
Abstract
In this chapter, we provide a comprehensive review of the recent developments and challenges associated with tuberculosis drug discovery. The chapter begins with an overview of the global TB burden with an emphasis on the high-burden countries such as India and the probable reasons associated with high disease burden. We have discussed the targets for the WHO End TB Strategy along with the requirements to achieve them. The chapter further provides an insight into the major obstacles of TB control, the problems associated with the current chemotherapy, the need for new anti-TB drugs and expectations from an ideal TB therapy. The chapter also provides a comprehensive review of the candidate drugs in the TB drug clinical pipeline with description of their identification, mechanistic action and in vitro and in vivo efficacy data along with clinical trial progress. We then provide details about the commonly employed approaches like whole cell phenotypic approach, target-based virtual screening and repurposing of drugs for TB drug discovery along with the advantages and major challenges associated with these approaches. In this regard, the success of whole cell-based phenotypic screening has been highlighted in view of discovery of the two recently FDA-approved anti-TB drugs, namely, bedaquiline and delamanid. The chapter also deals with another promising strategy for TB drug discovery based on rational drug design with a focus on some of the leads identified by this approach. We have also emphasized the recent advancements towards newer approaches like antisense RNA-based therapeutics, use of natural products, gene-editing tools such as CRISPR-CAS system and immunotherapy for the development of anti-TB molecules. Besides, the chapter also describes the development of methods to enhance the bioavailability of drugs such as novel delivery systems like nanoparticles/liposomes and devices for sustained release.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Tuberculosis
- Mycobacterium tuberculosis
- TB drugs
- Target-based virtual screening
- Whole cell phenotypic screening
- Repurposing approach
1 Global TB Scenario
Tuberculosis (TB) is a major global threat to public health. In spite of the presence of various intervention strategies against TB, the disease continues to persist and leads to loss of millions of human lives each year. TB is a complex disease due to multiple outcomes that can be manifested upon infection with the causative agent, Mycobacterium tuberculosis. The disease is caused by the inhalation of aerosol droplets containing the pathogen expelled from a diseased individual by coughing/sneezing.
TB is the ninth leading cause of human deaths worldwide and the leading cause of deaths from a single infectious agent, ranking above HIV/AIDS (WHO 2017). The epidemiology of the disease is indeed alarming and requires attention towards its urgent control. According to the WHO report on tuberculosis, 10.4 million people developed TB in 2016 worldwide, of which ~1.0 million were HIV positive (WHO 2017). 65% of these total TB incident cases were estimated to be in males and children accounted for 6.9% TB cases in the year 2016 (WHO 2017). Globally, 1.3 million HIV-negative people died of TB (down from 1.7 million in 2000) with an additional 0.37 million TB deaths observed in HIV-positive individuals in the year 2016 (WHO 2017). Drug-resistant TB continues to be a major threat with an estimated 0.6 million new cases resistant to rifampicin (RR-TB) in 2016 at a global level, of which 0.49 million had multidrug-resistant TB (MDR-TB). There were about 0.24 million deaths globally from MDR/RR-TB in 2016 (WHO 2017).

It has been observed over the years that India has been one of the highest TB-burden countries with high rate of incidence, mortality and resistant cases (WHO 2017). The reason for this can be largely attributed to the nature of the disease transmission, high population and overcrowding. Moreover, India also has major problems of malnutrition, poor hygienic conditions, poor supply of drugs and unregulated use of medicines, which have contributed in a big way to the high disease burden.
WHO End TB Strategy has proposed a target, with reference to the estimates of 2015, to achieve a 95% reduction in TB deaths and a 90% reduction in TB incidence (new cases per year) by 2035 (Uplekar et al. 2015). However, fulfilment of this goal demands providing TB care, preventive methods and awareness of health coverage at global level along with a deliberate collaboration among various stakeholders to tackle the socio-economic factors related to TB (WHO 2017). Moreover, it is of utmost importance to develop groundbreaking technological advancements in the next 5–7 years, whose implementation ought to result in reduction in the TB incidence rate at a level faster than in the past (WHO 2017).
2 Major Obstacles to TB Control
Since the discovery of M. tuberculosis by Robert Koch in 1882, as the causative agent of TB, the global TB epidemic till date remains persistent, highlighting the shortcomings of the current control measures available for combating tuberculosis. The bacteria spreads very easily through aerosols (a few droplet nuclei of 1–5 microns in diameter) which are coughed by an active TB patient and are inhaled by an uninfected person, and this easy transmission mode poses a big challenge to curtail the spread of this disease. BCG, the only vaccine available against TB, is highly effective in preventing childhood tuberculosis, but it is unsatisfactory in preventing pulmonary tuberculosis in adults showing a variable protective efficacy ranging from 0% to 80% (Colditz et al. 1994). The current diagnostic tests such as X-ray, Mantoux test, culture-based test and GeneXpert suffer from several limitations of being cost ineffective and time consuming and having shortcomings of sensitivity or specificity. Current efforts are being made towards the development of a better TB vaccine as well as an easy, rapid and effective diagnostic test.
The current chemotherapeutic regimen for the treatment of drug-susceptible tuberculosis consists of four first-line drugs, namely, rifampicin, isoniazid, pyrazinamide and ethambutol, administered for a span of 6 months (Guidelines for treatment of drug-susceptible tuberculosis and patient care (2017 update). http://apps.who.int/iris/bitstream/handle/10665/255052/9789241550000-eng.pdf;jsessionid=86C860DB7117D77D4A072A39ABCD6429?sequence=1). The standard treatment regimen for drug-susceptible TB comprises of 2 months of initiation phase consisting of all the four drugs (termed as 2HRZE), followed by a 4-month-long continuation phase consisting of rifampicin and isoniazid (termed as 4HR) (Guidelines for treatment of drug-susceptible tuberculosis and patient care (2017 update). http://apps.who.int/iris/bitstream/handle/10665/255052/9789241550000-eng.pdf;jsessionid=86C860DB7117D77D4A072A39ABCD6429?sequence=1). These drugs are administered in a daily dosing frequency, and the use of fixed-drug combination (FDC) has been recommended by WHO over separate drugs (Guidelines for treatment of drug-susceptible tuberculosis and patient care (2017 update). http://apps.who.int/iris/bitstream/handle/10665/255052/9789241550000-eng.pdf;jsessionid=86C860DB7117D77D4A072A39ABCD6429?sequence=1). This protracted therapy is one of the major reasons for the TB patients to default on the therapy, which leads to non-compliance and non-adherence. Thus, in spite of having an extremely effective treatment for treating active TB, the non-compliance to the therapy has led to an inexorable increase in the emergence of drug-resistant strains of the pathogen leading to drug-resistant TB cases (WHO guidelines for the programmatic management of drug-resistant tuberculosis. http://apps.who.int/iris/bitstream/handle/10665/130918/9789241548809_eng.pdf;jsessionid=BBA844744A619EF170C00783C655E148?sequence=1; Chiang et al. 2010). This kind of resistance can be classified as an acquired (secondary) drug resistance; however, primary drug resistance can also occur by infection with a drug-resistant strain of the pathogen (WHO guidelines for the programmatic management of drug-resistant tuberculosis. http://apps.who.int/iris/bitstream/handle/10665/130918/9789241548809_eng.pdf;jsessionid=BBA844744A619EF170C00783C655E148?sequence=1; Chiang et al. 2010). Resistant cases of TB can be divided into various categories depending on the kind of resistance to isoniazid (isoniazid-resistant TB), to rifampicin (RR-TB), to both rifampicin and isoniazid (MDR-TB), to any fluoroquinolone and to at least one of the injectable second-line drugs (amikacin, kanamycin, capreomycin) in addition to both rifampicin and isoniazid (XDR-TB) (WHO guidelines for the programmatic management of drug-resistant tuberculosis. http://apps.who.int/iris/bitstream/handle/10665/130918/9789241548809_eng.pdf;jsessionid=BBA844744A619EF170C00783C655E148?sequence=1; Chiang et al. 2010). Treatment of the drug-resistant strains requires the use of various combinations of the second-line drugs such as levofloxacin, moxifloxacin, gatifloxacin, amikacin, kanamycin, capreomycin, streptomycin, ethionamide, cycloserine, linezolid and clofazimine. The therapy for resistant cases may last anywhere from 9 to 24 months. The emergence of these multidrug-resistant (MDR) strains poses a serious challenge to the world’s health and towards the global control of this disease. It has been estimated that an active TB patient undergoing the treatment may transmit the infection to at least ten uninfected people suggesting the requirement of stringent control measures (www.who.int/mediacentre/factsheets/fs104/en/). Besides, the unpleasant side effects and a high pill burden of these drugs make the treatment of drug-resistant TB and compliance to the therapy an extremely daunting task. The prevalence of HIV makes the situation even more precarious due to an enhanced susceptibility of the HIV-infected immunocompromised people to TB infection. Additionally, the situation is complicated by the difficulty faced by using these anti-TB drugs along with antiretroviral therapy, which shows negative drug-drug interactions and, hence, precludes the use of these anti-TB drugs in HIV-positive patients (López-Cortés et al. 2002). Further, the key challenge is also to treat individuals who are subclinically infected with M. tuberculosis and are at a lifetime risk of reactivation TB due to various reasons such as HIV infection, anti-TNF therapy, diabetes, malnutrition or lowering of immunity (Narayanan et al. 2010; Gardam et al. 2003; Stevenson et al. 2007). The complexity also arises due to the fact that the pathogen has the ability to modulate the immune system thereby evading the immune surveillance. Its ability to persist in a latent and low metabolic state for years poses significant challenge like drug tolerance. Hence, latent TB disease requires several months of treatment for complete sterilization.
Thus, in view of the above challenges in the TB treatment, it is of utmost importance to develop novel chemotherapeutic regimens that are able to (i) reduce the duration of this long drawn therapy, (ii) reduce the pill burden, (iii) target the latent pathogen, (iv) target drug-resistant strains of the pathogen and (v) can be easily co-administered with HIV medication.

Although considerable efforts have been made in the field of TB drug discovery as is evident from a relatively filled drug pipeline now as compared to a few decades earlier, the progress of TB drug discovery program has been extremely slow with only two new anti-TB drugs, bedaquiline (marketed as Sirturo) and delamanid, receiving the FDA approval in the last 50 years that too with limited access and reserved only for the treatment of MDR-TB. Hence, looking at the high attrition rate in the TB drug discovery, it is important to develop robust approaches for identifying better candidate drug molecules that can be channelled for clinical assessment so that more anti-TB drugs can reach the market.
This chapter provides a comprehensive review of the recent developments in the field of TB drug discovery and addresses the major challenges associated with the current approaches being employed for the identification of new drugs.
3 TB Drug Clinical Pipeline: The Prospective Future Anti-TB Drugs
Although the TB drug pipeline remained almost empty for a long period of time till the 1990s, it is indeed optimistic to see that it is currently filled with more than 20 molecules, which are being assessed under different clinical phases (https://www.newtbdrugs.org/pipeline/clinical) (Fig. 25.1). Moreover, apart from the molecules in clinical trials, many more molecules are in the lead optimization phase or in the early preclinical stages and have the promise of entering the clinical pipeline (Fig. 25.1) (https://www.newtbdrugs.org/pipeline/clinical).

Candidate anti-TB drugs in various stages of clinical trials
The number of candidate molecules in the TB drug pipeline provides no assurance that these molecules may reach the advance stages of clinical development as is evident from the fact that only few molecules are presently being evaluated in phase III and almost 50% are being evaluated as combination regimens of these drugs. This highlights a high attrition rate and the necessity of more molecules to fill the pipeline. Besides, the failure of the current candidate molecules reflects the limitations and caveats of the existing drug discovery approaches and emphasizes the importance of devising innovative strategies and tools to develop new molecules or increase the efficacy of molecules that qualify for clinical trials.
The anti-TB molecules currently in the clinical pipeline belong to various classes like fluoroquinolones, diarylquinolines, nitroimidazoles, benzothiazinones, etc., targeting various proteins/enzymes/pathways of M. tuberculosis including cell wall biosynthesis enzymes, energy metabolism and protein synthesis. Most of these molecules have been identified either through whole cell phenotypic screening or by repurposing of the drugs already in use for other diseases. Candidate drugs in TB pipeline are mentioned below.
3.1 BTZ043
The antitubercular agent BTZ043 (belonging to the nitrobenzothiazinone (BTZ) class) specifically blocks the mycobacterial enzyme decaprenyl-phosphoribose-2′-epimerase (DprE1), responsible for the synthesis of a cell wall component D-arabinofuranose, and shows MIC in nanomolar range against the members of the M. tuberculosis complex (Makarov et al. 2009). BTZ043 showed superior antitubercular activity in comparison to isoniazid in vivo in mouse model with a low toxicological potential and was also well tolerated in rats and mini pigs (Kloss et al. 2017). The molecule is currently being evaluated in phase I trial (https://www.newtbdrugs.org/pipeline/clinical).
3.2 Contezolid (MRX-4/MRX-1)
Linezolid (LZD) belongs to oxazolidinone class of antibiotics that showed potent in vitro and in vivo activities against M. tuberculosis and was subsequently used in humans to treat drug-resistant TB; however, its use was restricted due to toxicity issues, including myelosuppression and peripheral and optic neuropathy (Mehta et al. 2016; Lee et al. 2012). Contezolid (MRX-1), a new oxazolidinone, was developed to treat gram-positive infections, such as methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci, and showed decreased toxicity as compared to LZD (Gordeev and Yuan 2014; Shoen et al. 2018). MRX-1 showed promising in vitro as well as in vivo activity against both drug-susceptible and drug-resistant M. tuberculosis in mice model and is currently being evaluated in phase III trials (https://www.newtbdrugs.org/pipeline/clinical, Gordeev and Yuan 2014).
3.3 OPC-167832
OPC-167832, a newly synthesized carbostyril derivative discovered by Otsuka healthcare, inhibits decaprenylphosphoryl-β-D-ribose 2′-oxidase (DprE1), essentially involved in cell wall biosynthesis of M. tuberculosis (http://www.cptrinitiative.org/wp-content/uploads/2017/05/Jeffrey_Hafkin_CPTR2017_JH.pdf). This anti-TB compound shows in vitro as well as in vivo efficacy against both laboratory and clinically isolated strains including multidrug-resistant and extensively drug-resistant M. tuberculosis (http://www.cptrinitiative.org/wp-content/uploads/2017/05/Jeffrey_Hafkin_CPTR2017_JH.pdf). Furthermore, OPC-167832, when administered along with delamanid, showed superior efficacy to the standard regimen RHZE (rifampicin + isoniazid + pyrazinamide + ethambutol) in mice (http://www.cptrinitiative.org/wp-content/uploads/2017/05/Jeffrey_Hafkin_CPTR2017_JH.pdf). OPC-167832 is presently being evaluated in phase I trial (https://www.newtbdrugs.org/pipeline/clinical).
3.4 GSK070
GSK070 is an oxaborole derivative that targets the leucyl-tRNA synthetase (LeuRS) required for charging the tRNALue with leucine, thereby inhibiting protein synthesis (Palencia et al. 2016; Rock et al. 2007). GSK70 was observed to demonstrate in vitro and in vivo efficacy against M. tuberculosis with potent enzyme inhibition of M. tuberculosis LeuRS (IC50 = 0.216 μM) (Palencia et al. 2016). GSK70 recently qualified for phase I clinical assessment (https://www.newtbdrugs.org/pipeline/clinical).
3.5 Macozinone (PBTZ169)
Lead optimization studies of the compound BTZ043 by using medicinal chemistry resulted in PBTZ169, which is a piperazinobenzothiazinone derivative. PBTZ169 covalently binds to DprE1, thereby inhibiting cell wall biosynthesis (Trefzer et al. 2010; Makarov et al. 2014). PBTZ169 has an MIC99 against M. tuberculosis in nanomolar range (0.3 ng/ml) and has shown additive effects with many TB therapeutic agents and has also demonstrated synergistic effects with bedaquiline and clofazimine in preclinical models (Makarov et al. 2014). Innovative Medicines for Tuberculosis (iM4TB) foundation (Lausanne, Switzerland) has initiated the phase I clinical study of PBTZ169 to investigate dose-related safety (https://www.newtbdrugs.org/pipeline/compound/macozinone-mcz-pbtz-169).
3.6 TBI-166
Clofazimine, which is a very potent anti-TB compound belonging to riminophenazine class of drugs, has shown extremely impressive bactericidal and sterilizing efficacy against TB both in vitro and in mouse models of the disease (Reddy et al. 1996). However, it has poor solubility and a long half-life which results in side effects including pronounced skin discoloration (Job et al. 1990; Levy and Randall 1970). Lead optimization of clofazimine led to the identification of TBI-166, which has shown improved physicochemical and pharmacokinetic properties with similar efficacy as the parent compound (Lu et al. 2011). Based on these preliminary, preclinical and toxicological studies, TBI-166 was approved for phase I clinical trials (https://www.newtbdrugs.org/pipeline/clinical).
3.7 TBA-7371
TBA-7371 is a novel molecule belonging to pyrazolopyridone class of inhibitors (1,4-azaindole series) identified by performing lead optimization studies of an imidazopyridine compound. This molecule inhibits DprE1 (decaprenylphosphoryl-β-D-ribose 2′-epimerase) by binding to it non-covalently and shows an IC50 value of 10 nM with an MIC range of 0.78–3.12 μM and demonstrates efficacy in a rodent model of tuberculosis (Shirude et al. 2013, 2014; Yuan and Sampson 2018). The TB Alliance has initiated its phase I trial to evaluate its safety, tolerability, pharmacokinetics and the pharmacokinetic interactions (https://www.newtbdrugs.org/pipeline/clinical).
3.8 Telacebec (Q203)
Q203 resulted from the lead optimization of imidazo[1,2-a]pyridine amides that target the respiratory cytochrome bc complex, which in turn disturbs the electron motive force (Pethe et al. 2013; Lu et al. 2018; Kang et al. 2014). The small molecule Q203 inhibits the growth of MDR and XDR M. tuberculosis clinical isolates in culture broth medium with MIC in the low nanomolar range as well as shows therapeutic efficacy in mice (Pethe et al. 2013; Lu et al. 2018; Kang et al. 2014). In addition, Q203 displayed good pharmacokinetic and safety profiles; hence, it was qualified for the phase I trial for a dose escalation study which revealed encouraging results. Phase II trial for early bactericidal activity (EBA) evaluation of Q203 will be soon initiated in South Africa (https://www.newtbdrugs.org/pipeline/compound/telacebec-q203).
3.9 Sutezolid (Previously Known as PNU-100480)
Sutezolid exhibited increased in vitro as well as in vivo antimycobacterial activity, when compared with linezolid against both drug-susceptible and drug-resistant TB and showed improved safety profile highlighting its potential as an anti-TB agent (Barbachyn et al. 1996; Cynamon et al. 1999; Shaw and Barbachyn 2011; Wallis et al. 2010, 2011; Alffenaar et al. 2011). In fact, recent studies also showed that the use of sutezolid along with standard therapy was able to shorten the treatment duration by preventing relapse, thus, suggesting that sutezolid may have sterilizing activity against drug-susceptible TB and MDR-TB (Barbachyn et al. 1996; Shaw and Barbachyn 2011). Sutezolid was well tolerated and safe up to a daily dose of 1200 mg up to 14 days and phase II trials showed early bactericidal activity. Hence, it is considered that sutezolid may show clinical efficacy in a larger phase II trial (https://www.newtbdrugs.org/pipeline/clinical, Wallis et al. 2010).
3.10 Delpazolid (LCB01-0371)
LCB01-0371 is also a new oxazolidinone compound similar to linezolid with cyclic amidrazone. In vitro activity of LCB01-0371 was found to be similar to linezolid with an improved safety profile (Zong et al. 2018; Kim et al. 2017; Jeong et al. 2010). In vivo activity of LCB01-0371 against systemic infections in mice was also evaluated, and it was found to be more active than linezolid against these systemic infections (Zong et al. 2018; Kim et al. 2017; Jeong et al. 2010). It is now in phase II trial for the evaluation of EBA studies (https://www.newtbdrugs.org/pipeline/clinical).
3.11 SQ109
SQ109 is a novel 1,2-ethylenediamine molecule having a novel mechanism of action targeting MmpL3, which is a mycolic acid transporter required for mycolic acid incorporation into the M. tuberculosis cell wall (Tahlan et al. 2012; Grzegorzewicz et al. 2012). SQ109 inhibited the growth of both drug-susceptible and multidrug-resistant M. tuberculosis strains, including extensively drug-resistant M. tuberculosis strains (Sacksteder et al. 2012; Protopopova et al. 2005). It also exhibited synergistic effect with no adverse pharmacokinetic (PK) parameters and also improved the overall efficacy of the regimen when given along with standard treatment in mice (Sacksteder et al. 2012; Chen et al. 2006; Nikonenko et al. 2007). Three phase I studies are completed for SQ109 in the USA along with two phase II studies in Africa in drug-sensitive TB patients (https://www.newtbdrugs.org/pipeline/compound/sq109).
3.12 Auranofin (Brand Name: Ridaura)
Auranofin is a gold complex FDA-approved drug to treat rheumatoid arthritis (Suarez-Almazor et al. 2000). The drug is able to reduce and improve arthritis-related symptoms like pain, tender and swollen joints and morning stiffness. Auranofin was identified by employing a cell-based screen under nutrient-deprivation conditions against M. tuberculosis (Harbut et al. 2015). It exhibits potent inhibition of the growth of both replicating and nonreplicating M. tuberculosis, which was found to be bactericidal in nature (Harbut et al. 2015). Phase II trial is initiated to study the efficacy of the auranofin against M. tuberculosis (https://www.newtbdrugs.org/pipeline/clinical).
3.13 Levofloxacin
Levofloxacin is a second-generation fluoroquinolone, which shows enhanced activity against gram-positive pathogens, including S. pneumoniae and S. aureus, and was shown to be effective in the treatment of upper and lower respiratory tract infections in adults (Peterson et al. 2009; Alsultan et al. 2015). Levofloxacin is one of the essential medicines listed by World Health Organization and may be used for the treatment of tuberculosis (https://www.newtbdrugs.org/pipeline/compound/levofloxacin). TBTC 32/NIAID OPTI-Q phase II studies will determine the levofloxacin dose and exposure required to achieve the maximal reduction in M. tuberculosis burden in Peru and South Africa (https://www.newtbdrugs.org/pipeline/compound/levofloxacin).
3.14 Nitazoxanide (NTZ)
NTZ is a synthetic nitrothiazolyl salicylamide prodrug that is deacetylated in the gastrointestinal tract to the active metabolite tizoxanide and is approved for the treatment of giardiasis and cryptosporidiosis (Aslam and Musher 2007). NTZ showed activity against other protozoa, helminths, rotavirus and hepatitis C (Aslam and Musher 2007; Stachulski et al. 2011; Adagu et al. 2002; Theodos et al. 1998; Darling and Fried 2009; Korba et al. 2008; Rossignol et al. 2008, 2010). It was also shown to kill both replicating and nonreplicating M. tuberculosis with a MIC of 16 μg/ml (de Carvalho et al. 2009). No resistant mutants were found on treatment of M. tuberculosis with various concentrations of NTZ suggesting that NTZ may have multiple targets (de Carvalho et al. 2009). It is currently undergoing phase II efficacy trial (https://www.newtbdrugs.org/pipeline/clinical).
3.15 Combination Regimens
The drugs that have successfully completed the phase II efficacy trials are now being evaluated in combination regimens in phase III trials to evaluate their therapeutic efficacy in shortening the treatment regimen.
3.16 Bedaquiline
Bedaquiline belongs to the class of diarylquinolines with a novel mechanism of action targeting the mycobacterial ATP synthase (Andries et al. 2005; Koul et al. 2007). It was found to exhibit activity against both drug-susceptible and drug-resistant strains of the pathogen having a strong bactericidal and sterilizing properties (Andries et al. 2005; Diacon et al. 2009). Bedaquiline got US FDA approval in 2012 as an anti-TB drug; however, its usage is reserved for the treatment of MDR-TB cases only (https://www.newtbdrugs.org/pipeline/compound/bedaquiline-0). It is now being evaluated in various combination regimens in which bedaquiline and pretomanid will be administered along with existing and repurposed anti-TB drugs for the treatment of biologically confirmed pulmonary multidrug-resistant TB (MDR-TB) (https://www.newtbdrugs.org/pipeline/compound/bedaquiline-0). Besides, another trial is scheduled to be carried out to assess its efficacy, when administered with delamanid (https://www.newtbdrugs.org/pipeline/compound/bedaquiline-0).
3.17 Rifapentine
Rifapentine is a semisynthetic derivative of rifamycin family with MIC99 value of 0.25 μg/ml in liquid medium (Sensi et al. 1959; Bemer-Melchior et al. 2000). It is currently approved for intermittent dosing in the treatment of TB. Also, it is currently included in combination regimen for phase III trials to test whether these regimens can shorten the treatment duration (https://www.newtbdrugs.org/pipeline/compound/rifapentine). The regimens being evaluated are (i) a single replacement of rifampin with rifapentine, initial 2 months of isoniazid, rifapentine, ethambutol and pyrazinamide, followed by another 2 months of isoniazid and rifapentine, and (ii) a double replacement of rifampin with rifapentine and ethambutol with moxifloxacin – initial 2 months of isoniazid, rifapentine, moxifloxacin and pyrazinamide, followed by another 2 months of isoniazid, rifapentine and moxifloxacin (https://www.newtbdrugs.org/pipeline/compound/rifapentine).
3.18 Delamanid
Delamanid (OPC-67683, Deltyba®) belongs to bicyclic nitroimidazole class of compounds, which showed a marked antituberculosis activity in vitro and a superior therapeutic efficacy in the chronic mouse model (Matsumoto et al. 2006; Tsubouchi et al. 2016). In 2014, European Medicines Agency (EMA) approved delamanid for the treatment of adult pulmonary MDR-TB (Yuan and Sampson 2018). Delamanid-resistant mutants revealed that it inhibits genes involved in F420-dependent deazaflavin nitroreductase bioactivation pathway (Fujiwara et al. 2018). In addition, in a phase IIb global trial, delamanid was found to increase the rate of 2-month sputum culture conversion, when it was added to an already optimized background regimen for the treatment of MDR-TB patients (Diacon et al. 2011; Gler et al. 2012). Clinical studies also revealed the efficacy of delamanid containing regimens in highly resistant TB patients that included cases with extensively drug-resistant TB (Skripconoka et al. 2013). Currently, for the evaluation of safety and efficacy of delamanid at a 200 mg oral daily dose, a phase III trial is being conducted (https://www.newtbdrugs.org/pipeline/compound/delamanid-0). Moreover, combined usage of delamanid and bedaquiline is also in progress to evaluate whether their combination can enhance the efficacy against MDR-TB (https://www.newtbdrugs.org/pipeline/compound/delamanid-0).
3.19 Clofazimine
Clofazimine, as described above, shows potent antitubercular activity (Reddy et al. 1996; Xu et al. 2012) and is now being evaluated in phase III trials for its efficacy, safety and tolerability, when it is administered in various regimens such as (i) TMC207 plus PA-824 plus pyrazinamide plus clofazimine, (ii) TMC207 plus PA-824 plus clofazimine, (iii) TMC207 plus pyrazinamide plus clofazimine and (iv) clofazimine alone, in adult patients with newly diagnosed, smear-positive pulmonary tuberculosis (https://www.newtbdrugs.org/pipeline/compound/clofazimine).
3.20 Pretomanid-Moxifloxacin-Pyrazinamide Regimen
Pretomanid (PA-824, Pa) belongs to nitroimidazo-oxazine class of compounds, which shows a very potent MIC of 0.125 μg/ml against M. tuberculosis and shows bactericidal activity during the initial and continuation phases of treatment in murine model with no issues of cross-resistance with other existing TB drugs (Stover et al. 2000; Tyagi et al. 2005). In addition, combination of PA-824, moxifloxacin and pyrazinamide (PaMZ) cured mice more rapidly than the first-line regimen of rifampin, isoniazid and pyrazinamide (Nuermberger et al. 2008; Tasneen et al. 2011).
PaMZ represents the first regimen to undergo clinical evaluation for multidrug TB treatment and has shown very positive results in phase II trial to conclude that it has potential of curing both the susceptible and resistant TB (https://www.newtbdrugs.org/pipeline/regimen/pretomanid-moxifloxacin-pyrazinamide-regimen). Besides, PaMZ also had better co-administration ability with the antiretrovirals, which is useful as an improved treatment option for HIV-TB co-infected patients (https://www.newtbdrugs.org/pipeline/regimen/pretomanid-moxifloxacin-pyrazinamide-regimen). TB Alliance is now focusing on advancing the BPaMZ regimen consisting of bedaquiline, PA-824, moxifloxacin and pyrazinamide (https://www.newtbdrugs.org/pipeline/regimen/pretomanid-moxifloxacin-pyrazinamide-regimen).
4 Advantages and Pitfalls of Various TB Drug Discovery Approaches
4.1 Target-Based Virtual Screening
The growing burden of antibiotic resistance propelled research towards the development of new antibiotics resulting in various drug discovery approaches. The genome sequence of M. tuberculosis was elucidated in the year 1998 (Cole et al. 1998), which gave a tremendous boost to the approaches such as target-based virtual screening, generation of gene knockout strains for the identification and validation of drug targets and system biology, etc. Target-/structure-based virtual screening relies on the use of computer-based software to identify potential inhibitors against the target structure by docking a library of small molecules into the active site and shortlisting the best binding molecules. An important prerequisite for this approach is the availability of a three-dimensional structure of the target protein, which gained its pace after the determination of genome sequence of M. tuberculosis. Another crucial step involved in virtual screening is the identification of an important drug target, which plays a key role in the pathogenesis of M. tuberculosis. This became possible as the knowledge about M. tuberculosis genes increased with the availability of its genome sequence, which also aided in developing gene deletion mutants. These knockout strains are then evaluated for their ability to grow in vitro, inside macrophages and subsequently in animal models for final validation; however, the major limitation of this technique, for a long time, has been a low frequency of site-specific recombination making it difficult to obtain the mutants and the inherent challenge of slow growth rate of M. tuberculosis. With newer methods of recombineering that were subsequently developed based on linear allelic exchange substrate, transposon mutagenesis and mycobacteriophage systems, generating knockout strains in M. tuberculosis became easier and the validation of essential and important drug targets gained a much faster pace (Van Kessel and Hatfull 2007; Sassetti et al. 2001). A study by Sassetti et al. revolutionized the drug discovery efforts by identifying several M. tuberculosis genes required for the in vitro and in vivo growth of the pathogen by using transposon mutagenesis, which led to a huge list of important drug targets along with the information on their essentiality for the growth of the pathogen (Sassetti et al. 2001; Sassetti and Rubin 2003). For instance, enzymes belonging to energy metabolism were shown to be essential for the growth of M. tuberculosis by Sassetti et al., and this information led to the identification of Q203 as an important small molecule inhibiting the cytochrome bc1 complex, which is currently being evaluated in clinical trials (https://www.newtbdrugs.org/pipeline/clinical, Kang et al. 2014). Additionally, many drug targets including UDP-galactopyranose mutase, 2C-methyl-D-erythritol 4-phosphate pathway and enzymes belonging to purine nucleotide biosynthetic pathway (GuaB2) were also shown to be essential by transposon mutagenesis by Sassetti et al. and have been employed as drug targets (Kincaid et al. 2015; Eoh et al. 2009; Singh et al. 2017). In addition, many other deletion mutants have been developed by using the above-mentioned methods, which have helped in the identification of several drug targets including MbtE, SapM, BioA, PptT, DrpE1, Rv3484, etc. (Reddy et al. 2013; Puri et al. 2013; Kar et al. 2017; Leblanc et al. 2012; Crellin et al. 2011; Malm et al. 2018). For instance, the mbtE mutant was developed by using linear AES method of homologous recombination, and MbtE was shown to be essential for the virulence and survival of the pathogen in broth culture and in macrophages. Besides, the mutant was also shown to be attenuated for its growth in guinea pigs as infection with it exhibited significantly reduced bacillary load and histopathological damage in the organs, in comparison to M. tuberculosis-infected animals (Reddy et al. 2013).
Target-based virtual screening has been employed extensively since the last decade, and till date many compounds have been screened against many important drug targets. A screening effort that evaluated 20,000 compounds against FtsZ, which plays an essential role in cell division, led to the identification of the inhibitor 297F, which showed inhibitory activity against M. tuberculosis in vitro growth (Lin et al. 2014). In another study, inhibitors were identified against M. tuberculosis thiamine phosphate synthase, an enzyme involved in the biosynthesis of thiamine by employing virtual screening resulting in promising inhibitors with IC50 of 34 μg/ml and MIC99-6 μg/ml (Khare et al. 2011). In a recent study by Singh et al., structure-based virtual screening was employed against the active site of BioA, an enzyme involved in biotin biosynthetic pathway, which is essential for M. tuberculosis in vivo survival. It resulted in a few hits with the most potent hit displaying an MIC90 of 20 μg/ml (Singh et al. 2018). In another study, virtual screening was carried out by employing NCI library at the active site of M. tuberculosis 4′-phosphopantetheinyl transferase (PptT), which is involved in phosphopantetheinylation of several important proteins in a post-translational manner (Rohilla et al. 2018). This study led to the identification of a number of molecules with potent inhibition of the PptT enzymatic activity (IC50 ≤ 10 μg/ml). Further, by employing a structure similarity approach based on chemoinformatics, a potent analogous molecule was identified with IC50 of 0.25 μg/ml, MIC90 of 10 μg/ml and negligible cytotoxicity (Rohilla et al. 2018). Recently, a study by Rohilla et al. showed that virtual screening of NCI library against IdeR, an essential iron regulatory transcriptional factor of M. tuberculosis, identified potent hits exhibiting IC50 values of 1–2 μg/ml (Rohilla et al. 2017). Major advantage of target-based screening is that there is a prior knowledge about the structure of the compound in question and the drug target, which makes it easier to elucidate the mechanism of action of the inhibition observed. Besides, the structural as well as the chemical knowledge about the compound helps in performing better lead optimization studies to obtain an improved antitubercular compound.
One of the major limitations of this strategy is the determination of the crystal structure of many M. tuberculosis proteins as they usually do not express well in E. coli and remain insoluble when expressed. Methods such as comparative homology modelling provide an alternative for the absence of crystal structures; however, they may not always result in potent inhibitors. Additionally, although this strategy holds promise in identifying a potent inhibitor against the target, many resulting enzyme inhibitors are unable to show a good MIC value in vitro (Singh et al. 2018; Rohilla et al. 2017, 2018; Kumar et al. 2017). M. tuberculosis is a complex pathogen comprising of a very strong cell wall which hampers the entry of most of the small inhibitor molecules, which are screened and shortlisted by employing target-based virtual screening. Even though the shortlisted molecules that are identified by screening chemical libraries against a validated target show high binding affinity for the target, their inability to enter the cell due to highly impermeable and hydrophobic cell wall of the pathogen might prevent them from arresting the cell growth. Besides, efflux of the compounds by the mycobacterial cells may also contribute to their poor MIC values (Kumar et al. 2017; Rodrigues et al. 2012; Zuniga et al. 2015; Kanji et al. 2016; Zhang et al. 2017; Parthasarathy et al. 2016). For instance, target-based virtual screening against many targets including PimA and PanC, both shown to be essential drug targets, resulted in the identification of several hits by screening many chemical libraries; however, none of the shortlisted molecules showed cellular activity emphasizing the possibility of their lack of entry into the cell (Kumar et al. 2017). Moreover, since M. tuberculosis is highly lipid rich, it is expected that a more lipophilic molecule will have a better permeability inside the mycobacterial cells and thus may be more potent as it has also been seen in the case of current anti-TB drugs, which are peculiarly more lipophilic than inhibitors against other bacteria (Kumar et al. 2017; Piccaro et al. 2015; Machado et al. 2018). However, in medicinal chemistry, the physicochemical characteristics such as high lipophilicity are not considered favourable for the development of a molecule into drug. This may explain the high attrition rate in the virtual screening approach with no identified compound reaching the TB drug pipeline, since most of the libraries selected for the screening are chosen as per the existing drug-likeness rules and exclude the lipophilic molecules. Hence, it seems more logical to reconsider the drug-likeness rules in the case of TB drug discovery, and new thinking beyond the existing dogma of Lipinski’s rule of five is needed to succeed (Piccaro et al. 2015; Machado et al. 2018).
4.2 Whole Cell Phenotype Screening
The fact that none of the molecules that are being evaluated in clinical pipeline are derived from structure-based screening, there was a paradigm shift from computational methods to whole cell phenotypic screening approach for the identification of new anti-TB agents having novel mechanism of action. The method involves direct screening of small molecule libraries against the growth of M. tuberculosis in broth culture. The use of this approach gained pace after the discovery of bedaquiline (TMC207) and the successful identification of its target ATP synthase through isolation of resistant colonies and whole genome sequencing (Andries et al. 2005). Moreover, the success of this strategy is evident from the number of drugs in the clinical pipeline, which have been identified by whole cell phenotypic screening. Apart from bedaquiline (TMC207), various molecules such as SQ109, OPC-67683, PBTZ-169 and Q203 have been identified by employing this approach exhibiting potent inhibition of M. tuberculosis growth and are in various clinical, preclinical or early drug discovery stages (Makarov et al. 2014; Pethe et al. 2013; Lu et al. 2018; Sacksteder et al. 2012; Protopopova et al. 2005; Matsumoto et al. 2006; Tsubouchi et al. 2016). The power of this approach is that it circumvents the problems associated with target-based screens like compound penetration, target redundancy, etc.; however, a major drawback of this approach is the lack of knowledge about the mode of action of the drug, and hence, no inputs can be obtained via structure activity relationship studies and medicinal chemistry for better and improved drug designing (Zuniga et al. 2015). In fact, many times, for example, in the case of multiple targets, it is not possible to isolate resistant colonies which makes target identification a hard task. However, genomic tools for the identification of mutations in resistant colonies and transcriptional profiling studies in the presence and absence of drug along with the advent of proteomics and metabolomics have been shown to be promising and valuable tools for finding the drug targets. In addition, if one is able to identify a potent inhibitor by using whole cell approach, it is sometimes difficult to understand its inhibitory activity in vivo due to a number of microenvironments the bacteria faces including hypoxic conditions, oxidative stress, acidic stress as well as the various metabolic states that the pathogen can acquire (Kumar et al. 2017; Koul et al. 2011). Hence, the development of better screening methods like carbon starvation model, hypoxic model and replicating and nonreplicating M. tuberculosis model systems that can mimic and represent the in vivo situation that M. tuberculosis encounters in human host will provide hope for improved drug designing (Kumar et al. 2017; Sala et al. 2010; Koul et al. 2008). More recently, TB investigators are focusing towards developing rapid and faster methods for screening inhibitors directly against the intraphagosomal growth of M. tuberculosis inside macrophages, the host niche where the bacteria resides, by employing GFP expressing strains of M. tuberculosis (Khare et al. 2013).

4.3 Screening of Natural Products
The limited chemical space and diversity provided by screening libraries of small molecules result in the identification of hits with limited target diversity, which also highlights why certain pathways are always the frequent hits (Kumar et al. 2017). Hence, considerable attention has been drawn towards the use of natural products as starting point for TB drug discovery that can provide diverse chemical space with novel scaffolds, which are not present in the pharmaceutical libraries used for target-based as well as whole cell screening methods (Kumar et al. 2017; Cragg and Newman 2013). Natural products from medicinal plants or antibacterial bioactive compounds have shown promising results in terms of growth inhibitory potential against M. tuberculosis (Rodrigues Felix et al. 2017; Hartkoorn et al. 2012; Pruksakorn et al. 2010; Steinmetz et al. 2007). These include secondary metabolites isolated from plants and other microbial sources such as bacteria, fungi, marine organisms and algae, belonging to various chemical types such as terpenes (sesquiterpenes, diterpenes, sesterterpenes, triterpenes), steroids (sterols), alkaloids (indole, quinoline, pyridoacridone, manzamine alkaloids, etc.) and aromatics (flavonoids, chalcones, coumarins, lignans, xanthones, anthracenes, anthraquinones, naphthalenes, chromones, etc.). Apart from the secondary metabolites, polyketides (acetylenic fatty acids, polycyclic esters, quinones, etc.) and peptides have also shown growth inhibitory potential. Moreover, these metabolites are bioactive in nature; their relative bioavailability is quite high which thereby increases their access to the site of action (Quan et al. 2017). One of the naturally occurring classes of compounds is phenazines that are biosynthetically produced by Actinobacteria phylum, which are used against bacteria and fungi as broad-spectrum antibiotics (Quan et al. 2017; Laursen and Nielsen 2004). There is a renewed interest in considering riminophenazines as lead compounds for TB drug discovery, especially after clofazimine belonging to the same class was shown to work very effectively against MDR-TB, when administered in combination with gatifloxacin, ethambutol, pyrazinamide, prothionamide, kanamycin and high-dose isoniazid for 9 months (Reddy et al. 1999; Van Deun et al. 2010). However, because of the toxicity observed with the use of clofazimine, analogues of this natural product were developed, and as mentioned above, TBI-166 has been identified as a promising compound (https://www.newtbdrugs.org/pipeline/clinical, Reddy et al. 1996; Job et al. 1990; Levy and Randall 1970; Lu et al. 2011). Likewise, piperidines is another class of naturally available molecules isolated from black pepper, which have been used as wide range of drugs such as neuroleptics, vasodilators, antipsychotics and opioids (Quan et al. 2017). SQ109, the drug that is currently in the clinical pipeline, is an adamantine-containing hydroxydipiperidine that exhibits potent growth inhibitory property against M. tuberculosis in vitro and in vivo (Sacksteder et al. 2012; Protopopova et al. 2005). Similarly, BTZ043 also belongs to piperidine-containing benzothiazinone displaying inhibitory activity against the clinical isolates of M. tuberculosis and drug-resistant strains (Makarov et al. 2009; Kloss et al. 2017; Quan et al. 2017). Notably, both SQ109 and BTZ043 were identified through screening of dipiperidines and sulphur containing heterocycle libraries, respectively, emphasizing the strength of using natural products for the identification of antitubercular compounds given that these metabolites/peptides comprise of broad plethora of diverse scaffolds and pharmacophores. Moreover, other molecules in the clinical pipeline also belong to various classes of secondary metabolites like mycins and quinolones. However, the reasons for apprehension in employing this approach and a lag observed in the success of using these natural products are (i) difficulties in extraction of high yields of purified compounds, (ii) complexity in determining the structure of the compound, (iii) accessibility of the source material reproducibly, (iv) lack of safety studies and (v) lack of information about mechanism of action. Hence, innovative research is required to overcome these shortcomings.
4.4 Repurposing of Drugs
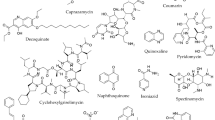
Another important strategy that has a lot of translational scope, which has gained focus and has resulted in promising molecules currently in the clinical pipeline, is the repurposing of the existing drugs also known as therapeutic switching or repositioning approach (Fig. 25.1) (Maitra et al. 2015). Repurposing drug approach employs the use of already existing drugs against various other diseases. Some of the drugs that are used to treat a particular condition can also interact with some other important target(s) and show its effect, which provides a window to analyse its therapeutic efficacy. Repurposing of the known drugs is promising as it is less time consuming in terms of the translation from preclinical work to the market, is less risky and is also less costly (Fig. 25.2).

The figure depicts various steps involved in the most commonly used drug discovery approaches for the identification of new anti-TB drugs
This approach benefits from the fact that these molecules are already well characterized in the context of its target validation, hit-to-lead optimization, in vivo metabolic studies and their safety and toxicity profiling (Maitra et al. 2015). Only 1 in 10,000 new chemical entities entering into pharmaceutical research actually makes it to the market; hence, the compounds already found to be safe in early-stage trials are less risky to begin with. It takes minimum a decade for a non-repurposed drug to reach the market as compared to only ~4 years for a repurposed drug. The most significant example of this approach is the use of sildenafil, which was used as an antihypertensive drug, also been shown to shorten the TB treatment in mouse model studies, when used along with standard drug regimen (Maiga et al. 2012). Moreover, three drugs, namely, clofazimine, linezolid and moxifloxacin, currently present in the TB drug clinical pipeline have resulted from repurposing approach (Van Deun et al. 2010; Till et al. 2002; Yanagihara et al. 2002; Alvirez-Freites et al. 2002). Clofazimine was initially used to treat leprosy and was shown to be successful in treating MDR- and XDR-TB; however, due to its side effects, it is being evaluated in combination with other TB drugs and further analogues are being prepared and tested such as TBI-166 (https://www.newtbdrugs.org/pipeline/clinical, Reddy et al. 1996; Job et al. 1990; Levy and Randall 1970; Lu et al. 2011; Xu et al. 2012; Van Deun et al. 2010; Garrelts 1991). Fluoroquinolones are potent broad-spectrum antibiotics that inhibit topoisomerases II and IV, in turn inhibiting DNA replication (Drlica and Zhao 1997). Moxifloxacin and gatifloxacin, which are the new generation fluoroquinolones, have shown sterilizing properties against M. tuberculosis in both in vitro and in vivo studies and are currently being used as second-line treatment for TB (Alvirez-Freites et al. 2002). Moxifloxacin is now being tested in phase III trials in combination with other drugs to evaluate its efficacy in shortening the treatment regimen (https://www.newtbdrugs.org/pipeline/clinical). Other classes of drugs such as members of the avermectin family, which are used for treating helminthic infections, are being tested for their activity against M. tuberculosis (Lim et al. 2013).
Efforts have also been made by the Indian scientists towards development of new drugs by using the repurposing approach. For example, Singhal et al. showed that the FDA-approved drug metformin used for treating diabetes was able to inhibit the intracellular growth of M. tuberculosis and enhance efficacy of the existing first-line TB drugs, thereby suggesting its use as part of adjunctive TB therapy (Singhal et al. 2014). Brindha et al. carried out virtual screening of FDA-approved drugs against the potential targets of M. tuberculosis, namely, TrpD and CoaA, and further screened the top ranking molecules for their ability to inhibit the in vitro growth of M. tuberculosis resulting into the identification of two potential inhibitors of susceptible as well as resistant strains of M. tuberculosis, namely, lymecycline and cefpodoxime (Brindha et al. 2017). Moreover, lymecycline and cefpodoxime exhibited synergistic activity with rifampin and isoniazid against M. tuberculosis, which suggests the potential of these drugs for the treatment of tuberculosis (Brindha et al. 2017). In another study, it was shown that administration of verapamil, an efflux pump inhibitor, as part of adjunctive therapy to infected mice was able to shorten the duration of standard TB regimen by accelerating the bacterial clearance and lowering down the relapse rates in comparison to mice that received only standard chemotherapy (Gupta et al. 2013). In addition, when verapamil was administered along with bedaquiline and clofazimine, it was able to sharply decrease the MIC of these drugs by 8- to 16-fold (Gupta et al. 2014). Another drug, namely, statin, which is used for lowering down of blood cholesterol, when given along with the first-line antitubercular drugs reduces the lung bacillary load in chronically infected mice (Dutta et al. 2016). Thus, drug repurposing approach is a very attractive strategy and provides alternatives for the treatment of drug-resistant cases.

4.5 New Approaches
-
a.
RNA-Based Therapeutics
Apart from the major challenges associated with the TB chemotherapy, the biggest concern that arises in developing new anti-TB agent is the emergence of drug resistance. Even if a new molecule is identified as a potent anti-TB agent targeting a novel protein or metabolic pathway, the problem of developing drug resistance against the newly identified drug still prevails, and thus, cutting-edge research and innovative technologies are required, which can circumvent the problem of emergence of resistant strains. One such strategy is the use of RNA-based therapeutics such as antisense oligonucleotides, which may represent antitubercular compounds of the future. Antisense oligonucleotides complementary to the target mRNA sequence are designed which lead to the formation of target mRNA/antisense oligo duplex (Bai and Luo 2012). This duplex recruits RNaseH and cleaves the target mRNA molecule resulting in silencing of the target gene (Bai and Luo 2012). One of the limitations, however, associated with the use of oligonucleotides can be their unstable nature and inefficient delivery. Recent studies have shown that modified oligonucleotides such as phosphorothioate oligodeoxynucleotide (PS-ODNs) have enhanced cellular stability, and the use of nanoparticles or liposomes results in better delivery, stability and increased bioavailability. In a study by Meng et al., a new formulation, which involved the use of anionic liposome for encapsulation and delivery of mecA-specific PS-ODNs to target methicillin-resistant Staphylococcus aureus, was employed (Meng et al. 2009). Infected mice when treated with the encapsulated PS-ODN833 downregulated mecA and rescued the animals from MRSA-caused septic death (Meng et al. 2009). Harth et al. investigated the effect of hairpin loop extensions by using a sequence-specific PS-ODNs having 3′ and 5′ hairpin loop extensions targeting the 30–32 kDa protein complex (antigen 85 complex) that is involved in the mycolic acid synthesis of M. tuberculosis (Harth et al. 2007). These PS-ODNs with hairpin loop extensions inhibited bacterial growth in broth culture, inside human macrophages, and also reduced target gene transcription by >90% and showed increased bacterial sensitivity to isoniazid (Harth et al. 2007). Although very preliminary studies have been conducted by employing antisense RNA as a therapeutic tool against M. tuberculosis, due to promising results witnessed in these studies, further research is needed to take this approach forward.
-
b.
Delivery Systems
-
b1.
Nanoparticles/Liposomes
-
b1.
Recent studies are also being focused on developing better delivery strategies to increase the bioavailability of the anti-TB agents. A drug either administered via intravenous route or given orally gets distributed throughout the body, and hence, there are a limited number of molecules that reach the target site (Nasiruddin et al. 2017). Moreover, there can be non-specific or adverse side effects as well. In the case of mycobacterial infection, the drugs need to further reach the bacteria residing in macrophages and inside granulomas. Besides, the short half-life and rapid clearance of the drug also limit its effectiveness (Greenblatt 1985). Hence, to overcome this challenge, either we need very effective drugs or better delivery methods that can enhance the effectiveness of the drug. To this end, nanoparticles or liposomal preparations encapsulating the TB drugs are being developed. Nanoparticles are taken up very efficiently by cells and have the property of controlled, slow and persistent drug release making them an attractive and promising tool for drug delivery. Many modified nanoparticles have been prepared, for example, PEGylated nanoparticles to increase the bioavailability of drugs (Pandey et al. 2003; Sharma et al. 2004). In a study, a single subcutaneous injection of PLG nanoparticles encapsulated with the first-line TB drugs, rifampicin, isoniazid and pyrazinamide resulted in sustained plasma drug levels for 32 days and in lungs and spleen for 36 days (Pandey and Khuller 2004). This led to complete sterilization of the organs of M. tuberculosis-infected mice and demonstrated better therapeutic efficacy as compared with daily oral intake of free drugs (Pandey and Khuller 2004). Liposomes offer an inherent advantage that they have fusogenic abilities to fuse with the macrophages and can efficiently release the drugs into the macrophage cells, which is the primary niche of mycobacteria. In fact, target-based delivery systems are also being developed to avoid non-specific interactions of the drug-encapsulated nanoparticles or liposomes by decorating them with molecules such as mannose residues or O-SAP (O-stearyl amylopectin), which can specifically target the macrophages (Mahajan et al. 2010; Vyas et al. 2004). Thus, the superior drug bioavailability can help enhance its therapeutic usefulness even at low doses of the formulation, which may further help in reducing the period of chemotherapy and patient’s compliance. In a study by Deol et al., it was demonstrated that liposome-encapsulated anti-TB drugs, isoniazid and rifampicin, showed better efficacy than free drugs against tuberculosis in mice model (Deol et al. 1997). Thus, advances should also be made towards improving various delivery systems, which can help in slow and sustained release of the drugs to finally increase the effectiveness and the associated therapeutic efficacy.
-
b2.
Devices for Sustained Release
One of the problems associated with the control of TB relates to poor adherence of the patients to the lengthy chemotherapy, which requires daily administration, high pill burden and frequent dosing. Hence, current research is also being focused on the development of orally administered devices, which can help in increasing the bioavailability of the drug along with sustained release, with holding capacities for close to a month’s pill dosage (Caffarel-Salvador et al. 2017). Such devices would make it feasible to provide the patient with the one time-large dose along with controlled release systems, which would help in getting rid of the needle injections and its associated complications. A similar device having these properties and ability to go and reside inside the GI tract is currently under preclinical evaluation. Such devices are advantageous over the existing injectable or oral drugs because of the ease of administration, a low immunological response and a greater accommodation of the drug inside the GI tract (Caffarel-Salvador et al. 2017). These devices would lead to drastic increase in the patient’s adherence to the therapy, which would prove a boon for the prevention of drug-resistant TB cases.
-
c.
Gene-Editing Tools
Recombineering techniques based on homologous recombination for the generation of deletion mutants are not a very efficient way to validate important drug targets due to high frequency of illegitimate recombinations and time-consuming procedure. Besides, multiple steps and specialized reagents are required, which make the procedure cost ineffective. As identification and validation of essential genes is a crucial requirement for the discovery of novel molecules, newer tools are needed that can speed up this process. CRISPR/CAS technology provides an alternative to the conventional method of gene silencing and has proven to be very useful in M. tuberculosis (Choudhary et al. 2015; Singh et al. 2016). The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) system is a novel genome-editing tool found in bacteria and archaea responsible for the adaptive immune system of prokaryotes providing them with resistance to invading foreign viruses or plasmids (Makarova et al. 2011). The type II CRISPR/Cas system comprises of two short RNA and the DNA endonuclease Cas9. The short RNAs direct Cas9 DNA endonuclease to the target DNA sequence called the protospacer on the target DNA next to the protospacer adjacent motif (PAM) for site-specific cleavage resulting in double-stranded breaks, which can be repaired either by (i) the efficient, however, error-prone non-homologous end joining (NHEJ) pathway or (ii) the high-fidelity but less efficient homology-directed repair (HDR) pathway (Jiang and Doudna 2017). The double-stranded DNA breaks that are repaired by NHEJ pathway result in premature stop codon within the ORF of the target gene by creating either deletions or insertions or frameshift mutations (Jiang and Doudna 2017). The desired result is a loss-of-function mutation within the target gene (Jiang and Doudna 2017). More recently, CRISPR/CAS interference method is developed, which utilizes a small guide RNA that directs the enzymatically inactive CAS endonuclease to specific gene target resulting in interference in the transcription (Marraffini and Sontheimer 2010). Recently, Choudhary et al. showed that by employing an optimized CRISPR/CAS interference system, complete repression of individual or multiple target genes in mycobacteria could be achieved, thus providing a simple, rapid and cost-effective tool for the selective loss of gene expression in mycobacteria (Choudhary et al. 2015). In another such study, prevention of expression of several essential M. tuberculosis genes including pknB was reported emphasizing the ability of the system to modulate the extent of transcription inhibition (Singh et al. 2016).
-
d.
Immunotherapeutic Approach
With an aim to shorten the treatment duration, newer strategies are being employed like the use of vaccines as an adjunct to standard chemotherapy. The basic rationale behind this approach is that vaccines may have the ability to alter the immune responses from unprotective Th2 type to protective Th1 type, which are typically needed for M. tuberculosis control. Hence, by administering the vaccine along with chemotherapy, it is believed that the combined effect exerted by both together might help in faster clearance of the bacteria from the host, which can have implications in reducing the duration of standard chemotherapy. For instance, immunotherapy with DNA expressing α-crystallin, an antigen associated with latency, was able to significantly reduce the chemotherapy period when compared with the chemotherapy alone (Chauhan et al. 2013). In another study, therapeutic vaccination of ID93/GLA-SE as an adjunct to chemotherapy decreased the bacillary load as well as improved the survival time of mice, when compared with mice that were given chemotherapy alone suggesting the possible benefits of adjunctive immunotherapy in shortening the treatment time (Coler et al. 2013). Vaccination with multivalent DNA vaccine encoding Ag85B, MPT-64 and MPT-83 in combination with isoniazid and pyrazinamide was effective in prevention of TB reactivation (Yu et al. 2008). Silva et al. demonstrated that immunotherapy with plasmid DNA encoding Mycobacterium leprae 65 kDa heat-shock protein (hsp65) in association with chemotherapy shortens the duration of treatment, improves the treatment of latent TB infection and is also effective against MDR-TB (Silva et al. 2005).

Figure 25.3 shows the problems and possible solutions involved in the TB drug discovery program.

TB drug discovery program: problems and possible solutions
Abbreviations
- TB:
-
Tuberculosis
- M. tuberculosis :
-
Mycobacterium tuberculosis
- WHO:
-
World Health Organization
- HIV:
-
Human immunodeficiency virus
- AIDS:
-
Acquired immunodeficiency syndrome
- RR-TB:
-
Rifampicin-resistant tuberculosis
- MDR-TB:
-
Multidrug-resistant tuberculosis
- XDR-TB:
-
Extremely drug-resistant tuberculosis
- FDA:
-
Food and Drug Administration
- BCG:
-
Bacillus Calmette-Guerin
- MIC:
-
Minimum inhibitory concentration
- IC:
-
Inhibitory concentration
- EBA:
-
Early bactericidal activity
- PK:
-
Pharmacokinetic
- NTZ:
-
Nitazoxanide
- LZD:
-
Linezolid
- NCI:
-
National Cancer Institute
- AES:
-
Allelic exchange substrate
- SAR:
-
Structure activity relationship
- GFP:
-
Green fluorescent protein
- PS-ODN:
-
Phosphorothioate oligodeoxynucleotide
- PLG:
-
Poly(lactide-co-glycolide)
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- Cas:
-
CRISPR-associated system
- PAM:
-
Protospacer adjacent motif
- NHEJ:
-
Non-homologous end joining
References
Adagu IS, Nolder D, Warhurst DC et al (2002) In vitro activity of nitazoxanide and related compounds against isolates of Giardia intestinalis, Entamoeba histolytica and Trichomonas vaginalis. J Antimicrob Chemother 49:103–111
Akhter M (2016) Identification of novel Mycobacterium tuberculosis dihydrofolate reductase inhibitors through rational drug design. Int J Mycobacteriol 5(Suppl 1):S96
Alffenaar JW, van der Laan T, Simons S et al (2011) Susceptibility of clinical Mycobacterium tuberculosis isolates to a potentially less toxic derivate of linezolid, PNU-100480. Antimicrob Agents Chemother 55:1287–1289
Alsultan A, An G, Peloquin CA (2015) Limited sampling strategy and target attainment analysis for levofloxacin in patients with tuberculosis. Antimicrob Agents Chemother 59:3800–3807
Alvirez-Freites EJ, Carter JL, Cynamon MH (2002) In vitro and in vivo activities of gatifloxacin against Mycobacterium tuberculosis. Antimicrob Agents Chemother 46:1022–1025
Andries K, Verhasselt P, Guillemont J et al (2005) A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227
Aslam S, Musher DM (2007) Nitazoxanide: clinical studies of a broad-spectrum anti-infective agent. Future Microbiol 2:583–590
Bai H, Luo X (2012) Antisense antibacterials: from proof-of-concept to therapeutic perspectives. In: Bobbarala V (ed) A search for antibacterial agents. IntechOpen, London, pp 319–344
Barbachyn MR, Hutchinson DK, Brickner SJ et al (1996) Identification of a novel oxazolidinone (U-100480) with potent antimycobacterial activity. J Med Chem 39:680–685
Bemer-Melchior P, Bryskier A, Drugeon HB (2000) Comparison of the in vitro activities of rifapentine and rifampicin against Mycobacterium tuberculosis complex. J Antimicrob Chemother 46:571–576
Brindha S, Sundaramurthi JC, Vincent S et al (2017) In silico and in vitro screening of FDA-approved drugs for potential repurposing against tuberculosis. bioRxiv: 228171
Caffarel-Salvador E, Abramson A, Langer R et al (2017) Oral delivery of biologics using drug-device combinations. Curr Opin Pharmacol 36:8–13
Chauhan P, Jain R, Dey B et al (2013) Adjunctive immunotherapy with α-crystallin based DNA vaccination reduces Tuberculosis chemotherapy period in chronically infected mice. Sci Rep 3:1821
Chen P, Gearhart J, Protopopova M et al (2006) Synergistic interactions of SQ109, a new ethylene diamine, with front-line antitubercular drugs in vitro. J Antimicrob Chemother 58:332–337
Chiang CY, Centis R, Migiori GB (2010) Drug-resistant tuberculosis: past, present, future. Respirology 15:413–432
Choudhary E, Thakur P, Pareek M et al (2015) Gene silencing by CRISPR interference in mycobacteria. Nat Commun 6:6267
Colditz GA, Brewer TF, Berkey CS et al (1994) Efficacy of BCG vaccine in the prevention of tuberculosis: meta-analysis of the published literature. JAMA 271:698–702
Cole ST, Brosch R, Parkhill J et al (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544
Coler RN, Bertholet S, Pine SO et al (2013) Therapeutic immunization against Mycobacterium tuberculosis is an effective adjunct to antibiotic treatment. J Infect Dis 207:1242–1252
Cragg GM, Newman DJ (2013) Natural products: a continuing source of novel drug leads. Biochim Biophys Acta 1830:3670–3695
Crellin PK, Brammananth R, Coppel RL (2011) Decaprenylphosphoryl-β-D-ribose 2′-epimerase, the target of benzothiazinones and dinitrobenzamides, is an essential enzyme in Mycobacterium smegmatis. PLoS One 6:e16869
Cynamon MH, Klemens SP, Sharpe CA et al (1999) Activities of several novel oxazolidinones against Mycobacterium tuberculosis in a murine model. Antimicrob Agents Chemother 43:1189–1191
Darling JM, Fried MW (2009) Nitazoxanide: beyond parasites toward a novel agent for hepatitis C. Gastroenterology 136:760–763
de Carvalho LP, Lin G, Jiang X et al (2009) Nitazoxanide kills replicating and nonreplicating Mycobacterium tuberculosis and evades resistance. J Med Chem 52:5789–5792
Deol P, Khuller GK, Joshi K (1997) Therapeutic efficacies of isoniazid and rifampin encapsulated in lung-specific stealth liposomes against Mycobacterium tuberculosis infection induced in mice. Antimicrob Agents Chemother 41:1211–1214
Diacon AH, Pym A, Grobusch M et al (2009) The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N Engl J Med 360:2397–2405
Diacon AH, Dawson R, Hanekom M et al (2011) Early bactericidal activity of delamanid (OPC-67683) in smear-positive pulmonary tuberculosis patients. Int J Tuberc Lung Dis 15:949–954
Drlica K, Zhao X (1997) DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev 61:377–392
Duckworth BP, Geders TW, Tiwari D et al (2011) Bisubstrate adenylation inhibitors of biotin protein ligase from Mycobacterium tuberculosis. Chem Biol 18:1432–1441
Dutta NK, Bruiners N, Pinn ML et al (2016) Statin adjunctive therapy shortens the duration of TB treatment in mice. J Antimicrob Chemother 71:1570–1577
Eoh H, Brennan PJ, Crick DC (2009) The Mycobacterium tuberculosis MEP (2C-methyl-d-erythritol 4-phosphate) pathway as a new drug target. Tuberculosis (Edinb) 89:1–11
Fujiwara M, Kawasaki M, Hariguchi N et al (2018) Mechanisms of resistance to delamanid, a drug for Mycobacterium tuberculosis. Tuberculosis (Edinb) 108:186–194
Gardam MA, Keystone EC, Menzies R et al (2003) Anti-tumour necrosis factor agents and tuberculosis risk: mechanisms of action and clinical management. Lancet Infect Dis 3:148–155
Garrelts JC (1991) Clofazimine: a review of its use in leprosy and Mycobacterium avium complex infection. DICP 25:525–531
Gler MT, Skripconoka V, Sanchez-Garavito E et al (2012) Delamanid for multidrug-resistant pulmonary tuberculosis. N Engl J Med 366:2151–2160
Gordeev MF, Yuan ZY (2014) New potent antibacterial oxazolidinone (MRX-I) with an improved class safety profile. J Med Chem 57:4487–4497
Greenblatt DJ (1985) Elimination half-life of drugs: value and limitations. Annu Rev Med 36:421–427
Grzegorzewicz AE, Pham H, Gundi VA et al (2012) Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat Chem Biol 8:334–341
Guidelines for treatment of drug-susceptible tuberculosis and patient care (2017 update). http://apps.who.int/iris/bitstream/handle/10665/255052/9789241550000-eng.pdf;jsessionid=86C860DB7117D77D4A072A39ABCD6429?sequence=1. Last assessed on 15 March 2019
Gupta S, Tyagi S, Almeida DV et al (2013) Acceleration of tuberculosis treatment by adjunctive therapy with verapamil as an efflux inhibitor. Am J Respir Crit Care Med 188:600–607
Gupta S, Cohen KA, Winglee K et al (2014) Efflux inhibition with verapamil potentiates bedaquiline in Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:574–576
Harbut MB, Vilchèze C, Luo X et al (2015) Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc Natl Acad Sci USA 112:4453–4458
Harth G, Zamecnik PC, Tabatadze D et al (2007) Hairpin extensions enhance the efficacy of mycolyl transferase-specific antisense oligonucleotides targeting Mycobacterium tuberculosis. Proc Natl Acad Sci USA 104:7199–7204
Hartkoorn RC, Sala C, Neres J et al (2012) Towards a new tuberculosis drug: pyridomycin–nature’s isoniazid. EMBO Mol Med 4:1032–1042
http://www.cptrinitiative.org/wp-content/uploads/2017/05/Jeffrey_Hafkin_CPTR2017_JH.pdf. Last accessed on 15 March 2019.
https://www.newtbdrugs.org/pipeline/clinical. Last accessed on 15 March 2019
https://www.newtbdrugs.org/pipeline/compound/bedaquiline-0. Last assessed on 15 March 2019
https://www.newtbdrugs.org/pipeline/compound/clofazimine. Last assessed on 15 March 2019
https://www.newtbdrugs.org/pipeline/compound/delamanid-0. Last assessed on 15 March 2019
https://www.newtbdrugs.org/pipeline/compound/levofloxacin. Last assessed on 15 March 2019
https://www.newtbdrugs.org/pipeline/compound/macozinone-mcz-pbtz-169. Last assessed on 15 March 2019
https://www.newtbdrugs.org/pipeline/compound/rifapentine. Last assessed on 15 March 2019
https://www.newtbdrugs.org/pipeline/compound/sq109. Last assessed on 15 March 2019
https://www.newtbdrugs.org/pipeline/compound/telacebec-q203. Last assessed on 15 March 2019
https://www.newtbdrugs.org/pipeline/regimen/pretomanid-moxifloxacin-pyrazinamide-regimen. Last assessed on 15 March 2019
Jeong JW, Jung SJ, Lee HH et al (2010) In vitro and in vivo activities of LCB01-0371, a new oxazolidinone. Antimicrob Agents Chemother 54:5359–5362
Jiang F, Doudna JA (2017) CRISPR–Cas9 structures and mechanisms. Annu Rev Biophys 46:505–529
Job CK, Yoder L, Jacobson RR et al (1990) Skin pigmentation from clofazimine therapy in leprosy patients: a reappraisal. J Am Acad Dermatol 23:236–241
Kang S, Kim RY, Seo MJ et al (2014) Lead optimization of a novel series of imidazo [1, 2-a] pyridine amides leading to a clinical candidate (Q203) as a multi-and extensively-drug-resistant anti-tuberculosis agent. J Med Chem 57:5293–5305
Kanji A, Hasan R, Zaver A et al (2016) Alternate efflux pump mechanism may contribute to drug resistance in extensively drug-resistant isolates of Mycobacterium tuberculosis. Int J Mycobacteriol 5(Suppl 1):S97–S98
Kar R, Nangpal P, Mathur S et al (2017) bioA mutant of Mycobacterium tuberculosis shows severe growth defect and imparts protection against tuberculosis in guinea pigs. PLoS One 12:e0179513
Khare G, Kar R, Tyagi AK (2011) Identification of inhibitors against Mycobacterium tuberculosis thiamin phosphate synthase, an important target for the development of anti-TB drugs. PLoS One 6:e22441
Khare G, Kumar P, Tyag AK (2013) Whole cell screening based identification of inhibitors against the intraphagosomal survival of Mycobacterium tuberculosis. Antimicrob Agents Chemother 57:6372–6377
Kim TS, Choe JH, Kim YJ et al (2017) Activities of LCB01-0371, a novel oxazolidinone, against Mycobacterium abscessus. Antimicrob Agents Chemother 61:pii:e02752-16
Kincaid VA, London N, Wangkanont K et al (2015) Virtual screening for UDP-galactopyranose mutase ligands identifies a new class of antimycobacterial agents. ACS Chem Biol 10:2209–2218
Kloss F, Krchnak V, Krchnakova A et al (2017) In Vivo dearomatization of the potent Antituberculosis agent BTZ043 via Meisenheimer complex formation. Angew Chem Int Ed Engl 56:2187–2191
Korba BE, Montero AB, Farrar K et al (2008) Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antiviral Res 77:56–63
Koul A, Dendouga N, Vergauwen K et al (2007) Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat Chem Biol 3:323–324
Koul A, Vranckx L, Dendouga N et al (2008) Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J Biol Chem 283:25273–25280
Koul A, Arnoult E, Lounis N et al (2011) The challenge of new drug discovery for tuberculosis. Nature 469:483–490
Kumar A, Chettiar S, Parish T (2017) Current challenges in drug discovery for tuberculosis. Expert Opin Drug Discov 12:1–4
Laursen JB, Nielsen J (2004) Phenazine natural products: biosynthesis, synthetic analogues, and biological activity. Chem Rev 104:1663–1686
Leblanc C, Prudhomme T, Tabouret G et al (2012) 4′-Phosphopantetheinyl transferase PptT, a new drug target required for Mycobacterium tuberculosis growth and persistence in vivo. PLoS Pathog 8:e1003097
Lee M, Lee J, Carroll MW et al (2012) Linezolid for treatment of chronic extensively drug-resistant tuberculosis. N Engl J Med 367:1508–1518
Levy L, Randall HP (1970) A study of skin pigmentation by clofazimine. Int J Lepr Other Mycobact Dis 38:404–416
Lim LE, Vilchèze C, Ng C et al (2013) Anthelmintic avermectins kill M. tuberculosis, including multidrug resistant clinical strains. Antimicrob Agents Chemother 57:1040–1046
Lin Y, Zhu N, Han Y et al (2014) Identification of anti-tuberculosis agents that target the cell-division protein FtsZ. J Antibiot (Tokyo) 67:671–676
López-Cortés LF, Ruiz-Valderas R, Viciana P et al (2002) Pharmacokinetic interactions between efavirenz and rifampicin in HIV-infected patients with tuberculosis. Clin Pharmokinet 41:681–690
Lu Y, Zheng M, Wang B et al (2011) Clofazimine analogs with efficacy against experimental tuberculosis and reduced potential for accumulation. Antimicrob Agents Chemother 55:5185–5193
Lu P, Asseri AH, Kremer M et al (2018) The anti-mycobacterial activity of the cytochrome bcc inhibitor Q203 can be enhanced by small-molecule inhibition of cytochrome bd. Sci Rep 8:2625
Machado D, Girardini M, Viveiros M et al (2018) Challenging the “drug-likeness” dogma for new drug discovery in tuberculosis. Front Microbiol 9:1367
Mahajan S, Prashant CK, Koul V et al (2010) Receptor specific macrophage targeting by mannose-conjugated gelatin nanoparticles-an in vitro and in vivo study. Curr Nanosci 6:413–421
Maiga M, Agarwal N, Ammerman NC et al (2012) Successful shortening of tuberculosis treatment using adjuvant host-directed therapy with FDA-approved phosphodiesterase inhibitors in the mouse model. PLoS One 7:e30749
Maitra A, Bates S, Kolvekar T et al (2015) Repurposing—a ray of hope in tackling extensively drug resistance in tuberculosis. Int J Infect Dis 32:50–55
Makarov V, Manina G, Mikusova K et al (2009) Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 324:801–804
Makarov V, Lechartier B, Zhang M et al (2014) Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol Med 6:372–383
Makarova KS, Haft DH, Barrangou R et al (2011) Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol 9:467–477
Malm S, Maaß S, Schaible UE et al (2018) In vivo virulence of Mycobacterium tuberculosis depends on a single homologue of the LytR-CpsA-Psr proteins. Sci Rep 8:3936
Marraffini LA, Sontheimer EJ (2010) CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet 11:181–190
Matsumoto M, Hashizume H, Tomishige T et al (2006) OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med 3:e466
Mehta S, Das M, Laxmeshwar C et al (2016) Linezolid-associated optic neuropathy in drug-resistant tuberculosis patients in Mumbai, India. PLoS One 11:e0162138
Meng J, Wang H, Hou Z et al (2009) Novel anion liposome-encapsulated antisense oligonucleotide restores susceptibility of methicillin-resistant Staphylococcus aureus and rescues mice from lethal sepsis by targeting mecA. Antimicrob Agents Chemother 53:2871–2878
Morgunova E, Meining W, Illarionov B et al (2005) Crystal structure of lumazine synthase from Mycobacterium tuberculosis as a target for rational drug design: binding mode of a new class of purinetrione inhibitors. Biochemistry 44:2746–2758
Narayanan S, Swaminathan S, Supply P et al (2010) Impact of HIV infection on the recurrence of tuberculosis in South India. J Infect Dis 201:691–703
Nasiruddin M, Neyaz M, Das S (2017) Nanotechnology-based approach in tuberculosis treatment. Tuberc Res Treat 2017:4920209
Nikonenko BV, Protopopova M, Samala R et al (2007) Drug therapy of experimental tuberculosis (TB): improved outcome by combining SQ109, a new diamine antibiotic, with existing TB drugs. Antimicrob Agents Chemother 51:1563–1565
Nuermberger E, Tyagi S, Tasneen R et al (2008) Powerful bactericidal and sterilizing activity of a regimen containing PA-824, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob Agents Chemother 52:1522–1524
Palencia A, Li X, Bu W et al (2016) Discovery of novel oral protein synthesis inhibitors of Mycobacterium tuberculosis that target leucyl-tRNA synthetase. Antimicrob Agents Chemother 60:6271–6280
Pandey R, Khuller GK (2004) Subcutaneous nanoparticle-based antitubercular chemotherapy in an experimental model. J Antimicrob Chemother 54:266–268
Pandey R, Zahoor A, Sharma S et al (2003) Nanoparticle encapsulated antitubercular drugs as a potential oral drug delivery system against murine tuberculosis. Tuberculosis (Edinb) 83:373–378
Parthasarathy K, Sivaraj A, Sundar R (2016) Identification of new efflux pump proteins from multidrug resistant Mycobacterium tuberculosis and screening for peptide based efflux pump inhibitors. Int J Mycobacteriol 45:404–405
Peterson J, Yektashenas B, Fisher AC (2009) Levofloxacin for the treatment of pneumonia caused by Streptococcus pneumoniae including multidrug-resistant strains: pooled analysis. Curr Med Res Opin 25:559–568
Pethe K, Bifani P, Jang J et al (2013) Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat Med 19:1157–1160
Piccaro G, Poce G, Biava M (2015) Activity of lipophilic and hydrophilic drugs against dormant and replicating Mycobacterium tuberculosis. J Antibiot (Tokyo) 68:711–714
Protopopova M, Hanrahan C, Nikonenko B et al (2005) Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1, 2-ethylenediamines. J Antimicrob Chemother 56:968–974
Pruksakorn P, Arai M, Kotoku N et al (2010) Trichoderins, novel aminolipopeptides from a marine sponge-derived Trichoderma sp., are active against dormant mycobacteria. Bioorg Med Chem Lett 20:3658–3663
Puri RV, Reddy PV, Tyagi AK (2013) Secreted acid phosphatase (SapM) of Mycobacterium tuberculosis is indispensable for arresting phagosomal maturation and growth of the pathogen in guinea pig tissues. PLoS One 8:e70514
Qiao C, Gupte A, Boshoff HI et al (2007) 5′-O-[(N-acyl) sulfamoyl] adenosines as antitubercular agents that inhibit MbtA: an adenylation enzyme required for siderophore biosynthesis of the mycobactins. J Med Chem 50:6080–6094
Quan D, Nagalingam G, Payne R et al (2017) New tuberculosis drug leads from naturally occurring compounds. Int J Infect Dis 56:212–220
Reddy VM, Nadadhur G, Daneluzzi D et al (1996) Antituberculosis activities of clofazimine and its new analogs B4154 and B4157. Antimicrob Agents Chemother 40:633–636
Reddy VM, O’Sullivan JF, Gangadharam PR (1999) Antimycobacterial activities of riminophenazines. J Antimicrob Chemother 43:615–623
Reddy PV, Puri RV, Chauhan P et al (2013) Disruption of mycobactin biosynthesis leads to attenuation of Mycobacterium tuberculosis for growth and virulence. J Infect Dis 208:1255–1265
Rock FL, Mao W, Yaremchuk A et al (2007) An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science 316:1759–1761
Rodrigues Felix C, Gupta R, Geden S et al (2017) Selective killing of dormant Mycobacterium tuberculosis by marine natural products. Antimicrob Agents Chemother 61: pii: e00743-17
Rodrigues L, Machado D, Couto I et al (2012) Contribution of efflux activity to isoniazid resistance in the Mycobacterium tuberculosis complex. Infect Genet Evol 12:695–700
Rohilla A, Khare G, Tyagi AK (2017) Virtual Screening, pharmacophore development and structure based similarity search to identify inhibitors against IdeR, a transcription factor of Mycobacterium tuberculosis. Sci Rep 7:4653
Rohilla A, Khare G, Tyagi AK (2018) A combination of docking and cheminformatics approaches for the identification of inhibitors against 4′ phosphopantetheinyl transferase of Mycobacterium tuberculosis. RSC Adv 8:328–341
Rossignol JF, Kabil SM, El-Gohary Y et al (2008) Clinical trial: randomized, double-blind, placebo-controlled study of nitazoxanide monotherapy for the treatment of patients with chronic hepatitis C genotype 4. Aliment Pharmacol Ther 28:574–580
Rossignol JF, Elfert A, Keeffe EB (2010) Treatment of chronic hepatitis C using a 4-week lead-in with nitazoxanide before peginterferon plus nitazoxanide. J Clin Gastroenterol 44:504–509
Sacksteder KA, Protopopova M, Barry CE 3rd et al (2012) Discovery and development of SQ109: a new antitubercular drug with a novel mechanism of action. Future Microbiol 7:823–837
Sala C, Dhar N, Hartkoorn RC et al (2010) Simple model for testing drugs against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 54:4150–4158
Sassetti CM, Rubin EJ (2003) Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci USA 100:12989–12994
Sassetti CM, Boyd DH, Rubin EJ (2001) Comprehensive identification of conditionally essential genes in mycobacteria. Proc Natl Acad Sci USA 98:12712–12717
Sensi P, Margalith P, Timbal MT (1959) Rifomycin, a new antibiotic; preliminary report. Farmaco Sci 14:146–147
Sharma A, Pandey R, Sharma S et al (2004) Chemotherapeutic efficacy of poly (DL-lactide-co-glycolide) nanoparticle encapsulated antitubercular drugs at sub-therapeutic dose against experimental tuberculosis. Int J Antimicrob Agents 24:599–604
Shaw KJ, Barbachyn MR (2011) The oxazolidinones: past, present, and future. Ann NY Acad Sci 1241:48–70
Shi C, Aldrich CC (2012) Design and synthesis of potential mechanism-based inhibitors of the aminotransferase BioA involved in biotin biosynthesis. J Org Chem 77:6051–6058
Shi C, Geders TW, Park SW et al (2011) Mechanism-based inactivation by aromatization of the transaminase BioA involved in biotin biosynthesis in Mycobacterium tuberculosis. J Am Chem Soc 133:18194–18201
Shirude PS, Shandil R, Sadler C et al (2013) Azaindoles: noncovalent DprE1 inhibitors from scaffold morphing efforts, kill Mycobacterium tuberculosis and are efficacious in vivo. J Med Chem 56:9701–9708
Shirude PS, Shandil RK, Manjunatha MR et al (2014) Lead optimization of 1, 4-azaindoles as antimycobacterial agents. J Med Chem 57:5728–5737
Shoen C, DeStefano M, Hafkin B et al (2018) In vitro and in vivo activity of contezolid (MRX-I) against Mycobacterium tuberculosis. Antimicrob Agents Chemother 62 pii: e00493-18
Silva CL, Bonato VL, Coelho-Castelo AA et al (2005) Immunotherapy with plasmid DNA encoding mycobacterial hsp65 in association with chemotherapy is a more rapid and efficient form of treatment for tuberculosis in mice. Gene Ther 12:281–287
Singh AK, Carette X, Potluri LP et al (2016) Investigating essential gene function in Mycobacterium tuberculosis using an efficient CRISPR interference system. Nucleic Acids Res 44:e143
Singh V, Donini S, Pacitto A et al (2017) The inosine monophosphate dehydrogenase, GuaB2, is a vulnerable new bactericidal drug target for tuberculosis. ACS Infect Dis 3:5–17
Singh S, Khare G, Bahal RK et al (2018) Identification of Mycobacterium tuberculosis BioA inhibitors by using structure-based virtual screening. Drug Des Devel Ther 12:1065–1079
Singhal A, Jie L, Kumar P et al (2014) Metformin as adjunct antituberculosis therapy. Sci Transl Med 6:263ra159
Skripconoka V, Danilovits M, Pehme L et al (2013) Delamanid improves outcomes and reduces mortality for multidrug-resistant tuberculosis. Eur Respir J 41:1393–1400
Stachulski AV, Pidathala C, Row EC et al (2011) Thiazolides as novel antiviral agents. 1. Inhibition of hepatitis B virus replication. J Med Chem 54:4119–4132
Steinmetz H, Irschik H, Kunze B et al (2007) Thuggacins, macrolide antibiotics active against Mycobacterium tuberculosis: isolation from myxobacteria, structure elucidation, conformation analysis and biosynthesis. Chemistry 13:5822–5832
Stevenson CR, Critchley JA, Forouhi NG et al (2007) Diabetes and the risk of tuberculosis: a neglected threat to public health? Chronic Illn 3:228–245
Stover CK, Warrener P, Van Devanter DR et al (2000) A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405:962–966
Suarez-Almazor ME, Spooner CH, Belseck E et al (2000) Auranofin versus placebo in rheumatoid arthritis. Cochrane Database Syst Rev 2:CD002048
Tahlan K, Wilson R, Kastrinsky DB et al (2012) SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob Agents Chemother 56:1797–1809
Tasneen R, Li SY, Peloquin CA et al (2011) Sterilizing activity of novel TMC207-and PA-824-containing regimens in a murine model of tuberculosis. Antimicrob Agents Chemother 55:5485–5492
Theodos CM, Griffiths JK, D’Onfro J et al (1998) Efficacy of nitazoxanide against cryptosporidium parvum in cell culture and in animal models. Antimicrob Agents Chemother 42:1959–1965
Till M, Wixson RL, Pertel PE (2002) Linezolid treatment for osteomyelitis due to vancomycin-resistant Enterococcus faecium. Clin Infect Dis 34:1412–1414
Trefzer C, Rengifo-Gonzalez M, Hinner MJ et al (2010) Benzothiazinones: prodrugs that covalently modify the decaprenylphosphoryl-β-D-ribose 2′-epimerase DprE1 of Mycobacterium tuberculosis. J Am Chem Soc 132:13663–13665
Tsubouchi H, Sasaki H, Ishikawa H et al (2016) Discovery of delamanid for the treatment of multidrug-resistant pulmonary Tuberculosis. In: Fischer J, Childers WE (eds) Successful drug discovery, vol 2. Wiley-VCH Verlag GmbH & Co, KGaA, Weinheim, pp 137–161
Tyagi S, Nuermberger E, Yoshimatsu T et al (2005) Bactericidal activity of the nitroimidazopyran PA-824 in a murine model of tuberculosis. Antimicrob Agents Chemother 49:2289–2293
Uplekar M, Weil D, Lonnroth K et al (2015) WHO’s new end TB strategy. Lancet 385:1799–1801
Van Deun A, Maug AK, Salim MA et al (2010) Short, highly effective, and inexpensive standardized treatment of multidrug-resistant tuberculosis. Am J Respir Crit Care Med 182:684–692
van Kessel JC, Hatfull GF (2007) Recombineering in Mycobacterium tuberculosis. Nat Methods 4:147–152
Vyas SP, Kannan ME, Jain S et al (2004) Design of liposomal aerosols for improved delivery of rifampicin to alveolar macrophages. Int J Pharm 269:37–49
Wallis RS, Jakubiec WM, Kumar V et al (2010) Pharmacokinetics and whole-blood bactericidal activity against Mycobacterium tuberculosis of single doses of PNU-100480 in healthy volunteers. J Infect Dis 202:745–751
Wallis RS, Jakubiec W, Kumar V et al (2011) Biomarker-assisted dose selection for safety and efficacy in early development of PNU-100480 for tuberculosis. Antimicrob Agents Chemother 55:567–574
WHO guidelines for the programmatic management of drug-resistant tuberculosis.. http://apps.who.int/iris/bitstream/handle/10665/130918/9789241548809_eng.pdf;jsessionid=BBA844744A619EF170C00783C655E148?sequence=1 (Last assessed on 6.09.2018)
WHO: Global Tuberculosis Report 2017. http://www.who.int/tb/publications/global_report/gtbr2017_main_text.pdf
www.who.int/mediacentre/factsheets/fs104/en/. Last accessed on 22 August 2018
Xu HB, Jiang RH, Xiao HP (2012) Clofazimine in the treatment of multidrug-resistant tuberculosis. Clin Microbiol Infect 18:1104–1110
Yanagihara K, Kaneko Y, Sawai T et al (2002) Efficacy of linezolid against methicillin-resistant or vancomycin-insensitive Staphylococcus aureus in a model of hematogenous pulmonary infection. Antimicrob Agents Chemother 46:3288–3291
Yu DH, Hu XD, Cai H (2008) Efficient tuberculosis treatment in mice using chemotherapy and immunotherapy with the combined DNA vaccine encoding Ag85B, MPT-64 and MPT-83. Gene Ther 15:652–659
Yuan T, Sampson NS (2018) Hit generation in TB drug discovery: from genome to granuloma. Chem Rev 118:1887–1916
Zhang Y, Zhang J, Cui P et al (2017) Identification of novel efflux proteins Rv0191, Rv3756c, Rv3008 and Rv1667c involved in Pyrazinamide resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 61:pii:e00940-17
Zong Z, Jing W, Shi J et al (2018) Comparison of in vitro activity and MIC distributions between the novel oxazolidinone delpazolid and linezolid against multidrug-resistant and extensively drug-resistant Mycobacterium tuberculosis in China. Antimicrob Agents Chemother 62:pii: e00165-18
Zuniga ES, Early J, Parish T (2015) The future for early-stage tuberculosis drug discovery. Future Microbiol 10:217–229
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Khare, G., Nangpal, P., Tyagi, A.K. (2019). Challenges and Advances in TB Drug Discovery. In: Hasnain, S., Ehtesham, N., Grover, S. (eds) Mycobacterium Tuberculosis: Molecular Infection Biology, Pathogenesis, Diagnostics and New Interventions. Springer, Singapore. https://doi.org/10.1007/978-981-32-9413-4_25
Download citation
DOI: https://doi.org/10.1007/978-981-32-9413-4_25
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-32-9412-7
Online ISBN: 978-981-32-9413-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)