
Abstract
Introduction
Duchenne muscular dystrophy (DMD) is induced by a wide spectrum of mutations such as exon deletions, duplications and small mutations in the dystrophin gene. This is the first study on the mutational spectrum in a cohort of DMD children from India, with an emphasis to compare the mutations in familial and sporadic forms.
Results
Multiplex ligation-dependent probe amplification (MLPA) and next-generation sequencing (NGS) identified 525 and 70 cases of DMD, respectively, while 11 cases showed absent dystrophin staining with no mutations detected. Families with two or more affected males contributed to 12% of the entire cohort. The mutations comprised of exonic deletions in 492/606 (81.2%), duplications in 33/606 (5.4%) and small mutations (point mutations and INDELs) in 70/606 (11.5%) cases. MLPA identified significantly more larger mutations in sporadic (88.2%) than in familial cases (75.3%). The mutations in NGS were: [nonsense = 40 (57.1%); frameshift = 17 (24.3%); splice variant = 12 (17.1%)]. Nonsense mutations were more common in familial than in sporadic cases: 17.8% vs 10.7%. The familial group reported an earlier onset of disease (2.8 ± 1.7 years) as compared to sporadic cases (3.8 ± 1.6 years).
Conclusion
MLPA could identify mutations in a high percentage of our DMD children. The preponderance of small mutations was noted to be distinctly higher in the familial group. Intriguingly, the familial form of DMD formed a small percentage of the entire cohort. The reasons could be increasing awareness among parents and physicians with early identification of DMD cases, genetic counseling and prenatal testing.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Duchenne muscular dystrophy (DMD) is a common X-linked recessive neuromuscular disorder with an universal prevalence of 1 in 3500 newborn boys [1]. Clinically, DMD is characterized by rapidly progressive limb girdle muscle weakness with calf muscle hypertrophy with the diagnosis generally being made before the age of 5 years. The dystrophin gene is the largest human gene, comprising of 79 exons occupying a genomic region of 2.3 Mb on X chromosome short arm [2]. DMD is the severe form of Xp21 mutation disorder either from a shift in the translational reading frame or a premature stop codon resulting in loss of functional dystrophin [3, 4]. The majority of DMD cases are caused by deletion (65%) or duplication (5% to 8%) of single or multiple exons [5]. About 25–30% cases are attributable to point mutations, and small insertion/deletions (INDELs) [4, 6].
Multiplex ligation-dependent probe amplification (MLPA) is a proven diagnostic tool for detection of exon deletions and duplications in DMD. MLPA is also extensively used in carrier detection and for prenatal diagnosis [5, 7, 8]. However, smaller mutations (point mutations and INDELs) require either direct Sanger sequencing of DMD gene or next-generation sequencing (NGS)-based testing [4]. Although mutations in DMD have been compiled in worldwide databases and studied elaborately in a few large cohorts (> 500 patients), information on mutational pattern in familial DMD is rarely described [1, 9,10,11,12,13]. Early genetic diagnosis and subsequent carrier analysis and genetic counseling can play a key role in prevention of new cases in the immediate family members and relatives. Although there are simple, reliable and affordable genetic tests available to diagnose DMD, the factors contributing to occurrence of familial forms need to be studied and measures to reduce the societal disease burden is critical. We have previously reported on various aspects including mutational aspects identified by multiplex polymerase chain reaction (mPCR) and MLPA, genotype–phenotype correlation, muscle magnetic resonance imaging (MRI) patterns, natural history and disease progression in hospital based studies of dystrophinopathy (DMD and BMD) patients from India [14,15,16,17,18]. In the current study, we have described and compared the pattern of mutations in sporadic cases and those with positive family history in a large cohort of 606 Indian DMD children who have undergone genetic testing by MLPA and NGS in a tertiary neurology hospital from South India.
Materials and methods
This is a retrospective study where patients were selected from the records of 804 suspected cases of dystrophinopathy registered at the Neuromuscular disorders’ clinic at a national referral center for neurological disorders. The study period was between October 2012 and May 2018. The case files contained detailed information on clinical history, neurological findings with muscle strength scoring chart and mutation data obtained by MLPA/NGS/muscle biopsy. The first molecular assay that is undertaken for all suspected cases of DMD is MLPA of the 79 DMD exons. If MLPA fails to identify any mutation, the sample is taken for NGS for genomic DNA-coding regions and/or muscle biopsy for dystrophin staining.
MLPA methodology
Exon deletion and duplication detection was performed using SALSA MLPA kit (MRC-Holland) with probe sets P034 and P035 for 79 exons of DMD gene as per the manufacturer protocol. Coffalyser MLPA analysis software (MRC-Holland) was used for analysis of the data.
NGS methodology
Sequencing for MLPA negative cases was performed on HiSeq and NextSeq 500 platforms (Illumina) following target capture with Sureselect XT method using custom DMD probe design (Agilent). Raw data analysis with sequence alignment and variant annotation was done using SureCall (Agilent) software.
Results
Entire DMD cohort
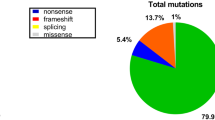
Among the 804 suspected cases, DMD diagnosis was confirmed in 606 children based on clinical features, raised CK level and a confirmatory genetic or immunohistochemical result. MLPA identified mutations in the DMD gene in 525 (86.6%) samples and was negative in 81 (13.4%) cases. The pattern of mutations was: exonic deletions in 492/606 (81.2%) cases with hot spot (45–54) region deletions in 365/492 (74.2%). Duplications occurred in 33/606 (5.4%) cases (Fig. 1). Out-of-frame mutations were found in 481/525 (91.6%) and in-frame in 44/525 (8.4%). There were 150 different deletion/duplication patterns identified. While exon 50 was most frequently deleted in 234/492 (47.6%), the most common mutation in the overall group was deletion of exon 45 in 38/525 (7.2%) cases. Single-exon deletions and duplications were observed in 120/492 (24.4%) and 7/33 (21.2%) patients, respectively. NGS-confirmed small mutations were seen in 70/606 (11.5%) [nonsense = 40 (57.1%); frameshift = 17 (24.3%); splice variant = 12 (17.1%)] cases (Fig. 1). Only one DMD patient in our entire cohort had a missense mutation. Small mutations showed random distribution with no hot spot predilection. Both MLPA and NGS did not identify any mutation in coding regions of DMD gene in eight patients (1.3%), and three patients did not undergo NGS but biopsy showed loss of dystrophin staining. Novel mutations were seen in 43/70 (61.4%) of cases. No hotspot region for duplications or small mutations was identified.

Bar diagram showing the comparative mutational spectrum in DMD cases
Familial group
Among the entire cohort of DMD cases, there were 73 families with more than 1 affected child. According to the history, a total of 117 affected family members were reported/identified: brothers of probands (n = 33), maternal uncles (n = 55), nephews (n = 7) cousins (n = 14), grand maternal uncles (n = 8). The 73 probands of the familial group comprised of 12% (73/606) of our total DMD cohort. Among this familial group, MLPA showed mutations in the DMD gene in 55/73 (75.3%) samples, while it was negative in 18/73 (24.7%) cases. Deletions were noted in 48/73 (65.7%) and duplications in 7/73 (9.6%) cases (Fig. 1). Thirty-three different deletion patterns were found (Fig. 2) in this cohort. Deletions in hot spot region were identified in 34/48 (70.8%) (Fig. 3) cases. All seven duplications were unique and uniformly distributed along the DMD gene (Table 1). Exon 50 was the most frequently deleted exon (24/48 = 50.0%) and single-exon 50 deletion was the single most common mutation occurring in 4/73 families. Single-exon deletion and duplication was present in 11/48 (22.9%) and 1/7 (14.3%), respectively. Out-of-frame mutations were present in 48/55 (87.3%) and in-frame in 7/55 (12.7%). NGS-confirmed mutations in 13/73 (17.8%) (Fig. 1) cases. Novel mutations occurred in 7/13 (53.8%) cases. Among the remaining five cases, all showed absent dystrophin staining; all were MLPA negative, two were NGS negative and three did not undergo NGS testing. The mean age at presentation was 6.8 ± 2.5 (2–12) years and age at onset was 2.8 ± 1.7 (1–7) years.

Bar diagram representing the deletion patterns in familial DMD cases

Bar diagram depicting the frequency of exon deletions in hot spot regions in familial DMD cases
Non-familial group
There were 533 non-familial cases and among this group MLPA was positive for mutations in the DMD gene in 470 (88.2%) and was negative in 63 (11.8%) samples. The pattern of mutation was: exonic deletions in 444/533 (83.3%) and exonic duplications in 26/533 (4.9%) cases (Fig. 1). There were 110 different deletion patterns and 24 duplication patterns (Supplementary Table 1A and 1B). One deletion (exons 1–29 and 40) and one duplication (exons 45–55, 63) were of non-contiguous pattern. Hot spot (45–54) deletions were present in 331/444 (74.5%), while no significant hot spot regions were identified for duplications (Fig. 4). Similar to the familial group, the most common exon deleted was exon 50 in 210/444 (47.3%), while exon 45 deletion was the most commonly repeated mutation (36 times). Single exon deletion and duplication was present in 109/444 (24.5%) and 6/26 (23.1%) cases, respectively. Out-of-frame mutations were observed in 433/470 (92.1%) patients while 37/470 (7.9%) had in-frame mutations. NGS-confirmed the presence of small mutations in 57/533 (10.7%) and no mutation in 6 (1.1%) samples (Fig. 1). Novel mutations were seen in 36/57 (63.1%) cases. The mean age at presentation was 7.6 ± 2.1 (3–14) years with onset of symptoms at 3.8 ± 1.6 (1–8) years.

Bar diagram illustrating the frequency of exon deletions showing hot spot region in sporadic DMD cases
Discussion
Comparison of familial and sporadic cases (Table 2)
In the current comparative study, in a large cohort of DMD children, we observed that identification of mutation by MLPA technique was significantly higher in the non-familial DMD group as compared to the familial group (p = 0.0025). Exonic deletions were significantly more frequent among the sporadic cases (p = 0.0003) while the proportion of duplications were more in the familial group. Subsequently, point mutations and INDELs identified by NGS were comparatively higher in the familial DMD group (~ 18%). This difference would have been greater considering that three of our MLPA negative familial cases with dystrophin loss on muscle biopsy did not undergo NGS due to non-availability of DNA samples. Point mutations are considered to be derived during spermatogenesis resulting in increased carrier frequency for these mutations in comparison to large deletions (≥ 1 exon) which can be predominantly de novo arising during oogenesis [19]. The large deletion pattern, hot spot involvement and single-exon mutations were comparable in both groups. The age at onset and presentation were significantly earlier in the familial group (p < 0.005) and could possibly be attributed to parents/family members identifying the motor disabilities at an early age, given the experience with previously affected family members. In comparison, the age at presentation of our DMD children is later than that of patients from western countries (4–5 years), and the delay increases further among sporadic cases (mean 7.6 years) [20, 21]. A common belief and also presumptions by the parents/guardians/general practitioners that the delayed motor milestones may be a normal variant, and leads to delay in investigating such boys for muscle diseases. In the current study, we found that positive family history was present in 12% of our entire DMD cohort, which is significantly less compared to the percentages of familial cases in the two large cohort studies (n > 500) that have reported the proportion of familial cases in DMD [11, 13]. Notably, comparative mutational spectrum in familial cases is lacking even in global DMD mutation database studies [1, 10]. Only an Italian study (n = 184) has presented a comparable percentage of familial cases. Studies from the USA and Asia mention percentages ranging from 21 to 43% [11, 13, 22,23,24] (Table 3). A study (n = 128) from South China [23] and a small cohort (n = 32) from Taiwan [24] have reported high percentage of familial forms of DMD, i.e., 31% and 43%, respectively. Yang et al. have reported 27.5% of familial cases in a large Chinese cohort of 1053 patients, but their study includes both DMD and BMD patients [13]. Our cohort comprised of only DMD patients. BMD cases are considered to have a higher positive family history due to transmission of mutations from affected males to daughters resulting in higher carrier frequency [19]. Yang et al. attributed the high incidence of familial cases to the lack of knowledge of DMD/BMD among primary care physicians, insufficient scientific information, and minimal awareness towards genetic counseling and prenatal testing in families with an affected boy and particularly with small DMD mutations [13]. In the western population, a study from Italy reported 9.2% patients out of 184 probands having a positive family history and this finding is similar to our cohort [22]. Flanigan et al. from the USA reported 203 (21%) familial cases in a large cohort of 1111 dystrophinopathy cases (from 967 families) including all phenotypes. However, they reported a selection bias as the study population was enriched with small mutations [11].
Our low percentage of familial cases might originate from the following aspects. On the one hand it might be caused by under-reporting because there might have been a few families who for unknown reasons did not reveal about other affected members. However, detailed, minimum three-generation pedigree diagrams are part of the clinical workup and missing out on familial forms is less likely. On the other hand, we could surmise from our findings that DMD cases were diagnosed early in our cohort and that swift genetic counseling helped in preventing subsequent cases in the same family. Also, as majority of our patients are from joint families, grandparents tend to recognize motor disability at a very early age and thus consult physicians while children are at a very young age. An earlier study from India indicates that prenatal diagnostic testing using mPCR method has been available since past 2 decades [25]. We are also aware that several centers in India are utilizing MLPA for prenatal diagnosis (PND) since it emerged as a routine screening tool for DMD patients and carriers [15, 26]. Although NGS has been undertaken for the last few years in India, its availability for DMD diagnosis and prenatal testing for our patients has been utilized form 2014 onwards. This could be considered as one of the reasons for the apparently increased number of point mutations in our familial cases. However, data on the usage of various tests for PND in India under the current scenario are lacking.
Other plausible factors that contributed to the low number of familial cases in our cohort would include easy accessibility to pediatricians and neurologists, as there is no referral system in India to consult specialists. It is also possible that our DMD cases may be more due to de novo mutations without mothers being carriers. Unfortunately, we could not perform carrier testing for this cohort.
Comparison with other large DMD cohorts (Table 3)
We have compared the mutational pattern of our entire DMD cohort with ten other large published global/country-wide studies and databases [1, 9,10,11,12,13, 22, 23, 27, 28] (Table 3). TREAT-NMD and Leiden DMD mutation databases are the largest global DMD registries with > 7000 and 4700 entries, respectively [1, 10]. In our group, the percentage of large deletions (81.2%) is higher, while duplications (5.4%) and small mutations (11.5%) are much lower compared to all other studies. Similar to the global registries and UMD database of French DMD mutations, exon 45 deletion is the most common exon deletion in our cohort [1, 9, 10]. In contrast, our familial group has exon 50 deletion as the most common mutation. The Remudy database from Japan [12], which includes a significant number of BMD cases (n = 295), has reported an in-frame deletion of exons 45–47 as the most common deletion while studies from Spain and Italy have found exon 51 and exons 45–52 deletions as most frequent mutations, respectively [12, 22, 28]. Deletion of exons 3–7 is the most common proximal deletion in our patients and this is similar to the French study [9]. While 74% of deletions occurred in the distal hot spot region between exons 45–54, we did not find any segregation of deletions and duplications in proximal exons as reported by previous studies [9, 10, 12]. Wang et al. have reported similar findings in 128 DMD patients from South China with no proximal hot spot for large rearrangements, but a distal hot spot for deletions at exons 45–54 was noted [23]. Exon 2 has been reported as the single most common duplication pattern in most of the databases available (Table 3). Interestingly, we did not identify isolated exon 2 duplication in any of our patients and only two patterns (duplication of exons 3–6 and 8–9) occurred more than once. The reading frame rule concordance is considered to be present in ~ 90% of DMD mutations [4]. In our entire cohort, out-of-frame mutations accounted for 91.6% which decreased slightly to 87.3% in the familial group. The significantly lower percentage of point mutations as observed in our patients is difficult to explain. It may be surmised that availability of NGS for the last 4 years only may be one of the reasons for detection of lesser number of point mutations. While the proportion of small mutations is significantly low in our patients in comparison to other studies, nonsense mutations resulting in stop codon constituted 57.0% of point mutations which increased to 61.5% when only familial cases were considered. Small INDELs resulting in frameshift accounted for 24.3% which decreased to 15.4% (2 patients only) in familial cases. Nonsense and frameshift mutations in previous large global studies accounted for about ~ 50.0% and ~ 32–34% of small mutations, respectively (Table 3). Truncating mutations in particular are considered to be more frequent in DMD than in BMD [9, 29]. The global mutational databases also include BMD cases which may explain the difference in the proportion of nonsense mutations in comparison to our cohort. Flanigan et al. have reported the highest proportion of nonsense mutations (65.0%) in a biased sample study group from USA and the lowest percentage (42.5%) was seen from a study from South China [11, 23]. The prevalence of splice-site and missense mutations seen in our cohort was comparable to previous studies. We did not find any hot spot region for small mutations unlike large deletions. Our overall mutation detection rate was 98.2% (595/606) with combined MLPA and NGS. In eight of our NGS negative cases, further data analysis and RNA sequencing might be required to look for any deep intronic mutations resulting in introduction of ‘pseudo-exon’ or other complex rearrangements/transpositions [9].
Novel therapeutic implications
An attempt was made to look for the number of DMD cases in our cohort who may be eligible for novel mutation-based therapies. While exon 51 skipping drug Eteplirsen has received accelerated approval by USFDA for further confirmatory studies, skipping agents of exons 53 and 45 are currently in phase 3 trials [30]. Skipping of single exons 51, 53 and 45 have been theoretically considered to be effective in 13%, 8.1% and 7.7%, respectively [31]. We identified that 38.6% (234/606) of our DMD patients are amenable for the top 3 single-exon skipping strategies (Table 4). Additionally, 40 patients (6.6%) with nonsense mutations resulting in stop codons can benefit from nonsense suppression treatment by compounds like Ataluren which enables read-through of premature stop codon [32]. This group of patients will be considered for Ataluren phase 3 clinical trials in India [Clinicaltrials.gov; CT/67/17-DCG(I)].
Conclusion
The results from this Indian single-center large cohort forms one of the largest well-established mutation databases. The current report is one among the very few reports in English literature that has studied the prevalence of familial forms of DMD and their mutation pattern. We have demonstrated distinct mutation pattern differences as compared to other Asian and Western populations. Our findings may have important implications in the genetic testing approach when familial cases are encountered in the Indian sub-continent. Although MLPA is considered to be the most powerful single technology to identify mutations in the huge dystrophin gene, it might be contemplated to use NGS as the first screening test in familial forms of DMD because of the higher possibility of obtaining a negative MLPA result when familial cases are encountered.
We also suggest that disease awareness in general population and primary physicians, introduction of new born screening for DMD in India would lead to early identification of sporadic cases with possibility of genetic counseling and subsequent pre-natal testing. In the light of upcoming novel therapies where early treatment is the key factor, early mutational screening is the need of the hour in India.
References
Aartsma-Rus A, Van Deutekom JCT, Fokkema IF et al (2006) Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 34:135–144. https://doi.org/10.1002/mus.20586
Tennyson CN, Klamut HJ, Worton RG (1995) The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat Genet 9:184–190. https://doi.org/10.1038/ng0295-184
Koenig M, Beggs AH, Moyer M et al (1989) The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet 45:498–506
Aartsma-Rus A, Ginjaar IB, Bushby K (2016) The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet 53:145–151. https://doi.org/10.1136/jmedgenet-2015-103387
Lalic T, Vossen RHAM, Coffa J et al (2005) Deletion and duplication screening in the DMD gene using MLPA. Eur J Hum Genet 13:1231–1234. https://doi.org/10.1038/sj.ejhg.5201465
Hegde MR, Chin ELH, Mulle JG et al (2008) Microarray-based mutation detection in the dystrophin gene. Hum Mutat 29:1091–1099. https://doi.org/10.1002/humu.20831
Gatta V, Scarciolla O, Gaspari AR et al (2005) Identification of deletions and duplications of the DMD gene in affected males and carrier females by multiple ligation probe amplification (MLPA). Hum Genet 117:92–98. https://doi.org/10.1007/s00439-005-1270-7
Itto AB, Hamzi K, Bellayou H et al (2013) Evolution of molecular diagnosis of Duchenne muscular dystrophy. J Mol Neurosci MN 50:314–316. https://doi.org/10.1007/s12031-013-9971-1
Tuffery-Giraud S, Béroud C, Leturcq F et al (2009) Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum Mutat 30:934–945. https://doi.org/10.1002/humu.20976
Bladen CL, Salgado D, Monges S et al (2015) The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat 36:395–402. https://doi.org/10.1002/humu.22758
Flanigan KM, Dunn DM, von Niederhausern A et al (2009) Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat 30:1657–1666. https://doi.org/10.1002/humu.21114
Okubo M, Goto K, Komaki H et al (2017) Comprehensive analysis for genetic diagnosis of Dystrophinopathies in Japan. Orphanet J Rare Dis 12:149. https://doi.org/10.1186/s13023-017-0703-4
Yang J, Li SY, Li YQ et al (2013) MLPA-based genotype–phenotype analysis in 1053 Chinese patients with DMD/BMD. BMC Med Genet 14:29. https://doi.org/10.1186/1471-2350-14-29
Swaminathan B, Shubha GN, Shubha D et al (2009) Duchenne muscular dystrophy: a clinical, histopathological and genetic study at a neurology tertiary care center in Southern India. Neurol India 57:734–738. https://doi.org/10.4103/0028-3886.59468
Manjunath M, Kiran P, Preethish-Kumar V et al (2015) A comparative study of mPCR, MLPA, and muscle biopsy results in a cohort of children with Duchenne muscular dystrophy: a first study. Neurol India 63:58. https://doi.org/10.4103/0028-3886.152635
Vengalil S, Preethish-Kumar V, Polavarapu K et al (2017) Duchenne muscular dystrophy and becker muscular dystrophy confirmed by multiplex ligation-dependent probe amplification: genotype–phenotype correlation in a large cohort. J Clin Neurol Seoul Korea 13:91–97. https://doi.org/10.3988/jcn.2017.13.1.91
Polavarapu K, Manjunath M, Preethish-Kumar V et al (2016) Muscle MRI in Duchenne muscular dystrophy: evidence of a distinctive pattern. Neuromuscul Disord 26:768–774
Singh R-J, Manjunath M, Preethish-Kumar V et al (2018) Natural history of a cohort of Duchenne muscular dystrophy children seen between 1998 and 2014: an observational study from South India. Neurol India 66:77
Lee T, Takeshima Y, Kusunoki N et al (2014) Differences in carrier frequency between mothers of Duchenne and Becker muscular dystrophy patients. J Hum Genet 59:46–50. https://doi.org/10.1038/jhg.2013.119
Flanigan KM (2014) Duchenne and Becker muscular dystrophies. Neurol Clin 32:671–688. https://doi.org/10.1016/j.ncl.2014.05.002 (viii)
van Ruiten HJA, Straub V, Bushby K, Guglieri M (2014) Improving recognition of Duchenne muscular dystrophy: a retrospective case note review. Arch Dis Child 99:1074–1077. https://doi.org/10.1136/archdischild-2014-306366
Magri F, Govoni A, D’Angelo MG et al (2011) Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J Neurol 258:1610–1623. https://doi.org/10.1007/s00415-011-5979-z
Wang D-N, Wang Z-Q, Yan L et al (2017) Clinical and mutational characteristics of Duchenne muscular dystrophy patients based on a comprehensive database in South China. Neuromuscul Disord NMD 27:715–722. https://doi.org/10.1016/j.nmd.2017.02.010
Liang W-C, Wang C-H, Chou P-C et al (2018) The natural history of the patients with Duchenne muscular dystrophy in Taiwan: a medical center experience. Pediatr Neonatol 59:176–183. https://doi.org/10.1016/j.pedneo.2017.02.004
Maheshwari M, Vijaya R, Kabra M et al (2000) Prenatal diagnosis of Duchenne muscular dystrophy. Natl Med J India 13:129–131
Verma PK, Dalal A, Mittal B, Phadke SR (2012) Utility of MLPA in mutation analysis and carrier detection for Duchenne muscular dystrophy. Indian J Hum Genet 18:91–94. https://doi.org/10.4103/0971-6866.96667
Juan-Mateu J, Gonzalez-Quereda L, Rodriguez MJ et al (2015) DMD mutations in 576 dystrophinopathy families: a step forward in genotype–phenotype correlations. PLoS One 10:e0135189. https://doi.org/10.1371/journal.pone.0135189
Vieitez I, Gallano P, González-Quereda L et al (2017) Espectro mutacional de la distrofia muscular de Duchenne en España: estudio de 284 casos. Neurología 32:377–385. https://doi.org/10.1016/j.nrl.2015.12.009
Flanigan KM, Dunn DM, von Niederhausern A et al (2011) Nonsense mutation-associated Becker muscular dystrophy: interplay between exon definition and splicing regulatory elements within the DMD gene. Hum Mutat 32:299–308. https://doi.org/10.1002/humu.21426
Crone M, Mah JK (2018) Current and emerging therapies for duchenne muscular dystrophy. Curr Treat Options Neurol 20:31. https://doi.org/10.1007/s11940-018-0513-6
Aartsma-Rus A, Fokkema I, Verschuuren J et al (2009) Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat 30:293–299. https://doi.org/10.1002/humu.20918
McDonald CM, Campbell C, Torricelli RE et al (2017) Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Lond Engl 390:1489–1498. https://doi.org/10.1016/S0140-6736(17)31611-2
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors have no conflict of interest.
Ethical standards
Approval from ‘Institutional Ethics Committee’ has been obtained.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Polavarapu, K., Preethish-Kumar, V., Sekar, D. et al. Mutation pattern in 606 Duchenne muscular dystrophy children with a comparison between familial and non-familial forms: a study in an Indian large single-center cohort. J Neurol 266, 2177–2185 (2019). https://doi.org/10.1007/s00415-019-09380-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-019-09380-3