
Abstract
The endothelium, inner layer of blood vessels, constitutes a metabolically active paracrine, endocrine, and autocrine organ, able to sense the neighboring environment and exert a variety of biological functions important to preserve the health of vasculature, tissues, and organs. Sphingolipids are both fundamental structural components of the eukaryotic membranes and signaling molecules regulating a variety of biological functions. Ceramide and sphingosine-1-phosphate (S1P), bioactive sphingolipids, have emerged as important regulators of cardiovascular functions in health and disease. In this review we discuss recent insights into the role of ceramide and S1P biosynthesis and signaling in regulating endothelial cell functions, in health and diseases. We also highlight advances into the mechanisms regulating serine palmitoyltransferase, the first and rate-limiting enzyme of de novo sphingolipid biosynthesis, with an emphasis on its inhibitors, ORMDL and NOGO-B. Understanding the molecular mechanisms regulating the sphingolipid de novo biosynthesis may provide the foundation for therapeutic modulation of this pathway in a variety of conditions, including cardiovascular diseases, associated with derangement of this pathway.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Sphingolipid de novo biosynthesis
- Serine palmitoyltransferase (SPT)
- Ceramide
- Sphingosine-1-phosphate (S1P)
- Endothelial cells
- Nogo-B
8.1 Endothelium: Structure and Functions
In 1628 William Harvey first described a network of vessels separating the blood from tissues. In 1661 Marcello Malpighi reported the existence of capillaries and red blood cells (RBC) by using the microscope. Only in 1800s Wilhelm His observed and coined the word “endothelium” to describe the inner layer lining of blood vessels and cavities. The endothelium has been initially considered as an “inert” physical barrier between blood and tissues. Over the years, the concept of endothelium gradually evolved from that of semipermeable barrier to a metabolically active paracrine, endocrine, and autocrine organ, able to sense the neighboring environment and exert a variety of biological functions important to maintain physiological homeostasis of the vasculature, tissues, and organs (Fig. 8.1).
Endothelial cell-to-cell adhesion is mainly maintained by tight and adheres junctions (AJ), which are transmembrane adhesive protein complex that interacts with intracellular cytoskeleton [1], providing a functional barrier regulating paracellular permeability and cellular polarity. As ultrastructure studies began to unfold in the 1960s, the endothelium appeared having a heterogeneous structure throughout the vascular tree, mirroring its diversified functions. For instance, the endothelium of the arteries and arterioles in the heart, brain, and muscle is non-fenestrated and continuous, with organized and tighter junctions than post-capillary venules. The latter are the primary site of plasma proteins and leukocytes extravasation during inflammation; therefore, the endothelial junctions are more loose and disorganized to better support their functions [2]. Discontinuous endothelium is characteristic of the liver sinusoids, containing large fenestrae (open pores across the cell lacking diaphragm), poorly formed basement membrane, and high endocytic capacity, supporting the metabolic clearance of the liver [3]. In tissue with high filtration rates such as glands and glomeruli, the endothelium of the capillaries is continuous and presents fenestrae, the majority of which is provided by diaphragms, subcellular structure [2, 4].
In resting state, the endothelium regulates the exchange of oxygen and nutrients between the bloodstream and tissues, controls blood flow, and refrains plasma proteins and leukocytes from extravasating [2, 5]. All endothelial cells (EC) of the vasculature preserve blood fluidity by providing an anti-thrombotic and anti-coagulant surface and factors [6]. For instance, the activation of thrombin and the formation of fibrin clot is inhibited by the tissue factor pathway inhibitors (TFPIs), anti-thrombin III bound to heparan sulfate proteoglycans, and by thrombomodulin, which converts the functions of thrombin from pro-coagulant to anti-coagulant (Fig. 8.1). The endothelium also contributes to the activation of the fibrinolytic system. For instance, EC can produce tissue-type and urokinase-type plasminogen activators (tPA and uPA), converting the inactive plasminogen to active plasmin, an enzyme able to degrade fibrin [7], forming the clot. EC also prevent platelet activation by sequestrating von Willebrand factor in Weibel–Palade bodies (intracellular granules), degrading adenosine-5′-triphosphate (ATP) via the membrane-bound ecto-adenosine diphosphate (ADP)-ases CD39 and CD73, and releasing factors such as endothelial nitric oxide synthase (eNOS)-derived nitric oxide (NO) and prostacyclin (PGI2), which are able to increase intracellular cGMP and cAMP in platelets and inhibit their activation [7].

Endothelial functions. (a) The endothelium preserves blood fluidity by providing an anti-thrombotic and anti-coagulant surface and factors, such as anti-thrombin III, thrombomodulin, and NO. (b) Adherens junctions provide a functional barrier regulating paracellular permeability. They are composed by the transmembrane proteins VE-cadherin, linked cytoplasmatically to a different intracellular binding partner, including p120-catenin, β-catenin, and γ-catenin, favoring the interaction of VE-cadherin complex with the cytoskeleton. Pro-permeability signals such as VEGF, thrombin, and histamine trigger VE-cadherin phosphorylation and Rho-dependent stress fibers formation, leading to barrier disruption. Signals that reinforce the endothelial barrier integrity, such as S1P, trigger VE-cadherin dephosphorylation and clustering and Rac-dependent cortical actin formation. (c) The endothelium regulates vascular tone and blood pressure, through the production of vasoactive molecules. NO is a potent endogenous vasodilator via cGMP-PKG pathway. S1P is a potent activator of eNOS-derived NO. NO nitric oxide, eNOS endothelial nitric oxide synthase, VEGF vascular endothelial growth factor, S1P sphingosine-1-phosphate, S1PR1 S1P receptor 1
One of the first functions recognized to the endothelium is the capacity to produce a variety of vasoactive molecules able to control vascular tone, therefore dictating the equilibrium between tissue metabolic demand and blood supply (Fig. 8.1). The initial observation from Furchgott and Zawadzki that the removal of the endothelium abolished the vasodilating effects of acetylcholine (Ach) [8] led to the identification of NO as the main endogenous regulator of vascular tone [9, 10], via cGMP [11, 12]. The association of endothelial eNOS with caveolin-1 keeps the enzyme in a low active state localized to the caveolae, which are invaginations of plasma membrane microdomains of lipid rafts composition [13]. The activity of eNOS is regulated by a complex and multilayered process. Following the activation of specific receptors on EC by agonists (i.e., Ach, bradykinin, vascular endothelial growth factor, or insulin), intracellular Ca2+ increases, and the complex Ca2+/calmodulin can interact with eNOS and displace caveolin-1, thereby enhancing its activity. Other proteins, such as heat shock protein 90 (HSP90), can interact with eNOS and enhance its activity [14]. Different phosphorylation sites have been identified in eNOS, which can modulate its activation. The Ser1176 of eNOS (Ser1177 in human and bovine) has been the most studied and when phosphorylated by phosphoinositide 3-kinase (PI3K)-Akt heightens eNOS activity [15, 16]. Other kinases can phosphorylate eNOS at Ser1176, including PKA (protein kinase activated by cAMP), AMPK (activated by AMP), and CaMKII (Ca2+/calmodulin-dependent protein kinase II). By using l-arginine as substrate, eNOS forms NO and the co-product citrulline [14]. NO is a free radical gas that can rapidly diffuse across biological membranes to the subjacent vascular smooth muscle cells of the media, where it induces vasorelaxation via cGMP-mediated mechanisms, including the decrease of intracellular Ca2+, and the activation of myosin light chain phosphatase, dephosphorylating myosin light chain [17].
The loss of the endothelium to accomplish any of these functions is defined “endothelial dysfunction” and represents an early event in the onset of cardiovascular diseases [18, 19]. For instance, pre-clinical and clinical studies have demonstrated that endothelial dysfunction precedes the onset of atherosclerosis and hypertension [20,21,22] and contributes not only to development of the lesions but also to later clinical complications.
8.2 Sphingolipid Pathway
Sphingolipids (SL) are a class of structurally and functionally diverse lipids, discovered in brain extracts in the 1870s. Sphingosine, from “Sphinx,” owes its name to J. L. W. Thudichum “in commemoration of the many enigmas which it presents to the inquirer.” Recent advances in mass spectrometry and lipidomics have enabled a sensitive and accurate characterization of the sphingolipidome, and to date the LIPID MAPS Lipidomics Gateway (www.lipidmaps.org) lists almost 5000 distinct chemical entities within this lipid class.
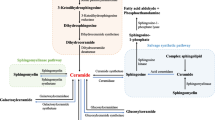
Structurally, SL are characterized by an 18-carbon amino-alcohol backbone called sphingoid base, N-acylated with fatty acids, that can be saturated or mono-saturated, with chain lengths ranging from 14 to 26 carbon atoms. Sphingolipid de novo biosynthesis starts in the cytosolic membrane of the endoplasmic reticulum (ER) with the condensation of l-serine and a saturated acyl-coenzyme A, typically palmitoyl-CoA, by serine palmitoyltransferase (SPT), first and rate-limiting enzyme of the pathway [23] (Fig. 8.2). In the ER ceramide synthases 1–6 N-acylates the sphingoid base with fatty acids to give rise to dihydroceramides [24], and dihydroceramide desaturase converts dihydroceramides in ceramides [25], central metabolites of the pathway. Ceramides can be modified in their head group to form complex SL. Sphingomyelins (SM), major SL in plasma membranes, are formed by the addition of phosphocholine or phosphoethanolamine to ceramides by sphingomyelin synthase. Addition of sugars by galactosyltransferase and glucosylceramide synthase forms glycosphingolipids (GLS) [26]. SM and GLS biosynthesis occurs in the Golgi. Because of their hydrophobicity, ceramides are transported to the cis-Golgi for SM synthesis mainly by CERT (ceramide transfer protein) [27] and by vesicular transport to the trans-Golgi for GSL synthesis [28]. All complex SL can be degraded to ceramide by the activity of specific hydrolases removing the head groups [26]. SM and GLS can be both catabolized to ceramide in the plasma membrane or internalized through the endosomal pathway and degraded by sphingomyelinases (SMase) and glucosidases to ceramide in the lysosomal compartment [26].

Sphingolipid biosynthetic pathways. SPT initiates de novo SL biosynthesis in the ER, where multiple enzymatic reactions generate ceramide. Ceramide can also derive from the catabolism (recycling pathway) of sphingomyelin and glycosphingolipid in the plasma membrane or in the lysosome. Ceramide can be degraded to sphingosine, which in turn is phosphorylated to S1P. S1P is then transported outside EC by SPNS2, where it can activate endothelial S1PRs or bind to its carrier, HDL-ApoM and albumin, to be transported into the circulation. SPT serine palmitoyltransferase, SL sphingolipids, S1P sphingosine-1-phosphate, S1PR S1P receptor, SM sphingomyelin, GSL glycosphingolipid, SPHK1,2 sphingosine kinase, SPNS2 spinster-2
Although ceramide is primarily converted into complex SL, it can also be phosphorylated by ceramide kinase in ceramide-1-phospate [29] or hydrolyzed to sphingosine and fatty acid by ceramidases [30]. Ceramide hydrolysis occurs in the lysosome or in the ER by acid or alkaline ceramidase, respectively. Sphingosine can be “salvaged” into the sphingolipid pathways, or phosphorylated to form sphingosine-1-phosphate (S1P), a potent bioactive lipid, by the two sphingosine kinases 1 and 2 (SPHK1 and 2) [31]. SPHK1 is mainly located in the cytoplasm, SPHK2 in the ER and in the nucleus [31]. S1P can be dephosphorylated by S1P phosphatase to sphingosine or degraded by S1P lyase to hexadecenal and phosphoethanolamine. The latter occurs in the cytosolic membrane of the ER and is the only enzymatic reaction in which a SL is converted into a non-SL molecule [32], thus constitutes the only exit of the pathway.
Functionally, SL are important building blocks of cell membranes [33]. Together with cholesterol, ceramides and SM are enriched in lipid rafts, small plasma membrane domains of nano- and microscale [34,35,36]. Lipid rafts are hypothesized to form by self-association of SL, favored by their long and mostly saturated hydrocarbon chain, that allow a tightly packing in the bilayer. Cholesterol molecules are believed to fill the voids between the associating SL, producing domains more ordered than the surrounding membranes [37, 38]. Lipid rafts are enriched in proteins post-translationally modified by glycosylphosphatidylinositol (GPI) anchor [35, 38], and protein–protein interactions are proposed to stabilize the small and dynamic rafts, leading to the formation of larger structures [39]. Ceramide-rich domains are another kind of SL-enriched domain in the plasma membrane, larger than the lipid rafts and devoid of cholesterol. Produced by the hydrolysis of SM to ceramide by SMase in response to external stimuli (i.e., TNFɑ), they are also postulated to exhibit higher order than the surrounding bilayer and to be enriched in GPI-anchored proteins [40]. Both lipid rafts and ceramide-rich domains are also platforms where tyrosine kinase receptors and G-protein couple receptors (GPCR) cluster [41, 42]. By modulating the membrane properties, SL can influence the activity of transmembrane proteins and their associated cellular processes, such as signal transduction, endocytosis, intracellular trafficking [34, 43].
SL function also as potent signaling molecules, and to date, the best characterized bioactive SL are S1P and ceramides [44]. More recently additional signaling functions have been ascribed to dihydroceramide [45], sphingosine [44], and ceramide-1-phosphate [46]. While S1P controls different cellular functions by activating five different GPCR, namely S1PR1–5 [44, 47], ceramides function mainly intracellularly as second messenger by modulating the activity of target proteins, such as protein phosphatases PP2A and PP1 [48, 49] and protein kinase Cζ [50]. Biological roles of S1P and ceramide in EC will be discussed later in this chapter.
8.3 S1P Biosynthesis, Secretion, Carriers
Sphingosine-1-phosphate (S1P), one of the most studied bioactive lipids, is generated by a two-step process: the hydrolysis of ceramide by ceramidases and the subsequential phosphorylation of sphingosine by SPHK1,2 [47]. In healthy state, erythrocytes [51, 52] and EC [53,54,55] are major sources of plasma S1P, while platelets represent an additional source in pathological conditions [56]. Once formed, endothelial S1P is rapidly transported outside by the transporter Spinster homolog 2 (SPNS2) [57, 58].
In mammals, S1P is spatially compartmentalized in blood (~1 μM) and lymph (~0.1 μM) circulation, while it is almost undetectable in interstitial tissue, including lymphoid organs (<1 nM) where S1P lyase activity is high, creating an S1P gradient across the endothelial barrier [59, 60]. This gradient is particularly important for immune cell trafficking. Lymphocytes egress into circulation by an S1PR1-dependent sensing of S1P gradients at the tissue–circulatory interface [61, 62]. Once in circulation, lymphocyte S1PR1 is mostly internalized and desensitized due to the high S1P concentration, allowing lymphocytes to overcome the S1P-dependent attraction to blood and return to lymphoid tissues [63].
Because of its lipophilic nature, plasmatic S1P is mainly carried by high density lipoprotein (HDL, ~65%) and albumin (~35%) [64]. In HDL, S1P is carried by apolipoprotein M (ApoM) in a lipid-binding pocket formed by eight antiparallel β-sheets [65, 66]. Interestingly, ligand-binding studies have demonstrated that S1P binds to ApoM with an IC50 of 0.9 μM, corresponding to plasma concentration of S1P [65, 66]. In ApoM knockout mice (ApoM−/−), plasma HDL-bound S1P is virtually absent, resulting in increased vascular permeability due to a disruption of the endothelial barrier function [66], and increased blood pressure (BP) due to diminished endothelial S1P-NO signaling activation [54]. Human studies reported an inverse correlation between the severity of sepsis with the plasma levels of ApoM and S1P [67, 68]. Similarly, in an E. coli-induced model of sepsis in baboons, ApoM and S1P plasma levels progressively dropped over time, reaching half the concentration at the 24 h time point [68]. Patients with stable coronary artery disease (CAD) and myocardial infarction (MI) have lower levels of circulating HDL-S1P than healthy individuals [69, 70], with an impaired capacity to stimulate eNOS-derived NO production [71]. While not associated with the extent of lesion stenosis or restenosis, HDL-bound S1P inversely correlates with the severity of CAD [72]. Interestingly, genetic variants of ApoM have been associated with CAD [73, 74].
While these findings suggest a role for ApoM-bound S1P signaling in maintaining cardiovascular health, further studies are needed to understand the pathological significance of altered levels and/or functions of HDL-bound S1P in disease settings such as atherosclerosis.
8.4 S1P and Ceramides in Endothelial Barrier Functions
Vascular permeability is governed by the tightness of the endothelial barrier (cell-to-cell contact), which ensures a proper compartmentalization between the blood and interstitial space, allows the exchange of small molecules (i.e., gases O2, CO2, sugar, salts), and refrains macromolecules (i.e., immunoglobulins, albumin) and immune cells from extravasating. The failure to maintain endothelial barrier integrity results in excessive increase of permeability, a hallmark of different inflammatory conditions [75, 76], including rheumatoid arthritis, healing wounds, tumors.
The core component of endothelial AJ is the transmembrane protein vascular endothelial cadherin (VE-cadherin, Fig. 8.1b). Its extracellular domains form trans-cadherin interactions between neighbor EC, while the intracellular domain mediates interactions with a complex of intracellular binding partners, including β-catenin, p120-catenin, and γ-catenin, essential for junctional stability, signaling, and cytoskeleton anchorage [77, 78]. Lee and colleagues demonstrated that S1P reinforces the assembly of AJ by increasing VE-cadherin and β-catenin localization at the cell-to-cell contact sites in HUVEC (human umbilical vein EC), through S1PR1 and S1PR3 activation [79]. The enhancement of AJ was coupled to stress fibers and cortical actin formation, mediated by Rac and Rho GTPases activation [79]. Rac activity is necessary to heighten the endothelial barrier, and S1PR1 is mainly accountable for Rac activation via Gαi-dependent PI3K signaling, although S1PR3 can also signal, in part, to Rac. A follow-up study by Garcia et al. showed that in different types of EC, S1P increases the trans-endothelial electrical resistance (TEER), a readout of the barrier strength. The increase in TEER was induced by the activation of PTX-sensitive S1PRs and Rho/Rac GTPases. They also showed that S1P enhances the re-establishment of the endothelial barrier following thrombin-induced disruption [80]. Other studies supported the role of S1P as endothelial barrier-enhancing factor and identified additional mechanisms by which S1P controls cytoskeleton remodeling [81]. Consistent with these findings, mice endothelial knockout for S1pr1 (S1pr1ECKO) subjected to reverse Arthus reaction, an established model of acute immunocomplex-mediated vascular injury and inflammation, showed increased pulmonary extravasation of albumin-bound Evans blue dye, increased lung weight, as well as a greater number of white and RBC in the bronchoalveolar lavage fluid compared to S1pr1f/f, index of an impaired endothelial barrier [82]. Exaggerated permeability and inflammation were also reported in the lung of S1pr1ECKO treated with LPS [83]. Furthermore, the increase of plasma S1P following LPS treatment was shown to enhance the expression of S1PR1, S1P metabolizing enzymes, and transporters (i.e., SPHK1 and SPNS2), hence favoring the re-establishment of endothelial barrier [83].
In vivo manipulation of S1P levels with different genetic approaches underlined the importance of circulating S1P in preserving the endothelial barrier functions and in refraining plasma proteins and leukocytes from extravasating. Mice global knockout for sphingosine kinase 1 (Sphk1−/−) showed an enhanced pulmonary edema in response to LPS and thrombin [84]. Moreover, SPHK1 is activated following thrombin stimulation and contributes to the recovery of the barrier following the breakdown. Camerer and colleagues demonstrated that mutant mice lacking erythrocyte-derived S1P (gene specific Sphk1/2−/− mouse model), lowering circulating S1P levels [56], presented vascular hyperpermeability associated with the formation of EC gaps, and an exaggerated leakage in response to platelet-activating factor (PAF) and histamine. Both the transfusion of mice with wild type (WT) RBC, restoring plasma S1P to control levels, and the acute administration of S1PR1 agonist, reduced vascular leakage and EC gaps in venules [85]. It has been also demonstrated that platelet-derived S1P enhances the endothelial barrier integrity in human and mice [86, 87] and contributes to reinforce the high endothelial venule cell barrier in lymph nodes during the immune response [88], underlying that both RBC and platelet-derived S1P exerts barrier-enhancing functions.
Endothelial-derived S1P is also important to maintain the integrity of the barrier. Murine lung EC isolated from Spns2−/− mice showed reduced TEER compared to control and downregulated SPNS2 expression following LPS or pro-inflammatory cytokines, suggesting that a reduction of endothelial-derived S1P during inflammation may contribute to barrier disruption. In support of the in vitro data, albumin leakage was significantly enhanced in bronchoalveolar lavage of Spns2−/− mice [89]. A recent study from Simmons et al., by specific deletion of Spns2 in lymphatic EC using Lyve1-Cre, demonstrated that endothelial-derived S1P is critical to preserve high endothelial venule barrier integrity, survival, and functions related to immune cell trafficking [90].
Christofferesen et al. showed the importance of ApoM of HDL-bound S1P in preserving the endothelial integrity [66, 91]. ApoM−/− mice, with about 50% reduction in plasma S1P, showed ca. 40% increase in lung vascular permeability in basal conditions compared to controls [66, 91]. Plasma reconstitution with ApoM-bound S1P, or treatment with an S1PR1 receptor agonist, restored the endothelial integrity and lowered lung permeability [91]. The importance of ApoM-bound S1P was also corroborated in a model of acute inflammation. Following a subplantar injection of carrageenan, a pro-inflammatory agent, ApoM−/− mice showed an exaggerated vascular leakage and leukocytes extravasation compared to WT. This phenotype was reversed by adenoviral-mediated ApoM overexpression [91].
Whereas much effort investigated the impact of S1P on endothelial barrier integrity, and its underlying mechanisms, the role of ceramide in healthy and disease states remains poorly defined. Several cellular stressors (i.e., pro-inflammatory cytokines, oxidative stress, ionizing radiations) activate neutral and/or acid SMase at the cell membrane and lysosome, respectively, resulting in an increase of ceramides [92, 93], linked to hyperpermeability. An early study by Goggel et al. showed that the activation of acid SMase and ceramide production contributes to the PAF-induced lung edema. Both SMase inhibitor ARC39 and genetic deletion of acid SMase in mice attenuated pulmonary edema formation [94], supporting a role for SM-derived ceramide in endothelial barrier disruption. Interestingly, acid SMase activity is elevated in a variety of pathological conditions associated with vascular hyperpermeability, such as acute lung injury [94] and sepsis [95] in animals, and in septic human patients [96].
By using bovine and human pulmonary EC in vitro, Lindner et al. showed that exogenous C6:0-ceramide (30 μM) significantly decreased TEER after 90 min [97]. Another study showed that in primary rat and human microvascular EC, cigarette smoke induced endothelial barrier disruption in a dose- and time-dependent manner, by a mechanism involving ROS-dependent activation of neutral SMase and ceramide elevation. The morphological changes included stress fiber formation, downregulation of zonula occludens-1 (ZO-1, forming TJ), and intercellular gap formation, which were ameliorated by neutral SMase inhibition. The addition of C6:0-ceramide (20 μM, 2 h) was able to recapitulate the disruption of endothelial barrier by cigarette smoke [98].
While these studies suggest an involvement of SMase-derived ceramide in the endothelial barrier disruption by a stressor, additional studies are necessary to underpin the mechanisms underlying these effects. For instance, ceramides can alter membrane biophysical properties and impact junctional strength, dynamics, and/or intracellular binding partners of the VE-cadherin complex, resulting in the breakdown of cell-to-cell contacts. Ceramides can also interact with intracellular targets, such as PP1, PP2A, PKCζ [48,49,50] which in turn signal to EC junctions and alter their stability. Lastly, ceramides can also impact oxidative stress [99] and induce cytoskeleton modification [100] to impact the barrier integrity.
8.5 S1P Signaling in Endothelial Control of Vascular Tone
The endothelium lines the interior walls of blood vessels, therefore it is in constant direct contact with circulating signals and can sense hemodynamic changes, such as shear stress of the flowing blood, in response to which it releases vasoactive factors including NO, PGI2, endothelin, and thromboxane [101]. S1P has been described as a potent activator of AKT-eNOS pathway [102, 103], comparable to other NO agonists (i.e., VEGF).
Several studies showed that low concentrations of exogenous S1P (0.1-100 nM) were able to vasodilate pre-constricted rodent resistance arteries ex vivo by activating endothelial NO production [104], mainly via S1PR1 [105]. It has been shown that HDL-bound S1P induces vasorelaxation of thoracic aorta rings ex vivo mainly via S1PR3 [106]. On the contrary, a recent study from our group revealed S1PR1 and not S1PR3 as main mediator of albumin-bound S1P-induced vasorelaxation in resistance arteries, which are relevant to BP regulation. In this study, second-order mesenteric arteries (MA) were mounted in the pressure myograph system using Krebs solution (containing salts and glucose), allowing to recapitulate hemodynamic conditions of blood flow and pressure as in vivo. In this setting, vasorelaxation in response to S1P was blunted in MA of S1pr1ECKO, but not S1pr3−/− mice, suggesting that in resistance arteries S1P vasorelaxation is mainly mediated by S1PR1 [105]. Considering the heterogeneity of EC, it is possible that S1PR distribution differs throughout the vascular tree (i.e., aorta vs. resistance arteries vs. capillaries), thus the role of S1PR1 and S1PR3 in mediating S1P vasodilation. Another consideration is that the carrier to which S1P is bound (albumin vs. HDL) might differentially activates S1P receptor subtypes [107]. FTY720 (Fingolimod, Gilenya) is an FDA-approved immunomodulator for the treatment of relapsing-remitting multiple sclerosis [108]. It blocks the egress of lymphocytes from lymphoid tissues by downregulating S1PR1 expression [109]. FTY720 is an analogue of S1P and is phosphorylated in vivo by SphK2 [110]. P-FTY720 targets 4 out of 5 S1P receptors, S1PR1 and S1PR3–5 [111], with highest affinity for S1PR1 [112]. Like S1P, P-FTY720 (1 nM-10 μM) induces eNOS-dependent vasodilation of murine thoracic aorta rings ex vivo [113]. However, when given chronically, FTY720 is a functional antagonist and induces the internalization and proteasomal degradation of S1PR1 [114]. In MA from mice treated chronically with FTY720 (0.3 mg/kg), vasodilation in response to S1P is suppressed, both in normotensive and hypertensive conditions [105]. Altogether, these findings underline that an important function of S1P is to control vascular tone mainly via endothelial S1PR1 activation, and in part via S1PR3, of eNOS-NO signaling.
8.6 Endothelial Ceramide in Vascular Tone Regulation
The elevation of circulating and local ceramide in certain pathological conditions, including obesity, type 2 diabetes (T2D), and atherosclerosis, has been linked to adverse vascular effects and endothelial dysfunction. Different eNOS-activating agonists, such as vascular endothelial growth factor, bradykinin, ATP, S1P, insulin, or fluid shear stress can induce the phosphorylation of eNOS at Ser1176, leading to NO production and vasodilation, which is impaired when endothelial dysfunction sets off.
Ceramides can directly interact with different intracellular targets and modify their functions and/or localization, including the repressive factor Inhibitor 2 (I2PP2A), disrupting its inhibitory interaction with protein phosphatase 2A (PP2A) [115]. Studies from the Symons’s lab demonstrated that in bovine aortic EC in vitro exposure to palmitate leads to ceramide accumulation and its binding to I2PP2A, resulting in PP2A activation, which in turn dephosphorylates Ser1177-eNOS [116, 117]. As consequence of PP2A-eNOS interaction, eNOS also dissociates from AKT and HSP90 complex, resulting in an overall reduced NO production [116, 117]. In vivo approaches using high fat diet (HFD) model of obesity-induced T2D showed that ceramides are increased in mouse aorta compared to standard chow diet-fed mice, and this correlated with worsen Ach-mediated vasodilation of aortic rings, eNOS phosphorylation, and hypertension [116, 117]. Both the inhibition of ceramide biosynthesis with the SPT inhibitor myriocin and the heterozygous deletion of the dihydroceramide desaturase (DES1) [116], or the inhibition of PP2A [117], ameliorated Ach-mediated vasodilation, eNOS phosphorylation, and improved hypertension in HFD-fed mice, suggesting that decreasing vascular ceramide content improves the functions. It is noteworthy to mention that in these studies ceramides have been measured in aorta and not in resistance arteries, accountable for BP regulation [26, 116, 117]. Furthermore, even if palmitate-induced ceramide accrual in EC in vitro has been demonstrated by different studies [116,117,118]; however, whether these changes occur also in EC in vivo in pathological settings is yet to be demonstrated, although inferred. In vivo, EC are exposed to a variety of plasma components, shear stress and circumferential stress due to the blood flow and pressure, which are not present in vitro. Further studies are needed to understand how ceramides change and impact on endothelial dysfunction and its sequela in vivo.
While different studies characterized how excessive ceramide contributes to endothelial dysfunction, the role of ceramide in the healthy endothelium received less attention. EC are bathed in plasma rich in SL. Cells can use sphingosine via recycling pathway to build ceramide and more complex SL [26]. Thus, it is conceivable to assume that endothelial de novo biosynthesis is dispensable to preserve sphingolipid homeostasis.
Recently, we showed that mice defective of endothelial de novo sphingolipid production, by specific Sptlc2 deletion (Sptlc2ECKO), have decreased plasma ceramide and S1P (ca. 30% and 50%, respectively), while SM were not affected, suggesting that the endothelium actively contributes to maintain circulating levels of ceramide an S1P. In liver EC deleted in vitro of Sptlc2, ceramide and hexosyl-ceramide were also significantly reduced, but not SM, at the time points assessed (48 h post-4-hydroxytamoxifen treatment). SM are the most abundant SL in the EC, and longer time points might be required to see an impact on their levels in vitro. Indeed, measurements of SL in MA demonstrated a significant decrease of SM, suggesting that endothelial de novo biosynthesis is required to maintain sphingolipid homeostasis. BP is significantly lower in Sptlc2ECKO compared to controls (Sptlc2f/f). S1P and ceramides function as rheostat of eNOS-derived NO production. While S1P is a potent activator of eNOS, ceramide levels inversely correlate with eNOS activity. In the absence of SPTLC2, ceramides levels are low, and I2PP2A remains bound to and inhibits PP2A, resulting in increased eNOS activity. Indeed, P-VASP, downstream target of NO-cGMP signaling, is significantly increased in the thoracic aorta of Sptlc2ECKO mice, supporting the lower BP. Although apparently beneficial, the decrease of ceramide below homeostatic levels impairs the endothelial control of vascular tone leading to dysfunction [119]. Specifically, endothelial-mediated vasorelaxation to VEGF, insulin, S1P, thrombin, PGI2 and flow was markedly impaired, except for Ach. These data clearly demonstrate that the activation of AKT-eNOS-cGMP axis was preserved in Sptlc2ECKO MA (i.e., vasorelaxation to Ach) and suggest that the decrease of endothelial ceramides, although heightened basal activation of eNOS, compromised the signal transduction. Indeed, VEGF-mediated phosphorylation of VEGFR2 and downstream AKT-eNOS signaling were impaired in EC lacking Sptlc2. The addition of C16:0-ceramide restored ceramide levels, as well as VEGF signaling in EC in vitro, suggesting that ceramides are necessary to preserve endothelial membrane signal transduction. It is likely that ceramides can modulate membrane biophysical properties, and/or the partition of receptors in specific membrane microdomains, and/or directly affect membrane receptor functions.
It is noteworthy to mention that multiple studies have used exogenous short chain ceramides (C2:0-, C6:0-ceramides), which are not present in mammals, to study how ceramide elevation impact vascular tone. Although it has been shown that short chain ceramides are deacylated and recycled into long-chain ceramides once administered [120], a process estimated to require ca. 1 h in lung epithelial cells (A549), how exogenous short chain ceramides change ceramide profile in vascular cells, and whether exogenous short chain or endogenous ceramides mediate the observed effects is often unknown. Thus, the results from studies using this approach need to be carefully interpreted.
The formation of reactive oxygen species (ROS) underlines endothelial dysfunction in diabetes and CV conditions [121]. In addition to a direct role in inhibiting eNOS activity, ceramides also promote ROS production by increasing NADPH oxidase activity [99, 122]. Li et al. have shown that treatment of HUVEC with C6:0-ceramide triggers ROS production in a concentration-dependent manner. These effects led to decreased bioactive NO, restored by the addition of tetrahydrobiopterin, a cofactor used by eNOS in the conversion of l-arginine to NO [99]. In a follow-up study, C2:0-ceramide was shown to increase superoxide production in bovine coronary arterial EC via activation of NADPH oxidase, followed by peroxynitrite accrual, findings that correlated with impaired bradykinin-mediated vasodilation of small bovine coronary arteries ex vivo. Mechanistically, ceramides induced the translocation of p47phox, a NADPH oxidase subunit, to the membrane, enhancing the enzyme activity [122]. It has been also proposed that ceramide-induced lipid rafts formation favors the aggregation of NADPH oxidase subunits, p47phox and gp91phox, to form the active enzyme producing ROS [123]. Similarly, carotid arteries from mice overexpressing CuZn superoxide dismutase, a ROS scavenger, were resistant to ceramide-reduced vasodilation to Ach compared to controls [124]. Growing evidence implicate altered mitochondrial dynamics and mitophagy in endothelial dysfunction, particularly in diabetes [125]. Although how ceramides contribute to impaired mitochondrial dynamics and mitophagy is an ongoing area of research, ceramide impact on mitochondria biology in EC remains poorly investigated.
Interestingly, it has been shown that in patients with CAD the endothelial mediator of flow-induced dilation shifts from NO to mitochondrial-derived hydrogen peroxide (H2O2) [126, 127]. Despite both being vasorelaxant factors, whereas NO elicits anti-inflammatory and anti-thrombotic pathways, H2O2 has pro-inflammatory and prothrombotic effects [128]. Recently, Freed et al. demonstrated that C2:0-ceramide treatment of healthy adipose arterioles triggered mitochondrial H2O2 production, which mediated the vasodilation in response to flow. Inhibition of neutral SMase with GW4869 restored endothelial NO as flow-mediated dilator in arterioles from adipose tissue and atria of patients with CAD [129]. A complementary study corroborated the role of ceramide in triggering H2O2-mediated vasodilation to flow in arterioles isolated from human adipose tissue and treated with neutral ceramidase or SPHKs inhibitors, both increasing ceramides [130]. These data revealed another mechanism of action of ceramide accrual in the onset of endothelial dysfunction.
Altogether, these studies support the concept that physiological ceramide levels need to be tightly regulated within a narrow window to preserve endothelial functions, and that both, ceramides decrease and accrual, are “deleterious” and lead to dysfunction.
8.7 Endothelial-Derived S1P: A Mechanotransduction Signaling in Blood Flow Regulation
The half-life of S1P in the plasma is about 15 min [55], suggesting the presence of high-capacity cellular sources to preserve plasma S1P levels in the low micromolar range. In addition to RBC [56] the endothelium is an important source of plasma S1P [54, 55, 119]. Changes in shears stress due to the flowing blood stimulate the release of S1P from the endothelium [55]. However, the biological significance of endothelial-derived S1P is not limited to sustain the circulating carrier-bound S1P, but is critical in regulating blood flow. EC can sense and respond to changes in fluid stress, which is the frictional force per unit area from flowing blood acting on the endothelium. Blood flow regulation is important not only to match the metabolic requirements of a tissue by providing nutrients, oxygen, and removing waste, but also for vascular morphogenesis and physiology.
A recent study from our group provided direct evidence of this S1P function. The increase of flow led to vasorelaxation of WT MA mounted in a pressure myograph system via activation of eNOS and NO production. However, genetic deletion of endothelial Spns2 markedly suppressed the vasorelaxation to flow [54], directly implicating the endothelial-derived S1P in the activation of S1PR1-eNOS signaling in blood flow regulation. This phenotype was supported by a significant reduction of phosphorylated-VASP (vasodilator-stimulated phosphoprotein), a downstream target of the NO-cGMP pathway, in thoracic aorta [131]. These results suggest that endothelial-derived S1P and autocrine S1PR1-signaling is necessary to preserve vascular functions and blood flow regulation [54].
Both S1PR1 and S1PR3, expressed by the healthy endothelium, can activate eNOS via PI3K-AKT pathway [102]. However, vasorelaxation in response to flow was dramatically suppressed in S1pr1ECKO, but not S1pr3−/− MA, and this phenotype correlated with reduced plasma NO levels [105]. Similar results were obtained in MA lacking Sptlc2. Flow-induced vasorelaxation was significantly reduced in Sptlc2ECKO MA [119]. Furthermore, ex vivo treatment of MA with W146, a competitive antagonist of S1PR1, suppressed flow-mediated vasodilation and eNOS activation in concentration-dependent manner [53]. These findings clearly identified endothelial-derived S1P and the autocrine S1PR1-eNOS-signaling as critical mechanotransduction pathway regulating blood flow.
An elegant study of Jung et al. demonstrated that the genetic deletion of endothelial S1pr1 resulted in misalignment of the aortic endothelium with blood flow, compared to control morphological alignment of EC lacking S1pr1 [132]. In vivo, the deletion of endothelial S1PR1 led to an abnormal hypersprouting and poorly perfused vasculature in the retina [132].
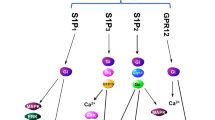
Altogether these studies propose S1PR1 as a critical mechanotransduction signaling. Flow induces endothelial S1P production, which is transported outside of the cell via SPNS2, and can induce vasodilation by activating S1PR1-eNOS-NO signaling (Fig. 8.3). These findings also suggest that a deranged endothelial S1P production/degradation, and/or transport by SPNS2, may impair vascular functions and contribute to the pathogenesis of atherosclerosis, hypertension, and metabolic-related cardiovascular disorders.

Endothelial-derived S1P: a mechanotransduction signaling in blood flow regulation. Changes in blood flow stimulate the synthesis of S1P by EC. Once produced, S1P is transported outside of the endothelial cells by SPNS2. Endothelial-derived S1P can act in an autocrine or paracrine manner on S1PRs. Endothelial S1PR1 activation is critical in blood flow-mediated vasodilation via eNOS-derived NO production. S1P sphingosine-1-phosphate, S1PR S1P receptor, SPNS2 spinster-2, eNOS endothelial nitric oxide synthase, NO nitric oxide
8.8 S1P and Ceramide Rheostat in Blood Pressure Homeostasis
BP is determined by cardiac output, defined as the volume of blood ejected by the heart per minute, and total vascular resistance, which is the resistance exerted by all the vasculature on the flowing blood. BP is very dynamic and regulated by the interplay of heart (i.e., morphology and function); kidney, controlling extracellular fluid and blood volume via the pressure-natriuresis mechanism; neuroendocrine system (i.e., the sympathetic nervous system, renin-angiotensin-aldosterone system); and peripheral vascular resistance. In this regard, generalized vasodilation usually leads to a decreased systemic BP, but may also occur in specific tissues causing a local augmentation of blood flow. Endothelial dysfunction, mainly defined as the loss of the endothelium to induce a NO-mediated vasodilation in response to changes in flow, Ach, or other NO-stimulating agents, comprises the impairment of all the endothelial properties, including barrier integrity, anti-thrombotic and anti-inflammatory functions. In the past three decades endothelial dysfunction emerged as a pivotal and initial event in the onset of hypertension, atherosclerosis, and other CV diseases [18,19,20]. Dysfunctional endothelium is no longer able to meet an organ’s needs and to preserve its functions, and over time contributes to end-organ damage, such as kidney and heart failure, cognitive impairments, stroke, etc. [133].
In in vivo anesthetized rats, acute administration of S1P (0.2 mg/kg) evoked a rapid decline of heart rate and mean arterial BP, followed by a transient hypertension concomitant to the increase in heart rate [134]. While the acute activation of endothelial S1PR1 and S1PR3 and downstream NO signaling is most likely responsible for the initial drop in BP, the following elevation in BP might be due, in part, to the functional antagonism of S1PR1 downregulating eNOS activation [102, 103] and NO bioavailability, and by activation of smooth muscle S1PR2/3 leading to vasoconstriction via Rho-kinase activation [135, 136]. However, mice systemically lacking S1PR3 [105] and S1PR2 [137] did not show any difference in BP at baseline. Consistently, when challenged with chronic infusion of AngII, S1pr3−/− mice developed hypertension to the same extent of control mice [105], arguing against the role of S1PR3 in hypertension. It is possible that in absence of S1PR3, smooth muscle S1PR2 may play a compensatory role in systemic BP regulation. Nonetheless, myogenic tone, which is the vasoconstriction of resistance arteries in response to pressure increase, was significantly reduced in S1pr3−/− MA [105]. Thus, although dispensable in systemic BP regulation, it is possible that S1PR3 controls blood flow in specific vascular beds, such as MA [105] and cerebral arteries [135, 136]. On the contrary, S1PR1 signaling is critical for BP homeostasis. In a recent study we reported that mice S1pr1ECKO have a higher BP compared to mice S1pr1f/f, both in physiological and hypertensive conditions. Vasodilation of S1pr1ECKO MA in response to S1P and flow was significantly reduced, as the downstream eNOS-derived NO signaling [105]. Interestingly, chronic administration of FTY720, which dramatically downregulates endothelial S1PR1 in blood vessels, significantly elevated BP in healthy and in AngII-induced hypertensive mice [105]. This hypertensive phenotype correlated with impaired endothelial-dependent vasodilation of MA to Ach, S1P and flow, supporting vasculo-protective functions of endothelial S1PR1 signaling. Additional studies underlined the role of S1PR1 in BP regulation. A single dose of FTY720 increased the BP of spontaneously hypertensive rats after 24h [138], while chronic administration of FTY720 significantly raised BP in normotensive rats in a dose-dependent manner [139]. Clinical studies using FTY720 corroborated the role of S1PR1 in BP regulation. In patients, a sustained elevation of BP (≈4–5 mmHg) was observed after 2 months of FTY720 treatment [140]. Altogether, animal and human studies underline the importance of the S1P-S1PR1 pathway in maintaining vascular and BP homeostasis [105, 140].
Endothelial S1PR1 is activated by both circulating carrier-bound S1P and endothelial-derived S1P in autocrine manner. Our group demonstrated that endothelial deletion of Spns2 (Spns2ECKO) leads to a significant elevation in BP and impairment of BP dipping [54], which is the difference between the diurnal and nocturnal mean BP. Altered BP dipping has been associated with a higher risk of CV events [141, 142].
Because plasma S1P increases in hypertension [54, 143, 144], it was postulated a pro-hypertensive role. When challenged with chronic infusion of AngII, Spns2ECKO mice developed an exaggerated hypertension, although plasma S1P remained ca. 40% reduced in Spns2ECKO mice compared to control (Spns2f/f), arguing against a pro-hypertensive role for plasma S1P [54]. Interestingly, cardiac hypertrophy was also worsened in hypertensive Spns2ECKO. Both the increase in systemic BP, which causes a hemodynamic stress on the heart, and the decrease of local endothelial-derived S1P and NO, exerting cardioprotective functions [145, 146], might contribute to aggravate the cardiac hypertrophy in Spns2ECKO mice. Interestingly, Spns2ECKO mice showed a decrease not only in S1P, but also in ceramides in both EC and circulation, and lowered systemic BP compared to controls [119]. In these conditions, the impact of ceramide prevails on S1P. Specifically, the decrease of ceramide-dependent PP2A activation is accountable for the increased eNOS phosphorylation and NO production at baseline, hence the lower BP [119].
ApoM-bound S1P also contributes for BP homeostasis. Mice ApoM−/− showed higher systolic BP at baseline and following chronic infusion of AngII, correlating with worsen endothelial dysfunction and cardiac hypertrophy compared to control [54]. These data underline the importance of endogenous ApoM-bound S1P in preserving endothelial functions and BP homeostasis and, suggest that ApoM-bound S1P could be exploited therapeutically to treat CV conditions such as hypertension. In a follow-up study with Hla’s group, we reported that administration of an engineered ApoM, named ApoM-Fc, which does not need to bind HDL and has longer half-life than native ApoM, leads to a sustained anti-hypertensive effect in mice lasting ca. 7 days. ApoM-Fc-bound S1P can activate S1PR1-NO signaling and lower BP, as demonstrated by the treatment with W146, an S1PR1 antagonist, abolishing its BP lowering effects [147].
To understand the role of S1P in BP homeostasis, initial studies used mice Sphk1 or Sphk2 knockout, of a combined deletion of their alleles, since the deletion of both genes is embryonically lethal [148]. However, these studies led to confusing outcomes for different reasons. First, the degradation of S1P by S1P lyase is the only catabolic exit of sphingolipid pathway; therefore, the deletion of Sphks results in the loss of S1P formation and a backlog of ceramides, adding a confounding factor to the interpretation of the phenotypes. Second, whereas circulating levels of S1P are reduced in Sphk1−/− [143, 149] and Sphk1+/− Sphk2−/− [56, 85] mice, they are increased in Sphk2−/− [150, 151] mice, probably for an overcompensation by SPHK1 [151]. At baseline, BP was not affected in mice Sphk1−/− [143, 152,153,154,155,156] and Sphk2−/− [152, 154, 155], nor in mice overexpressing SPHK1 [157]. Several groups studied the role of SPHKs in regulating BP in response to acute AngII administration in vivo. Furuya et al. showed that AngII administration (i.p.) induced higher BP elevation in Sphk2−/− but not in Sphk1−/− mice compared to controls. However, it is difficult to ascribe this protective effect to SPHK2-derived S1P or to the excessive elevation of plasma S1P when Sphk2 is deleted [152]. On the contrary, Wilson et al. showed that BP elevation following AngII was reduced in mice Sphk1−/−, and considering that Sphk1−/− mice have lower BP at baseline than WT, the BP increase between the two groups is of similar magnitude [156].
More consistent results have been obtained with chronic infusion of AngII. Siedlinski et al. reported that SphK1 mRNA and protein expression was upregulated in MA and aorta of AngII-treated WT mice [143], and AngII-induced BP elevation was significantly lower in Sphk1−/− mice WT mice [143, 156]. Meissner et al. showed that both Sphk1−/− and Sphk2−/− mice are protected from AngII-induced hypertension, with a more profound effect in the Sphk2−/− [154]. Furthermore, administration of the SPHK2 antagonists ABC294640 or K-145, but not the SPHK1 antagonist N,N-dimethylsphingosine, significantly reduced BP in hypertensive mice, suggesting that SPHK2 contributes to the onset of hypertension [154]. Altogether these data suggest a role for SPHK2, and in part SPHK1, in the pathogenesis of hypertension. Data in Sphk2−/− support an anti-hypertensive rather than pro-hypertensive role of plasma S1P, considering that is higher in Sphk2−/− mice [150, 151]. Furthermore, the backlog of ceramide can also contribute to some of the observed phenotypes. However, all the studies have been performed in mice global knockout for Sphks, and to better dissect the specific roles of SPHK1 and SPHK2 in BP homeostasis (i.e., vasculature, immune system) further studies will be required in tissue-specific knockout mice.
Although additional studies are required to elucidate how S1P signaling is impaired in hypertensive conditions, collectively these studies provide significant evidence of its role in the regulation of BP homeostasis.
8.9 Endothelial Fine-Tuning of Sphingolipid De Novo Biosynthesis: Role of Nogo-B and ORMDLs
Localized in the cytosolic leaflet of the smooth ER, SPT is formed by two major subunits, SPTLC1 and SPTLC2, ubiquitously expressed [158,159,160]. A third subunit, SPTLC3, with tissue-specific distribution, was recently identified and can form an active complex with SPTLC1 [160]. The active site is located at the interface of the two SPT subunits [161,162,163]. Importantly, SPTLC3 has a higher activity with lauryl- and myristoyl-CoA, generating C14- and C16-sphingoid bases [164], whereas SPTLC2 forms mainly C18- and C20-sphingoid bases.
Altered sphingolipid homeostasis has been implicated in different pathological conditions, including CV [165, 166], metabolic [167] and inflammatory [93] diseases, and cancer [168]. Yet, under normal physiology, sphingolipid levels are tightly regulated. Mechanisms governing cellular sensing of SL and accordingly sphingolipid biosynthesis remain poorly understood. SPT small subunits a and b (ssSPTa and ssSPTb) have been identified by the Dunn’s group as enhancer of SPT activity through their single transmembrane domain [169, 170]. In 2010, Weissman’s group reported ORMDL1–3 proteins (Orm proteins in yeast) as negative regulator of SPT activity [171]. ORMDLs are a conserved family of ER membrane proteins, ubiquitously expressed in fetal and adult tissues. ORMDL1, 2, and 3 share more than 80% of identity in the same species, and 96–99% of homology between human and mouse [172]. Orthologues of the ORMDLs have been reported in yeast, plants, microsporidia, urochordates, and invertebrates. Identity between mammal and yeast ORMDLs/Orms is limited to 30–40% [172]. Structurally, ORMDLs are anchored to the ER membrane by 4 transmembrane domains [173,174,175], with both N- and C-terminus facing the cytosol [176]. Breslow et al. demonstrated that lowering SL with myriocin triggers the phosphorylation of Orms, which releases its interaction with SPT, hence heightening sphingolipid de novo biosynthesis [171]. However, this regulatory mechanism is not shared by mammalian ORMDLs as they lack the N-terminus hosting the phosphosites in yeast [172]. Nonetheless, in eukaryotic cell lines, the absence of ORMDLs increases the activity of SPT [171, 177], corroborating their inhibitory function on SPT. How ORMDLs interaction with SPT is regulated in mammals has attracted much attention. ORMDLs expression can be induced both at transcriptional level by allergenic and inflammatory stimuli (i.e., OVA, LPS, IL-4, IL-13) [178], and at translational level by increased expression of catalytically active SPT [179], although the underlying molecular mechanisms remain unknown. Studies from Wattenberg’s group using cell-free isolated membranes and permeabilized cells showed that non-physiological ceramides (C6:0- and C8:0-ceramide), and endogenously produced ceramides, can acutely downregulate the activity of SPT, probably by interacting with SPT-ORMDLs complex [173, 177]. Although this proposed mechanism has been demonstrated in HeLa cells, whether it is physiologically relevant is yet to be demonstrated. Two recent crystallographic studies have elegantly showed that ORMDL3 directly inhibits SPT activity by inserting its N-terminus in the opening between SPTLC1 and SPTLC2, therefore preventing substrates access to the catalytic domain [174, 175]. How ORMDL3 inhibition of SPT is dynamically regulated by SL levels remains a standing question. In this regard, our group recently discovered that ORMDLs are constitutively hydroxylated at Pro137, which is an obligatory step for their ubiquitination and proteasomal degradation. This mechanism preserves SPT activity at steady state by refraining ORMDL levels. Moreover, we identified S1P as the key sphingolipid sensed by EC via S1PRs. S1P-activation of S1PR1,3 signaling stabilizes ORMDLs to negatively regulate SPT and maintain sphingolipid homeostasis [180]. Interestingly, genetic disruption of S1P-S1PRs signaling pathway at different levels, by the deletion of SphK1,2, or S1pr1,3 or Spns2, relieved the inhibition on SPT with consequent increase of ceramide levels. In particular, the deletion of Spns2 highlighted the importance of endothelial-derived autocrine S1P signaling to maintain not only the vessels response to blood flow, but also sphingolipid homeostasis. Spns2 deletion caused ceramide accrual, mitochondrial dysfunction, and impaired signal transduction, all leading to endothelial dysfunction, an early event in the onset of cardio- and cerebrovascular diseases [180] (Fig. 8.4). It is noteworthy to mention that, while S1P is protective for many of the endothelial functions discussed in this chapter, an excessive increase may lead to a deleterious inhibition of sphingolipid biosynthesis via S1PRs-ORMDLs. It is likely that the disruption of this newly described S1P-ORMDL-SPT signaling in EC might be implicated in the pathogenesis of several conditions characterized by deranged SL metabolism, such as diabetes, cardiomyopathies, and neurodegeneration.

Endothelial fine-tuning of sphingolipid de novo biosynthesis: role of Nogo-B and ORMDLs. (a) In absence of Spns2 (Spns2ECKO), S1P-S1PRs dependent stabilization of ORMDL3 is disrupted, leading to an upregulation of SPT activity. Ceramide build up whereas S1P is no longer transported outside of the cells, thus autocrine/paracrine endothelial S1P signaling is lost. In these conditions, the S1P/ceramide rheostat shifts towards ceramides, resulting in generalized endothelial dysfunction, including barrier impairment, mitochondrial dysfunction, apoptosis, impaired signal transduction, dysregulation of vascular tone, hypertension, and severe cardiac hypertrophy. (b) In absence of Nogo-B (Nogo-A/B−/−), the upregulation of SPT activity leads to the beneficial increase of both ceramide and S1P levels, within a window of opportunity. The rheostat S1P/ceramide shifts towards S1P beneficial effects, resulting in reduced pathological cardiac hypertrophy, vascular dysfunction, hypertension, leucocyte extravasation, and permeability. S1P sphingosine-1-phosphate, S1PR S1P receptor, SPT serine palmitoyltransferase, SM sphingomyelin, GSL glycosphingolipid, SPHK1,2 sphingosine kinase, SPNS2 spinster-2
ORMDL3 has been the most studied among the ORMDL isoforms, especially considering that single nucleotide polymorphisms (SNP) increasing its expression have been associated with asthma (rs8076131 and rs4065275) [181, 182]. Consistently, global overexpression of human ORMDL3 in mice led to airway remodeling, inflammation, and hyperresponsiveness to methacholine versus control mice [183]. Vice versa, deletion of Ormdl3 specifically in lung epithelial cells protected the mice from airway hyperresponsiveness, inflammation, mucus secretion, and collagen deposition [184]. Finally, mice Ormdl1/3−/− manifested SL accrual in the sciatic nerves and reduced level of myelinization [185], underlying the importance of ORMDL proteins in preserving sphingolipid homeostasis in higher eukaryotes.
Later in 2015, our group discovered Reticulon-4B (RTN4B, aka NOGO-B) as a negative regulator of SPT activity in mammalian cells (Fig. 8.4) [53]. NOGO-B belongs to the family of reticulon proteins, localized in the membrane of the tubular ER (aka smooth ER), contributing to the tubulogenesis of the ER network [186]. RTN proteins are characterized by two transmembrane domains separated by a loop of 66-aa, named reticulon-homology domain (RHD) [186, 187]. Rtn4 gene gives rise to three major splicing isoforms, namely NOGO-A, NOGO-B, and NOGO-C. While NOGO-A and -C are mainly expressed in the central nervous system [187], and low levels of NOGO-C are found the skeletal muscle [188], NOGO-B is highly expressed, but not exclusively, in the blood vessels [53]. In physiological conditions, NOGO-B is also expressed in bone marrow cells [189] and in cardiomyocytes of the atria but not ventricles [190]. In this regard, growing evidence demonstrated an upregulation of myocardial RTN4 expression in cardiomyopathy in mice and humans [191,192,193], implicating a role for NOGO in heart diseases. In EC the most abundant isoform is NOGO-B, while NOGO-A is undetectable [190].
8.9.1 NOGO-B Regulates BP and Vascular Tone Via SPT
NOGO-B binds to and downregulates SPT activity of ca. 40% in both murine and human EC in vitro and in murine lung microsomes. In the absence of NOGO-B, SPT activity and sphingolipid levels, particularly S1P, are increased [53]. However, the molecular basis governing the dynamic interaction of NOGO-B/SPT, for instance in response to changes in sphingolipid levels, remains to be elucidated. Mice lacking Nogo-A/B globally (Nogo-A/B−/−) and specifically in EC (Nogo-A/BECKO) are born at Mendelian ratios. In physiological conditions, they only present a significant lower systolic BP than controls (ca. 90 vs. 115 mmHg, respectively). Strikingly, both Nogo-A/B−/− and Nogo-A/BECKO mice were protected from developing high BP following chronic infusion of AngII, suggesting a pro-hypertensive role of NOGO-B. These in vivo findings were supported by vascular reactivity studies ex vivo. The loss of NOGO-B preserved vasorelaxation of MA in response to Ach and flow, which correlated with higher cGMP levels in thoracic aorta, index of endothelial NO production [53]. Mechanistically, in the absence of NOGO-B endothelial-derived S1P was markedly upregulated, hence the S1PR1-NO signaling. Interestingly, treatment of Nogo-A/B−/− mice with myriocin restored BP to pathological levels (from ca. 110 to 140 mmHg in AngII-treated mice), due to the downregulation of S1P-S1PR1-NO signaling. Although in the absence of NOGO-B, SPT activity increases and results in the elevation of both ceramide and S1P, this upregulation is within a “window of opportunity” that upregulates endothelial S1P and S1PR1-NO signaling without activating PP2A via ceramide. Overall, these findings indicate the Nogo-B downregulation of SPT activity is relevant to vascular diseases and endothelial dysfunction.
8.9.2 NOGO-B and Barrier Function
In addition to eNOS activation, S1P can enhance the endothelial barrier function via Rac-mediated cytoskeleton remodeling [80] and VE-cadherin dephosphorylation, as discussed in Sect. 8.4 [79]. The loss of NOGO-B enhances endothelial S1P production in vitro, which correlates with higher TEER enhanced barrier function [193]. Plasma S1P is also significantly elevated in Nogo-A/B−/− mice. Interestingly Nogo-A/B−/− mice were protected from myocardial permeability and inflammation following pressure overload (induced by transverse aortic constriction), whereas myriocin treatment abolished these beneficial effects. On the contrary, SEW2871, an agonist of S1PR1, was able to reduce inflammation following pressure overload [193], suggesting that endothelial S1P-S1PR1-mediated barrier enhancement in absence of NOGO-B refrains plasma proteins and leukocytes from extravasating and damaging the heart. It is noteworthy that while the loss of NOGO-B did not affect EC adhesion molecules expression (ICAM-1 and CD31) [194], exogenous addition of S1P was able to induce a significant and dose-dependent increase in VCAM-1 and E-selectin [195]. This difference may be due to the fact that in EC lacking NOGO-B the increase in S1P is into physiological levels, whereas exogenous S1P was added to EC in the μM range (1–5 μM), significantly higher than concentrations used for in vitro studies (100-300 nM), thus might overcome beneficial effects of S1P. In models of acute inflammation, such as carrageenan-induced paw edema and zymosan air-pouch, the enhanced barrier function in Nogo-A/B−/− resulted in decreased vascular leakage and extravasation of inflammatory cells, particularly neutrophils and monocytes [194]. Studies in vitro of endothelial–leukocyte interactions under flow demonstrated that paracellular trans-endothelial migration of neutrophils and monocytes was markedly suppressed in Nogo-A/B−/− EC, supporting the enhanced barrier [194]. Mechanistically, in the absence of NOGO-B the upregulation of local S1P production counterbalances VE-cadherin phosphorylation and stress fiber formation induced by ICAM-1 cross-linking [194], thus restraining the opening of the endothelial junctions during inflammation, in line with similar S1P antagonizing effects reported for VEGF and thrombin-mediated barrier disruption [80]. Accordingly, mice overexpressing S1PR1 specifically in the endothelium showed a decreased expression of ICAM-1, confirming that S1P-S1PR1 signaling suppresses the expression of inflammatory genes.
8.9.3 Endothelial NOGO-B and Pathological Cardiac Hypertrophy
In absence of endothelial NOGO-B, SPT activity is upregulated. Nogo-A/B−/− mice are protected from cardiac hypertrophy, fibrosis, and failure following chronic pressure overload [193]. Myriocin treatment inhibits SPT and reinstates the pathological phenotype in Nogo-A/B−/−, most luckily by suppressing local S1P-S1PR1 signaling. Interestingly, Nogo-A/BECKO mice are protected from developing pathological cardiac hypertrophy, recapitulating the phenotype observed in Nogo-A/B−/−. Mechanistically, the upregulation of S1P/eNOS/NO pathway in the absence of NOGO-B exerts a protective effect on the pressure overloaded heart by different means, including the reduction of permeability and inflammation by enhancing the EC barrier and cardioprotective effects of endothelial S1P [146], as well as of NO [145, 196, 197], on cardiomyocytes.
Altered sphingolipid metabolism has been implicated in cancer, and CV, immune and neurodegenerative diseases [198]. Thus, it is not surprising that more than one regulatory mechanism is in place regulating the de novo biosynthesis to preserve sphingolipid homeostasis in response to metabolic and environmental stimuli. This pathway is dynamical regulated in response to the changes of the microenvironment (i.e., inflammatory stimuli, radiations, blood flow, hemodynamic stress in the heart, etc.). We are just beginning to understand how sphingolipid levels are sensed by the cells and the multilayered regulatory mechanisms maintaining sphingolipid homeostasis. The stabilization of ORMDL by S1P signaling identified in EC might be also implemented in other cell types. Unraveling the regulatory mechanisms of sphingolipid biosynthesis may provide the framework for therapeutics to restore homeostasis in disease settings.
8.10 Plasma Ceramide as Biomarker for Major Cardiovascular Events and Other Conditions
Traditional serum lipid biomarkers of CV diseases are triglycerides and cholesterol. In the recent years, multiple studies demonstrated a strong relationship between the levels of circulating ceramides and the incidence of adverse CV events, such as CAD, MI, hypertension, and stroke, both with non-fatal and fatal outcomes [144, 199,200,201,202,203,204,205,206,207].
Most of these studies identified three major ceramide species, C16:0-, C18:0-, and C24:1-ceramide, positively associated with CV disease incidence [199], secondary CV events [201, 205], and CV mortality [200, 202, 203]; whereas C24:0-ceramide negatively correlated with CV mortality [200, 202, 203]. C24:0- and C24:1-ceramide positively correlated with the severity of hypertension [144]. Levels of C16:0-, C22:0-, and C24:0-ceramide associated with increased risk and severity of ischemic stroke [206]. Another study positively correlated C20:0- and negatively correlated C24:1-ceramide with acute ischemic stroke. Moreover, the ratios S1P/C24:1-ceramide and C24:0/C24:1-ceramide were particularly increased in acute stroke patients, suggesting a potential use as biomarkers [207]. CERT1 (cardiac event risk test 1) was developed for clinical practice and is currently in use at Mayo Clinic. Based on the concentrations of C16:0-, C18:0-, and C24:1-ceramide, alone or in their ratio to C24:0-ceramide, CERT1 stratifies patients into four CV risk categories [201]. A second risk score, CERT2, was developed taking into consideration that also phosphatidylcholine has shown prognostic values for CV events, but is not currently used in clinical practice [201, 208]. However, the inclusion of these scores in clinical practice would allow a more aggressive interventions in patients at higher risk. A recent study of Poss and colleagues evaluated a more comprehensive panel of 32 SL species in the plasma of patients with CAD, including dihydroceramides, ceramides, glucosylceramides, dihydroSM, SM, sphingosine, and sphinganine. Thirty out of 32 SL measured were elevated in patients affected by familial CAD versus healthy controls and correlated with the severity of the disease [204]. They proposed a new risk score, named SIC (sphingolipid-inclusive CAD), including C18:0-dihydroceramide, C18:0-, C22:0-, and C24:0-ceramide, C24:1-dihydroSM, C18:0- and C24:0-SM, and sphingosine, expected to outperform CERT1 in the stratification of patients with CAD [204].
In addition to CV diseases, plasma C18:0-, C20:0-, and C24:1-ceramide are also significantly elevated in obese patients with T2D compared to lean healthy individuals and correlated with the severity of insulin resistance [209]. Interestingly, the ratio C18:0/C16:0-ceramide was predictive of T2D 10 years before the diagnosis [210], and C16:0-, C22:0-, C24:0-, C24:1-dihydroceramides were increased and predictive of T2D 9 years before the diagnosis [211]. While it is known that EC and RBC are major sources of plasma S1P [53, 54, 212], little is known on the cellular sources of ceramides in circulation. We have recently demonstrated that endothelial deletion of SPTLC2 leads to ca. 50% and 30% reduction in plasma S1P and ceramides, respectively, but not in SM [119]. A significant reduction in plasma S1P (ca. 40%) was also achieved by genetic deletion of endothelial SPNS2, supporting the role of the endothelium as source of both plasma S1P and ceramide [54, 58]. Based on clinical data, it is conceivable that the endothelium contributes, at least in part, to remodel plasma ceramide landscape in patients affected by CV and metabolic diseases, including CAD, heart failure, and diabetes. Thus, in addition to predict major CV events, plasma ceramides might also function as biomarkers of the diseases state of blood vessel. It is noteworthy to mention that how SL change in the diseased versus healthy endothelium, and their impact on the vascular pathology, remains poorly understood. The increase of endothelial ceramide in CV diseases and diabetes has been inferred by in vitro findings but never measured in vivo. Thus, further studies are needed to understand how sphingolipid metabolism changes and contributes to endothelial dysfunction, culprit event in the pathogenesis of cardiovascular and cerebrovascular diseases.
8.11 Gene Variants of Sphingolipid Pathway and Cardiovascular Diseases
Several rare monogenic disorders have been associated with mutations impairing the activity of proteins of sphingolipid metabolism, and most of them manifest neurological disorders, like in hereditary sensory and autonomic neuropathy type 1, and Niemann–Pick, Gaucher, Krabbe, Fabry, and Farber diseases [213]. Growing literature reported the association between single nucleotide polymorphism (SNP) of sphingolipid metabolism genes and CV diseases. In 2009, Hicks et al. conducted a Genome-Wide Association Study (GWAS) between 318,237 SNPs and the levels of circulating SL species in five different European populations. They found SNPs in SPTLC3, CERS4, SGPP1 (S1P phosphatase), and FADS1–3 (fatty acid desaturase) associated with circulating sphingolipid levels. Moreover, SNP in SPTLC3 correlated with the incidence of MI [214]. A recent study identified 28 SNPs close to the SPTLC3 locus that significantly associated with reduction in plasma C22:0- and C24:0-ceramide [215], and increased levels of these ceramide species have been linked to CAD and heart failure. A genetic variant in dihydroceramide desaturase (DEGS1), leading to L175Q mutation (rs191144864), causes a partial loss-of-function [216]. Patients with this mutation have increased dihydroceramides and a modest decrease in three established ceramide ratios (C16:0-, C20:0-, and C24:1- in ratio with C24:0-ceramide) linked to CV diseases, suggesting that this SNP in DEGS1 may have protective functions [216]. Moreover, egs Degs1−/− mice on leptin-deficient background or fed an obesogenic diet have reduced hepatic steatosis and insulin resistance. Chaurasia et al. [217], suggesting that DEGS1 L175Q may protect from obesity and diabetes. A SNP in CERS2 (rs267738) leading to a partial loss-of-function [218], and three in CERS6 (rs3845724, rs4668102, and rs4668089) mapping in an intronic region [219], have been associated with metabolic disease and T2D, respectively. Hla’s group characterized the functional consequences of several SNPs in S1PR1 [220]. R120P mutation (rs149198314) abrogates the ability of S1PR1 to bind S1P, and therefore its endocytosis and downstream signaling. I45T (rs148977042) and G305C (rs146890331) mutations impair S1PR1 desensitization process. Thus, the efficacy of FTY720 treatment might be affected in individuals carrying these S1PR1 mutations. Finally, R13G mutation in S1PR1 was found enriched in a high CV risk population, but it appeared to protect from CAD [220], although in vitro data of S1PR1 R13G mutant showed no functional differences with the native receptor. SNPs SphK1 [221] associated with increased CV events; SNPs in NOGO-B (rs17046570) and ORMDL3 (rs7216389 and rs9303277) positively correlated with CAD [212, 222].
Circulating concentrations of several key components of sphingolipid metabolism are under genetic control, and it is conceivable to think that other variants in their loci can be accountable for the disruption in the SL homeostasis seen in the cardiometabolic diseases.
References
Bazzoni, G., & Dejana, E. (2004). Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiological Reviews, 84, 869–901.
Aird, W. C. (2007). Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circulation Research, 100, 158–173.
Poisson, J., et al. (2017). Liver sinusoidal endothelial cells: Physiology and role in liver diseases. Journal of Hepatology, 66, 212–227.
Tse, D., & Stan, R. V. (2010). Morphological heterogeneity of endothelium. Seminars in Thrombosis and Hemostasis, 36, 236–245.
Pober, J. S., & Sessa, W. C. (2014). Inflammation and the blood microvascular system. Cold Spring Harbor Perspectives in Biology, 7, a016345.
van Hinsbergh, V. W. (2012). Endothelium—role in regulation of coagulation and inflammation. Seminars in Immunopathology, 34, 93–106.
Arnout, J., Hoylaerts, M. F., & Lijnen, H. R. (2006). Haemostasis. In Handbook of experimental pharmacology (pp. 1–41).
Furchgott, R. F., & Zawadzki, J. V. (1980). The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature, 288, 373–376.
Ignarro, L. J., Byrns, R. E., Buga, G. M., & Wood, K. S. (1987). Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circulation Research, 61, 866–879.
Palmer, R. M., Ferrige, A. G., & Moncada, S. (1987). Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature, 327, 524–526.
Arnold, W. P., Mittal, C. K., Katsuki, S., & Murad, F. (1977). Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proceedings of the National Academy of Sciences of the United States of America, 74, 3203–3207.
Rapoport, R. M., & Murad, F. (1983). Agonist-induced endothelium-dependent relaxation in rat thoracic aorta may be mediated through cGMP. Circulation Research, 52, 352–357.
Parton, R. G., & del Pozo, M. A. (2013). Caveolae as plasma membrane sensors, protectors and organizers. Nature Reviews. Molecular Cell Biology, 14, 98–112.
Forstermann, U., & Sessa, W. C. (2012). Nitric oxide synthases: Regulation and function. European Heart Journal, 33(829–837), 837a–837d.
Fulton, D., et al. (1999). Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature, 399, 597–601.
Dimmeler, S., et al. (1999). Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature, 399, 601–605.
Vanhoutte, P. M., Zhao, Y., Xu, A., & Leung, S. W. (2016). Thirty years of saying NO: Sources, fate, actions, and misfortunes of the endothelium-derived vasodilator mediator. Circulation Research, 119, 375–396.
Harrison, D. G., Freiman, P. C., Armstrong, M. L., Marcus, M. L., & Heistad, D. D. (1987). Alterations of vascular reactivity in atherosclerosis. Circulation Research, 61, 1174–1180.
Panza, J. A., Quyyumi, A. A., Brush, J. E., Jr., & Epstein, S. E. (1990). Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. The New England Journal of Medicine, 323, 22–27.
Taddei, S., et al. (1996). Defective L-arginine-nitric oxide pathway in offspring of essential hypertensive patients. Circulation, 94, 1298–1303.
Celermajer, D. S., et al. (1992). Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet, 340, 1111–1115.
Balletshofer, B. M., et al. (2000). Endothelial dysfunction is detectable in young normotensive first-degree relatives of subjects with type 2 diabetes in association with insulin resistance. Circulation, 101, 1780–1784.
Merrill, A. H., Jr., & Wang, E. (1986). Biosynthesis of long-chain (sphingoid) bases from serine by LM cells. Evidence for introduction of the 4-trans-double bond after de novo biosynthesis of N-acylsphinganine(s). The Journal of Biological Chemistry, 261, 3764–3769.
Pewzner-Jung, Y., Ben-Dor, S., & Futerman, A. H. (2006). When do lasses (longevity assurance genes) become CerS (ceramide synthases)?: Insights into the regulation of ceramide synthesis. The Journal of Biological Chemistry, 281, 25001–25005.
Michel, C., et al. (1997). Characterization of ceramide synthesis. A dihydroceramide desaturase introduces the 4,5-trans-double bond of sphingosine at the level of dihydroceramide. The Journal of Biological Chemistry, 272, 22432–22437.
Gault, C. R., Obeid, L. M., & Hannun, Y. A. (2010). An overview of sphingolipid metabolism: From synthesis to breakdown. Advances in Experimental Medicine and Biology, 688, 1–23.
Hanada, K., et al. (2003). Molecular machinery for non-vesicular trafficking of ceramide. Nature, 426, 803–809.
Watson, P., & Stephens, D. J. (2005). ER-to-Golgi transport: Form and formation of vesicular and tubular carriers. Biochimica et Biophysica Acta, 1744, 304–315.
Sugiura, M., et al. (2002). Ceramide kinase, a novel lipid kinase. Molecular cloning and functional characterization. The Journal of Biological Chemistry, 277, 23294–23300.
Mao, C., & Obeid, L. M. (2008). Ceramidases: Regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochimica et Biophysica Acta, 1781, 424–434.
Wattenberg, B. W., Pitson, S. M., & Raben, D. M. (2006). The sphingosine and diacylglycerol kinase superfamily of signaling kinases: Localization as a key to signaling function. Journal of Lipid Research, 47, 1128–1139.
Ikeda, M., Kihara, A., & Igarashi, Y. (2004). Sphingosine-1-phosphate lyase SPL is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5′-phosphate binding domain exposed to the cytosol. Biochemical and Biophysical Research Communications, 325, 338–343.
van Meer, G., Voelker, D. R., & Feigenson, G. W. (2008). Membrane lipids: Where they are and how they behave. Nature Reviews. Molecular Cell Biology, 9, 112–124.
Kolesnick, R. N., Goni, F. M., & Alonso, A. (2000). Compartmentalization of ceramide signaling: Physical foundations and biological effects. Journal of Cellular Physiology, 184, 285–300.
Lingwood, D., & Simons, K. (2010). Lipid rafts as a membrane-organizing principle. Science, 327, 46–50.
Pike, L. J. (2006). Rafts defined: A report on the keystone symposium on lipid rafts and cell function. Journal of Lipid Research, 47, 1597–1598.
Rietveld, A., & Simons, K. (1998). The differential miscibility of lipids as the basis for the formation of functional membrane rafts. Biochimica et Biophysica Acta, 1376, 467–479.
Simons, K., & Ikonen, E. (1997). Functional rafts in cell membranes. Nature, 387, 569–572.
Nyholm, T. K. (2015). Lipid-protein interplay and lateral organization in biomembranes. Chemistry and Physics of Lipids, 189, 48–55.
Bollinger, C. R., Teichgraber, V., & Gulbins, E. (2005). Ceramide-enriched membrane domains. Biochimica et Biophysica Acta, 1746, 284–294.
Insel, P. A., et al. (2005). Compartmentation of G-protein-coupled receptors and their signalling components in lipid rafts and caveolae. Biochemical Society Transactions, 33, 1131–1134.
Pike, L. J. (2005). Growth factor receptors, lipid rafts and caveolae: An evolving story. Biochimica et Biophysica Acta, 1746, 260–273.
Lippincott-Schwartz, J., & Phair, R. D. (2010). Lipids and cholesterol as regulators of traffic in the endomembrane system. Annual Review of Biophysics, 39, 559–578.
Hannun, Y. A., & Obeid, L. M. (2008). Principles of bioactive lipid signalling: Lessons from sphingolipids. Nature Reviews. Molecular Cell Biology, 9, 139–150.
Siddique, M. M., Li, Y., Chaurasia, B., Kaddai, V. A., & Summers, S. A. (2015). Dihydroceramides: From bit players to Lead actors. The Journal of Biological Chemistry, 290, 15371–15379.
Presa, N., Gomez-Larrauri, A., Dominguez-Herrera, A., Trueba, M., & Gomez-Munoz, A. (2020). Novel signaling aspects of ceramide 1-phosphate. Biochimica et Biophysica Acta, Molecular and Cell Biology of Lipids, 1865, 158630.
Spiegel, S., & Milstien, S. (2003). Sphingosine-1-phosphate: An enigmatic signalling lipid. Nature Reviews. Molecular Cell Biology, 4, 397–407.
Chalfant, C. E., et al. (1999). Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. The Journal of Biological Chemistry, 274, 20313–20317.
Dobrowsky, R. T., Kamibayashi, C., Mumby, M. C., & Hannun, Y. A. (1993). Ceramide activates heterotrimeric protein phosphatase 2A. The Journal of Biological Chemistry, 268, 15523–15530.
Lozano, J., et al. (1994). Protein kinase C zeta isoform is critical for kappa B-dependent promoter activation by sphingomyelinase. The Journal of Biological Chemistry, 269, 19200–19202.
Xiong, Y., Yang, P., Proia, R. L., & Hla, T. (2014). Erythrocyte-derived sphingosine 1-phosphate is essential for vascular development. The Journal of Clinical Investigation, 124, 4823–4828.
Hanel, P., Andreani, P., & Graler, M. H. (2007). Erythrocytes store and release sphingosine 1-phosphate in blood. FASEB Journal, 21, 1202–1209.
Cantalupo, A., et al. (2015). Nogo-B regulates endothelial sphingolipid homeostasis to control vascular function and blood pressure. Nature Medicine, 21, 1028–1037.
Del Gaudio, I., et al. (2021). Endothelial Spns2 and ApoM regulation of vascular tone and hypertension via Sphingosine-1-phosphate. Journal of the American Heart Association, 10, e021261.
Venkataraman, K., et al. (2008). Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circulation Research, 102, 669–676.
Pappu, R., et al. (2007). Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science, 316, 295–298.
Fukuhara, S., et al. (2012). The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. The Journal of Clinical Investigation, 122, 1416–1426.
Hisano, Y., Kobayashi, N., Yamaguchi, A., & Nishi, T. (2012). Mouse SPNS2 functions as a sphingosine-1-phosphate transporter in vascular endothelial cells. PLoS One, 7, e38941.
Hla, T., Venkataraman, K., & Michaud, J. (2008). The vascular S1P gradient-cellular sources and biological significance. Biochimica et Biophysica Acta, 1781, 477–482.
Schwab, S. R., et al. (2005). Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science, 309, 1735–1739.
Matloubian, M., et al. (2004). Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature, 427, 355–360.
Pham, T. H., Okada, T., Matloubian, M., Lo, C. G., & Cyster, J. G. (2008). S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity, 28, 122–133.
Lo, C. G., Xu, Y., Proia, R. L., & Cyster, J. G. (2005). Cyclical modulation of sphingosine-1-phosphate receptor 1 surface expression during lymphocyte recirculation and relationship to lymphoid organ transit. The Journal of Experimental Medicine, 201, 291–301.
Murata, N., et al. (2000). Interaction of sphingosine 1-phosphate with plasma components, including lipoproteins, regulates the lipid receptor-mediated actions. The Biochemical Journal, 352(Pt 3), 809–815.
Sevvana, M., et al. (2009). Serendipitous fatty acid binding reveals the structural determinants for ligand recognition in apolipoprotein M. Journal of Molecular Biology, 393, 920–936.
Christoffersen, C., et al. (2011). Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proceedings of the National Academy of Sciences of the United States of America, 108, 9613–9618.
Kumaraswamy, S. B., Linder, A., Akesson, P., & Dahlback, B. (2012). Decreased plasma concentrations of apolipoprotein M in sepsis and systemic inflammatory response syndromes. Critical Care, 16, R60.
Frej, C., et al. (2016). Sphingosine 1-phosphate and its carrier apolipoprotein M in human sepsis and in Escherichia coli sepsis in baboons. Journal of Cellular and Molecular Medicine, 20(6), 1170–1181.
Sattler, K. J., et al. (2010). Sphingosine 1-phosphate levels in plasma and HDL are altered in coronary artery disease. Basic Research in Cardiology, 105, 821–832.
Argraves, K. M., et al. (2011). S1P, dihydro-S1P and C24:1-ceramide levels in the HDL-containing fraction of serum inversely correlate with occurrence of ischemic heart disease. Lipids in Health and Disease, 10, 70.
Gomaraschi, M., et al. (2013). Inflammation impairs eNOS activation by HDL in patients with acute coronary syndrome. Cardiovascular Research, 100, 36–43.
Sattler, K., et al. (2014). HDL-bound sphingosine 1-phosphate (S1P) predicts the severity of coronary artery atherosclerosis. Cellular Physiology and Biochemistry, 34, 172–184.
Jiao, G. Q., et al. (2007). A prospective evaluation of apolipoprotein M gene T-778C polymorphism in relation to coronary artery disease in Han Chinese. Clinical Biochemistry, 40, 1108–1112.
Xu, W. W., et al. (2008). A genetic variant of apolipoprotein M increases susceptibility to coronary artery disease in a Chinese population. Clinical and Experimental Pharmacology & Physiology, 35, 546–551.
Claesson-Welsh, L., Dejana, E., & McDonald, D. M. (2021). Permeability of the endothelial barrier: Identifying and reconciling controversies. Trends in Molecular Medicine, 27, 314–331.
Mehta, D., Ravindran, K., & Kuebler, W. M. (2014). Novel regulators of endothelial barrier function. American Journal of Physiology. Lung Cellular and Molecular Physiology, 307, L924–L935.
Giannotta, M., Trani, M., & Dejana, E. (2013). VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Developmental Cell, 26, 441–454.
Hartsock, A., & Nelson, W. J. (2008). Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochimica et Biophysica Acta, 1778, 660–669.
Lee, M. J., et al. (1999). Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell, 99, 301–312.
Garcia, J. G., et al. (2001). Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. The Journal of Clinical Investigation, 108, 689–701.
Xiong, Y., & Hla, T. (2014). S1P control of endothelial integrity. Current Topics in Microbiology and Immunology, 378, 85–105.
Burg, N., Swendeman, S., Worgall, S., Hla, T., & Salmon, J. E. (2018). Sphingosine 1-phosphate receptor 1 signaling maintains endothelial cell barrier function and protects against immune complex-induced vascular injury. Arthritis & Rhematology, 70, 1879–1889.
Akhter, M. Z., et al. (2021). Programming to S1PR1(+) endothelial cells promotes restoration of vascular integrity. Circulation Research, 129, 221–236.
Tauseef, M., et al. (2008). Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circulation Research, 103, 1164–1172.
Camerer, E., et al. (2009). Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. Journal of Clinical Investigation, 119(7), 1871–1879.
Schaphorst, K. L., et al. (2003). Role of sphingosine-1 phosphate in the enhancement of endothelial barrier integrity by platelet-released products. American Journal of Physiology. Lung Cellular and Molecular Physiology, 285, L258–L267.
Gazit, S. L., et al. (2016). Platelet and erythrocyte sources of S1P are redundant for vascular development and homeostasis, but both rendered essential after plasma S1P depletion in anaphylactic shock. Circulation Research, 119, e110–e126.
Herzog, B. H., et al. (2013). Podoplanin maintains high endothelial venule integrity by interacting with platelet CLEC-2. Nature, 502, 105–109.
Jeya Paul, J., et al. (2020). Inflammatory conditions disrupt constitutive endothelial cell barrier stabilization by alleviating autonomous secretion of sphingosine 1-phosphate. Cell, 9, 928.
Simmons, S., et al. (2019). High-endothelial cell-derived S1P regulates dendritic cell localization and vascular integrity in the lymph node. eLife, 8, e41239.
Christensen, P. M., et al. (2016). Impaired endothelial barrier function in apolipoprotein M-deficient mice is dependent on sphingosine-1-phosphate receptor 1. FASEB Journal, 30(6), 2351–2359.
Jernigan, P. L., Makley, A. T., Hoehn, R. S., Edwards, M. J., & Pritts, T. A. (2015). The role of sphingolipids in endothelial barrier function. Biological Chemistry, 396, 681–691.
Maceyka, M., & Spiegel, S. (2014). Sphingolipid metabolites in inflammatory disease. Nature, 510, 58–67.
Goggel, R., et al. (2004). PAF-mediated pulmonary edema: A new role for acid sphingomyelinase and ceramide. Nature Medicine, 10, 155–160.
Wong, M. L., et al. (2000). Acute systemic inflammation up-regulates secretory sphingomyelinase in vivo: A possible link between inflammatory cytokines and atherogenesis. Proceedings of the National Academy of Sciences of the United States of America, 97, 8681–8686.
Claus, R. A., et al. (2005). Role of increased sphingomyelinase activity in apoptosis and organ failure of patients with severe sepsis. FASEB Journal, 19, 1719–1721.
Lindner, K., Uhlig, U., & Uhlig, S. (2005). Ceramide alters endothelial cell permeability by a nonapoptotic mechanism. British Journal of Pharmacology, 145, 132–140.
Schweitzer, K. S., et al. (2011). Mechanisms of lung endothelial barrier disruption induced by cigarette smoke: Role of oxidative stress and ceramides. American Journal of Physiology. Lung Cellular and Molecular Physiology, 301, L836–L846.
Li, H., et al. (2002). Dual effect of ceramide on human endothelial cells: Induction of oxidative stress and transcriptional upregulation of endothelial nitric oxide synthase. Circulation, 106, 2250–2256.
Zeidan, Y. H., Jenkins, R. W., & Hannun, Y. A. (2008). Remodeling of cellular cytoskeleton by the acid sphingomyelinase/ceramide pathway. The Journal of Cell Biology, 181, 335–350.
Furchgott, R. F., & Vanhoutte, P. M. (1989). Endothelium-derived relaxing and contracting factors. The FASEB Journal, 3, 2007–2018.
Igarashi, J., Bernier, S. G., & Michel, T. (2001). Sphingosine 1-phosphate and activation of endothelial nitric-oxide synthase. Differential regulation of Akt and MAP kinase pathways by EDG and bradykinin receptors in vascular endothelial cells. The Journal of Biological Chemistry, 276, 12420–12426.
Morales-Ruiz, M., et al. (2001). Sphingosine 1-phosphate activates Akt, nitric oxide production, and chemotaxis through a Gi protein/phosphoinositide 3-kinase pathway in endothelial cells. The Journal of Biological Chemistry, 276, 19672–19677.
Dantas, A. P., Igarashi, J., & Michel, T. (2003). Sphingosine 1-phosphate and control of vascular tone. American Journal of Physiology. Heart and Circulatory Physiology, 284, H2045–H2052.
Cantalupo, A., et al. (2017). S1PR1 (Sphingosine-1-phosphate receptor 1) signaling regulates blood flow and pressure. Hypertension, 70, 426–434.
Nofer, J. R., et al. (2004). HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. The Journal of Clinical Investigation, 113, 569–581.
Galvani, S., et al. (2015). HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Science Signaling, 8, ra79.
Kappos, L., et al. (2010). A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. The New England Journal of Medicine, 362, 387–401.
Mandala, S., et al. (2002). Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science, 296, 346–349.
Paugh, S. W., Payne, S. G., Barbour, S. E., Milstien, S., & Spiegel, S. (2003). The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS Letters, 554, 189–193.
Brinkmann, V., et al. (2002). The immune modulator FTY720 targets sphingosine 1-phosphate receptors. The Journal of Biological Chemistry, 277, 21453–21457.
Camm, J., Hla, T., Bakshi, R., & Brinkmann, V. (2014). Cardiac and vascular effects of fingolimod: Mechanistic basis and clinical implications. American Heart Journal, 168, 632–644.
Tolle, M., et al. (2005). Immunomodulator FTY720 induces eNOS-dependent arterial vasodilatation via the lysophospholipid receptor S1P3. Circulation Research, 96, 913–920.
Oo, M. L., et al. (2007). Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. The Journal of Biological Chemistry, 282, 9082–9089.
Mukhopadhyay, A., et al. (2009). Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. The FASEB Journal, 23, 751–763.
Zhang, Q. J., et al. (2012). Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes, 61, 1848–1859.
Bharath, L. P., et al. (2015). Ceramide-initiated protein phosphatase 2A activation contributes to arterial dysfunction in vivo. Diabetes, 64, 3914–3926.
Mehra, V. C., et al. (2014). Ceramide-activated phosphatase mediates fatty acid-induced endothelial VEGF resistance and impaired angiogenesis. The American Journal of Pathology, 184, 1562–1576.
Cantalupo, A., et al. (2020). Endothelial sphingolipid De novo synthesis controls blood pressure by regulating signal transduction and NO via ceramide. Hypertension, 75, 1279–1288.
Ogretmen, B., et al. (2002). Biochemical mechanisms of the generation of endogenous long chain ceramide in response to exogenous short chain ceramide in the A549 human lung adenocarcinoma cell line. Role for endogenous ceramide in mediating the action of exogenous ceramide. The Journal of Biological Chemistry, 277, 12960–12969.
Incalza, M. A., et al. (2018). Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascular Pharmacology, 100, 1–19.
Zhang, D. X., Zou, A. P., & Li, P. L. (2003). Ceramide-induced activation of NADPH oxidase and endothelial dysfunction in small coronary arteries. American Journal of Physiology. Heart and Circulatory Physiology, 284, H605–H612.
Zhang, A. Y., Yi, F., Zhang, G., Gulbins, E., & Li, P. L. (2006). Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension, 47, 74–80.
Didion, S. P., & Faraci, F. M. (2005). Ceramide-induced impairment of endothelial function is prevented by CuZn superoxide dismutase overexpression. Arteriosclerosis, Thrombosis, and Vascular Biology, 25, 90–95.
Ikeda, Y., et al. (2015). Molecular mechanisms mediating mitochondrial dynamics and mitophagy and their functional roles in the cardiovascular system. Journal of Molecular and Cellular Cardiology, 78, 116–122.
Liu, Y., et al. (2003). Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circulation Research, 93, 573–580.
Sato, A., Sakuma, I., & Gutterman, D. D. (2003). Mechanism of dilation to reactive oxygen species in human coronary arterioles. American Journal of Physiology. Heart and Circulatory Physiology, 285, H2345–H2354.
Cai, H. (2005). Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovascular Research, 68, 26–36.
Freed, J. K., Beyer, A. M., LoGiudice, J. A., Hockenberry, J. C., & Gutterman, D. D. (2014). Ceramide changes the mediator of flow-induced vasodilation from nitric oxide to hydrogen peroxide in the human microcirculation. Circulation Research, 115, 525–532.
Schulz, M. E., Katunaric, B., Hockenberry, J. C., Gutterman, D. D., & Freed, J. K. (2019). Manipulation of the sphingolipid rheostat influences the mediator of flow-induced dilation in the human microvasculature. Journal of the American Heart Association, 8, e013153.
Oelze, M., et al. (2000). Vasodilator-stimulated phosphoprotein serine 239 phosphorylation as a sensitive monitor of defective nitric oxide/cGMP signaling and endothelial dysfunction. Circulation Research, 87, 999–1005.
Jung, B., et al. (2012). Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Developmental Cell, 23, 600–610.
Versari, D., Daghini, E., Virdis, A., Ghiadoni, L., & Taddei, S. (2009). Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care, 32(Suppl 2), S314–S321.
Forrest, M., et al. (2004). Immune cell regulation and cardiovascular effects of sphingosine 1-phosphate receptor agonists in rodents are mediated via distinct receptor subtypes. The Journal of Pharmacology and Experimental Therapeutics, 309, 758–768.
Coussin, F., Scott, R. H., Wise, A., & Nixon, G. F. (2002). Comparison of sphingosine 1-phosphate-induced intracellular signaling pathways in vascular smooth muscles: Differential role in vasoconstriction. Circulation Research, 91, 151–157.
Salomone, S., et al. (2003). S1P3 receptors mediate the potent constriction of cerebral arteries by sphingosine-1-phosphate. European Journal of Pharmacology, 469, 125–134.
Lorenz, J. N., Arend, L. J., Robitz, R., Paul, R. J., & MacLennan, A. J. (2007). Vascular dysfunction in S1P2 sphingosine 1-phosphate receptor knockout mice. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 292, R440–R446.
Spijkers, L. J., Alewijnse, A. E., & Peters, S. L. (2012). FTY720 (fingolimod) increases vascular tone and blood pressure in spontaneously hypertensive rats via inhibition of sphingosine kinase. British Journal of Pharmacology, 166, 1411–1418.
Fryer, R. M., et al. (2012). The clinically-tested S1P receptor agonists, FTY720 and BAF312, demonstrate subtype-specific bradycardia (S1P(1)) and hypertension (S1P(3)) in rat. PLoS One, 7, e52985.
Kappos, L., et al. (2006). Oral fingolimod (FTY720) for relapsing multiple sclerosis. The New England Journal of Medicine, 355, 1124–1140.
Di Raimondo, D., Musiari, G., & Pinto, A. (2020). Nocturnal blood pressure patterns and cardiac damage: There is still much to learn. Hypertension Research, 43, 246–248.
Yang, W. Y., et al. (2019). Association of office and ambulatory blood pressure with mortality and cardiovascular outcomes. JAMA, 322, 409–420.
Siedlinski, M., et al. (2017). Vascular transcriptome profiling identifies sphingosine kinase 1 as a modulator of angiotensin II-induced vascular dysfunction. Scientific Reports, 7, 44131.
Spijkers, L. J., et al. (2011). Hypertension is associated with marked alterations in sphingolipid biology: A potential role for ceramide. PLoS One, 6, e21817.
Jones, S. P., & Bolli, R. (2006). The ubiquitous role of nitric oxide in cardioprotection. Journal of Molecular and Cellular Cardiology, 40, 16–23.
Keul, P., et al. (2016). Sphingosine-1-phosphate receptor 1 regulates cardiac function by modulating Ca2+ sensitivity and Na+/H+ exchange and mediates protection by ischemic preconditioning. Journal of the American Heart Association, 5, e003393.
Swendeman, S. L., et al. (2017). An engineered S1P chaperone attenuates hypertension and ischemic injury. Science Signaling, 10, eaal2722.
Mizugishi, K., et al. (2005). Essential role for sphingosine kinases in neural and vascular development. Molecular and Cellular Biology, 25, 11113–11121.
Allende, M. L., et al. (2004). Mice deficient in sphingosine kinase 1 are rendered lymphopenic by FTY720. The Journal of Biological Chemistry, 279, 52487–52492.
Kharel, Y., et al. (2012). Sphingosine kinase type 2 inhibition elevates circulating sphingosine 1-phosphate. The Biochemical Journal, 447, 149–157.
Olivera, A., et al. (2007). The sphingosine kinase-sphingosine-1-phosphate axis is a determinant of mast cell function and anaphylaxis. Immunity, 26, 287–297.
Furuya, H., et al. (2013). Effect of sphingosine kinase 1 inhibition on blood pressure. FASEB Journal, 27, 656–664.
Gorshkova, I. A., et al. (2013). Inhibition of sphingosine-1-phosphate lyase rescues sphingosine kinase-1-knockout phenotype following murine cardiac arrest. Life Sciences, 93, 359–366.
Meissner, A., et al. (2017). Sphingosine-1-phosphate signalling-a key player in the pathogenesis of angiotensin II-induced hypertension. Cardiovascular Research, 113, 123–133.
Olivera, A., et al. (2010). Sphingosine kinase 1 and sphingosine-1-phosphate receptor 2 are vital to recovery from anaphylactic shock in mice. The Journal of Clinical Investigation, 120, 1429–1440.
Wilson, P. C., et al. (2015). Inhibition of sphingosine kinase 1 ameliorates angiotensin II-induced hypertension and inhibits transmembrane calcium entry via store-operated Calcium Channel. Molecular Endocrinology, 29, 896–908.
Takuwa, N., et al. (2010). S1P3-mediated cardiac fibrosis in sphingosine kinase 1 transgenic mice involves reactive oxygen species. Cardiovascular Research, 85, 484–493.
Nagiec, M. M., Lester, R. L., & Dickson, R. C. (1996). Sphingolipid synthesis: Identification and characterization of mammalian cDNAs encoding the Lcb2 subunit of serine palmitoyltransferase. Gene, 177, 237–241.
Weiss, B., & Stoffel, W. (1997). Human and murine serine-palmitoyl-CoA transferase—cloning, expression and characterization of the key enzyme in sphingolipid synthesis. European Journal of Biochemistry, 249, 239–247.
Hornemann, T., Richard, S., Rutti, M. F., Wei, Y., & von Eckardstein, A. (2006). Cloning and initial characterization of a new subunit for mammalian serine-palmitoyltransferase. The Journal of Biological Chemistry, 281, 37275–37281.
Gable, K., et al. (2002). Mutations in the yeast LCB1 and LCB2 genes, including those corresponding to the hereditary sensory neuropathy type I mutations, dominantly inactivate serine palmitoyltransferase. The Journal of Biological Chemistry, 277, 10194–10200.
McCampbell, A., et al. (2005). Mutant SPTLC1 dominantly inhibits serine palmitoyltransferase activity in vivo and confers an age-dependent neuropathy. Human Molecular Genetics, 14, 3507–3521.
Beattie, A. E., et al. (2013). The pyridoxal 5′-phosphate (PLP)-dependent enzyme serine palmitoyltransferase (SPT): Effects of the small subunits and insights from bacterial mimics of human hLCB2a HSAN1 mutations. BioMed Research International, 2013, 194371.
Hornemann, T., et al. (2009). The SPTLC3 subunit of serine palmitoyltransferase generates short chain sphingoid bases. The Journal of Biological Chemistry, 284, 26322–26330.
Choi, R. H., Tatum, S. M., Symons, J. D., Summers, S. A., & Holland, W. L. (2021). Ceramides and other sphingolipids as drivers of cardiovascular disease. Nature Reviews. Cardiology, 18(10), 701–711.
Sasset, L., Zhang, Y., Dunn, T. M., & Di Lorenzo, A. (2016). Sphingolipid de novo biosynthesis: A rheostat of cardiovascular homeostasis. Trends in Endocrinology and Metabolism, 27, 807–819.
Green, C. D., Maceyka, M., Cowart, L. A., & Spiegel, S. (2021). Sphingolipids in metabolic disease: The good, the bad, and the unknown. Cell Metabolism, 33, 1293–1306.
Ogretmen, B. (2018). Sphingolipid metabolism in cancer signalling and therapy. Nature Reviews. Cancer, 18, 33–50.
Han, G., et al. (2009). Identification of small subunits of mammalian serine palmitoyltransferase that confer distinct acyl-CoA substrate specificities. Proceedings of the National Academy of Sciences of the United States of America, 106, 8186–8191.
Harmon, J. M., et al. (2013). Topological and functional characterization of the ssSPTs, small activating subunits of serine palmitoyltransferase. The Journal of Biological Chemistry, 288, 10144–10153.
Breslow, D. K., et al. (2010). Orm family proteins mediate sphingolipid homeostasis. Nature, 463, 1048–1053.
Hjelmqvist, L., et al. (2002). ORMDL proteins are a conserved new family of endoplasmic reticulum membrane proteins. Genome Biology, 3, RESEARCH0027.
Davis, D., Suemitsu, J., & Wattenberg, B. (2019). Transmembrane topology of mammalian ORMDL proteins in the endoplasmic reticulum as revealed by the substituted cysteine accessibility method (SCAM). Biochimica et Biophysica Acta—Proteins and Proteomics, 1867, 382–395.
Li, S., Xie, T., Liu, P., Wang, L., & Gong, X. (2021). Structural insights into the assembly and substrate selectivity of human SPT-ORMDL3 complex. Nature Structural & Molecular Biology, 28, 249–257.
Wang, Y., et al. (2021). Structural insights into the regulation of human serine palmitoyltransferase complexes. Nature Structural & Molecular Biology, 28, 240–248.
Cantero-Recasens, G., Fandos, C., Rubio-Moscardo, F., Valverde, M. A., & Vicente, R. (2010). The asthma-associated ORMDL3 gene product regulates endoplasmic reticulum-mediated calcium signaling and cellular stress. Human Molecular Genetics, 19, 111–121.
Siow, D. L., & Wattenberg, B. W. (2012). Mammalian ORMDL proteins mediate the feedback response in ceramide biosynthesis. The Journal of Biological Chemistry, 287, 40198–40204.
Miller, M., et al. (2012). ORMDL3 is an inducible lung epithelial gene regulating metalloproteases, chemokines, OAS, and ATF6. Proceedings of the National Academy of Sciences of the United States of America, 109, 16648–16653.
Gupta, S. D., et al. (2015). Expression of the ORMDLS, modulators of serine palmitoyltransferase, is regulated by sphingolipids in mammalian cells. The Journal of Biological Chemistry, 290, 90–98.
Sasset, L., et al. (2021). S1P controls endothelial sphingolipid homeostasis via ORMDL. bioRxiv, 2021.2010.2022.465503.
Moffatt, M. F., et al. (2007). Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature, 448, 470–473.
Schedel, M., et al. (2015). Polymorphisms related to ORMDL3 are associated with asthma susceptibility, alterations in transcriptional regulation of ORMDL3, and changes in TH2 cytokine levels. The Journal of Allergy and Clinical Immunology, 136, 893–903e814.
Miller, M., et al. (2014). ORMDL3 transgenic mice have increased airway remodeling and airway responsiveness characteristic of asthma. Journal of Immunology, 192, 3475–3487.
Wang, H., Liu, Y., Shi, J., & Cheng, Z. (2019). ORMDL3 knockdown in the lungs alleviates airway inflammation and airway remodeling in asthmatic mice via JNK1/2-MMP-9 pathway. Biochemical and Biophysical Research Communications, 516, 739–746.
Clarke, B. A., et al. (2019). The Ormdl genes regulate the sphingolipid synthesis pathway to ensure proper myelination and neurologic function in mice. eLife, 8, e51067.
Voeltz, G. K., Prinz, W. A., Shibata, Y., Rist, J. M., & Rapoport, T. A. (2006). A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell, 124, 573–586.
GrandPre, T., Nakamura, F., Vartanian, T., & Strittmatter, S. M. (2000). Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature, 403, 439–444.
Huber, A. B., Weinmann, O., Brosamle, C., Oertle, T., & Schwab, M. E. (2002). Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. The Journal of Neuroscience, 22, 3553–3567.
Yu, J., et al. (2009). Reticulon 4B (Nogo-B) is necessary for macrophage infiltration and tissue repair. Proceedings of the National Academy of Sciences of the United States of America, 106, 17511–17516.
Acevedo, L., et al. (2004). A new role for Nogo as a regulator of vascular remodeling. Nature Medicine, 10, 382–388.
Bullard, T. A., et al. (2008). Identification of Nogo as a novel indicator of heart failure. Physiological Genomics, 32, 182–189.
Sarkey, J. P., et al. (2011). Nogo-A knockdown inhibits hypoxia/reoxygenation-induced activation of mitochondrial-dependent apoptosis in cardiomyocytes. Journal of Molecular and Cellular Cardiology, 50, 1044–1055.
Zhang, Y., et al. (2016). Endothelial Nogo-B regulates sphingolipid biosynthesis to promote pathological cardiac hypertrophy during chronic pressure overload. JCI Insight, 1, e85484.
Di Lorenzo, A., Manes, T. D., Davalos, A., Wright, P. L., & Sessa, W. C. (2011). Endothelial reticulon-4B (Nogo-B) regulates ICAM-1-mediated leukocyte transmigration and acute inflammation. Blood, 117, 2284–2295.
Xia, P., et al. (1998). Tumor necrosis factor-alpha induces adhesion molecule expression through the sphingosine kinase pathway. Proceedings of the National Academy of Sciences of the United States of America, 95, 14196–14201.
Jones, S. P., et al. (1999). Myocardial ischemia-reperfusion injury is exacerbated in absence of endothelial cell nitric oxide synthase. The American Journal of Physiology, 276, H1567–H1573.
Jones, S. P., et al. (2004). Endothelial nitric oxide synthase overexpression attenuates myocardial reperfusion injury. American Journal of Physiology. Heart and Circulatory Physiology, 286, H276–H282.
Cartier, A., & Hla, T. (2019). Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science, 366, eaar5551.
de Carvalho, L. P., et al. (2018). Plasma ceramides as prognostic biomarkers and their arterial and myocardial tissue correlates in acute myocardial infarction. JACC Basic to Translational Science, 3, 163–175.
Havulinna, A. S., et al. (2016). Circulating ceramides predict cardiovascular outcomes in the population-based FINRISK 2002 cohort. Arteriosclerosis, Thrombosis, and Vascular Biology, 36, 2424–2430.
Hilvo, M., et al. (2020). Development and validation of a ceramide- and phospholipid-based cardiovascular risk estimation score for coronary artery disease patients. European Heart Journal, 41, 371–380.
Laaksonen, R., et al. (2016). Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. European Heart Journal, 37, 1967–1976.
Peterson, L. R., et al. (2018). Ceramide remodeling and risk of cardiovascular events and mortality. Journal of the American Heart Association, 7, e007931.
Poss, A. M., et al. (2020). Machine learning reveals serum sphingolipids as cholesterol-independent biomarkers of coronary artery disease. The Journal of Clinical Investigation, 130, 1363–1376.
Wang, D. D., et al. (2017). Plasma ceramides, Mediterranean diet, and incident cardiovascular disease in the PREDIMED trial (Prevencion con Dieta Mediterranea). Circulation, 135, 2028–2040.
Gui, Y. K., et al. (2020). Plasma levels of ceramides relate to ischemic stroke risk and clinical severity. Brain Research Bulletin, 158, 122–127.
Fiedorowicz, A., Kozak-Sykala, A., Bobak, L., Kalas, W., & Strzadala, L. (2019). Ceramides and sphingosine-1-phosphate as potential markers in diagnosis of ischaemic stroke. Neurologia i Neurochirurgia Polska, 53, 484–491.
Mundra, P. A., et al. (2018). Large-scale plasma lipidomic profiling identifies lipids that predict cardiovascular events in secondary prevention. JCI Insight, 3, e121326.
Haus, J. M., et al. (2009). Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes, 58, 337–343.
Hilvo, M., et al. (2018). Ceramide stearic to palmitic acid ratio predicts incident diabetes. Diabetologia, 61, 1424–1434.
Wigger, L., et al. (2017). Plasma dihydroceramides are diabetes susceptibility biomarker candidates in mice and humans. Cell Reports, 18, 2269–2279.
Ma, X., et al. (2015). ORMDL3 contributes to the risk of atherosclerosis in Chinese Han population and mediates oxidized low-density lipoprotein-induced autophagy in endothelial cells. Scientific Reports, 5, 17194.
Kolter, T., & Sandhoff, K. (2006). Sphingolipid metabolism diseases. Biochimica et Biophysica Acta, 1758, 2057–2079.
Hicks, A. A., et al. (2009). Genetic determinants of circulating sphingolipid concentrations in European populations. PLoS Genetics, 5, e1000672.
Cresci, S., et al. (2020). Genetic architecture of circulating very-long-chain (C24:0 and C22:0) ceramide concentrations. Journal of Lipid and Atherosclerosis, 9, 172–183.
Blackburn, N. B., et al. (2019). Rare DEGS1 variant significantly alters de novo ceramide synthesis pathway. Journal of Lipid Research, 60, 1630–1639.
Chaurasia, B., et al. (2019). Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science, 365, 386–392.
Nicholson, R. J., et al. (2021). Characterizing a common CERS2 polymorphism in a mouse model of metabolic disease and in subjects from the Utah CAD study. The Journal of Clinical Endocrinology and Metabolism, 106, e3098–e3109.
Good, D. A., et al. (2019). Noncoding variations in the gene encoding ceramide synthase 6 are associated with type 2 diabetes in a large indigenous Australian pedigree. Twin Research and Human Genetics, 22, 79–87.
Obinata, H., et al. (2014). Individual variation of human S1P(1) coding sequence leads to heterogeneity in receptor function and drug interactions. Journal of Lipid Research, 55, 2665–2675.
Shendre, A., et al. (2017). Local ancestry and clinical cardiovascular events among African Americans from the atherosclerosis risk in communities study. Journal of the American Heart Association, 6, e004739.
Domarkiene, I., et al. (2013). RTN4 and FBXL17 genes are associated with coronary heart disease in genome-wide association analysis of lithuanian families. Balkan Journal of Medical Genetics, 16, 17–22.
Funding
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health grants R01 HL126913 and R01 HL152195 to A. Di Lorenzo.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive licence to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Sasset, L., Di Lorenzo, A. (2022). Sphingolipid Metabolism and Signaling in Endothelial Cell Functions. In: Jiang, XC. (eds) Sphingolipid Metabolism and Metabolic Disease. Advances in Experimental Medicine and Biology, vol 1372. Springer, Singapore. https://doi.org/10.1007/978-981-19-0394-6_8
Download citation
DOI: https://doi.org/10.1007/978-981-19-0394-6_8
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-0393-9
Online ISBN: 978-981-19-0394-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)