
Abstract
Objective: Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is thought to be a promising anti-neoplastic agent because of its ability to selectively induce apoptosis in cancer cells. However, some cancer cells are resistant to TRAIL. The mechanisms underlying this resistance are unclear. The aim of this study was to explore the role of programmed cell death 4 (PDCD4) in regulating TRAIL sensitivity in gastric cancer cells.
Methods: PDCD4 complementary DNA and PDCD4-specific short-hairpin RNA (shRNA) fragments were transfected into TRAIL-sensitive and -resistant gastric cancer cells. Expression of PDCD4 and Akt was detected via western blot. Cell survival and apoptosis were measured using 3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT) and flow cytometry (FCM) assays.
Results: We found that upregulation of PDCD4 enhanced TRAIL sensitivity in gastric cancer cells. Downregulation of PDCD4 decreased TRAIL sensitivity. Inhibition of Akt by the phosphoinositide 3-kinase (PI3K) inhibitor LY294002 induced PDCD4 activity and enhanced TRAIL sensitivity in TRAIL-resistant gastric cancer cells.
Conclusion: We demonstrated that PDCD4 regulates TRAIL sensitivity in gastric cancer cells by inhibiting the PI3K/Akt signaling pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) selectively induces apoptosis in a wide variety of cancer cells but not in normal cells.[1,2] However, many cancer cells are resistant to TRAIL or develop resistance during therapy.[3] Different molecular factors have been proposed to confer TRAIL resistance, including decoy receptors, FLICE-inhibitory protein (FLIP), nuclear factor (NF)-κB, and activation of anti-apoptotic kinases.[4] It has been reported that TRAIL resistance in many types of cancer cells can be reversed by treatment with chemotherapeutic agents,[5] RNA synthesis inhibitors, and protein synthesis inhibitors.[6] Thus a combinatorial treatment using TRAIL or TRAIL receptor agonists and TRAIL-sensitizing agents could make up the molecular basis of a successful cancer treatment regimen.
Programmed cell death 4 (PDCD4) is a novel tumor suppressor that inhibits tumorigenesis, tumor progression, and tumor invasion.[7–9] It has also been found that PDCD4 inhibits neoplastic transformation.[10] Overexpression of PDCD4 inhibits 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced transformation in P+ cells and development of a tumor phenotype in transformed JB6 cells.[11,12] Expression of PDCD4 protein and/or messenger RNA is downregulated in advanced carcinomas in the lung, breast, colon, prostate,[13] brain,[14] and liver,[15] compared with adjacent normal tissues. Loss of PDCD4 in advanced tumors correlates with pathological grading and prognosis.[13,16] In addition, PDCD4 suppresses carcinoma cell invasion by inhibiting transactivation of the transcription factor activator protein-1 (AP1). It also promotes transforming growth factor-β (TGFβ)-induced apoptosis in the Huh7 human hepatocarcinoma cell line[15] and inhibits growth of tumor cells by suppressing carbonic anhydrase type II.[13,17] Furthermore, PDCD4 acts as a target for tumor therapy by inducing apoptosis, inhibiting angiogenesis, and enhancing sensitivity to antitumor drugs or radiotherapy.[18,19] However, the effect of PDCD4 on apoptosis induction in TRAIL-resistant gastric cell lines is poorly understood.
There are many factors contributing to resistance to TRAIL-induced apoptosis. Akt (protein kinase B), a serine/threonine protein kinase, is one of the important survival factors that contributes to TRAIL resistance.[20–22] Akt is activated by a phosphoinositide-3-kinase (PI3K)-dependent translocation to the cell membrane and subsequent phosphorylation at amino acids Thr308 and Ser473.[23,24] Akt then specifically phosphorylates Ser67 and Ser457 of PDCD4. Phosphorylation of PDCD4 by Akt causes nuclear translocation of PDCD4.[25]
The PI3K/Akt pathway regulates a number of normal cellular processes — including cell proliferation, survival, and motility — through phosphorylation of multiple downstream targets.[26] Dysregulation of the PI3K/Akt pathway has been found to be involved in the pathogenesis of several human cancers, such as breast, colon, ovarian, pancreas, and prostate cancer.[27–29] PI3K/Akt-dependent signaling pathways serve to regulate hypoxia-induced epithelial-mesenchymal transition in hepatocellular carcinoma cells.[30] Dysregulation of the PI3K/Akt/PTEN pathway is relevant to prognoses in node-negative breast carcinoma.[31]
The effects of PI3K/Akt signaling on PDCD4-mediated regulation of TRAIL-induced apoptosis in gastric cancer cells have not been studied. Here, we demonstrate that PDCD4 sensitizes TRAIL-resistant BGC823 gastric cancer cells to TRAIL-induced apoptosis. In contrast, TRAIL sensitivity is reduced after short-hairpin RNA (shRNA) knockdown of PDCD4. Furthermore, Akt inhibition by the PI3K inhibitor LY294002 enhances PDCD4 activity. This study is the first to demonstrate PDCD4-mediated regulation of TRAIL-induced apoptosis in TRAIL-resistant BGC823 gastric cancer cells, probably through inactivation of the PI3K/Akt pathway. This model may provide a novel framework for overcoming TRAIL resistance in other cancers.
Materials and Methods
Cell Lines and Antibodies
Human gastric carcinoma cell lines MKN28, BGC823, and SGC7901 were maintained in Dulbecco’s Modified Eagle Medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum at 37°C in a humidified 5% CO2 atmosphere. Soluble recombinant human (rh)TRAIL was purchased from ProSpec-Tany TechnoGene (Rehovot, Israel). PI3K inhibitor (LY294002) was obtained from Sigma-Aldrich. Anti-PDCD4, anti-TRAIL, and anti-phosphorylated-Akt (anti-p-Akt) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-b-actin and anti-rabbit horseradish peroxidase (HRP)-coupled antibody, nitrocellulose membranes, anti-mouse HRP-coupled antibody, and enhanced chemiluminescence (ECL) solutions were obtained from Boster Biological Technology (Wuhan, China).
Plasmid Construction and Transfection
PDCD4 complementary DNA (cDNA) was excised from the pBluescriptR-PDCD4 vector (Invitrogen, Carlsbad, CA, USA) using BamHI and EcoRI. The resulting fragment was sub-cloned into a pIRES-EGFP plasmid and transfected into DH5α Escherichia coli and maintained on LB plates supplemented with 30 µg/mL kanamycin, 20 µL x-gal, and 2 µL IPTG. Single white clones were selected and expanded in LB broth. Plasmid DNA was extracted, sequenced, and transfected into gastric cancer cells with Lipofactamine2000 according to the manufacturer’s protocol. PDCD4-overexpressing cell lines were established after screening with G418 (800µg/mL) for 3 weeks.
Targeted Downregulation of Programmed Cell Death 4 (PDCD4) by Short-Hairpin RNA
An shRNA fragment targeting PDCD4 (5′GTGCTTCTG AGTTCTA3′) was obtained from a database of shRNA gene-silencing constructs.[32] ShRNA was subcloned into the p-RNAT-U6.1/Neo carrier and transfected into MKN28 cells with Lipofactamine2000. Uninfected cells and control shRNA-infected cells were used as negative controls. After screening for 3 weeks with G418, a PDCD4-knockdown cell line was established.
Western Blot Analysis
Cells were lysed on ice for 30 minutes in lysis buffer (50mmol/L Tris-HCL, 150mmol/L NaCl, 5mmol/L EDTA, 1 mmol/L phenylmethylsulfonyl fluoride, and protease inhibitor). Approximately 50 µg of whole protein was denatured in 2×loading buffer at 100°C for 5 minutes, separated on an SDS-PAGE gel, and transferred to nitrocellulose membranes. Proteins were detected using specific antibodies and appropriate secondary antibodies, and visualized using ECL with X-films.
Cell Viability Assays and Assessment of Apoptosis
Cell viability was determined by 3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay. Gastric carcinoma cells were plated on 96-well plates at 15×103 cells per well. After the indicated treatments, cells were incubated for 2–3 hours with 0.5 mg/mL of MTT reagent and lysed with dimethyl sulfoxide. Absorbance was measured at 490 nm in a microplate reader (Bio-Rad, Richmond, CA, USA). Cell survival rates were determined by dividing the absorbance of treated cells by that of untreated cells. Tumor cells were grown and stained with annexin V/propidium iodide (PI). The apoptotic index was detected by flow cytometry (FCM) analysis according to the manufacturer’s protocol. Briefly, cells from the medium supernatant were collected and pelleted at 1200U/min. The pellets were washed twice with cold PBS and then re-suspended in a binding buffer at a concentration of 1×106 cells/mL, and 100 µL of the solution (1×105 cells) was transferred to each of two 5 mL culture tubes. Five microliters of Annexin V-FITC and 5 µL of PI were added into each 100 µL solution, and the cells were gently vortexed and incubated for 15 minutes at room temperature in the dark. Four hundred microliters of 1×binding buffer was added to the sample and analyzed by FCM within 1 hour.
Statistical Analysis
The data were expressed as means ± SDs of three or more independent experiments. Statistical analysis was performed using a two-tailed Student’s t-test for paired data. A p-value of < 0.05 was considered statistically significant.
Results
PDCD4 Expression Correlates with the Sensitivity of Gastric Cancer Cells to rhTRAIL
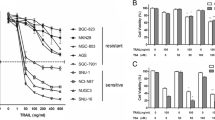
First, we investigated PDCD4 expression in the BGC823, SGC7901, and MKN28 gastric carcinoma cell lines by western blot analysis. We found that PDCD4 expression was lowest in BGC823 cells but highest in MKN28 cells (figure 1a). We also tested the cytotoxic effects of TRAIL on the three cell lines by MTT assay. We found that BGC823 cells were resistant to TRAIL in a dose- and time-dependent manner, whereas MKN28 cells were sensitive to TRAIL (figure 1b). To ascertain whether cell death occurred via apoptosis, we used AnnexinV/PI staining with FCM to determine the apoptotic index. The data showed that MKN28 cells were sensitive to TRAIL-induced apoptosis, but BGC823 were resistant in a time- and dose-dependent manner (figure 1c).

Expression of programmed cell death 4 (PDCD4) protein and sensitivity of gastric cancer cells (MKN28, SGC7901, and BGC823) to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). (a) Western blot analyses were performed with anti-PDCD4. Anti-β-actin was used as a control, (b) MKN28 cells were sensitive to TRAIL in a time- and dose-dependent manner, while BGC823 cells were resistant to TRAIL. Cell survival was measured by 3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay at different times or with different doses of TRAIL. The data are presented as means ± SDs. (c) TRAIL induces apoptosis in a time- and dose-dependent manner in MKN28 cells but not in BGC823cells. The cell apoptotic index (AI) was detected by flow cytometry. The data are presented as means ± SDs.
Upregulation of PDCD4 Enhances the Sensitivity of BGC823 Cells to TRAIL
Having demonstrated that PDCD4 expression was lower in BGC823 cells than in the other two cell lines, we trans-fected pIRES2-PDCD4 into BGC823 cells. After screening for 3 weeks with G418, a single-cell clone was selected to generate a cell line that stably overexpressed PDCD4 (figure 2a and figure 2b). After TRAIL treatment, the viability of transfected BGC823 cells was tested by MTT assay. Surprisingly, we found that PDCD4-overexpressing BGC823 cells were more sensitive to TRAIL than control cells (figure 2c). Moreover, the AI was significantly increased in transfected BGC823 cells (figure 2d). These data suggest that PDCD4 overexpression enhances the sensitivity of BGC823 cells to TRAIL.

BGC823 cells transfected with programmed cell death 4 (PDCD4) showed increased sensitivity to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). (a) BGC823 cells were observed by fluorescence microscopy. PDCD4-expressing cells display detectable levels of green fluorescent protein (GFP). (b) Expression of PDCD4 is upregulated in transfected BGC823 cells. Western blot analyses were performed with anti-PDCD4 antibody. Anti-β-actin was used as a control. (c) Overexpression of PDCD4 enhances TRAIL sensitivity of BGC823 cells in a time- and dose-dependent manner. Cell survival was measured by 3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay at different times or with different doses of TRAIL. The data are presented as means ± SDs. (d) Overexpression of PDCD4 enhances TRAIL-induced apoptosis in BGC823 cells. The apoptotic index (AI) was measured by flow cytometry. The data are presented as means ± SDs.
Inhibition of PDCD4 by shRNA Knockdown Reduces the Sensitivity of MKN28 Cells to TRAIL
Next, we used shRNA to specifically inhibit PDCD4 expression. We transfected PDCD4-specific shRNA into TRAIL-sensitive MKN28 cells, which express high levels of PDCD4, using the pRNAT-U6.1/Neo carrier vector. A cell line stably expressing PDCD4-specific shRNA was established (figure 3a and figure 3b). After TRAIL treatment, cell viability was determined by MTT assay, and the AI was measured by FCM. We found that TRAIL sensitivity and the AI of shRNA-transfected MKN28 cells were reduced (figure 3c and figure 3d). These data demonstrated that inhibition of PDCD4 expression inhibited the sensitivity of MKN28 cells to TRAIL.

Inhibition of programmed cell death 4 (PDCD4) by PDCD4-specific short hairpin RNA (shRNA) decreases tumor necrosis factor-related apop-tosis-inducing ligand (TRAIL) sensitivity in MKN28 cells. (a) MKN28 cells were observed by fluorescence microscopy. Transfected cells (shPDCD4) show detectable levels of green fluorescent protein (GFP). (b) PDCD4 expression is downregulated in transfected MKN28 cells. Western blot analyses were performed with anti-PDCD4 antibody. Anti-β-actin was used as a control. (c) Inhibition of PDCD4 decreases TRAIL sensitivity in MKN28 in a time- and dose-dependent manner. Cell survival was measured by 3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT) assay at different times or with different doses of TRAIL. The data are presented as means± SDs. (d) Downregulation of PDCD4 inhibits TRAIL-induced apoptosis in MKN28 cells. The apoptotic index (AI) was measured by flow cytometry. The data are presented as means ± SDs.
PDCD4 is Regulated by the PI3K/Akt Signaling Pathway in Gastric Cancer Cells
Finally, we attempted to identify regulators upstream of PDCD4. First, we examined Akt activity by western blot, using phospho-specific anti-Akt antibody in BGC823, SGC7901, and MKN28 gastric cancer cells. BGC823 cells showed maximal expression of active Akt among the cell lines tested, SGC7901 cells showed moderate expression, and MKN28 cells had minimal active Akt expression (figure 4a). We found that Akt activity was negatively correlated with PDCD4 expression in gastric cancer cells. Since BGC823 cells possess high levels of active Akt but low levels of PDCD4, we sought to examine whether inhibition of Akt could enhance PDCD4 expression and make cancer cells sensitive to TRAIL. To this end, we examined PDCD4 activity following inhibition of Akt activity using a PI3K inhibitor (LY294002). We found that PDCD4 expression was enhanced following Akt inhibition (figure 4b). These data suggest that expression of PDCD4 is regulated by the PI3K/Akt signaling pathway.

Programmed cell death 4 (PDCD4) expression is regulated by the phosphoinositide 3-kinase (PI3K)/Akt signaling pathway in gastric cancer cells (MKN28, SGC7901, and BGC823). (a) Akt expression varies in gastric cancer cell lines. Western blot analyses were performed with anti-PDCD4 and antiphosphorylated-Akt antibodies. Anti-β-actin was used as a control. (b) Inhibition of Akt activity enhances PDCD4 expression. Western blot analyses were performed after adding PI3K inhibitor LY294002. Anti-β-actin was used as a control.
Discussion
Although TRAIL is a potent inducer of apoptosis in cancer cells, many cancers are resistant to TRAIL. Many successful approaches have been devised to overcome this resistance, such as combining TRAIL treatment with reagents such as DNA-damaging agents,[33,34] ionizing radiation,[35] or viruses expressing wild-type tumor protein p53 (TP53).[36] In this study, we tested the effect of PDCD4 on TRAIL-induced apoptosis in TRAIL-resistant gastric cancer cell lines. First, we assessed the expression of PDCD4 and TRAIL sensitivity in different gastric cancer cells. We found that MKN28 cells were most sensitive to TRAIL and possessed the highest level of PDCD4; however, BGC823 cells were resistant to TRAIL and expressed minimal amounts of PDCD4. We found that restoring PDCD4 expression can sensitize TRAIL-resistant BGC823 cells to TRAIL-induced apoptosis. However, inhibition of PDCD4 expression by PDCD4-specific shRNA decreased TRAIL sensitivity in TRAIL-sensitive MKN28 cells. Our results suggest that PDCD4 is important in regulating the TRAIL sensitivity of gastric cancer cells and may be a suitable target molecule for drug-resistant gastric cancers. However, the mechanism by which PDCD4 enhances the sensitivity of gastric cancer cells to TRAIL-induced apoptosis is not clear.
The present study demonstrates that PDCD4 is down-regulated in TRAIL-resistant gastric cancer cells. This down-regulation is not only a marker of drug sensitivity but also contributes to decreased TRAIL sensitivity in BGC823 gastric cancer cells. TRAIL sensitivity increased significantly in TRAIL-resistant BGC823 cells expressing PDCD4 cDNA. Increased TRAIL sensitivity was accompanied by enhanced cell cytotoxicity and apoptosis. One primary mode of PDCD4 inactivation in gastric cancer appears to involve downregulated protein expression. This downregulation results in decreased sensitivity to TRAIL cytotoxicity. Thus upregulating PDCD4 expression may be promising for TRAIL-based combination therapy.
The Akt pathway is an important intracellular signal transduction pathway involved in drug resistance.[37] Increased expression of phosphorylated Akt (p-Akt) was frequently detected in gastric cancer and was correlated with increased vascular endothelial growth factor (VEGF) expression, angiogenesis, and poor prognosis.[38–40] In the present study, we showed that Akt activity differs in gastric cancer cells with different levels of TRAIL sensitivity. The activity of Akt in TRAIL-resistant BGC823 cells is much lower than in TRAIL-sensitive MKN28 cells, suggesting that Akt is a regulator of TRAIL sensitivity in gastric cancer.
PI3K is a major signaling component downstream of many growth factor receptor-associated tyrosine kinases.[41] PI3K activation leads to phosphorylation of Akt, which regulates a wide range of target proteins that control cell proliferation, survival, and cell growth.[23,29] Some previous studies have demonstrated that PDCD4 was phosphorylated by Akt,[42,43] and suppression of Akt enhanced PDCD4 expression.[44–47] Moreover, another study showed that expression of PDCD4 correlated inversely with p-Akt in colorectal cancer.[48] In this study, we found that PDCD4 expression can be inhibited by the PI3K/Akt signaling pathway and that inhibition of PI3K by LY294002 induces PDCD4 expression, indicating that PDCD4 enhances TRAIL sensitivity in gastric cancer cells by inhibiting the PI3K/Akt signaling pathway.[49]
Conclusion
Our study provides insight into the association between the PI3K/Akt pathway and TRAIL sensitivity in human gastric cancer cells. Inhibition of this pathway by LY294002, an inhibitor of PI3K, results in increased sensitivity to TRAIL. Additionally, we also demonstrated that PDCD4 enhances TRAIL sensitivity in gastric cancers by inhibiting the PI3K/Akt signaling pathway. Better understanding of the mechanism that regulates TRAIL sensitivity may help identify targets for gastric cancer therapy.
References
Yagita H, Takeda K, Hayakawa Y, et al. TRAIL and its receptors as targets for cancer therapy. Cancer Sci 2004; 95: 777–83
Kruyt FA. TRAIL and cancer therapy. Cancer Lett 2008; 263: 14–25
Ozoren N, El-Deiry WS. Cell surface death receptor signaling in normal and cancer cells. Semin Cancer Biol 2003; 13: 135–47
Panner A, Parsa AT, Pieper RO. Translational regulation of TRAIL sensitivity. Cell Cycle 2006; 5: 147–50
Ashkenazi A, Pai RC, Fong S, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest 1999; 104: 155–62
Griffith TS, Lynch DH. TRAIL: a molecule with multiple receptors and control mechanisms. Curr Opin Immunol 1998; 10: 559–63
Jansen AP, Camalier CE, Colburn NH. Epidermal expression of the translation inhibitor programmed cell death 4 suppresses tumorigenesis. Cancer Res 2005; 65: 6034–41
Hilliard A, Hilliard B, Zheng SJ, et al. Translational regulation of autoimmune inflammation and lymphoma genesis by programmed cell death 4. J Immunol 2006; 177: 8095–102
Yang HS, Matthews CP, Clair T, et al. Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion. Mol Cell Biol 2006; 26: 1297–306
Cmarik JL, Min H, Hegamyer G, et al. Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation. Proc Natl Acad Sci U S A 1999; 96: 14037–42
Yang HS, Jansen AP, Nair R, et al. A novel transformation suppressor, Pdcd4, inhibits AP-1 transactivation but not NF-kappaB or ODC transactivation. Oncogene 2001; 20: 669–76
Yang HS, Knies JL, Stark C, et al. Pdcd4 suppresses tumor phenotype in JB6 cells by inhibiting AP-1 transactivation. Oncogene 2003; 22: 3712–20
Goke R, Barth P, Schmidt A, et al. Programmed cell death protein 4 suppresses CDK1/cdc2 via induction of p21(Waf1/Cip1). Am J Physiol Cell Physiol 2004; 287: C1541–6
Gao F, Zhang P, Zhou C, et al. Frequent loss of PDCD4 expression in human glioma: possible role in the tumorigenesis of glioma. Oncol Rep 2007; 17: 123–8
Zhang H, Ozaki I, Mizuta T, et al. Involvement of programmed cell death 4 in transforming growth factor-beta1-induced apoptosis in human hepatocellular carcinoma. Oncogene 2006; 25: 6101–12
Chen Y, Knosel T, Kristiansen G, et al. Loss of PDCD4 expression in human lung cancer correlates with tumour progression and prognosis. J Pathol 2003; 200: 640–6
Lankat-Buttgereit B, Gregel C, Knolle A, et al. Pdcd4 inhibits growth of tumor cells by suppression of carbonic anhydrase type II. Mol Cell Endocrinol 2004; 214: 149–53
Jin H, Kim TH, Hwang SK, et al. Aerosol delivery of urocanic acid-modified chitosan/programmed cell death 4 complex regulated apoptosis, cell cycle, and angiogenesis in lungs of K-ras null mice. Mol Cancer Ther 2006; 5: 1041–9
Jansen AP, Camalier CE, Stark C, et al. Characterization of programmed cell death 4 in multiple human cancers reveals a novel enhancer of drug sensitivity. Mol Cancer Ther 2004; 3: 103–10
Chen X, Thakkar H, Tyan F, et al. Constitutively active Akt is an important regulator of TRAIL sensitivity in prostate cancer. Oncogene 2001; 20: 6073–83
Thakkar H, Chen X, Tyan F, et al. Pro-survival function of Akt/protein kinase B in prostate cancer cells: relationship with TRAIL resistance. J Biol Chem 2001; 276: 38361–9
Lane D, Robert V, Grondin R, et al. Malignant ascites protect against TRAIL-induced apoptosis by activating the PI3K/Akt pathway in human ovarian carcinoma cells. Int J Cancer 2007; 121: 1227–37
Bellacosa A, Chan TO, Ahmed NN, et al. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene 1998; 17: 313–25
Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem 1999; 68: 965–1014
Palamarchuk A, Efanov A, Maximov V, et al. Akt phosphorylates and regulates Pdcd4 tumor suppressor protein. Cancer Res 2005; 65: 11282–6
Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell 2003; 4: 257–62
Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci U S A 2001; 98: 10983–5
Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal 2002; 14: 381–95
Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer 2002; 2: 489–501
Yan W, Fu Y, Tian D, et al. PI3 kinase/Akt signaling mediates epithelial-mesenchymal transition in hypoxic hepatocellular carcinoma cells. Biochem Biophys Res Commun 2009; 382: 631–6
Capodanno A, Camerini A, Orlandini C, et al. Dysregulated PI3K/Akt/PTEN pathway is a marker of a short disease-free survival in node-negative breast carcinoma. Hum Pathol 2009; 40: 1408–17
Olson A, Sheth N, Lee JS, et al. RNAi Codex: a portal/database for short-hairpin RNA (shRNA) gene-silencing constructs. Nucleic Acids Res 2006; 34: D153–7
Nagane M, Pan G, Weddle JJ, et al. Increased death receptor 5 expression by chemotherapeutic agents in human gliomas causes synergistic cytotoxicity with tumor necrosis factor-related apoptosis-inducing ligand in vitro and in vivo. Cancer Res 2000; 60: 847–3
Kim K, Fisher MJ, Xu SQ, et al. Molecular determinants of response to TRAIL in killing of normal and cancer cells. Clin Cancer Res 2000; 6: 335–46
Chinnaiyan AM, Prasad U, Shankar S, et al. Combined effect of tumor necrosis factor-related apoptosis-inducing ligand and ionizing radiation in breast cancer therapy. Proc Natl Acad Sci U S A 2000; 97: 1754–9
Kim K, Takimoto R, Dicker DT, et al. Enhanced TRAIL sensitivity by p53 overexpression in human cancer but not normal cell lines. Int J Oncol 2001; 18: 241–7
Oki E, Baba H, Tokunaga E, et al. Akt phosphorylation associates with LOH of PTEN and leads to chemoresistance for gastric cancer. Int J Cancer 2005; 117: 376–80
Kobayashi I, Semba S, Matsuda Y, et al. Significance of Akt phosphorylation on tumor growth and vascular endothelial growth factor expression in human gastric carcinoma. Pathobiology 2006; 73: 8–17
Lin HL, Yang MH, Wu CW, et al. 2-Methoxyestradiol attenuates phosphatidylinositol 3-kinase/Akt pathway-mediated metastasis of gastric cancer. Int J Cancer 2007; 121: 2547–55
Ang KL, Shi DL, Keong WW, et al. Upregulated Akt signaling adjacent to gastric cancers: implications for screening and chemoprevention. Cancer Lett 2005; 225: 53–9
Cantley LC. The phosphoinositide 3-kinase pathway. Science 2002; 296: 1655–7
Dorrello NV, Peschiaroli A, Guardavaccaro D, et al. S6K1-and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006; 314: 467–71
Schmid T, Jansen AP, Baker AR, et al. Translation inhibitor Pdcd4 is targeted for degradation during tumor promotion. Cancer Res 2008; 68: 1254–60
Ozpolat B, Akar U, Steiner M, et al. Programmed cell death-4 tumor suppressor protein contributes to retinoic acid-induced terminal granulocytic differentiation of human myeloid leukemia cells. Mol Cancer Res 2007; 5: 95–108
Carayol N, Katsoulidis E, Sassano A, et al. Suppression of programmed cell death 4 (PDCD4) protein expression by BCR-ABL-regulated engagement of the mTOR/p70 S6 kinase pathway. J Biol Chem 2008; 283: 8601–10
Lankat-Buttgereit B, Muller S, Schmidt H, et al. Knockdown of Pdcd4 results in induction of proprotein convertase 1/3 and potent secretion of chromogranin A and secretogranin II in a neuroendocrine cell line. Biol Cell 2008; 100: 703–15
Woodard J, Sassano A, Hay N, et al. Statin-dependent suppression of the Akt/mammalian target of rapamycin signaling cascade and programmed cell death 4 up-regulation in renal cell carcinoma. Clin Cancer Res 2008; 14: 4640–9
Mudduluru G, Medved F, Grobholz R, et al. Loss of programmed cell death 4 expression marks adenoma-carcinoma transition, correlates inversely with phosphorylated protein kinase B, and is an independent prognostic factor in resected colorectal cancer. Cancer 2007; 110: 1697–707
Li D, Qu X, Hou K, et al. PI3K/Akt is involved in bufalin-induced apoptosis in gastric cancer cells. Anticancer Drugs 2009; 20: 59–64
Acknowledgments
Wei-Qiang Wang and Hao Zhang contributed equally to this work. The work was financially supported by the Scientific Research Foundation of the Chinese People’s Liberation Army, during the 10th Five-Year Plan Period, No. 01MA172.
The authors have no conflicts of interest that are directly relevant to the content of this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wang, WQ., Zhang, H., Wang, HB. et al. Programmed Cell Death 4 (PDCD4) Enhances the Sensitivity of Gastric Cancer Cells to TRAIL-Induced Apoptosis by Inhibiting the PI3K/Akt Signaling Pathway. Mol Diag Ther 14, 155–161 (2010). https://doi.org/10.1007/BF03256368
Published:
Issue Date:
DOI: https://doi.org/10.1007/BF03256368