
Abstract
Dendrobium is widely used in traditional Chinese medicine, and Dendrobium species show high diversity in potential adaptation to drought stress. Although miRNAs play crucial roles in regulating drought responses in plants, their functions in the drought response in Dendrobium remain unclear. To investigate the roles of miRNAs and their targets during drought stress, a small-RNA library from drought-treated Dendrobium huoshanense was constructed and sequenced. A total of 211 miRNAs were identified, comprising 115 known miRNAs and 96 novel miRNAs. Among them, 36 conserved and 33 novel miRNAs were characterized as differentially expressed miRNAs between drought and control samples. Target prediction, Gene Ontology (GO)-based functional classification, and Kyoto Encyclopedia of Genes and Genomes (KEGG)-based functional enrichment showed that drought-regulated miRNAs might play roles in the response to drought stress by targeting a series of stress-related genes. Importantly, integrated analysis of the miRNA transcriptome and qRT-PCR detection were performed to identify miR156, miR157d, and miR160a-5p and their targets (auxin response factor, cytokinin receptor, two-component response regulator, and DELLA protein gene). The results indicated that drought-regulated miRNAs might regulate their targets to respond to hormone signaling, thereby regulating the morphological and physiological responses of the plant to drought stress. Our results provide new insights into the complex regulatory network of medicinal plant responses to drought stress involving plant hormone signal transduction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Drought is one of the major abiotic factors and destructive environmental stresses that restricts the growth, development and yield of plants; in the meantime, some signaling pathways in plants would been activated during the drought stress response (Ashraf 2010). Plants have developed a variety of defense strategies against drought at the morphological, physiological, biochemical, cellular, and molecular levels (Farooq et al. 2012). At the morphological level, to avoid desiccation, plants lower their growth rates, deepen their rooting systems, and modify their root to shoot ratios; at the physiological level, and they induce stomatal closure and antioxidant accumulation (Tátrai et al. 2016). At the molecular level, regulation of gene expression (enzymes and drought-related proteins) and epigenetic alterations (methylation, histone modifications and posttranscriptional alterations) are important mechanisms in the plant response to drought (Bhargava and Sawant 2013). Research to date shows that the plant drought stress responses, tolerance mechanisms and the genetic control of tolerance are complex.
MicroRNA (miRNA)-mediated regulatory mechanisms play crucial roles in plants, including regulating several plant developmental processes, such as leaf development, flowering time, and organ polarity, and responses to biotic and abiotic stresses (Chen 2005; Jeong and Green 2013). Many miRNAs have been found to participate in the response to drought stress in plants via signal transduction pathways, such as auxin signaling, and via ABA-mediated regulation, osmoprotectant biosynthesis, and scavenging of antioxidants (Candar-Cakir et al. 2016; Ding et al. 2013). Drought-related miRNAs and their targets have been identified in several plants, such as Arabidopsis, cotton, and tomato (Candar-Cakir et al. 2016; Liu et al. 2008; Xie et al. 2015b). For example, in a study employing small-RNA and degradome deep sequencing in tomato, 699-conserved miRNAs were identified, and some of those miRNAs associated with the drought response involving plant hormone signal transduction and drought-tolerant tomato breeding (Candar-Cakir et al. 2016). Furthermore, an analysis entailing citationRank-based literature mining in cotton led to the identification of a total of 337 miRNAs, and miRNAs associated with the expression of important genes related to drought stress were found (Xie et al. 2015b). However, most research on drought-related miRNAs has been focused on crop plants, and the potential functions of these miRNAs in medicinal plants are unknown.
Recently, some important drought-responsive miRNAs were identified in plants. miR169, miR398, and miR5505 were candidates with potential in developing drought tolerance in six Oryza sativa genotypes based on the genome-wide analysis (Xia et al. 2020). miR169 was found commonly present under drought stress but with different outputs depending on the species: downregulated in A. thaliana (Li et al. 2016) and N. tabacum (Yin et al. 2014) and upregulated in O. sativa (Xia et al. 2020). In N. tabacum, miR160 regulates the expression of auxin response factor genes; auxins are vital phytohormones for plant growth and development also involved in tolerance to drought stress (Li et al. 2016). miR156 and miR172 are other transcription factor-related regulators commonly involved in moderation of abiotic stresses (Pagano et al. 2021). Moreover, three conserved miRNAs (miR156, miR159, and miR319) were identified and played important roles in the seedlings of a drought-tolerant inbred Z. mays and N. tabacum (Li et al. 2013).
Dendrobium huoshanense is a famous perennial medical herb of the family Orchidaceae and is extensively used in China to remedy symptoms such as chronic superficial gastritis and throat phlegmonosis, which enhance immunity and achieve other benefits (Bao et al. 2001; Liu et al. 2018). D. huoshanense contains a variety of chemical components, such as polysaccharides and flavonoids, which have numerous pharmacological activities, including antioxidant, immunomodulation, antifatigue, digestion-promoting, salivary secretion-stimulating, hypoglycemic, antihypertension, and antitumor activities (Li et al. 2015). Although several studies have revealed the adverse impacts of drought stress on medicinal plants, such as negative effects on plant growth, development and yield of natural products, the effects of drought stress on the quality of medicinal plants are multilayered and very complex, and no study focused on the genome-wide identification of drought-responsive miRNAs in D. huoshanense has been conducted (Selmar and Kleinwächter 2013).
The current study attempts to identify expression pattern of miRNAs in response to drought stress in D. huoshanense through high-throughput sequencing and identifies the miRNA predicted target gene functions. We sequenced the genome of D. huoshanense. The total length of its assembly was 1.29 Gb and 20,879 protein-coding genes were identified. In this study, characteristics of small regulatory microRNAs (miRNAs) in the response to drought stress in D. huoshanense were investigated. A total of 115 conserved miRNAs and 96 novel miRNAs were identified from different libraries, of which at least 8 miRNAs were drought specific. Evidence from miRNA-target prediction, Gene Ontology (GO) term classification, Kyoto Encyclopedia of Genes and Genomes (KEGG)-based pathway analysis, and quantitative real-time PCR (qRT-PCR) results further suggested that these identified miRNAs may contribute to the response to drought stress in D. huoshanense. Together, these findings could increase our understanding of the complex regulatory network that underlies the draught tolerance of D. huoshanense.
Materials and Methods
Plant Material and RNA Extraction
Specimens of D. huoshanense were collected from a wild population in northern Yunnan Province, China, and available from the College of Biological and Pharmaceutical Engineering, West Anhui University, Lu’an, China. D. huoshanense plants were maintained in a greenhouse as previously reported (Yang et al. 2014; Zou et al. 2018).
Seeds of D. huoshanense were surface sterilized with 70% ethanol and HgCl2 and then transferred to solid MS medium supplemented with 0.5 mg/L 6-benzylaminopurine and 0.5 mg/L 1-naphthaleneacetic acid (NAA). The plants were grown at 27 °C under a relative humidity of 50%. After 9 months of cultivation, 10-month-old individuals were chosen and transferred to plastic pots (20.0 cm in diameter) filled with a substrate mix of composted pine bark and small stones. The plants were maintained at 27/22 °C (day/night), a photoperiod of 12/12 hr (day/night), under a relative humidity of 50/70% (day/night) and were watered every two days. After 2 months, strong individuals were selected and divided into two groups. Plants without watering for 7 days, then leaf water content was measured with the fourth mature leaves and the effects of drought stress were divided into mild, moderate, and severe drought stress with leaf water content of > 78%, ~ 75%, and < 72%, moderate drought stress-treated D. huoshanense was chosen as the drought-treated group. Plants watering every two days without undergoing drought or waterlogging as the control group. The fourth mature leaves from the apex of each individual were harvested and placed in liquid nitrogen for later RNA extraction.
Small-RNA Library Construction and Sequencing
Total RNA of D. huoshanense was extracted as previously described (Liu et al. 2018). The RNA was treated with DNase I (Invitrogen) to remove any potential genomic DNA contamination. RNA degradation and contamination were monitored on 1% agarose gels. The quality and quantity of RNAs were measured with a NanoDrop ND-2000 spectrophotometer (Thermo Scientific, Wilmington, DE). RNA concentration was measured using a Qubit® RNA assay kit and a Qubit® 2.0 fluorometer (Life Technologies, Pleasanton, CA, USA). Small RNAs (15–30 nt) were extracted from total RNA on a 15% denaturing polyacrylamide gel and ligated to specific 5′ adaptor and 3′ adaptor samples. Two sRNA libraries (two independent biological replicates) from the same treatment were, respectively, used as samples, and an Illumina HiSeq 4000 platform was used for high-throughput sequencing.
Data Analysis and Identification of miRNA Genes
The raw reads were processed, and miRNA genes were identified as in our previous study with minor modifications (Shao et al. 2019). Briefly, high-quality small-RNA reads were obtained from the raw reads by filtering out poor-quality reads and removing adaptor sequences using the FASTX toolkit with the default settings (Blankenberg et al. 2010). Adaptor-trimmed unique sequences were aligned to the D. huoshanense genome with Bowtie (Langmead et al. 2009). After removal of known noncoding RNAs (rRNAs, tRNAs, snRNAs, and snoRNAs) by BLASTall, BLASTn, and Bowtie, the unannotated small RNAs were used for novel miRNA prediction by using mireap (Li et al. 2012). Then, for annotation of the remaining sequences, conserved miRNAs were mapped to the miRBase database (v22.1 on March 26th, 2020; http://www.mirbase.org/ftp.shtml). The raw abundances of the miRNAs were normalized according to transcripts per million (TPM) normalization: small RNAs were mapped back onto the reference genome, and read count for each small RNA was obtained from the mapping results (t Hoen et al. 2008). PsRobot (https://tools4mirs.org/software/target_prediction/psrobot/) and TargetFinder (http://plantgrn.noble.org/psRNATarget/) were used for the prediction of miRNAs and their targets according to plant-like target interactions and based on previously described methods.
Expression Analysis of miRNAs and Target Genes
The stem-loop quantitative real-time PCR method was used to quantitate miRNA expression in this study using 5S rRNA as the internal control for each sample (Candar-Cakir et al. 2016). In brief, stem-loop reverse transcription was carried out using the TaqMan® MicroRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). The expression of target genes was detected as mentioned above, and the Actin gene was used as the internal control for each sample. qRT-PCRs were carried out using the CFX96™ Real-Time PCR System (Bio-Rad) with SYBR® Premix Ex Taq™ II (TaKaRa). The fold changes were calculated using 2−ΔΔCT values, and six independent experiments were performed. All data are presented as the mean ± SE of six replicates.
Statistical Analysis
Data were analyzed by one-way analysis of variance, followed by Dunnett’s multiple comparison and Tukey’s test using the SPSS v23.0 software. All results were expressed as the mean ± standard error of the mean (SD). p < 0.05 was considered statistically significant.
Results
Identification of miRNAs in D. huoshanense
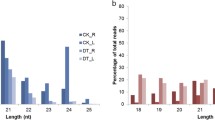
A total of 84,908,472 raw reads were obtained from the small-RNA libraries generated from the drought treatment (51,851,851) and the control treatment (33,056,621), from which a total of 58,588,717 clean reads were obtained. After alignment against the D. huoshanense genome, 25,049,100 and 26,455,704 clean reads from the drought and control samples, respectively, were mapped to the plant genome. A total of 211 miRNAs were identified from the different samples of D. huoshanense (Table S1). miRNAs are well known to be highly conserved across plant species. A total of 115 known plant miRNAs were identified in D. huoshanense after the alignment of all clean reads from the two libraries against all known plant miRNAs in miRBase (Table S1). We identified 96 putative novel miRNA gene loci in D. huoshanense based on the methods described in a previous study (Fig. 1a and Table S1) (Mutum et al. 2016). According to mature sequence length, 67.3% of all miRNAs were of canonical length (20–23 bp), and the largest percentage of miRNAs, 37.91%, were in the 21-nt class, followed by the 20-nt class (15.64%) (Fig. 1b). The distribution of 5’ terminal bases differed among miRNAs of different lengths; those 20–23 bp in length had a strong preference for “U”, in contrast to miRNAs with lengths of 24–28 bp (Fig. 1c).

Identification and characteristics of D. huoshanense miRNAs. a Examples of the secondary structure of locus-specific miRNA precursors. The green sequence represents mature miRNA. b Size distribution of miRNAs. c 5′-terminal nucleotides of miRNAs
miRNA Expression in Response to Drought Treatment
Among the 211 identified miRNAs, 8 miRNAs were expressed exclusively in the drought samples, whereas 190 were expressed in both drought and control samples (Fig. 2a). The abundance of miRNAs was normalized according to transcripts per million (TPM) normalization, and we used an absolute value of the log2 ratio ≥ 1 (p < 0.05) as the threshold to determine significant differences in miRNA expression. A total of 69 miRNAs were found to have significantly different expressions between the drought and control samples: 38 miRNAs were upregulated while the remainder were downregulated in drought samples relative to control samples (Fig. 2a and Table S1). To gain more insight into the characteristics of miRNA expression, a heatmap of the differentially expressed miRNAs was constructed (Fig. 2c). Compared to their expression following control treatment, numerous miRNAs were upregulated during the drought response, such as miR156, miR167d_1, miR160-5p, and miR396a-5p, which have been identified as upregulated in several plant species under drought stress (Gentile et al. 2015). These significantly differentially expressed miRNAs may play crucial roles in the drought response in D. huoshanense.

Expression levels of miRNAs in the control and drought samples. a Venn diagram of the number of miRNAs from different samples. b The differentially expressed miRNAs. c Heatmaps of D. huoshanense miRNAs in the control and drought samples
Identification and Functional Prediction of miRNA Targets
Plant miRNAs directly affect their target genes by cleaving them or repressing their translation. To understand the biological functions triggered by miRNAs in the response to drought treatment in D. huoshanense, the target genes of these miRNAs were identified. After strict filtration with a series of miRNA-target features, a total of 852 unique coding genes were predicted to be targets of these miRNAs, and a total of 1458 miRNA-target pairs were identified (Table S2). Gene ontology (GO) functional analysis (Conesa et al. 2005) and Kyoto Encyclopedia of Genes and Genome (KEGG) pathway enrichment analysis were carried out to identify the potential function of each putative target gene. Based on sequence homology, the target genes were categorized into 30 functional groups (p < 0.05). “Regulation of transcription, DNA-templated,” “Regulation of nucleic acid-templated transcription,” “Regulation of RNA biosynthetic process,” and “Regulation of RNA metabolic process” were the most enriched terms in the GO term categories (Fig. 3a). Their target genes were enriched in 12 KEGG pathways (p < 0.05) (Fig. 3b). Of these pathways, “plant hormone signal transduction” was the most enriched pathway term, with 24 target genes being annotated to this term. Plant hormone signaling machinery modulates plant growth, development, and defense, suggesting that miRNAs may regulate D. huoshanense responses to drought via the plant hormone signal transduction pathway (Wang et al. 2015).

Significantly enriched GO (a) and KEGG (b) terms for the target genes of differentially expressed miRNAs
miRNA Predicted Targeted Genes Involved in the Plant Hormone Signal Transduction Pathway
To further explore the interaction between drought-responsive miRNAs and their potential target genes involved in the plant hormone signal transduction pathway, a KEGG pathway map was constructed for the plant hormone signal transduction pathway (Fig. 4a). The auxin response factor (ARF), cytokinin receptor (CRE1), two-component response regulator (B-ARR), DELLA protein (DELLA), and brassinosteroid insensitive protein 1 (BRI1) genes, which play crucial roles in plant growth, shoot initiation, and induced germination, were targeted by drought responsive, differentially expressed miRNAs. To understand and visualize the regulation network between miRNAs and their possible targets involved in the plant hormone signal transduction pathway, regulatory network analysis was performed; the results are shown in Fig. 4b. A total of 11 miRNAs were predicted to target 27 genes involved in plant hormone signal transduction pathways (Table S3). The miR156 family had connections with 9 target genes, and the miR171 family had connections with 6 target genes, suggesting that these families likely function in the response to drought by targeting a number of different genes in D. huoshanense.

Network analysis of drought-responsive miRNAs and their potential targets in hormone signaling pathways. a Target genes mapped for the plant hormone signal transduction pathway. b Network analysis of target genes involved in plant hormone signal transduction
To validate the regulatory roles of miRNAs in plant hormone signal transduction during the drought response, we analyzed the expression profiles of the 11 miRNAs and their corresponding targets with qRT-PCR (Fig. 5a). The expression profiles of all 11 miRNAs were consistent with the sequencing data and showed significant differential expression between the drought and control samples (Fig. 5b). Among these 27 genes, 24 genes were detected with qRT-PCR in drought and control samples. Nine of these genes were significant differentially expressed, with 6 being upregulated while three being downregulated in the drought samples (Fig. 5a). Based on “Plant miRNAs could directly affect their target genes with cleavage,” we searched the miRNAs and their target genes that showed opposing expression patterns. Eight miRNAs and eight target genes, representing 13 miRNA-target pairs, were found.

The proposed model of the miRNA-mediated regulatory network associated with hormone signal transduction. a qRT-PCR analysis of expression profiles of miRNAs and their corresponding targets. b Correlation of qRT-PCR and sequencing data of miRNAs. c The proposed model of the miRNA-mediated regulatory network involved in hormone signal transduction. red, upregulation; blue, downregulation
In Arabidopsis thaliana, Nicotiana tabacum, Triticum aestivum, Zea mays, and O. sativa, expression of miR156 is known to be induced by different abiotic stresses, helping plants resist adverse environments through regulation of the squamosa promoter-binding protein-like target (Pagano et al. 2021); miR160-directed regulation of Arabidopsis auxin response factor is essential for the tolerance to various abiotic stresses (Li et al. 2016); Moreover, miR171 and miR172 family miRNAs could be specifically involved in the chilling stress response by regulating transcript targets influencing development (Chen et al. 2016). In our study, Genes encoding squamosa promoter-binding family protein (Dhu-21537) and auxin response factor family proteins (Dhu-20362 and Dhu-11346) were negatively regulated by drought-upregulated miRNAs (miR156, miR157d, and miR160a-5p), whereas genes encoding other squamosa promoter-binding family proteins (Dhu-20470, Dhu-18859, and Dhu-08742), a nodulation-signaling pathway protein (Dhu-16621), and a two-component response regulator protein (Dhu-21930) were negatively regulated by drought-upregulated miRNAs (miR156e_1, miR156a-5p, miR171I-3p, miR171f-3, and miR172e-3p_1). Based on information from our and previous studies, a schematic of the relationships between these miRNAs and plant hormone signal transduction is presented in Fig. 5c.
Discussion
Dendrobium is widely used in traditional Chinese medicine and contains many kinds of active ingredients. Some Dendrobium species, such as D. huoshanense, not only are adapted to mid- or high-land moist areas at elevations up to 800 m dpl but can also grow in dry lowland regions (Xiang et al. 2016). Knowledge of their strategies for adaptation is essential for their conservation and wide cultivation, and drought stress can affect the growth and secondary metabolites of Dendrobium (Metusala et al. 2017; Wu et al. 2016). Compared with D. arcuatum, D. capra shows more specialized anatomical features for adapting to drought and dry conditions (Metusala et al. 2017). miRNAs, as endogenous small noncoding regulatory RNAs, play important roles in abiotic and biotic stress responses by targeting plant mRNAs for cleavage or translational repression (Bartel 2004; Hajyzadeh et al. 2015). However, no genome-wide study on drought-responsive miRNAs in Dendrobium has been reported to date. In this research, miRNAs that exhibit differential expression upon drought stress in D. huoshanense were determined.
Drought-responsive miRNAs have been identified in many plant species through microarray or deep-sequencing technologies, such as Arabidopsis, wheat, tobacco, potato, and barley (Candar-Cakir et al. 2016; Gentile et al. 2015; Jeong and Green 2013; Xie et al. 2015a). Studies have shown that the expression of miRNAs is altered in the response to drought stress; miRNAs, as gene regulators, are expected to participate in gene regulation in plants (Gentile et al. 2015). Based on the obtained transcriptome datasets, 36 conserved and 33 novel miRNAs were characterized as differentially expressed miRNAs in drought and control samples. In our study, miR156 and miR394a_1 were upregulated under drought stress while miR168a-3p_1 was downregulated. These results are similar to results from Arabidopsis, whereas the expression of these three miRNAs showed opposite patterns in rice and maize, revealing that the expression level or drought responsiveness of a miRNA is species dependent (Liu et al. 2008; Wei et al. 2009; Zhou et al. 2010).
Both drought-upregulated and drought-downregulated miRNAs are potentially relevant for engineering plant drought tolerance (Gentile et al. 2015). Hormones play a central role in regulating plant growth, development, and defense (Wang et al. 2015). In this study, ARF, CRE1, B-ARR, DELLA, and BRI1-coding genes, as key genes in plant hormone signal transduction pathways, were targeted by drought responsive, differentially expressed miRNAs in D. huoshanense. The qRT-PCR results demonstrated that miRNAs may regulate the expression of their target genes involved in hormone signaling and thereby control the growth and development of D. huoshanense during the drought response.
In previous studies, miR156 contributes to plants to drought environment through regulation of the squamosa promoter-binding protein-like target (Pagano et al. 2021); miR160-directed regulation of Arabidopsis auxin response factor is essential for the tolerance to various abiotic stresses (Li et al. 2016); Moreover, miR171 and miR172 family miRNAs could be specifically involved in the chilling stress response by regulating transcript targets influencing development (Chen et al. 2016). Importantly, in our study, genes encoding squamosa promoter-binding family protein and auxin response factor family proteins were negatively regulated by miR156 and miR160a-5p, whereas genes encoding other squamosa promoter-binding family proteins, a nodulation-signaling pathway protein, and a two-component response regulator protein were negatively regulated by miR171I-3p, miR171f-3, and miR172e-3p_1, further confirmed that conserved roles of miRNAs from miR156, miR160, and miR171 families in different crop species (Pagano et al. 2021).
In conclusion, genome-wide investigation led to the identification of drought-responsive miRNAs in the medical plant, D. huoshanense, and these miRNAs might regulate plant growth and development by altering their targets in response to hormone signaling. These findings highlight the importance of detailed characterization of stress-responsive miRNAs in plants.
References
Ashraf M (2010) Inducing drought tolerance in plants: recent advances. Biotechnol Adv 28:169–183. https://doi.org/10.1016/j.biotechadv.2009.11.005
Bao XS, Shun QS, Chen LZ (2001) The medicinal plants of Dendrobium (SHI-HU) in China. A Coloured Atlas. Shanghai: Press of Fudan University and Press of Shanghai Medical University (In Chinese).
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297. https://doi.org/10.1016/S0092-8674(04)00045-5
Bhargava S, Sawant K (2013) Drought stress adaptation: metabolic adjustment and regulation of gene expression. Plant Breeding 132:21–32. https://doi.org/10.1111/pbr.12004
Blankenberg D, Gordon A, Von Kuster G, Coraor N, Taylor J, Nekrutenko A, Team G (2010) Manipulation of FASTQ data with Galaxy. Bioinformatics 26:1783–1785. https://doi.org/10.1093/bioinformatics/btq281
Candar-Cakir B, Arican E, Zhang B (2016) Small RNA and degradome deep sequencing reveals drought-and tissue-specific micrornas and their important roles in drought-sensitive and drought-tolerant tomato genotypes. Plant Biotechnol J 14:1727–1746. https://doi.org/10.1111/pbi.12533
Chen L, Han J, Deng X, Tan S, Li L, Li L, Zhou J, Peng H, Yang G, He G, Zhang W (2016) Expansion and stress responses of AP2/EREBP superfamily in Brachypodium Distachyon. Sci Rep-UK 6:1–14. https://doi.org/10.1038/srep21623
Chen XM (2005) microRNA biogenesis and function in plants. Febs Lett 579:5923–5931. https://doi.org/10.1016/j.febslet.2005.07.071
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676. https://doi.org/10.1093/bioinformatics/bti610
Ding YF, Tao YL, Zhu C (2013) Emerging roles of microRNAs in the mediation of drought stress response in plants. J Exp Bot 64:3077–3086. https://doi.org/10.1093/jxb/ert164
Farooq M, Hussain M, Wahid A, Siddique K (2012) Drought stress in plants: an overview. Plant responses to drought stress. Springer, Berlin, pp 1–33
Gentile A, Dias LI, Mattos RS, Ferreira TH, Menossi M (2015) MicroRNAs and drought responses in sugarcane. Front Plant Sci 6:58. https://doi.org/10.3389/fpls.2015.00058
Hajyzadeh M, Turktas M, Khawar KM, Unver T (2015) miR408 overexpression causes increased drought tolerance in chickpea. Gene 555:186–193. https://doi.org/10.1016/j.gene.2014.11.002
Jeong DH, Green PJ (2013) The role of rice microRNAs in abiotic stress responses. J Plant Biol 56:187–197. https://doi.org/10.1007/s12374-013-0213-4
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. https://doi.org/10.1186/gb-2009-10-3-r25
Li F, Cui SH, Zha XQ, Bansal V, Jiang YL, Asghar MN, Wang JH, Pan LH, Xu BF, Luo JP (2015) Structure and bioactivity of a polysaccharide extracted from protocorm-like bodies of Dendrobium huoshanense. Int J Biol Macromol 72:664–672. https://doi.org/10.1016/j.ijbiomac.2014.08.026
Li JS, Fu FL, An M, Zhou SF, She YH, Li WC (2013) Differential expression of micrornas in response to drought stress in Maize. J Integr Agric 12:1414–1422. https://doi.org/10.1016/S2095-3119(13)60311-1
Li SC, Xie ZZ, Hu CG, Zhang JZ (2016) A review of auxin response factors (ARFs) in plants. Front Plant Sci 7:47. https://doi.org/10.3389/fpls.2016.00047
Li Y, Zhang Z, Liu F, Vongsangnak W, Jing Q, Shen B (2012) Performance comparison and evaluation of software tools for microRNA deep-sequencing data analysis. Nucleic Acids Res 40:4298–4305. https://doi.org/10.1093/nar/gks043
Liu HH, Tian X, Li YJ, Wu CA, Zheng CC (2008) Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 14:836–843. https://doi.org/10.1261/rna.895308
Liu L, Han R, Yu N, Zhang W, Xing L, Xie D, Peng D (2018) A method for extracting high-quality total RNA from plant rich in polysaccharides and polyphenols using Dendrobium huoshanense. PLoS ONE 13:e0196592. https://doi.org/10.1371/journal.pone.0196592
Metusala D, Supriatna J, Nisyawati SD (2017) Comparative leaf and root anatomy of two Dendrobium species (Orchidaceae) from different habitat in relation to their potential adaptation to drought. InAIP Conference Proceedings 1862:030118
Mutum RD, Kumar S, Balyan S, Kansal S, Mathur S, Raghuvanshi S (2016) Identification of novel miRNAs from drought tolerant rice variety Nagina. SCI Rep-UK 6:30786. https://doi.org/10.1038/srep30786
Pagano L, Rossi R, Paesano L, Marmiroli N, Marmiroli M (2021) miRNA regulation and stress adaptation in plants. Environ Exp Bot 184:104369. https://doi.org/10.1016/j.envexpbot.2020.104369
Selmar D, Kleinwächter M (2013) Influencing the product quality by deliberately applying drought stress during the cultivation of medicinal plants. Ind Crop Prod 42:558–566. https://doi.org/10.1016/j.indcrop.2012.06.020
Shao Y, Tang J, Chen S, Wu Y, Wang K, Ma B, Zhou Q, Chen A, Wang Y (2019) milR4 and milR16 mediated fruiting body development in the medicinal fungus Cordyceps militaris. Front Microbiol 10:83. https://doi.org/10.3389/fmicb.2019.00083
t Hoen PA, Ariyurek Y, Thygesen HH, Vreugdenhil E, Vossen RH, de Menezes RX, Boer JM, van Ommen GJ, den Dunnen JT (2008) Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res 36:e141. https://doi.org/10.1093/nar/gkn705
Tátrai ZA, Sanoubar R, Pluhár Z, Mancarella S, Orsini F, Gianquinto G (2016) Morphological and physiological plant responses to drought stress in Thymus citriodorus. Int J Agronomy 2016:1–8. https://doi.org/10.1155/2016/4165750
Wang C, Liu Y, Li SS, Han GZ (2015) Insights into the origin and evolution of the plant hormone signaling machinery. Plant Physiol 167:872–886. https://doi.org/10.1104/pp.114.247403
Wei LY, Zhang DF, Xiang F, Zhang ZX (2009) Differentially expressed mirnas potentially involved in the regulation of defense mechanism to drought stress in maize seedlings. Int J Plant Sci 170:979–989. https://doi.org/10.1086/605122
Wu X, Yuan J, Luo A, Chen Y, Fan Y (2016) Drought stress and re-watering increase secondary metabolites and enzyme activity in Dendrobium moniliforme. Ind Crop Prod 94:385–393. https://doi.org/10.1016/j.indcrop.2016.08.041
Xia H, Yu S, Kong D, Xiong J, Ma X, Chen L, Luo L (2020) Temporal responses of conserved miRNAs to drought and their associations with drought tolerance and productivity in rice. BMC Genom 21:1–16. https://doi.org/10.1186/s12864-020-6646-5
Xiang XG, Mi XC, Zhou HL, Li JW, Chung SW, Li DZ, Huang WC, Jin WT, Li ZY, Huang LQ, Jin XH (2016) Biogeographical diversification of mainland Asian Dendrobium (Orchidaceae) and its implications for the historical dynamics of evergreen broad-leaved forests. J Biogeogr 43:1310–1323. https://doi.org/10.1111/jbi.12726
Xie F, Wang Q, Sun R, Zhang B (2015a) Deep sequencing reveals important roles of microRNAs in response to drought and salinity stress in cotton. J Exp Bot 66:789–804. https://doi.org/10.1093/jxb/eru437
Yang Z, Yang D, Ding X, Gao Y, Li D, Xu T (2014) MicroRNA expression profiles in conventional and micropropagated Dendrobium officinale. Genes Genom 37:315–325. https://doi.org/10.1007/s13258-014-0257-y
Yin F, Gao J, Liu M, Qin C, Zhang W, Yang A (2014) Genome-wide analysis of water-stress-responsive microRNA expression profile in tobacco roots. Funct Integr Genom 14:319–332. https://doi.org/10.1007/s10142-014-0365-4
Zhou LG, Liu YH, Liu ZC, Kong DY, Duan M, Luo LJ (2010) Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J Exp Bot 61:4157–4168. https://doi.org/10.1093/jxb/erq237
Zou LH, Wan X, Deng H, Zheng BQ, Li BJ, Wang Y (2018) RNA-seq transcriptomic profiling of crassulacean acid metabolism pathway in Dendrobium catenatum. Sci Data 5:180252. https://doi.org/10.1038/sdata.2018.252
Acknowledgements
This work was supported by Postdoctoral Research Project of West Anhui University (WXBSH2019003) and Anhui Province (2020B453), National Natural Science Foundation of China (31901383, 21705119 and C0031750), Nature Science Research Project of Anhui Province (1808085QB33), Natural Science Fund of Education Department of Anhui Province (KJ2017A408 and KJ2019A0628).
Author information
Authors and Affiliations
Contributions
The authors have made the following declarations about their contributions: Conceived and designed the experiments: YC, BH, and YW. Performed the experiments: YW, JD, RC, CS, PW, and YW. Analyzed the data: YW, JD, RC, CS, PW, and YW. Wrote the paper: YC, BH, and YW.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflict of interest.
Additional information
Handling Editor: Rhonda Peavy.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, Y., Dai, J., Chen, R. et al. miRNA-Based Drought Regulation in the Important Medicinal Plant Dendrobium huoshanense. J Plant Growth Regul 41, 1099–1108 (2022). https://doi.org/10.1007/s00344-021-10366-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00344-021-10366-7