
Abstract
Two acid mine drainage (AMD) samples were studied by a PCR-based cloning approach, which were from Yunfu sulfide mine in Guangdong province, China. A total of 15 operational taxonomic units (OTUs) were obtained from the two AMD samples. The percentage of overlapped OTUs in two AMD samples was 42.1%. Phylogenetic analysis revealed that the bacterium in the two samples fell into four putative divisions, which were Nitrospira, α-Proteobacteria, β-Proteobacteria, and γ-Proteobacteria four families. Organisms of genuses Acidithiobacillus and Gallionella, which were in γ-Proteobacteria family and β-Proteobacteria family, respectively, were dominant in two samples. The proportions of clones affiliated with Gallionella in each sample were 47.2% (G2) and 16.9% (G1). The result suggested that organisms of Gallionella were a very important composition in microbial communities of the two AMD samples we studied. In addition, the PCR amplification of archaeal 16S rDNA genes form these two AMD samples have been performed with two sets of archaea-specific primers, but no PCR product found.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Dissolution of sulfide ores exposed to oxygen, water, and microorganisms results in acid production and environmentally detrimental acid mine drainage (AMD) (Nordstrom and Alpers 1999). Microorganisms that are able to develop under extreme conditions, especially iron-oxidizing chemolithotrophics are responsible for the solubilization of metals from sulfide minerals in acidic environments via direct action of their enzymes or indirectly through chemical action of their metabolic products (Southam and Beveridge 1992; Wulf-Durand et al. 1997; Wielinga et al. 1999).
Because of the limited types of substrates available in mining environments, the biotopes were initially expected to be extremely poor with respect to the diversity of the microbial flora. However, cultivation-based studies revealed a great diversity of the microbial community in AMD (Johnson 1998; Hallberg and Johonson 2001). The presence of various bacterial species including common prokaryotic chemolithotrophs other than Acidithiobacillus ferrooxidans, such as A. thiooxidans, A. caldus, and Leptospirillum ferrooxidans and so on, has been reported in mining environments. The presence of archaea including a group of sulfur and/or iron-oxidizers, such as Sulfolobus, Acidianus, Metallosphaera, Sulfurisphaera, and Ferroplasma has also been reported in acidic environments (Fuchs et al. 1995, 1996; Kurosawa et al. 1998; Edwards et al. 2000; Golyshina et al. 2000).
Cultivation-based analysis is not considered a suitable method for characterizing microbial communities (Ward et al. 1990; Delong 1992), and researchers have used methods based on the analysis of 16S rDNA sequences to study microbial communities of acidic environments (Edwards et al. 1999; Bond et al. 2000; Burton and Norris 2002; Simmons and Norris 2002; Baker and Banfield 2003).
To better understand the compositions and structures of microorganisms in different AMD environments, a PCR-based cloning approach was used in this work. Two AMD samples from Yunfu sulfide mine were studied. The Yunfu sulfide mine was in Guangdong province, China, which began to mine pyrite for sulfuric acid manufacture since 1988, for its high concentration of sulfur and low concentrations of other elements. The microbial communities in AMD samples from Yunfu sulfide mine have not been investigated to date yet.
Materials and methods
Sites description and samples collection
Samples were colleted from Yunfu sulfide mine, in Guangdong province, China. The mine had mainly produced pyrite for sulfuric acid manufacture since 1988, for its high concentration of sulfur and low concentrations of other elements.
There were three aqueous AMD samples collected from two separated sites in Yunfu sulfide mine. pH and temperature were same in two samples, which were 2.5 and 25.0°C, respectively. One of two samples was named as G1, from which the clones with 16S rDNA inserts were given the prefix GY. Similarly, the other sample was named as G2 and from which clones with 16S rDNA inserts were given the prefix G.
Ten-liter water sample was colleted from each position. Samples were processed within 24 h after collection. Two water samples were filtered through 0.22 μm hyper filtration membrane with vacuum pump, respectively. The sediments on the membrane were washed by sterile deionized water twice. Then the sediments were stored at −70°C for reservation. The filtered water samples were prepared for chemical analysis.
Chemical analysis of water samples
The element analysis of filtered water samples was carried out by Inductively Coupled Plasma-Atomic Emission Spectrometry (ICP-AES).
Twenty-nine elements were tested in each water sample. They were as following: Hg, As, P, Ni, Co, Cr, Be, Ti, W, Zn, In, Mg, Mn, Ca, S, Mo, Bi, Au, Fe, Si, Cu, Sn, Sb, Cd, Ga, Pt, Al, Ag.
DNA extraction and purification
Extraction of nucleic acids was according to procedure described by Zhou et al. (1996). Five gram of sediment was mixed with 13.5 ml extraction buffer (0.1 M phosphate [pH 8.0], 0.1 M EDTA, 0.1 M EDTA, 1.5 M NaCl, 1% CTAB) and 50 μl protenase K (10 mg/ml) in 50 ml centrifuge tube, then incubated at 37°C for 30 min. 1.5 ml of 20% SDS was added and mixed gently, then incubated at 65°C for 2 h. The mixture was centrifuged and the supernatant was transferred into a new 50 ml of centrifuge tube. The soil pellet was resuspended with extraction buffer, and 0.5 ml 20% SDS was added. The mixture was incubated at 65°C for 15 min, then centrifuged and the supernatant was collected and combined with the previous supernatant. The combined supernatant was extracted with chloroform. 2-Isopropanol was added to the supernatant collected and then mixed gently. The mixture was kept at the room temperature for an hour or overnight, then centrifuged. The pellet was washing with 70% ethanol and dissolved with 200–500 μl sterile water. By using combined methods that included grinding, freezing and thawing, and treatment with sodium dodecyl sulfate, various types of bacterial could been effectively lysed. The crude DNA was purified by using Wizard plus sv Minipreps DNA purification system (Promega Corporation, USA) and quantified by ethidium bromide-UV detection on an agarose gel.
PCR and fractionation of 16S rDNA genes
Bacterial 16S rDNA genes were amplified with the primer set was 1492R (5′-CGGCTACCTTGTTACGACTT-3′), and 27F (5′-AGAGTTTGATCCTGGCTC AG-3′) (Lane 1991). A gene amp (Biometra, T-Grandient, Genman) was used to incubate reactions through an initial denaturation at 94°C for 2 min, followed by 35 cycles of 94°C for 40 s, 55°C for 30 s, and 72°C for 1 min, and completed with an extension period of 10 min at 72°C. Products from the amplification reactions of expected size (about 1,500 bp) were pooled and purified before ligation later.
PCR amplification of archaeal 16S rDNA genes was carried out following the PCR reactions described as above with two different sets of archaea-specific primers, which were as follows: S-D-Arch-0025-a-S-17 (5′-CTGGT TGATCCTGCCAG-3′) (Robb et al. 1995) or S-D-Arch-0344-a-S-20 (5′-ACGGGGCGCAGCAGGCGCGA-3′) (Weisburg et al. 1991) with S-*-Univ-1517-a-A-21 (5′-ACGGCTACCTTGTTACGACTT-3′) (Raskin et al. 1994) to yield 1,500 or 1,120 bp PCR products, respectively.
Cloning, RFLP, and sequencing
The purified PCR products were ligated to the vector PGEM-T (Promega Corporation), and used to transform DH5α competent host cells. About 120 white colonies were randomly selected from each library. The transformation efficiency in our study was around 5 × 108 cfu/μg DNA. And we used an external control to track efficiency, offered by the PGEM-T vector system (Promega company). For restriction fragment length polymorphism (RFLP) and sequencing, the inserted fragments were amplified with the vector-specific T7 and SP6 primers. These unpurified PCR products were digested with two restriction endonucleases AfaI and MspI (TaKaRa Biotechnology Co, Ltd.), incubated at 37°C for 3 h. The restricted fragments were separated by gel electrophoresis in 3.0% agarose with ethidium bromide staining and observed on UV illumination. RFLP patterns were identified and grouped, and representative clones were selected for nucleotide sequencing.
Phylogenetic analysis
Phylogenetic affiliations of the partial sequences were initially estimated using the program BLAST (Basic alignment search tool) (Bond et al. 2000). Similarity of partial sequences was determined using ARB (a software environment for sequence data) (Strunk and Ludwig 1995). The initial phylogenetic trees were based on all available sequences and were constructed by using the DNA distance program Neighbor-Joining with Felsenstein Correction in ARB (Smith et al. 1994). Based on the initial phylogenetic results, appropriate subsets of 16S rDNA sequences were selected and subjected to a final phylogenetic analysis with CLUSTAL X.
Statistical methods
The rarefaction analysis was performed with SigmaPlot software. An exponential model, y = a × [1 − exp(−b × x)], was used with SigmaPlot 8.0 nonlinear regression software to fit the clone distribution data.
Nucleotide sequence accession numbers
Sequences have been submitted to GenBank with accession numbers are as follow: DQ480487 (G77), DQ480488 (GY28), DQ480486 (G74), DQ480485 (G51-19), DQ480484 (G31), DQ480483 (G24), DQ480481 (G5), DQ480482 (G71), DQ480479 (G28), DQ480480 (G44), DQ480478 (G51-12), DQ480476 (G27), DQ480477 (G66), DQ480475 (G52), DQ480474 (G8).
Results
Biogeochemical properties of two AMD samples
Although pH and temperature in two AMD samples both were 2.5 and 25.0°C, respectively, elements’ concentration of them was very different by analysis of ICP-AES. Sample G2 had higher elements’ concentration than those in sample G1, except element copper. The data of 29 elements’ concentration are shown in Table 1.
RFLP analysis of 16S rDNA clone libraries
Two sets of archaea-specific primers used to amplify archaeal 16S rDNA from two AMD samples both failed, while the bacterial-specific 16S rDNA primer set 27F and 1492R worked.
Consequently, the primer set 27F and 1492R was used to amplify bacterial 16S rDNA gene from two AMD samples in Yunfu sulfide mine. The PCR products formed a single band approximately 1,500 bp in length. After T-A cloning, 120 clones containing 16S rDNA inserts were obtained from each sample. The profiles of RFLP in two samples are shown in Figs. 1, 2.

Restriction fragment length profiles of 16S rDNA fragments amplified from sample G1 in Yunfu sulfide mine. The 16S rDNA fragments were amplified using the primer set 27F and 1492R, digested with the restriction endonucleases AfaI and MspI, and then analyzed by 3.0% ararose gel electrophoresis. The clones tested were shown in (a–e). M: 100 bp DNA ladder plus

Restriction fragment length profiles of 16S rDNA fragments amplified from sample G2 in Yunfu sulfide mine. The 16S rDNA fragments were amplified using the primer set 27F and 1492R, digested with the restriction endonucleases AfaI and MspI, and then analyzed by 3.0% ararose gel electrophoresis. The clones tested were shown in (a–g). M: 100 bp DNA ladder plus
The rarefaction analysis was used in RFLP analysis. The results are shown in Fig. 3. Nonlinear regression suggested that saturations were at 70 clones and 40 clones for samples G2 and G1, respectively. The result also suggested that the clones tested in the experiment were sufficient to detect the level of microbial communities’ diversity and infer the level of distribution within communities of two samples.

Evaluation of the representation of clones obtained from two AMD samples in Yunfu sulfide mine. G1: one AMD sample in Yunfu sulfide mine, G2: the other AMD sample in Yunfu sulfide mine
The RFLP analysis revealed extensive diversity of 16S rDNA for two AMD samples. A total of 15 OTUs (unique RFLP patterns) were obtained. There were 14 OTUs in sample G2; 5 OTUs were detected in sample G1. The distributions of OTUs, which were ranked in the order of abundance in each sample, were shown in Fig. 4.

Distributions of OTUs in clone libraries in two AMD samples. G1: one AMD sample in Yunfu sulfide mine, G2: the other AMD sample in Yunfu sulfide mine. Clones GY10 and G66: the RFLP patterns were same, with 99% similarity to Acidithiobacillus ferrooxidans strain QXS-1; clones GY8 and G28: the RFLP patterns were same, with 99% similarity to Acidithiobacillus ferrooxidans strain YTW; clones GY81 and G8: the RFLP patterns were same, with 99% similarity to uncultured bacterium clone 69-8H; clone GY21: the RFLP patterns were same, uncultured bacterium Tui3-12; GY28: with 96% similarity to Acinetobacter sp. An9; clone G52: with 99% similarity to Acidithiobacillus albertensis strain DSM 14366; clone G27: with 93% similarity to Nitrosospira sp. III7; clone G51-12: with 99% similarity to uncultured Proteobacterium clone EV221H2111601SAH95; clone G44: with 99% similarity to Leptospirillum ferrooxidans strain CF12; clone G5: with 99% similarity to uncultured bacterium clone EV818SWSAP4
The RFLP patterns of clones GY10, GY8, GY81, GY21, and GY28 represented 48.2, 33.7, 8.4, 8.4, and 1.2% of the total clone populations in AMD sample G1, respectively. In the other AMD sample G2, the RFLP patterns of clones G8, G52, G27, G66, G51-12, G28, G44, and G5 represented 31.5, 17.6, 10.2, 10.2, 10.2, 5.6, 3.7, and 3.7% of the total clone populations, respectively. Some RFLP patterns in the two AMD samples were overlapped with each other, such as OTUs GY10 and G66, OTUs GY8 and G28, and so on. The percentage of overlapped OTUs between samples G1 and G2 was 42.1%. There were only five OTUs detected in sample G1, four of which could be also detected in sample G2.
Phylogenetic analysis
To determine the phylogenetic diversity, representative OTUs that occurred more than once in libraries, as well as representatives of the unique OTUs, were fully sequenced. The results of similarity among the sequences are shown in Table 2. Sequences’ comparison showed that the clones in two AMD samples had high similarity, which was 72–98% similar.
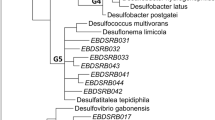
The phylogenetic analysis in two samples was established with a bootstrap neighbor-joining method with the sequences. The phylogenetic tree is shown in Fig. 5. Of note is that there were 15 OTUs with RFLP analysis, corresponding to 12 kinds of particular organisms. The accuracy of RFLP screening was only about 80% evaluated by sequencing representative clones, which may be due to the comparatively simplicity of microbial community in AMD, and this method has been commonly used for it is comparatively accurate and fast.

Phylogenetic tree based on comparative analysis of 16S rDNA sequence data from 15 OTUs and their close relatives. The sequences obtained in this study are indicated in bold
These 16S rDNA sequences fell into four putative phylogenetic divisions. They were Nitrospira, α-Proteobacteria, β-Proteobacteria, and γ-Proteobacteria four families. In sample G2, α-Proteobacteria, β-Proteobacteria, and γ-Proteobacteria, Nitrospira four families were all detected, proportions of which were 0.9, 48.1, 37.1, and 13.9%, respectively. In sample G1, there were only two families detected, which were β-Proteobacteria (16.9%) and γ-Proteobacteria (83.1%). The distributions of the four families divided by phylogenetic analysis in two AMD samples are shown in Fig. 6.

Distributions of four families divided by phylogenetic analysis in two samples. G1: one AMD sample in Yunfu sulfide mine, G2: the other AMD sample in Yunfu sulfide mine. Nit: Nitrospira family, Alp: α-Proteobacteria family, Bet: β-Proteobacteria family, Gam: γ-Proteobacteria family. a Proportions of four families in AMD sample G1, b proportions of four families in AMD sample G2
γ-Proteobacteria family was a predominant one in two AMD samples. There were two groups in the family. One group was clustered with the genus Acidithiobacillus, including OTUs G52, G28, and G66. The other group was divided into two sub-groups. One with OTUs GY28 and G24 was clustered with Acinetobacter sp. The other with OTUs G51-19 and G77 was clustered with uncultured bacterium.
β-Proteobacteria family was the other predominant family in two AMD samples. It also fell into two groups. One group including OTUs G27, G71, G5, and G8 was clustered with the genus Gallionella. The other group was clustered with Pseudomonastestosterone, just including one OUT, G31.
Nitrospira family was only detected in sample G2, in which there were two OTUs G44 and G51-12. The two OTUs were both affiliated with the genus Leptospirillum. G44 was affiliated with L. ferrooxidans, while G51-12 was affiliated with the bacterium of the group III of Leptospirillum.
The last family was α-Proteobacteria family, which was also just detected in AMD sample G2. Only one OTU G74 was detected in this family. It had 99% similarity to Hyphomicrobium vulgare strain IFAM MC-750.
Discussion
Currently, AMD environments have been set as model systems for analysis of biogeochemical interactions and microbial community structure and function (Hallberg and Lindström 1994; Baker and Banfield 2003). And any AMD system has certain microbial niches for variations in environmental data (Druschel et al. 1999; Bond et al. 2000).
In this work, culture-independent molecular methods were employed, including 16S rDNA clone library analysis and sequence determination. Microbial community structures in two AMD samples from Yunfu sulfide mine have been identified, which have not been investigated to date. According to the RFLP analysis, there were 15 OTUs detected in two samples. They fell into four putative families, which were α-Proteobacteria, β-Proteobacteria, γ-Proteobacteria, and Nitrospira four families. The organisms of genuses Acidithiobacillus and Gallionella, which were in γ-Proteobacteria and β-Proteobacteria, respectively, were dominant in two AMD samples we studied.
γ-Proteobacteria was a ubiquitous family in two AMD samples, with which clones clustered were mostly affiliated with the genus Acidithiobacillus. The proportions of clones affiliated with Acidithiobacillus in samples G1 and G2 were 81.9 and 33.4%, respectively.
Acidithiobacillus spp. has been extensively studied. Acidithiobacillus is widely considered to be the microorganism that controls the rate of generation of AMD and A.ferrooxidans has been used as model microbe in bioleaching research (Lane 1991; Boon and Heijnen 1993; Fowler et al. 1999; Kelly and Wood 2000; Baker and Banfield 2003). Scherenk et al. (1998) used FISH probes to show that Acidithiobacillus spp. were abundant in environments with pH >1.5 and temperature <30°C at the Richmond mine. Acidithiobacillus spp. was also found to be the dominant iron-oxidizer stream draining the King’s copper mine (pH 2.5) in Roeros, Norway (Johnson et al. 2001). In our study, we also found the genus Acidithiobacillus was dominant in the two AMD samples we studied. The reason should be that the environments in two samples were suitable for the growth of Acidithiobacillus. The pH and temperature in two samples were 2.5 and 25.0°C, respectively, which were around the optimum of pH and temperature for the growth of Acidithiobacillus. And concentration of poisonous ions was very low in two samples for Acidithiobacillus. Previous researches reported that Acidithiobacillus were very susceptive with concentration of arsenic ion, especially arsenic ion (III) (Goebel and Stackebrandt 1994). The concentrations of arsenic in samples G1and G2 were only 2.5 and 5.9 mg/l, respectively, which were much lower than the maximal withstanding concentration of element arsenic for Acidithiobacillus.
β-Proteobacteria family was the other ubiquitous family in two samples. The clones in the family were mostly affiliated with the genus Gallionella. The proportions of clones affiliated with Gallionella in samples G1 and G2 were 16.9 and 47.2%, respectively. Organisms of genus Gallionella were neutrophilic iron-oxidizing and chemolithotrophic, which lived in a relatively poor-nutrition environment containing reduced iron. And oxidization of iron might be mainly caused by organisms of Gallionella in low-oxygen environments (Halbach et al. 2001). Interestingly, organisms of Gallionella, neutrophilic microbe, were dominant in two AMD samples we studied with pH around 2.5. The same result recently appeared in the study by Bruneel et al. that Gallionlla was dominant in Fe–As-rich AMD waters of Carnoulès, France (Bruneel et al. 2006).
Nitrospira family was only detected in sample G2. The clones clustered `with the family were all affiliated with the genus Leptospirillum. The organisms of Leptospirillum have been reported to be dominant in AMD environments (Druschel et al. 1999; Bond et al. 2000; Hippe 2000; Coram and Rawlings 2002), but they were not dominant in the two AMD samples we studied. The reason might be that the environments in the two samples were not suitable for the genus Leptospirillum. First of all, nutrition in two samples was too poor for the growth of Leptospirillum. Leptospirillum spp. just fed on iron and could not use sulfur as energy resource. In two samples studied, the concentrations of element iron were only 1.07 g/l (G1) and 4.0 g/l (G2). Then the temperature and pH in two samples were not also suitable for the growth of Leptospirillum. Reports of previous studies by Schrenk et al. (1998) with respect to Leptospirillum suggested that Leptospirillum spp. might share more natural distribution in low pH and high temperature. Oligonucleotide probe-based studies within the Richmond deposit indicated that Leptospirillum strains often dominated microbial communities growing at temperatures up to 50°C and in solutions with pH values as low as 0.5 (Edwards et al. 1999; Druschel et al. 1999; Bond et al. 2000).
Some other microorganisms related to the AMD environments were also detected in two AMD samples. Although their presence with low number, they were also significant in AMD environments. For example, Hyphomicrobium sp. in α-Proteobacteria family was detected in AMD sample G2, which could live in acidic environment and had contribution to the AMD generation by oxidizing iron to get energy (Pelczar et al. 1993).
In addition, the PCR amplification of archaeal 16S rDNA genes from these two AMD samples have also been performed with two sets of archaea-specific primers described as above. But there were no PCR products detected with these two primer sets. The same experimental procedures have been carried out on some other AMD samples, and several archaeal lineages, reported from AMD environments, have been detected (data not shown here). Therefore, it suggested that there might have no achaea in microbial communities in the two AMD samples from Yunfu sulfide mine.
References
Baker BJ, Banfield JF (2003) Microbial communities in acid mine drainage. FEMS Microbiol Ecol 44:139–152
Bond PL, Smriga SP, Banfield JF (2000) Phylogeny of microorganisms populating a thick, subaerial, predominantly lithotrophic biofilm at an extreme acid mine drainage site. Appl Environ Microbiol 66:3842–3849
Boon M, Heijnen JJ (1993) Mechanisms and rate limiting steps in bioleaching of biohydrometallurgy technologies. In: Torma AE, Wey JI, Lakshmanan VL (eds) Bioleaching processes, p 217
Bruneel O, Duran R, Casiot C, Elbaz PF, Personné JC (2006) Diversity of microorganisms in Fe–As-rich acid mine drainage waters of Carnoulès, France. Appl Environ Microbiol 72:551–556
Burton NR, Norris PR (2002) Microbiology of acidic, geothermal springs of Montserrat: environmental rDNA analysis. Extremophiles 4:315–320
Coram NJ, Rawlings DE (2002) Molecular relationship between two groups of the genus Leptospirillum and the finding that Leptospirillum ferriphilum sp. nov. dominates South African commercial biooxidation tanks that operate at 40°C. Appl Environ Microbiol 68:838–845
Delong EF (1992) Archaea in coastal marine environments. Proc Natl Acad Sci USA 89:5685–5689
Druschel GK, Baker BJ, Gihring T, Banfield JF (1999) Acid mine drainage biogeochemistry at Iron Mounta in California 65:3627
Edwards KJ, Gihring TM, Banfield JF (1999) Seasonal variations in microbial populations and environmental conditions in an extreme acid mine drainage environment. Appl Environ Microbiol 65:3627–3632
Edwards KJ, Bond PL, Gihring TM, Banfield JF (2000) An archaeal iron-oxidizing extreme acidophile important in acid mine drainage. Science 279:1796–1799
Fowler TA, Holmes PR, Crundwell FK (1999) Mechanism of pyrite dissolution in the presence of Thiobacillus ferrooxidans. Appl Environ Microbiol 65:2987–2993
Fuchs T, Huber H, Teiner K, Burggraf S, Stetter KO (1995) Metallosphaera prunae, sp. nov., a novel metal-mobilizing, thermoacidophilic archaeum, isolated from a uranium mine in Germany. Syst Appl Microbiol 18:560–566
Fuchs T, Huber H, Burggraf S, Stetter KO (1996) 16S rDNA-based phylogeny of the archaeal order Sulfolobales and reclassification of Desulfurobus ambivalens as Acidianus ambivalens comb. nov. Syst Appl Microbiol 19:56–60
Goebel BM, Stackebrandt E (1994) Cultural and phylogenetic analysis of mixed microbial populations found in natural and commercial bioleaching environments. Appl Environ Microbiol. 60:1614–1621
Golyshina OV, Pivovarova TA, Karavaiko GI, Kondrateva TF, Moore ER, Abraham WR, Lunsdorf H, Timmis KN, Yakimov MM, Golyshin PN (2000) Ferroplasma acidiphilum gen. nov., sp. nov., an acidophilic, autotrophic, ferrous-iron-oxidizing, cell-wall-lacking, mesophilic member of the Ferroplasmaceae fam. nov., comprising a distinct lineage of the Archaea. Int J Syst Evol Microbiol 50:997–1006
Halbach M, Koschinsky A, Halbach P (2001) Report on the discovery of Gallionella ferruginea from an active hydrothermal field in the deep sea. Inter Ridge News 10(1):18–20
Hallberg KB, Johonson DB (2001) Biodiversity of acidophilic prokaryotes. Adv Appl Microbiol 49:37–84
Hallberg KB, Lindström EB (1994) Characterization of Thiobacillus caldus, sp. nov., a moderately thermophilic acidophile. Microbiology 140:3451–3456
Hippe H (2000) Leptospirillum gen nov (ex Markosyan 1972), nom rev including Leptospirillum ferrooxidans sp. nov (ex Markosyan 1972), nom rev and Leptospirillum. Int J Syst Evol Microbiol 2:501–503
Johnson DB (1998) Biodiversity and ecology of acidophilic microorganisms. FEMS Microbiol Ecol 27:307–317
Johnson DB, Rolfe S, Hallberg KB, Iversen E (2001) Isolation and phylogenetic characterization of acidophilic microorganisms indigenous to acidic drainage waters at an abandoned Norwegian copper mine. Environ Microbiol 3(10):630–637
Kelly DP, Wood AP (2000) Reclassification of some species of Thiobacillus to the newly designated genera Acidithiobacillus gen nov Halothiobacillus gen nov and Thermithiobacillus gen nov. Int J Syst Bacteriol 50:511–516
Kurosawa N, Itoh YH, Iwai T, Sugai A, Uda I, Kimura N, Horiuchi T, Itoh T (1998) Sulfurisphaera ohwakuensis gen nov sp nov a novel extremely thermophilic acidophile of the order Sulfolobales. Int J Syst Bacteriol 48:451–456
Lane DJ (1991) 16S/23S rDNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. Wiley, Chichester, pp 115–175
Nordstrom DK, Alpers CN (1999) Negative pH, efflorescent mineralogy, and consequences for environmental restoration at the Iron Mountain Superfund site, California. Proc Natl Acad Sci USA 96:3455–3462
Pelczar MJ, Chan CS, Krieg NR (1993) Microbiology, 5th edn. McGraw-Hill Book Co., New York
Raskin L, Stromley JM, Rittmann BE, Stahl DA (1994) Groupspecific 16S rRNA hybridization probes to describe natural communities of methanogens. Appl Environ Microbiol 60:1232–1240
Robb FT, Sowers KR, DasSharma S, Place AR, Schreier HJ (1995) Thermophiles. In: Fleischmann EM (ed) Archaea: a laboratory manual. Cold Spring Harbor Press, Cold Spring Harbor, pp 202–203
Schrenk MO, Edwards KJ, Goodman RM, Hamers RJ, Banfield JF (1998) Distribution of Thiobacillus ferrooxidans and Leptospirillum ferrooxidans: implications for generation of acid mine drainage. Science 279:1519–1522
Simmons S, Norris PR (2002) Acidophiles of saline water at thermal vents of Vulcano, Italy. Extremophiles 6:201–207
Smith SW, Overbeek R, Woese CR, Gilbert W, Gillevet PM (1994) The genetic data environment: an expandable GUI for multiple sequenceanalysis. Comput Appl Biosci 10:671–675
Southam G, Beveridge TJ (1992) Enumeration of thiobacilli within pH-neutral and acidic mine tailings and their role in the development of secondary mineral soil. Appl Environ Microbiol 58:1904–1912
Strunk O, Ludwig W (1995) ARB—a software environment for sequence data. Department of Microbiology, Technical University of Munich, Munich
Ward DM, Weller R, Bateson MM (1990) 16S rRNA sequences reveal numerous uncultured microorganisms in a natural community. Nature 345:63–65
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol 173:697–703
Wielinga B, Lucy JK, Moore JN, Seastone OF, Gannon JE (1999) Microbiological and geochemical characterization of fluvially deposited sulfidic mine tailings. Appl Environ Microbiol 65:1548–1555
Wulf-Durand DP, Bryant LJ, Sly LI (1997) PCR mediated detection of acidophilic bioleaching-associated bacteria. Appl Environ Microbiol 63:2944–2948
Zhou J, Bruns MA, Tiedje JM (1996) DNA recovery from soils of diverse composition. Appl Environ Microbiol 62:316–322
Acknowledgments
This work was supported by National Basic Research Program of China (2004CB619204) and National Natural Science Foundation of China (30400010, 30570047).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by K. Horikoshi.
Zhiguo He, Shengmu Xiao, Xuehui Xie, and Hui Zhong equally contributed to this work.
Rights and permissions
About this article
Cite this article
He, Z., Xiao, S., Xie, X. et al. Molecular diversity of microbial community in acid mine drainages of Yunfu sulfide mine. Extremophiles 11, 305–314 (2007). https://doi.org/10.1007/s00792-006-0044-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-006-0044-z