DM et al (2019) Age dependent dysfunction of mitochondrial and ROS metabolism induced by mitonuclear mismatch. Front Genet 10:130. https://doi.org/10.3389/fgene.2019.00130
Wu NN, Zhang Y, Ren J (2019) Mitophagy, mitochondrial dynamics, and homeostasis in cardiovascular aging. Oxidative Med Cell Longev 2019:9825061. https://doi.org/10.1155/2019/9825061
Uittenbogaard M, Chiaramello A (2014) Mitochondrial biogenesis: a therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr Pharm Des 20(35):5574–5593. https://doi.org/10.2174/1381612820666140305224906
Gottschalk WK, Lutz MW, He YT, Saunders AM, Burns DK, Roses AD, Chiba-Falek O (2014) The broad impact of TOM40 on neurodegenerative diseases in aging. J Parkinsons Dis Alzheimers Dis 1(1):12. https://doi.org/10.13188/2376-922X.1000003
Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK (2006) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci 26(35):9057–9068. https://doi.org/10.1523/JNEUROSCI.1469-06.2006
Herman AM, Khandelwal PJ, Stanczyk BB, Rebeck GW, Moussa CE (2011) Beta-amyloid triggers ALS-associated TDP-43 pathology in AD models. Brain Res 1386:191–199. https://doi.org/10.1016/j.brainres.2011.02.052
Raskin J, Cummings J, Hardy J, Schuh K, Dean RA (2015) Neurobiology of Alzheimer's disease: integrated molecular, physiological, anatomical, biomarker, and cognitive dimensions. Curr Alzheimer Res 12(8):712–722. https://doi.org/10.2174/1567205012666150701103107
Franco-Iborra S, Cuadros T, Parent A, Romero-Gimenez J, Vila M, Perier C (2018) Defective mitochondrial protein import contributes to complex I-induced mitochondrial dysfunction and neurodegeneration in Parkinson's disease. Cell Death Dis 9(11):1122. https://doi.org/10.1038/s41419-018-1154-0
Zarei S, Carr K, Reiley L, Diaz K, Guerra O, Altamirano PF, Pagani W, Lodin D et al (2015) A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int 6:171. https://doi.org/10.4103/2152-7806.169561
Dupuis L, Gonzalez de Aguilar JL, Oudart H, de Tapia M, Barbeito L, Loeffler JP (2004) Mitochondria in amyotrophic lateral sclerosis: a trigger and a target. Neurodegener Dis 1(6):245–254. https://doi.org/10.1159/000085063
Foerster BR, Welsh RC, Feldman EL (2013) 25 years of neuroimaging in amyotrophic lateral sclerosis. Nat Rev Neurol 9(9):513–524. https://doi.org/10.1038/nrneurol.2013.153
Plowman EK, Watts SA, Tabor L, Robison R, Gaziano J, Domer AS, Richter J, Vu T et al (2016) Impact of expiratory strength training in amyotrophic lateral sclerosis. Muscle Nerve 54(1):48–53. https://doi.org/10.1002/mus.24990
Lehmer C, Schludi MH, Ransom L, Greiling J, Junghanel M, Exner N, Riemenschneider H, van der Zee J et al (2018) A novel CHCHD10 mutation implicates a Mia40-dependent mitochondrial import deficit in ALS. EMBO Mol Med 10(6):e8558. https://doi.org/10.15252/emmm.201708558
Mordas A, Tokatlidis K (2015) The MIA pathway: a key regulator of mitochondrial oxidative protein folding and biogenesis. Acc Chem Res 48(8):2191–2199. https://doi.org/10.1021/acs.accounts.5b00150
Yano H, Baranov SV, Baranova OV, Kim J, Pan Y, Yablonska S, Carlisle DL, Ferrante RJ et al (2014) Inhibition of mitochondrial protein import by mutant huntingtin. Nat Neurosci 17(6):822–831. https://doi.org/10.1038/nn.3721
Cowan K, Anichtchik O, Luo S (2019) Mitochondrial integrity in neurodegeneration. CNS Neurosci Ther 25(7):825–836. https://doi.org/10.1111/cns.13105
Yablonska S, Ganesan V, Ferrando LM, Kim J, Pyzel A, Baranova OV, Khattar NK, Larkin TM et al (2019) Mutant huntingtin disrupts mitochondrial proteostasis by interacting with TIM23. Proc Natl Acad Sci U S A 116(33):16593–16602. https://doi.org/10.1073/pnas.1904101116
Dagda RK (2018) Role of mitochondrial dysfunction in degenerative brain diseases, an overview. Brain Sci 8(10):178. https://doi.org/10.3390/brainsci8100178
Pfanner N, Warscheid B, Wiedemann N (2019) Mitochondrial proteins: from biogenesis to functional networks. Nat Rev Mol Cell Biol 20(5):267–284. https://doi.org/10.1038/s41580-018-0092-0
Jucker M, Walker LC (2018) Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci 21(10):1341–1349. https://doi.org/10.1038/s41593-018-0238-6
Johri A, Beal MF (2012) Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther 342(3):619–630. https://doi.org/10.1124/jpet.112.192138
Vos M, Lauwers E, Verstreken P (2010) Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci 2:139. https://doi.org/10.3389/fnsyn.2010.00139
Sokol AM, Sztolsztener ME, Wasilewski M, Heinz E, Chacinska A (2014) Mitochondrial protein translocases for survival and wellbeing. FEBS Lett 588(15):2484–2495. https://doi.org/10.1016/j.febslet.2014.05.028
Wiedemann N, Pfanner N (2017) Mitochondrial machineries for protein import and assembly. Annu Rev Biochem 86:685–714. https://doi.org/10.1146/annurev-biochem-060815-014352
Wang W, Zhao F, Ma X, Perry G, Zhu X (2020) Mitochondria dysfunction in the pathogenesis of Alzheimer's disease: recent advances. Mol Neurodegener 15(1):30. https://doi.org/10.1186/s13024-020-00376-6
Nicolas E, Tricarico R, Savage M, Golemis EA, Hall MJ (2019) Disease-associated genetic variation in human mitochondrial protein import. Am J Hum Genet 104(5):784–801. https://doi.org/10.1016/j.ajhg.2019.03.019
Luo Y, Hoffer A, Hoffer B, Qi X (2015) Mitochondria: a therapeutic target for Parkinson's disease? Int J Mol Sci 16(9):20704–20730. https://doi.org/10.3390/ijms160920704
Yadav A, Agarwal S, Tiwari SK, Chaturvedi RK (2014) Mitochondria: prospective targets for neuroprotection in Parkinson's disease. Curr Pharm Des 20(35):5558–5573. https://doi.org/10.2174/1381612820666140305224545
Chaturvedi RK, Flint Beal M (2013) Mitochondrial diseases of the brain. Free Radic Biol Med 63:1–29. https://doi.org/10.1016/j.freeradbiomed.2013.03.018
Johri A, Chaturvedi RK, Beal MF (2011) Hugging tight in Huntington's. Nat Med 17(3):245–246. https://doi.org/10.1038/nm0311-245
Chaturvedi RK, Calingasan NY, Yang L, Hennessey T, Johri A, Beal MF (2010) Impairment of PGC-1alpha expression, neuropathology and hepatic steatosis in a transgenic mouse model of Huntington's disease following chronic energy deprivation. Hum Mol Genet 19(16):3190–3205. https://doi.org/10.1093/hmg/ddq229
Agarwal S, Yadav A, Chaturvedi RK (2017) Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem Biophys Res Commun 483(4):1166–1177. https://doi.org/10.1016/j.bbrc.2016.08.043
Tiwari SK, Chaturvedi RK (2014) Peptide therapeutics in neurodegenerative disorders. Curr Med Chem 21(23):2610–2631. https://doi.org/10.2174/0929867321666140217125857
Chaturvedi RK, Beal MF (2013) Mitochondria targeted therapeutic approaches in Parkinson's and Huntington's diseases. Mol Cell Neurosci 55:101–114. https://doi.org/10.1016/j.mcn.2012.11.011
Chaturvedi RK, Hennessey T, Johri A, Tiwari SK, Mishra D, Agarwal S, Kim YS, Beal MF (2012) Transducer of regulated CREB-binding proteins (TORCs) transcription and function is impaired in Huntington's disease. Hum Mol Genet 21(15):3474–3488. https://doi.org/10.1093/hmg/dds178
Chaturvedi RK, Adhihetty P, Shukla S, Hennessy T, Calingasan N, Yang L, Starkov A, Kiaei M et al (2009) Impaired PGC-1alpha function in muscle in Huntington's disease. Hum Mol Genet 18(16):3048–3065. https://doi.org/10.1093/hmg/ddp243
Chaturvedi RK, Beal MF (2008) Mitochondrial approaches for neuroprotection. Ann N Y Acad Sci 1147:395–412. https://doi.org/10.1196/annals.1427.027
Chaturvedi RK, Beal MF (2008) PPAR: a therapeutic target in Parkinson's disease. J Neurochem 106(2):506–518. https://doi.org/10.1111/j.1471-4159.2008.05388.x
Li PA, Hou X, Hao S (2017) Mitochondrial biogenesis in neurodegeneration. J Neurosci Res 95(10):2025–2029. https://doi.org/10.1002/jnr.24042
Schmidt O, Pfanner N, Meisinger C (2010) Mitochondrial protein import: From proteomics to functional mechanisms. Nat Rev Mol Cell Biol 11(9):655–667. https://doi.org/10.1038/nrm2959
Grevel A, Pfanner N, Becker T (2019) Coupling of import and assembly pathways in mitochondrial protein biogenesis. Biol Chem 401(1):117–129. https://doi.org/10.1515/hsz-2019-0310
Kausar S, Wang F, Cui H (2018) The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells 7(12):274. https://doi.org/10.3390/cells7120274
Stanga S, Caretto A, Boido M, Vercelli A (2020) Mitochondrial dysfunctions: a red thread across neurodegenerative diseases. Int J Mol Sci 21(10):3719. https://doi.org/10.3390/ijms21103719
Monzio Compagnoni G, Di Fonzo A, Corti S, Comi GP, Bresolin N, Masliah E (2020) The role of mitochondria in neurodegenerative diseases: the lesson from Alzheimer's disease and Parkinson's disease. Mol Neurobiol 57(7):2959–2980. https://doi.org/10.1007/s12035-020-01926-1
Vazquez-Calvo C, Suhm T, Buttner S, Ott M (2020) The basic machineries for mitochondrial protein quality control. Mitochondrion 50:121–131. https://doi.org/10.1016/j.mito.2019.10.003
Lu B, Guo S (2020) Mechanisms linking mitochondrial dysfunction and Proteostasis failure. Trends Cell Biol 30(4):317–328. https://doi.org/10.1016/j.tcb.2020.01.008
Shiota T, Traven A, Lithgow T (2015) Mitochondrial biogenesis: cell-cycle-dependent investment in making mitochondria. Curr Biol 25(2):R78–R80. https://doi.org/10.1016/j.cub.2014.12.006
Carlton JG, Jones H, Eggert US (2020) Membrane and organelle dynamics during cell division. Nat Rev Mol Cell Biol 21(3):151–166. https://doi.org/10.1038/s41580-019-0208-1
Mishra P, Chan DC (2014) Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol 15(10):634–646. https://doi.org/10.1038/nrm3877
Pradelli LA, Beneteau M, Ricci JE (2010) Mitochondrial control of caspase-dependent and -independent cell death. Cell Mol Life Sci 67(10):1589–1597. https://doi.org/10.1007/s00018-010-0285-y
Rebelo-Guiomar P, Powell CA, Van Haute L, Minczuk M (2019) The mammalian mitochondrial epitranscriptome. Biochim Biophys Acta Gene Regul Mech 1862(3):429–446. https://doi.org/10.1016/j.bbagrm.2018.11.005
Valera-Alberni M, Canto C (2018) Mitochondrial stress management: a dynamic journey. Cell Stress 2(10):253–274. https://doi.org/10.15698/cst2018.10.158
Cenini G, Voos W (2019) Mitochondria as potential targets in Alzheimer disease therapy: an update. Front Pharmacol 10:902. https://doi.org/10.3389/fphar.2019.00902
Putti R, Sica R, Migliaccio V, Lionetti L (2015) Diet impact on mitochondrial bioenergetics and dynamics. Front Physiol 6:109. https://doi.org/10.3389/fphys.2015.00109
Agarwal S, Yadav A, Tiwari SK, Seth B, Chauhan LK, Khare P, Ray RS, Chaturvedi RK (2016) Dynamin-related protein 1 inhibition mitigates bisphenol A-mediated alterations in mitochondrial dynamics and neural stem cell proliferation and differentiation. J Biol Chem 291(31):15923–15939. https://doi.org/10.1074/jbc.M115.709493
Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337(6098):1062–1065. https://doi.org/10.1126/science.1219855
Horten P, Colina-Tenorio L, Rampelt H (2020) Biogenesis of mitochondrial metabolite carriers. Biomolecules 10(7):1008. https://doi.org/10.3390/biom10071008
Diaz F, Moraes CT (2008) Mitochondrial biogenesis and turnover. Cell Calcium 44(1):24–35. https://doi.org/10.1016/j.ceca.2007.12.004
Seager R, Lee L, Henley JM, Wilkinson KA (2020) Mechanisms and roles of mitochondrial localisation and dynamics in neuronal function. Neuronal Signal 4(2):NS20200008. https://doi.org/10.1042/NS20200008
Topf U, Uszczynska-Ratajczak B, Chacinska A (2019) Mitochondrial stress-dependent regulation of cellular protein synthesis. J Cell Sci 132(8):jcs226258. https://doi.org/10.1242/jcs.226258
Melber A, Haynes CM (2018) UPR (mt) regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res 28(3):281–295. https://doi.org/10.1038/cr.2018.16
Jiang YF, Lin HL, Wang LJ, Hsu T, Fu CY (2020) Coordinated organization of mitochondrial lamellar cristae and gain of COX function during mitochondrial maturation in Drosophila. Mol Biol Cell 31(1):18–26. https://doi.org/10.1091/mbc.E19-08-0450
Stojanovski D, Bohnert M, Pfanner N, van der Laan M (2012) Mechanisms of protein sorting in mitochondria. Cold Spring Harb Perspect Biol 4(10):a011320. https://doi.org/10.1101/cshperspect.a011320
Ferramosca A, Zara V (2013) Biogenesis of mitochondrial carrier proteins: molecular mechanisms of import into mitochondria. Biochim Biophys Acta 1833(3):494–502. https://doi.org/10.1016/j.bbamcr.2012.11.014
Herrmann JM, Riemer J (2012) Mitochondrial disulfide relay: redox-regulated protein import into the intermembrane space. J Biol Chem 287(7):4426–4433. https://doi.org/10.1074/jbc.R111.270678
Straub SP, Stiller SB, Wiedemann N, Pfanner N (2016) Dynamic organization of the mitochondrial protein import machinery. Biol Chem 397(11):1097–1114. https://doi.org/10.1515/hsz-2016-0145
Todd LR, Damin MN, Gomathinayagam R, Horn SR, Means AR, Sankar U (2010) Growth factor erv1-like modulates Drp1 to preserve mitochondrial dynamics and function in mouse embryonic stem cells. Mol Biol Cell 21(7):1225–1236. https://doi.org/10.1091/mbc.E09-11-0937
Stojanovski D, Muller JM, Milenkovic D, Guiard B, Pfanner N, Chacinska A (2008) The MIA system for protein import into the mitochondrial intermembrane space. Biochim Biophys Acta 1783(4):610–617. https://doi.org/10.1016/j.bbamcr.2007.10.004
Napoli E, Wong S, Hung C, Ross-Inta C, Bomdica P, Giulivi C (2013) Defective mitochondrial disulfide relay system, altered mitochondrial morphology and function in Huntington's disease. Hum Mol Genet 22(5):989–1004. https://doi.org/10.1093/hmg/dds503
Becker T, Pfannschmidt S, Guiard B, Stojanovski D, Milenkovic D, Kutik S, Pfanner N, Meisinger C et al (2008) Biogenesis of the mitochondrial TOM complex: Mim1 promotes insertion and assembly of signal-anchored receptors. J Biol Chem 283(1):120–127. https://doi.org/10.1074/jbc.M706997200
Doan KN, Grevel A, Martensson CU, Ellenrieder L, Thornton N, Wenz LS, Opalinski L, Guiard B et al (2020) The mitochondrial import complex MIM functions as main translocase for alpha-helical outer membrane proteins. Cell Rep 31(4):107567. https://doi.org/10.1016/j.celrep.2020.107567
Lionello S, Marzaro G, Martinvalet D (2020) SAM50, a side door to the mitochondria: the case of cytotoxic proteases. Pharmacol Res. https://doi.org/10.1016/j.phrs.2020.105196
Qiu J, Wenz LS, Zerbes RM, Oeljeklaus S, Bohnert M, Stroud DA, Wirth C, Ellenrieder L et al (2013) Coupling of mitochondrial import and export translocases by receptor-mediated supercomplex formation. Cell 154(3):596–608. https://doi.org/10.1016/j.cell.2013.06.033
Chacinska A, van der Laan M, Mehnert CS, Guiard B, Mick DU, Hutu DP, Truscott KN, Wiedemann N et al (2010) Distinct forms of mitochondrial TOM-TIM supercomplexes define signal-dependent states of preprotein sorting. Mol Cell Biol 30(1):307–318. https://doi.org/10.1128/MCB.00749-09
Alcazar-Fabra M, Navas P, Brea-Calvo G (2016) Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim Biophys Acta 1857(8):1073–1078. https://doi.org/10.1016/j.bbabio.2016.03.010
Wenz LS, Opalinski L, Schuler MH, Ellenrieder L, Ieva R, Bottinger L, Qiu J, van der Laan M et al (2014) The presequence pathway is involved in protein sorting to the mitochondrial outer membrane. EMBO Rep 15(6):678–685. https://doi.org/10.1002/embr.201338144
Song J, Tamura Y, Yoshihisa T, Endo T (2014) A novel import route for an N-anchor mitochondrial outer membrane protein aided by the TIM23 complex. EMBO Rep 15(6):670–677. https://doi.org/10.1002/embr.201338142
Wollweber F, von der Malsburg K, van der Laan M (2017) Mitochondrial contact site and cristae organizing system: a central player in membrane shaping and crosstalk. Biochim Biophys Acta Mol Cell Res 1864(9):1481–1489. https://doi.org/10.1016/j.bbamcr.2017.05.004
Schmidt O, Harbauer AB, Rao S, Eyrich B, Zahedi RP, Stojanovski D, Schonfisch B, Guiard B et al (2011) Regulation of mitochondrial protein import by cytosolic kinases. Cell 144(2):227–239. https://doi.org/10.1016/j.cell.2010.12.015
Lucero M, Suarez AE, Chambers JW (2019) Phosphoregulation on mitochondria: Integration of cell and organelle responses. CNS Neurosci Ther 25(7):837–858. https://doi.org/10.1111/cns.13141
Martensson CU, Priesnitz C, Song J, Ellenrieder L, Doan KN, Boos F, Floerchinger A, Zufall N et al (2019) Mitochondrial protein translocation-associated degradation. Nature 569(7758):679–683. https://doi.org/10.1038/s41586-019-1227-y
Eldeeb MA, Ragheb MA, Esmaili M (2020) How does protein degradation regulate TOM machinery-dependent mitochondrial import? Curr Genet 66(3):501–505. https://doi.org/10.1007/s00294-020-01056-0
Eldeeb MA, MacDougall EJ, Ragheb MA, Fon EA (2019) Beyond ER: regulating TOM-complex-mediated import by Ubx2. Trends Cell Biol 29(9):687–689. https://doi.org/10.1016/j.tcb.2019.07.003
Ng S, De Clercq I, Van Aken O, Law SR, Ivanova A, Willems P, Giraud E, Van Breusegem F et al (2014) Anterograde and retrograde regulation of nuclear genes encoding mitochondrial proteins during growth, development, and stress. Mol Plant 7(7):1075–1093. https://doi.org/10.1093/mp/ssu037
Lionaki E, Gkikas I, Tavernarakis N (2016) Differential protein distribution between the nucleus and mitochondria: implications in aging. Front Genet 7:162. https://doi.org/10.3389/fgene.2016.00162
Ryan MT, Hoogenraad NJ (2007) Mitochondrial-nuclear communications. Annu Rev Biochem 76:701–722. https://doi.org/10.1146/annurev.biochem.76.052305.091720
Yang D, Kim J (2019) Mitochondrial retrograde signalling and metabolic alterations in the tumour microenvironment. Cells 8(3):275. https://doi.org/10.3390/cells8030275
Kasai S, Yamazaki H, Tanji K, Engler MJ, Matsumiya T, Itoh K (2019) Role of the ISR-ATF4 pathway and its cross talk with Nrf2 in mitochondrial quality control. J Clin Biochem Nutr 64(1):1–12. https://doi.org/10.3164/jcbn.18-37
Rius-Perez S, Torres-Cuevas I, Millan I, Ortega AL, Perez S (2020) PGC-1alpha, inflammation, and oxidative stress: an integrative view in metabolism. Oxidative Med Cell Longev 2020:1452696. https://doi.org/10.1155/2020/1452696
Munch C (2018) The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol 16(1):81. https://doi.org/10.1186/s12915-018-0548-x
Ge P, Dawson VL, Dawson TM (2020) PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson's disease. Mol Neurodegener 15(1):20. https://doi.org/10.1186/s13024-020-00367-7
Baker MJ, Palmer CS, Stojanovski D (2014) Mitochondrial protein quality control in health and disease. Br J Pharmacol 171(8):1870–1889. https://doi.org/10.1111/bph.12430
Aerts L, Craessaerts K, De Strooper B, Morais VA (2015) PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J Biol Chem 290(5):2798–2811. https://doi.org/10.1074/jbc.M114.620906
Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S et al (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189(2):211–221. https://doi.org/10.1083/jcb.200910140
Bertolin G, Ferrando-Miguel R, Jacoupy M, Traver S, Grenier K, Greene AW, Dauphin A, Waharte F et al (2013) The TOMM machinery is a molecular switch in PINK1 and PARK2/PARKIN-dependent mitochondrial clearance. Autophagy 9(11):1801–1817. https://doi.org/10.4161/auto.25884
Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496(7445):372–376. https://doi.org/10.1038/nature12043
Butterfield DA, Palmieri EM, Castegna A (2016) Clinical implications from proteomic studies in neurodegenerative diseases: lessons from mitochondrial proteins. Expert Rev Proteomics 13(3):259–274. https://doi.org/10.1586/14789450.2016.1149470
Burchell VS, Gandhi S, Deas E, Wood NW, Abramov AY, Plun-Favreau H (2010) Targeting mitochondrial dysfunction in neurodegenerative disease: Part II. Expert Opin Ther Targets 14(5):497–511. https://doi.org/10.1517/14728221003730434
Mucke L (2009) Neuroscience: Alzheimer's disease. Nature 461(7266):895–897. https://doi.org/10.1038/461895a
Pinho CM, Teixeira PF, Glaser E (2014) Mitochondrial import and degradation of amyloid-beta peptide. Biochim Biophys Acta 1837(7):1069–1074. https://doi.org/10.1016/j.bbabio.2014.02.007
Winner B, Winkler J (2015) Adult neurogenesis in neurodegenerative diseases. Cold Spring Harb Perspect Biol 7(4):a021287. https://doi.org/10.1101/cshperspect.a021287
Tiwari SK, Seth B, Agarwal S, Yadav A, Karmakar M, Gupta SK, Choubey V, Sharma A et al (2015) Ethosuximide induces hippocampal neurogenesis and reverses cognitive deficits in an amyloid-beta toxin-induced Alzheimer rat model via the phosphatidylinositol 3-kinase (PI3K)/Akt/Wnt/beta-catenin pathway. J Biol Chem 290(47):28540–28558. https://doi.org/10.1074/jbc.M115.652586
Tandon A, Singh SJ, Gupta M, Singh N, Shankar J, Arjaria N, Goyal S, Chaturvedi RK (2020) Notch pathway up-regulation via curcumin mitigates bisphenol-A (BPA) induced alterations in hippocampal oligodendrogenesis. J Hazard Mater 392:122052. https://doi.org/10.1016/j.jhazmat.2020.122052
Moreno-Jimenez EP, Flor-Garcia M, Terreros-Roncal J, Rabano A, Cafini F, Pallas-Bazarra N, Avila J, Llorens-Martin M (2019) Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer's disease. Nat Med 25(4):554–560. https://doi.org/10.1038/s41591-019-0375-9
Crews L, Adame A, Patrick C, Delaney A, Pham E, Rockenstein E, Hansen L, Masliah E (2010) Increased BMP6 levels in the brains of Alzheimer's disease patients and APP transgenic mice are accompanied by impaired neurogenesis. J Neurosci 30(37):12252–12262. https://doi.org/10.1523/JNEUROSCI.1305-10.2010
Kempermann G, Gage FH, Aigner L, Song H, Curtis MA, Thuret S, Kuhn HG, Jessberger S et al (2018) Human adult neurogenesis: evidence and remaining questions. Cell Stem Cell 23(1):25–30. https://doi.org/10.1016/j.stem.2018.04.004
Zhao C, Deng W, Gage FH (2008) Mechanisms and functional implications of adult neurogenesis. Cell 132(4):645–660. https://doi.org/10.1016/j.cell.2008.01.033
Castellani RJ, Rolston RK, Smith MA (2010) Alzheimer disease. Dis Mon 56(9):484–546. https://doi.org/10.1016/j.disamonth.2010.06.001
Bartolome F, de la Cueva M, Pascual C, Antequera D, Fernandez T, Gil C, Martinez A, Carro E (2018) Amyloid beta-induced impairments on mitochondrial dynamics, hippocampal neurogenesis, and memory are restored by phosphodiesterase 7 inhibition. Alzheimers Res Ther 10(1):24. https://doi.org/10.1186/s13195-018-0352-4
Yoo SM, Park J, Kim SH, Jung YK (2020) Emerging perspectives on mitochondrial dysfunction and inflammation in Alzheimer's disease. BMB Rep 53(1):35–46. https://doi.org/10.5483/BMBRep.2020.53.1.274
Weidling IW, Swerdlow RH (2020) Mitochondria in Alzheimer's disease and their potential role in Alzheimer's proteostasis. Exp Neurol 330:113321. https://doi.org/10.1016/j.expneurol.2020.113321
Lim JW, Lee J, Pae AN (2020) Mitochondrial dysfunction and Alzheimer's disease: prospects for therapeutic intervention. BMB Rep 53(1):47–55. https://doi.org/10.5483/BMBRep.2020.53.1.279
Penke B, Toth AM, Foldi I, Szucs M, Janaky T (2012) Intraneuronal beta-amyloid and its interactions with proteins and subcellular organelles. Electrophoresis 33(24):3608–3616. https://doi.org/10.1002/elps.201200297
Pavlov PF, Wiehager B, Sakai J, Frykman S, Behbahani H, Winblad B, Ankarcrona M (2011) Mitochondrial gamma-secretase participates in the metabolism of mitochondria-associated amyloid precursor protein. FASEB J 25(1):78–88. https://doi.org/10.1096/fj.10-157230
Lopez Sanchez MIG, van Wijngaarden P, Trounce IA (2019) Amyloid precursor protein-mediated mitochondrial regulation and Alzheimer's disease. Br J Pharmacol 176(18):3464–3474. https://doi.org/10.1111/bph.14554
Mossmann D, Vogtle FN, Taskin AA, Teixeira PF, Ring J, Burkhart JM, Burger N, Pinho CM et al (2014) Amyloid-beta peptide induces mitochondrial dysfunction by inhibition of preprotein maturation. Cell Metab 20(4):662–669. https://doi.org/10.1016/j.cmet.2014.07.024
Backes S, Hess S, Boos F, Woellhaf MW, Godel S, Jung M, Muhlhaus T, Herrmann JM (2018) Tom70 enhances mitochondrial preprotein import efficiency by binding to internal targeting sequences. J Cell Biol 217(4):1369–1382. https://doi.org/10.1083/jcb.201708044
Pagani L, Eckert A (2011) Amyloid-Beta interaction with mitochondria. Int J Alzheimers Dis 2011:925050. https://doi.org/10.4061/2011/925050
Li Y, Lu J, Cao X, Zhao H, Gao L, Xia P, Pei G (2020) A newly synthesized rhamnoside derivative alleviates Alzheimer's amyloid-beta-induced oxidative stress, mitochondrial dysfunction, and cell senescence through upregulating SIRT3. Oxidative Med Cell Longev 2020:7698560. https://doi.org/10.1155/2020/7698560
Picone P, Nuzzo D, Caruana L, Scafidi V, Di Carlo M (2014) Mitochondrial dysfunction: different routes to Alzheimer's disease therapy. Oxidative Med Cell Longev 2014:780179. https://doi.org/10.1155/2014/780179
Cenini G, Rub C, Bruderek M, Voos W (2016) Amyloid beta-peptides interfere with mitochondrial preprotein import competence by a coaggregation process. Mol Biol Cell 27(21):3257–3272. https://doi.org/10.1091/mbc.E16-05-0313
Devi L, Anandatheerthavarada HK (2010) Mitochondrial trafficking of APP and alpha synuclein: Relevance to mitochondrial dysfunction in Alzheimer's and Parkinson's diseases. Biochim Biophys Acta 1802(1):11–19. https://doi.org/10.1016/j.bbadis.2009.07.007
Takei N, Miyashita A, Tsukie T, Arai H, Asada T, Imagawa M, Shoji M, Higuchi S et al (2009) Genetic association study on in and around the APOE in late-onset Alzheimer disease in Japanese. Genomics 93(5):441–448. https://doi.org/10.1016/j.ygeno.2009.01.003
Roses A, Sundseth S, Saunders A, Gottschalk W, Burns D, Lutz M (2016) Understanding the genetics of APOE and TOMM40 and role of mitochondrial structure and function in clinical pharmacology of Alzheimer's disease. Alzheimers Dement 12(6):687–694. https://doi.org/10.1016/j.jalz.2016.03.015
Peng Y, Gao P, Shi L, Chen L, Liu J, Long J (2020) Central and peripheral metabolic defects contribute to the pathogenesis of Alzheimer's disease: targeting mitochondria for diagnosis and prevention. Antioxid Redox Signal 32(16):1188–1236. https://doi.org/10.1089/ars.2019.7763
Hu W, Wang Z, Zheng H (2018) Mitochondrial accumulation of amyloid beta (Abeta) peptides requires TOMM22 as a main Abeta receptor in yeast. J Biol Chem 293(33):12681–12689. https://doi.org/10.1074/jbc.RA118.002713
Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, Ito A et al (2008) The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A 105(35):13145–13150. https://doi.org/10.1073/pnas.0806192105
Sato TK, Kawano S, Endo T (2019) Role of the membrane potential in mitochondrial protein unfolding and import. Sci Rep 9(1):7637. https://doi.org/10.1038/s41598-019-44152-z
Sirk D, Zhu Z, Wadia JS, Shulyakova N, Phan N, Fong J, Mills LR (2007) Chronic exposure to sub-lethal beta-amyloid (Abeta) inhibits the import of nuclear-encoded proteins to mitochondria in differentiated PC12 cells. J Neurochem 103(5):1989–2003. https://doi.org/10.1111/j.1471-4159.2007.04907.x
Dores-Silva PR, Barbosa LR, Ramos CH, Borges JC (2015) Human mitochondrial Hsp70 (mortalin): shedding light on ATPase activity, interaction with adenosine nucleotides, solution structure and domain organization. PLoS One 10(1):e0117170. https://doi.org/10.1371/journal.pone.0117170
Zeitlow K, Charlambous L, Ng I, Gagrani S, Mihovilovic M, Luo S, Rock DL, Saunders A et al (2017) The biological foundation of the genetic association of TOMM40 with late-onset Alzheimer's disease. Biochim Biophys Acta Mol basis Dis 1863(11):2973–2986. https://doi.org/10.1016/j.bbadis.2017.07.031
Chai YL, Xing H, Chong JR, Francis PT, Ballard CG, Chen CP, Lai MKP (2018) Mitochondrial translocase of the outer membrane alterations may underlie dysfunctional oxidative phosphorylation in Alzheimer's disease. J Alzheimers Dis 61(2):793–801. https://doi.org/10.3233/JAD-170613
Liu W, Duan X, Fang X, Shang W, Tong C (2018) Mitochondrial protein import regulates cytosolic protein homeostasis and neuronal integrity. Autophagy 14(8):1293–1309. https://doi.org/10.1080/15548627.2018.1474991
Wang X, Chen XJ (2015) A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature 524(7566):481–484. https://doi.org/10.1038/nature14859
Wrobel L, Topf U, Bragoszewski P, Wiese S, Sztolsztener ME, Oeljeklaus S, Varabyova A, Lirski M et al (2015) Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 524(7566):485–488. https://doi.org/10.1038/nature14951
Singh S, Mishra A, Mohanbhai SJ, Tiwari V, Chaturvedi RK, Khurana S, Shukla S (2018) Axin-2 knockdown promote mitochondrial biogenesis and dopaminergic neurogenesis by regulating Wnt/beta-catenin signaling in rat model of Parkinson's disease. Free Radic Biol Med 129:73–87. https://doi.org/10.1016/j.freeradbiomed.2018.08.033
Pahuja R, Seth K, Shukla A, Shukla RK, Bhatnagar P, Chauhan LK, Saxena PN, Arun J et al (2015) Trans-blood brain barrier delivery of dopamine-loaded nanoparticles reverses functional deficits in parkinsonian rats. ACS Nano 9(5):4850–4871. https://doi.org/10.1021/nn506408v
Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, Zharkovsky A, Kaasik A (2011) Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 286(12):10814–10824. https://doi.org/10.1074/jbc.M110.132514
Emamzadeh FN (2016) Alpha-synuclein structure, functions, and interactions. J Res Med Sci 21:29. https://doi.org/10.4103/1735-1995.181989
Vicario M, Cieri D, Brini M, Cali T (2018) The close encounter between alpha-synuclein and mitochondria. Front Neurosci 12:388. https://doi.org/10.3389/fnins.2018.00388
Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK (2008) Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem 283(14):9089–9100. https://doi.org/10.1074/jbc.M710012200
Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, Hu X, McCoy J et al (2016) Alpha-synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson's disease. Sci Transl Med 8(342):342ra378. https://doi.org/10.1126/scitranslmed.aaf3634
Rahmani F, Kamalian A, Aarabi MH (2017) New evidence comes to light: How is alpha-synuclein aggregation related to mitochondrial protein import in Parkinson's disease? Mov Disord 32(1):107. https://doi.org/10.1002/mds.26889
Koo JH, Cho JY, Lee UB (2017) Treadmill exercise alleviates motor deficits and improves mitochondrial import machinery in an MPTP-induced mouse model of Parkinson's disease. Exp Gerontol 89:20–29. https://doi.org/10.1016/j.exger.2017.01.001
Joseph AM, Ljubicic V, Adhihetty PJ, Hood DA (2010) Biogenesis of the mitochondrial Tom40 channel in skeletal muscle from aged animals and its adaptability to chronic contractile activity. Am J Phys Cell Phys 298(6):C1308–C1314. https://doi.org/10.1152/ajpcell.00644.2008
Joseph AM, Hood DA (2012) Plasticity of TOM complex assembly in skeletal muscle mitochondria in response to chronic contractile activity. Mitochondrion 12(2):305–312. https://doi.org/10.1016/j.mito.2011.11.005
Jacoupy M, Hamon-Keromen E, Ordureau A, Erpapazoglou Z, Coge F, Corvol JC, Nosjean O, Mannoury la Cour C et al (2019) The PINK1 kinase-driven ubiquitin ligase Parkin promotes mitochondrial protein import through the presequence pathway in living cells. Sci Rep 9(1):11829. https://doi.org/10.1038/s41598-019-47352-9
Sekine S, Youle RJ (2018) PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol 16(1):2. https://doi.org/10.1186/s12915-017-0470-7
Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85(2):257–273. https://doi.org/10.1016/j.neuron.2014.12.007
Nardin A, Schrepfer E, Ziviani E (2016) Counteracting PINK/Parkin deficiency in the activation of mitophagy: a potential therapeutic intervention for Parkinson's disease. Curr Neuropharmacol 14(3):250–259. https://doi.org/10.2174/1570159x13666151030104414
Sekine S, Wang C, Sideris DP, Bunker E, Zhang Z, Youle RJ (2019) Reciprocal roles of Tom7 and OMA1 during mitochondrial import and activation of PINK1. Mol Cell 73(5):1028–1043 e1025. https://doi.org/10.1016/j.molcel.2019.01.002
Neethling A, Engelbrecht L, Loos B, Kinnear C, Theart R, Abrahams S, Niesler T, Mellick GD et al (2019) Wild-type and mutant (G2019S) leucine-rich repeat kinase 2 (LRRK2) associate with subunits of the translocase of outer mitochondrial membrane (TOM) complex. Exp Cell Res 375(2):72–79. https://doi.org/10.1016/j.yexcr.2018.12.022
Kazlauskaite A, Muqit MM (2015) PINK1 and Parkin - mitochondrial interplay between phosphorylation and ubiquitylation in Parkinson's disease. FEBS J 282(2):215–223. https://doi.org/10.1111/febs.13127
Ando M, Fiesel FC, Hudec R, Caulfield TR, Ogaki K, Gorka-Skoczylas P, Koziorowski D, Friedman A et al (2017) The PINK1 p.I368N mutation affects protein stability and ubiquitin kinase activity. Mol Neurodegener 12(1):32. https://doi.org/10.1186/s13024-017-0174-z
Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ et al (2011) PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 20(5):867–879. https://doi.org/10.1093/hmg/ddq526
Pellegrino MW, Haynes CM (2015) Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol 13:22. https://doi.org/10.1186/s12915-015-0129-1
Martinez BA, Petersen DA, Gaeta AL, Stanley SP, Caldwell GA, Caldwell KA (2017) Dysregulation of the mitochondrial unfolded protein response induces non-apoptotic dopaminergic neurodegeneration in C. elegans models of Parkinson's disease. J Neurosci 37(46):11085–11100. https://doi.org/10.1523/JNEUROSCI.1294-17.2017
Kojima W, Kujuro Y, Okatsu K, Bruno Q, Koyano F, Kimura M, Yamano K, Tanaka K et al (2016) Unexpected mitochondrial matrix localization of Parkinson's disease-related DJ-1 mutants but not wild-type DJ-1. Genes Cells 21(7):772–788. https://doi.org/10.1111/gtc.12382
Yaguchi T, Aida S, Kaul SC, Wadhwa R (2007) Involvement of mortalin in cellular senescence from the perspective of its mitochondrial import, chaperone, and oxidative stress management functions. Ann N Y Acad Sci 1100:306–311. https://doi.org/10.1196/annals.1395.032
Banerjee R, Gladkova C, Mapa K, Witte G, Mokranjac D (2015) Protein translocation channel of mitochondrial inner membrane and matrix-exposed import motor communicate via two-domain coupling protein. Elife 4:e11897. https://doi.org/10.7554/eLife.11897
Callegari S, Cruz-Zaragoza LD, Rehling P (2020) From TOM to the TIM23 complex - handing over of a precursor. Biol Chem 401(6–7):709–721. https://doi.org/10.1515/hsz-2020-0101
Geissler A, Rassow J, Pfanner N, Voos W (2001) Mitochondrial import driving forces: enhanced trapping by matrix Hsp70 stimulates translocation and reduces the membrane potential dependence of loosely folded preproteins. Mol Cell Biol 21(20):7097–7104. https://doi.org/10.1128/MCB.21.20.7097-7104.2001
Burbulla LF, Schelling C, Kato H, Rapaport D, Woitalla D, Schiesling C, Schulte C, Sharma M et al (2010) Dissecting the role of the mitochondrial chaperone mortalin in Parkinson's disease: functional impact of disease-related variants on mitochondrial homeostasis. Hum Mol Genet 19(22):4437–4452. https://doi.org/10.1093/hmg/ddq370
Larsen SB, Hanss Z, Kruger R (2018) The genetic architecture of mitochondrial dysfunction in Parkinson's disease. Cell Tissue Res 373(1):21–37. https://doi.org/10.1007/s00441-017-2768-8
Kaushik S, Cuervo AM (2018) The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 19(6):365–381. https://doi.org/10.1038/s41580-018-0001-6
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS et al (2013) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16(4):394–406. https://doi.org/10.1038/nn.3350
Singh A, Zhi L, Zhang H (2019) LRRK2 and mitochondria: recent advances and current views. Brain Res 1702:96–104. https://doi.org/10.1016/j.brainres.2018.06.010
Sironi L, Restelli LM, Tolnay M, Neutzner A, Frank S (2020) Dysregulated interorganellar crosstalk of mitochondria in the pathogenesis of Parkinson's disease. Cells 9(1):233. https://doi.org/10.3390/cells9010233
Cui J, Yu M, Niu J, Yue Z, Xu Z (2011) Expression of leucine-rich repeat kinase 2 (LRRK2) inhibits the processing of uMtCK to induce cell death in a cell culture model system. Biosci Rep 31(5):429–437. https://doi.org/10.1042/BSR20100127
Shen Z, Ye C, McCain K, Greenberg ML (2015) The role of cardiolipin in cardiovascular health. Biomed Res Int 2015:891707. https://doi.org/10.1155/2015/891707
Fox TD (2012) Mitochondrial protein synthesis, import, and assembly. Genetics 192(4):1203–1234. https://doi.org/10.1534/genetics.112.141267
Paradies G, Paradies V, Ruggiero FM, Petrosillo G (2019) Role of cardiolipin in mitochondrial function and dynamics in health and disease: molecular and pharmacological aspects. Cells 8(7):728. https://doi.org/10.3390/cells8070728
Lu YW, Claypool SM (2015) Disorders of phospholipid metabolism: an emerging class of mitochondrial disease due to defects in nuclear genes. Front Genet 6:3. https://doi.org/10.3389/fgene.2015.00003
Malhotra K, Modak A, Nangia S, Daman TH, Gunsel U, Robinson VL, Mokranjac D, May ER et al (2017) Cardiolipin mediates membrane and channel interactions of the mitochondrial TIM23 protein import complex receptor Tim50. Sci Adv 3(9):e1700532. https://doi.org/10.1126/sciadv.1700532
Chu CT, Bayir H, Kagan VE (2014) LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: implications for Parkinson disease. Autophagy 10(2):376–378. https://doi.org/10.4161/auto.27191
Kowalska M, Piekut T, Prendecki M, Sodel A, Kozubski W, Dorszewska J (2020) Mitochondrial and nuclear DNA oxidative damage in physiological and pathological aging. DNA Cell Biol 39(8):1410–1420. https://doi.org/10.1089/dna.2019.5347
Cai Q, Tammineni P (2017) Mitochondrial aspects of synaptic dysfunction in Alzheimer's disease. J Alzheimers Dis 57(4):1087–1103. https://doi.org/10.3233/JAD-160726
Popov LD (2020) Mitochondrial biogenesis: an update. J Cell Mol Med 24(9):4892–4899. https://doi.org/10.1111/jcmm.15194
Manganas P, MacPherson L, Tokatlidis K (2017) Oxidative protein biogenesis and redox regulation in the mitochondrial intermembrane space. Cell Tissue Res 367(1):43–57. https://doi.org/10.1007/s00441-016-2488-5
MacKenzie JA, Payne RM (2007) Mitochondrial protein import and human health and disease. Biochim Biophys Acta 1772(5):509–523. https://doi.org/10.1016/j.bbadis.2006.12.002
Akbar M, Essa MM, Daradkeh G, Abdelmegeed MA, Choi Y, Mahmood L, Song BJ (2016) Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain Res 1637:34–55. https://doi.org/10.1016/j.brainres.2016.02.016
Martin LJ (2012) Biology of mitochondria in neurodegenerative diseases. Prog Mol Biol Transl Sci 107:355–415. https://doi.org/10.1016/B978-0-12-385883-2.00005-9
Grunewald A, Kumar KR, Sue CM (2019) New insights into the complex role of mitochondria in Parkinson's disease. Prog Neurobiol 177:73–93. https://doi.org/10.1016/j.pneurobio.2018.09.003
Acknowledgments
The authors are thankful to the Director. CSIR-IITR manuscript communication number-3658.
Funding
This work was supported by the Lady Tata Memorial-UK Young Scientist Grant, Science and Engineering Research Board (SERB), New Delhi, EMR Grant (EMR/2016/001933), Department of Biotechnology, New Delhi Research Grant (BT/PR15819/MED/31/322/2015), Department of Biotechnology, National Bioscience Award Research grant (BT/HRD/NBA/38/07/2018), and CSIR Network Projects (BSC-0115 and BSC-0111) to RKC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Goyal, S., Chaturvedi, R.K. Mitochondrial Protein Import Dysfunction in Pathogenesis of Neurodegenerative Diseases. Mol Neurobiol 58, 1418–1437 (2021). https://doi.org/10.1007/s12035-020-02200-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02200-0