
Abstract
The recent discovery of new innate lymphoid cells (ILCs) has revolutionized the field of allergies. Since most allergic diseases induce a type 2 immune response, Th2 cells, which produce IL-4, IL-5, and IL-13 in an antigen-dependent manner, in addition to basophils and mast cells which are activated by antigen-specific IgE, are thought to play a major role in the pathogenesis. However, since group 2 innate lymphoid cells (ILC2s) produce type 2 cytokines (i.e., IL-2, IL-4, IL-5, IL-6, IL-9, IL-13, GM-CSF, and amphiregulin) in response to various cytokines, including IL-33 in the surrounding environment, the possibility has emerged that there are two types of allergies: allergies induced in an antigen-dependent manner by Th2 cells and allergies induced in an antigen-independent manner by ILC2s. In order to make an impact on the increasing incidence of allergic diseases in the world, it is essential to research and develop new treatments that focus not only on Th2 cells but also on ILC2s. In this chapter, the role of ILCs in allergic diseases, which has rapidly changed with the discovery of ILCs, is discussed, focusing mainly on ILC2s.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Asthma
- Atopic dermatitis
- Contact hypersensitivity
- Allergic rhinitis
- Chronic rhinosinusitis
- Food allergy
- Allergic conjunctivitis
6.1 Introduction
The word “allergy” is coined from the Greek words “allos” (changed) and “ergon” (reaction), meaning that the immune response which evolved to prevent disease turns into a harmful reaction. It also means that since the distant past, people have been aware that an allergy is a strange reaction different from the common immune response, but they had no way of knowing how it had changed. One of the most beneficial roles of the type 2 immune response in organisms is in eliminating helminths. Most helminths are much larger than bacteria and viruses, and are beyond the range of phagocytosis. Therefore, our body has evolved its own defense mechanisms against helminths. IL-5 induces eosinophils to weaken the parasite, and IL-13 induces mucus production from goblet cells to flush helminths out of the body. In this case, since it is difficult for dendritic cells (DCs) to phagocytose a target that is too large, Th2 cells, which are activated based on antigen presentation by DCs, are unable to exert their full power. On the other hand, the larger the target, the more the tissue that will be physically destroyed, and the necrotic epithelial cells will release large amounts of IL-33. In addition to destroying tissue, proteases, which are a component of helminth shells, contribute significantly to IL-33 production by directly inducing cell death in epithelial cells. ILC2s are cells that highly express ST2/IL-1RL1 (the IL-33 receptor) and are located near IL-33-producing cells. ILC2s can quickly respond to IL-33 and produce type 2 cytokines such as IL-5 and IL-13 to flush helminths out of the mouth by sputum or out of the anus by diarrhea in cases of pulmonary or intestinal infection, respectively. This mechanism is primitive compared to the antigen-specific response of acquired immunity, but it is indispensable for humans who coexist with helminths in an uneasy balance. However, our living environment has changed drastically in the last half century, and parasitic infections have decreased dramatically in developed countries.
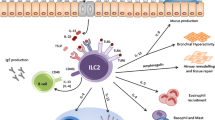
In countries that are no longer infected with parasites, strange reactions are beginning to occur, just like “allos” plus “ergon.” According to the World Allergy Organization (WAO), 30–40% of the world’s population suffers from one or more allergy symptoms, making it a serious health problem. The reason for the increase in allergic diseases is not only the decrease in parasitic infections, but also that improved sanitation decreases bacterial and viral infections, which leads to a decrease in the type 1 immune response used to suppress the type 2 immune response. However, if we focus on the fact that parasite infections have decreased, then type 2 immune responses, which are the main cause of allergies, should have also decreased along with the decrease in infections. What needs to be considered here is the mechanism common to both parasitic infections and allergies, whereby ILC2s recognize IL-33 and produces type 2 cytokines. Unlike Th2 cells, ILC2s cannot specifically recognize antigens or distinguish whether IL-33 is produced by a parasite or an allergen that is harmless to the body. As well as parasites, various allergenic antigens such as dust mites, pollen, fungi, and fruits contain proteases which directly induce necrosis of the epithelium, and stimulate it to release IL-33. The days when ILC2s maintain an exquisite coexistence with parasites are long gone, but ILC2s deep inside the body may still be unaware of the changing times and are needlessly initiating type 2 immune responses, relying on IL-33 as an alarm signal (Fig. 6.1).

Mechanism of allergy induced by Th2 cells and ILC2s. In antigen-dependent allergy, DCs that have phagocytosed the antigen migrate to lymph nodes and present the antigen to Th0 cells via MHC class II and TCR. Th0 cells differentiate into Th2 cells over a period of about 5 days, and mature Th2 cells migrate to peripheral tissues and produce type 2 cytokines. In contrast, in antigen-independent allergy, the protease activity of the allergen directly causes necrosis in epithelial cells. Since ILC2s are tissue-resident cells, they immediately respond to IL-33 and produce type 2 cytokines. IL-25 secreted by tuft cells and TSLP produced by epithelial cells, stromal cells, and DCs are also involved in the activation of ILC2s
6.2 Innate Lymphoid Cells in the Lungs
Since the lungs are vital organs for the gas exchange of oxygen and carbon dioxide, they are constantly exposed to various allergens and viruses through breathing. The airway epithelial cells are covered with a layer of mucus to capture allergens and viruses, and ciliated cells eliminate them by ciliary movement. However, when the allergens and viruses are not eliminated and reach airway epithelial cells, cell-derived cytokines are rapidly released and directly activate ILCs and trigger innate immune responses. Among ILCs in the lungs, ILC2s in particular have been reported to be involved in allergic diseases by inducing type 2 inflammation through the production of IL-5 and IL-13.
The proportion of lung ILCs is small, accounting for only about 0.4–1% of lung cells in naïve mice [1]. Among ILCs, ILC2s are the most abundant (>60%), followed by ILC1s (<20%) and ILC3s (<20%) [2]. On the other hand, the frequency of ILCs in the human lung is only about 0–0.1% of CD45+ cells, and conventional NK (cNK) cells are the major ILCs, followed by ILC1s and ILC3s, while ILC2s are rare [2, 3]. However, the proportion of ILCs has been reported to change dynamically due to plasticity, and the presence of inflammatory cytokines such as IL-1β in the lung due to smoking and obesity increases the frequency of ILC1s and ILC3s [4, 5]. Thus, although human lung ILC2s are relatively rare cell populations, they express high levels of IL13 as well as epithelial cell-derived cytokine receptors IL1RL1 and IL17RB, compared to blood and tonsil ILC2s [6], and are suggested to have a significant impact on the induction of type 2 inflammation.
6.2.1 Mechanism of IL-33-Induced Asthma
Various allergens and pathogens, such as helminths, house dust mites [7], Alternaria alternata [8], papain [9], chitin [10], α-GalCer [11], rhinovirus [12], and influenza virus [13], induce epithelial cell-derived cytokines such as IL-33 via cytotoxicity and ATP release [14] (Fig. 6.2). The mechanism by which IL-33 is released is ingenious considering that allergen-derived proteases not only damage epithelial cells to release IL-33, but also play a role in cleaving full-length IL-33 into an activated form [15]. IL-33 stimulates NF-κB and MAPK signaling pathways in ILC2s, and activates the transcription factor GATA3 via p38 MAPK, which strongly induces cell proliferation and production of type 2 cytokines such as IL-5 and IL-13 [16, 17]. IL-5 promotes eosinophil activation, migration, and survival, and IL-13 promotes goblet cell hyperplasia and mucus hypersecretion in the lungs [18]. This IL-33/ILC2-mediated immune response is referred to as “nonallergic eosinophilic inflammation” since it occurs in an antigen-independent manner without acquired immunity [19].

IL-33 induces activation of ILC2s in diverse pathological conditions. In helminth infection, IL-33 induces IL-5 and IL-13 production from ILC2s, which work to expel helminths from the body via worm attack by eosinophils and mucus production by goblet cells, respectively. In viral infections, ILC2s activated by IL-33 play a protective role by producing IL-13 and amphiregulin. IL-13 acts to promote epithelial cell regeneration and amphiregulin promotes tissue repair. In allergy, IL-33 activates ILC2s to produce a variety of cytokines, including IL-4, IL-5, IL-13, and amphiregulin. IL-5 induces eosinophilic inflammation, and IL-13 promotes mucus production by goblet cells, resulting in runny nose and sputum. It has been suggested that IL-4 induces IgE production from B cells, and IL-4, IL-13, and amphiregulin may be involved in fibrosis
Asthma is a chronic respiratory disease characterized by chronic airway inflammation and reversible airway obstruction, and is considered to be a heterogeneous syndrome consisting of a variety of pathologies. Most people with asthma, except for those with neutrophilic asthma, have type 2 inflammation, and Th2 cells have been considered to play a vital role in inflammation. However, accumulating evidence suggests that IL-33 and ILC2s are also involved in the pathophysiology of asthma. Genome-wide association studies (GWAS) have reported that single-nucleotide polymorphisms (SNPs) in IL33 and its receptor, IL1RL1, are associated with asthma [20], and IL-33 expression is increased in the airways and bronchoalveolar lavage (BAL) fluid in patients with asthma, and is correlated with disease severity and lung function [21,22,23]. Similarly, the number of ILC2s is increased in the sputum and BAL of patients with asthma, especially in those with severe asthma [22, 24]. However, there are no definitive results on the number of ILC2s in the peripheral blood of patients with asthma [24, 25], which suggests that it is difficult to use the blood to monitor the status of ILC2s in the lungs. Furthermore, house dust mites and rhinoviruses induced IL-33 and increased the number of ILC2s in sputum and BAL, but decreased the number of ILC2s in the blood [26,27,28]. These findings suggest that even though ILC2s are recognized as tissue-resident cells in mice, a portion of ILC2s are transferred from the peripheral blood to the airways in humans [29, 30]. Until the discovery of ILC2, asthma was thought to be an antigen-specific disease, but it is now clear that IL-33/ILC2-mediated immune responses play an important role, especially in the pathogenesis of virus-induced asthma exacerbation, uncontrolled asthma, and severe asthma [24, 27, 31].
6.2.2 Cytokines and Lipid Mediators that Regulate the Function of ILC2s
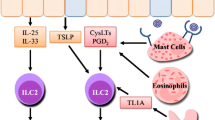
IL-33 rapidly and potently activates ILC2s and induces type 2 inflammation, but other epithelial cell-derived cytokines also activate ILC2s in the lungs. For example, IL-25 is an epithelial cell-derived cytokine that activates murine and human ILC2s in combination with IL-2 in vitro [32, 33], and tuft cells in the intestinal tract and bronchi have been reported as IL-25-producing cells [34]. However, IL-25 is less potent than IL-33 in inducing type 2 inflammation and airway contraction in mice [35]. Thymic stromal lymphopoietin (TSLP) is also an epithelial cell-derived cytokine that has modest effects on the activation of ILC2s, but it is rather important in altering the properties of ILC2s; TSLP induces corticosteroid resistance via phosphorylation of STAT5 and expression of Bcl-xL in ILC2s, leading to corticosteroid-resistant type 2 inflammation in mice [36] (Fig. 6.3). Indeed, TSLP expression is increased in the airways of patients with severe asthma [37], and TSLP concentration in BAL correlated with corticosteroid resistance in ILC2s in humans [31]. Although IL-33, IL-25, and TSLP are often described in parallel in reviews as epithelial cell-derived cytokines, more research on the cytokine production mechanism for IL-25 and TSLP is required to understand the regulation of ILC2 by these three cytokines.

IL-33 and TSLP cooperatively induce steroid resistance in ILC2. Under steady-state and IL-33 stimulation, ILC2s are sensitive to steroids, but under simultaneous stimulation of IL-33 and TSLP, they become resistant to steroid. TSLP induces Bcl-xL expression by phosphorylation of STAT5, resulting in steroid resistance in ILC2s
ILC2s do not express antigen-specific receptors such as Th2 cells, but ILC2s express various receptors for cytokines and lipid mediators, which are influenced by the surrounding environment. For example, proteases in house dust mites and papain induce basophils to produce IL-4, which increases the responsiveness of ILC2s to IL-33 in mice [38]. TNF-like ligand 1A (TL1A, TNFSF15), glucocorticoid-induced TNF receptor ligand (GITRL, TNFSF18), leukotriene, and prostaglandin D2 (PGD2) have been reported to activate ILC2s synergistically with IL-33 in mice and humans [25, 39,40,41,42,43,44,45,46,47]. Cysteinyl leukotrienes and PGD2 bind to G protein-coupled receptors, which increase the intracellular Ca2+ concentrations of ILC2s and promote nuclear transfer of NFAT [48]. Since this signaling pathway is different from that induced by IL-33 or IL-25, lipid mediators can activate ILC2s synergistically with IL-33 and induce IL-4 production from ILC2s [43]. Some patients with asthma have aspirin-exacerbated respiratory disease (AERD), which is characterized by swelling of the mucous membranes of the sinuses, nasal passages, and airways; these symptoms are exacerbated by the ingestion of cyclooxygenase-1 (COX-1) inhibitors, including aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) [49]. In patients with AERD, the administration of COX-1 inhibitors increases the production of lipid mediators, such as leukotriene and PGD2, and induces the accumulation of ILC2s in the nasal mucosa, whereas the number of ILC2s is decreased in the blood of patients with AERD [50]. These findings suggest that ILC2s contribute to the pathogenesis of AERD through the production of lipid mediators.
In contrast, some cytokines and lipid mediators inhibit the activation of murine and human ILC2s and suppress type 2 inflammation. Interferons (IFNs) and IL-27 produced by NK cells, T cells, plasmacytoid dendritic cells, and interstitial macrophages suppress ILC2 activation in a STAT1-dependent manner [30, 51,52,53,54]. These cytokines are involved in the resolution of ILC2-mediated type 2 inflammation, and without these cytokines, type 2 inflammation would persist for a long time. The bronchial epithelium in patients with asthma has been reported to produce less IFN-β when infected with rhinoviruses [55]. Therefore, replenishment of IFN-β and other suppressive cytokines may be a treatment option for virus-induced exacerbation of asthma [56, 57]. In addition, other lipid mediators such as PGE2, PGI2, and lipoxin A4 have been reported to inhibit the production of IL-5 and IL-13 from ILC2s via cAMP activation [58,59,60,61].
Finally, since ILC2s have plasticity, the type of cytokines they produce changes dynamically depending on the surrounding cytokine environment. IL-1β, IL-18, and IL-12 induce IFN-γ production, and retinoic acid induces IL-10 production in murine and human ILC2s [4, 62,63,64]. Viral infection and smoking provoke inflammatory cytokines, including IL-12, and the frequency of IFN-γ-producing ILC2s is increased in patients with chronic obstructive pulmonary disease [4]. Thus, various regulatory mechanisms exist for ILC2-mediated type 2 inflammation, and a disruption of these regulatory mechanisms results in the exacerbation and progression of allergen-independent type 2 inflammation in vivo.
6.2.3 Neuroimmune Interaction in ILC2s
Neuronal and immune systems have close bidirectional interactions. Neuron-derived neuropeptides and neurotransmitters regulate immune cell functions, whereas inflammatory mediators produced by immune cells enhance neuronal activation [65]. In recent years, several neuropeptides have been shown to directly affect ILC2s. Pulmonary neuroendocrine cells (PNECs) are rare airway epithelial cells that sense various stimulations, including oxygen, stretch, and chemicals, and release neuropeptides, such as calcitonin gene-related peptide (CGRP). ILC2s have been reported to express CGRP receptors and co-localize with PNECs in the lungs of mice [66]. However, the effect of CGRP on ILC2s is complicated; CGRP has induced IL-5 production in murine ILC2s, but constrained IL-13 production and proliferation [67]. ILC2s have also expressed neuromedin U receptor 1 (NmUR1), while neuromedin U (NMU) had a potent effect on inducing the proliferation and cytokine production of IL-5, IL-9, IL-13, and amphiregulin from ILC2s in mice [68,69,70]. In addition, pulmonary sensory nerve-derived vasoactive intestinal peptide (VIP) activated ILC2s and CD4 T cells to enhance type 2 inflammation in ovalbumin (OVA)- and house dust mite (HDM)-induced asthma mouse models [71]. Therefore, genetic ablation of Nav1.8-positive sensory nerves reduces immune cell infiltration and airway hyperreactivity in OVA-induced asthma mouse models [71].
In contrast, cholinergic neurons produce acetylcholine and α7nAChR, an acetylcholine receptor, which suppresses type 2 cytokine production from murine ILC2s [72]. Furthermore, sympathetic nerves release noradrenalin while β-adrenoceptor stimulations suppress ILC2 proliferation and ILC2-mediated inflammation in mice [73]. These data suggest that the neural system, in addition to the interactions with epithelial cells and other immune cells, may regulate ILC2-induced type 2 inflammation. At this stage, it is not clear how these neural factors are involved in the pathogenesis of asthma, but they may be important factors in understanding the full picture of ILC2-dependent asthma.
6.2.4 The Role of ILC2s in Trained Immunity and Acquired Immunity
ILC2s have been implicated not only in innate immune responses, but also in trained and acquired immunity. The increased number of ILC2s in the lungs and mediastinal lymph nodes of mice treated with IL-33 or papain was maintained even after 4 weeks, indicating that ILC2s are long-lived cells. Furthermore, ILC2s responded more strongly to restimulation and induced more severe type 2 inflammation, suggesting that ILC2s have a memory mechanism [74]. Experiments with Rag1−/− mice treated with Alternaria allergen extract have shown that ICOS+ST2+ ILC2 generates memory in asthma through epigenetic changes [75].
ILC2s also enhance acquired immune responses. This effect is limited to localized reactions in the lungs and is dispensable in systemic reactions. ILC2s enhance Th2 cell responses but has no effect on Th1 or Th17 cells [76]. Murine ILC2s express MHC class 2, OX40L, CD80, and CD86 and can directly activate CD4+ T cells, which may promote the induction of acquired immunity [77,78,79,80]. ILC2-derived IL-13 promotes the migration of dendritic cells in the lung to the lymph node, where they promote naïve T cells to differentiate into Th2 cells during allergic lung inflammation [81]. ILC2-derived IL-13 promotes the expression of IL-33 in airway epithelial cells, creating a positive feedback loop and disrupting tight junctions between the cells [82], thereby increasing the penetration of allergens into the epithelium, which may allow for higher penetration of allergens across the epithelium. In summary, ILC2s, which induce innate immune responses, may enhance acquired immune responses and exacerbate asthma.
6.2.5 Treatment of ILC2-Mediated Type 2 Inflammation in the Lungs
Although new asthma therapies targeting ILC2s are expected, few drugs have proven inhibitory effects on ILC2s in patients with asthma. Corticosteroids and leukotriene receptor antagonists, generally used to treat asthma, appear to be somewhat effective against ILC2-mediated type 2 inflammation. Indeed, inhaled corticosteroid treatment (i.e., budesonide) decreased the number of ILC2s in peripheral blood and suppressed type 2 cytokine production from ILC2s [83]. However, other studies have demonstrated that ILC2s in the BAL are resistant to corticosteroids, and continue to produce type 2 cytokines even in the presence of dexamethasone [31]. Importantly, TSLP induces corticosteroid resistance of ILC2s and promotes innate type 2 inflammation, and also induces acquired immunity via DCs and CD4+ T cells in asthma model mice [84]. Suppression of TSLP is important for resolving ILC2-induced steroid resistance, while tezepelumab, an anti-TSLP human monoclonal antibody, has been reported to effectively suppress a wide range of inflammation [85,86,87]. IL-33 is the most potent cytokine that activates ILC2s, and antibodies against IL-33 or its receptor, ST2/IL-1RL1, are currently under development [88]. In addition, among the biologics currently in use, dupilumab, an anti-IL-4/13R antibody, has been suggested to suppress ILC2s in humans [89]. Since ILC2s have a major impact on type 2 inflammation in the lungs, the development of drugs targeting ILC2-mediated inflammation is expected to progress in the future (Fig. 6.4).

Biologics targeting type 2 immunity. Approved indications vary by country. EGPA eosinophilic granulomatosis with polyangiitis, HES hypereosinophilic syndrome
6.3 Innate Lymphoid Cells in the Skin
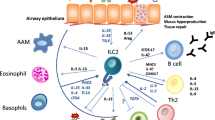
The skin is the largest organ and provides a barrier to protect the body from bacterial and viral invasion, UV, and external stress damage. Immune cells are strategically placed in the skin to create efficient protective immunity through cellular communication. Similarly to the respiratory and intestinal tracts, the skin also harbors tissue-resident ILCs. Skin ILCs not only have essential functions in maintaining tissue homeostasis, but also play an important role in the pathogenesis of skin diseases, including allergies. The skin consists of three layers: epidermis composed of four different keratinocytes, collagen-rich dermis, and adipocyte-rich subcutaneous tissue. ILCs, which account for 5% of immune cells in murine skin, are present in all three skin layers, but in different proportions. Transcriptome analysis revealed layer-specific heterogeneity of skin ILC subsets (Fig. 6.5) [90]. The subcutaneous tissue is rich in typical GATA3+ ILC2s, which express Sca-1 and IL-33R and produce IL-5 and IL-13, while the epidermal and dermal tissues contain ILCs with mixed phenotypes of ILC3s and ILC2s. Epidermal and dermal ILCs express ICOS and CCR6, but do not express Sca-1 and IL-33R. They express both GATA3 and RORγt and produce IL-13, IL-22, TNF, and lymphotoxins [90]. Skin ILCs exhibit distinct tissue-resident patterns, with ILC1s continuously migrating between the circulation and peripheral lymph nodes in a CD62L- and CCR7-dependent manner, while ILC2s and ILC3s remain in the tissue [91]. With regard to allergic diseases of the skin, ILC2s are thought to play an important role in atopic dermatitis, while ILC1s are thought to cause contact dermatitis together with NK cells.

Skin ILCs with distinct transcriptome landscapes. (a) Single RNA-seq analysis revealed transcriptome heterogeneity of skin ILC subsets. The subcutaneous ILCs are GATA3+ ILC2s, while the epidermal and dermal tissues contain ILCs with mixed phenotypes of ILC3s and ILC2s. (b) Bulk RNA-seq analysis further showed layer-specific identities of skin ILCs. IL-5 is mainly produced by subcutaneous and Sca1+ dermal ILCs, while epidermal ILCs highly express IL-13. (c) Distribution of ILCs in the skin. (Fig. 5a and b are adapted from Kobayashi et al., Cell 2019, with permission from Elsevier)
6.3.1 ILC2s in Atopic Dermatitis
Atopic dermatitis is a chronic inflammatory skin disease in which the patient’s quality of life is severely impaired due to endless itching. Since atopic dermatitis is associated with high levels of antigen-specific IgE in the serum and high T cell infiltration in the skin lesions, acquired immunity is thought to be important in the pathogenesis of this disease. In recent years, however, attention has begun to focus on the important role of innate immunity in initiating and promoting atopic inflammation, considering that patients often do not exhibit antigen-specific reactions that are seen in antigen-restricted allergic disorders such as pollen and food allergies. Repeated scratching is known to aggravate symptoms of atopic dermatitis. Tissue damage caused by scratching stimulates the production of cytokines and chemokines in the epidermis, which in turn recruits and activates immune cells, thereby aggravating the condition. From this perspective, alarmins such as IL-33, TSLP, and IL-25 produced by epithelium upon tissue damage that directly activate ILC2s may be deeply involved in the pathogenesis of atopic dermatitis. GWAS have identified susceptibility genes for atopic dermatitis, including genes encoding IL-33R, IL-18R, IL-7R, IL-2R, TSLP, IL-4, and IL-13 [92, 93], all of which are associated with ILC2 activation and effector functions. Studies on patients with atopic dermatitis have reported that ILC2s, both in the lesional skin and peripheral blood, are increased compared to ILC2 populations in healthy subjects [94, 95].
To understand the mechanism by which ILC2s are activated and involved in the pathogenesis of atopic dermatitis, an atopic-like dermatitis mouse model with topical application of calcipotriol (MC903), a vitamin D3 analog, has been used. Although dermatitis is known to occur in T cell-deficient mice, depletion of T cells and ILCs by intraperitoneal administration of anti-CD25 or anti-CD90.2 antibodies alleviates dermatitis. On the other hand, dermatitis was not alleviated in mice lacking IL-33(Il33−/−) or IL-25R (Il17rb−/−), whereas TSLP receptor-deficient mice (Tslpr−/−) showed impaired responsiveness to ILC2s and improved dermatitis, suggesting that TSLP is important for the activation of skin ILC2s [94]. In addition, IL-13-producing ILC2s interact with mast cells in the dermis of mouse skin and are involved in eosinophil infiltration in response to IL-2 [96].
On the other hand, the importance of IL-33 and IL-25 has also been reported. While the above studies used mice of the C57BL/6 strain background, studies using the BALB/c strain showed that MC903 induced atopic-like dermatitis was reduced in mice lacking IL-25R (Il17rb−/−) and IL-33R (Il1rl1−/−) [95]. It has been shown that transgenic mice with forced expression of IL-33, specifically in epidermal keratinocytes, have increased IL-33R+ ILC2s and spontaneous development of atopic-like dermatitis [97]. In these mice, depletion of ILC2s or basophils resulted in an improvement of dermatitis, suggesting that ILC2s and basophils activated by IL-33 are involved in the pathogenesis of atopic inflammation [98]. In the abovementioned study, IL-33R+ ILC2s were enriched in skin lesions of patients with atopic dermatitis, and in vitro stimulation of IL-33 induced the production of type 2 cytokines from human skin-derived ILC2s, further highlighting the role of IL-33 in skin ILC2 activation [95]. The role of IL-25 in ILC2 activation has been shown in an OVA-induced atopic-like dermatitis model. Either IL-25 deficiency, IL-13 deficiency, or ILC2-specific IL-25R deficiency (Rora-Cre Il17rbflox/flox) suppressed dermatitis, suggesting a mechanism by which epidermal derived IL-25 promotes ILC2s to produce IL-13, which leads to chemokine production such as CCL17 and CCL22 from the epidermis and recruits T cells and further promotes dermatitis [99].
It is difficult to conclude which cytokines are most important for the activation of skin ILC2s, because different mouse models of atopic dermatitis and different strains of mice can affect the results. Nonetheless, there is no doubt that type 2 cytokines are central to the pathogenesis of atopic dermatitis, as monoclonal antibodies that block IL-4 and IL-13 signaling markedly improve clinical symptoms of atopic dermatitis [100, 101]. If atopic dermatitis is a more antigen-dependent disease, Th2 cells are likely to be the source of these type 2 cytokines; however, patients often demonstrate increased pruritus by sweating, stress, and diurnal rhythms, but not when exposed to specific antigens, suggesting that ILC2s are a possible source of type 2 cytokines. New therapeutic targets may be identified by studying the pathogenesis of atopic dermatitis from the perspective of ILC2s, which are activated by cytokines produced in response to tissue damage such as scratching.
6.3.2 NK and ILC1 in Contact Hypersensitivity
Allergic contact dermatitis is an inflammatory skin disease caused by the penetration of low-molecular-weight chemicals and metals. In hapten-induced contact hypersensitivity (CHS) in mice [102], antigen-specific or nonspecific innate immune memory may contribute to the induced response. Tissue-resident NK cells in the liver, currently referred to as ILC1s, induce hapten-specific memory responses independent of T and B lymphocytes in the CHS model, suggesting memory-like properties of NK cells or ILC1 [103, 104]. In contrast to the tissue residency of ILC2s and ILC3s, IL-7R+ ILC1s acquire hapten-specific memory in skin-draining lymph nodes, and are recruited to the liver via CXCR6 and maintained by IL-7R signaling [105]. Although hapten-specific memory responses have not been demonstrated in human ILC1s or NK cells, NK cells accumulate in the skin of patients with allergic contact dermatitis, and NK cells release IFN-γ in the presence of T lymphocytes that produce IL-2 in vitro [106]. These studies suggest the existence of an innate memory response (or trained immunity); however, the underlying mechanism for the long-term persistence of the immune response by ILCs requires further investigation.
6.4 Innate Lymphoid Cells in the Nasal Mucosa
The olfactory system is not only a chemosensory organ, but also a sophisticated immune system that acts as the first line of defense against infections, due to its constant exposure to the open air containing abundant immunogens. ILCs are known to be present in the nasal mucosa, but in healthy individuals, ILC1s and ILC2s are found in less than 0.1% of CD45-positive cells, and considerably less than 0.01% that of ILC3s. However, in inflammatory conditions such as chronic sinusitis, ILC2s are increased by more than 100-fold. Since the nasal mucosa is constantly invaded by antigens from the outside world, it is prone to various allergic symptoms; hence, allergic rhinitis and chronic sinusitis are well-known allergic inflammations of the nose. The discovery of ILC2s has accelerated our understanding of the pathogenesis of these diseases, which is thought to be mainly caused by Th2 cells. Since the nasal polyps, which form in the paranasal sinuses, were one of the first tissues in which human ILC2s were discovered, research on the pathogenesis of nasal allergies, with a focus on ILC2s, has been conducted mainly in humans.
6.4.1 Allergic Rhinitis and ILC2s
Allergic rhinitis is classified as an IgE-mediated type 1 allergy, in which allergen exposure causes a runny nose, sneezing, and nasal congestion. Within 30 min of exposure to an allergen, the cross-linking of IgE and allergen induces histamine release from mast cells, triggering the initial allergic reaction. Subsequently, 6–24 h after allergen exposure, eosinophilic infiltration occurs and triggers tissue destruction and remodeling. Patients with allergic rhinitis have elevated levels of IL-25, IL-33, and TSLP in the serum or nasal lavage fluid [107,108,109]. GWAS revealed that SNPs in the IL33 gene are associated with Japanese cedar pollinosis, suggesting that IL-33 is strongly involved in the pathogenesis of allergic rhinitis [109]. IL-33-deficient mice display reduced symptoms of allergic rhinitis [110, 111]. In addition to IL-33, a number of GWAS analyses have demonstrated that SNPs in the TSLP gene are also correlated with allergic rhinitis [112,113,114,115,116], and the expression of TSLP in the nasal cavity is associated with the severity of the symptoms [117, 118]. In a mouse model of allergic rhinitis, the roles of TSLP and IL-33 were different between acute and chronic models. In the acute allergic rhinitis model, both TSLP and IL-33 contributed to the initial sneeze, and the subsequent eosinophilic infiltration depended on IL-33, whereas in the chronic model, the sneeze response or eosinophilic infiltration depended on TSLP or TSLP and IL-33, respectively. Furthermore, IL-25 was also detected in the nasal lavage fluid of allergic rhinitis patients, and the expression of Il25 was enhanced in the nasal mucosa of OVA-induced allergic rhinitis model mice [119]. However, in a mouse model of HDM-induced allergic rhinitis, IL-25 deficiency had no effect on the symptoms [111]. Since IL-25 enhances the expression of TSLP in nasal epithelial cells, further analysis of the possible involvement of IL-25 in the pathogenesis of allergic rhinitis is needed [107].
Recently, ILC2s were analyzed in patients with allergic rhinitis. The frequency of ILC2s in peripheral blood was significantly higher in patients with allergic rhinitis than that in healthy subjects [120, 121]. Interestingly, there seems to be a difference in the reactivity of ILC2s depending on the type of allergen: patients with allergic rhinitis to HDM had the highest frequency of ILC2s in the peripheral blood, while patients with allergic rhinitis to other allergens, such as wormwood, had higher ILC2s than those of healthy subjects, but predominantly lower than those of patients with allergic rhinitis to HDM [121, 122]. However, in all patients, there was a correlation between the severity of symptoms and the percentage of ILC2s in the peripheral blood [121], suggesting that ILC2s play an important role in the pathogenesis of allergic rhinitis. Since allergic rhinitis is one of the most antigen-dependent allergic diseases, it is necessary to clarify how ILC2s contribute to its pathogenesis via IL-25, IL-33, and TSLP.
6.4.2 Chronic Rhinosinusitis and ILC2s
Chronic rhinosinusitis (CRS) is an inflammatory disease of the nasal mucosa triggered by upper respiratory tract inflammation, such as from the common cold that spreads to the mucosa and persists for more than 3 months. It has been demonstrated that in CRS with nasal polyps (CRSwNPs), type 2 inflammation, including IgE, IL-5, and IL-13 production, and eosinophil infiltration occur, suggesting that Th2 cells are strongly involved in the pathogenesis. Human ILC2s were first isolated from the nasal polyps of CRSwNP patients in Netherlands, along with the lungs and gut, and ILC2s were found to accumulate in polyps [33]. Subsequently, the accumulation of ILC2s in nasal polyps was confirmed in the United States [123]. It has been proposed that CRSwNPs can be divided into eosinophilic (ECRS) and non-eosinophilic (NECRS) subgroups [124], and ILC2s have been shown to accumulate in nasal polyps in ECRS but not in NECRS in various countries [125,126,127], suggesting that the mechanism of ILC2-induced ECRS is common worldwide. Despite the accumulation of ILC2s in nasal polyps, there was no difference in ILC2s in peripheral blood in all studies, consistent with the view that ECRS is a local inflammatory disease. Other ILC subsets, including ILC1s and ILC3s, were also analyzed in polyps of CRS; however, while ILC2s were selectively accumulated in polyps and increased 100-fold compared to the ILC2s in the sinus mucosa of healthy subjects, other ILC subsets were unchanged in the polyps [128], suggesting that ILC2s predominantly contribute to polyp development in CRS.
Polyp-derived ILC2s respond to IL-25, IL-33, and TSLP and produce IL-4, IL-5, IL-9, IL-13, and GM-CSF. Many studies have shown that the expression of IL-33 is comparable between the sinus mucosa of CRSwNPs, CRS without nasal polyps (CRSsNPs), and healthy subjects [33, 123, 129]. However, the expression of IL-33 was increased in epithelial cells derived from recurrent CRSwNPs compared with that from first-onset CRSwNP, suggesting that alteration of IL-33 expression in nasal epithelial cells may contribute to the pathogenesis [130]. In contrast, TSLP was shown to be upregulated in the nasal mucosa of CRSwNPs and ECRS [131,132,133,134]. Since the expression of TSLP in the nasal mucosa correlated with polyp scores, and GWAS analysis showed that SNPs in the TSLP gene were associated with CRSwNPs, TSLP may play an important role in the pathogenesis of CRSwNPs [135]. The increased expression of IL-25 in CRSwNPs is controversial and therefore inconclusive. However, it has been reported that polyp-derived ILC2s respond to IL-25 [33] and that IL-25 induces differentiation of fibroblasts into myofibroblasts and contributes to polyp remodeling [136], suggesting that IL-25 expression may change with disease progression, or that ECRS pathogenesis is a heterogeneous disease that can be further divided by endotypes.
Recently, the dramatic effect of dupilumab on ECRS has revealed that IL-4 and IL-13 are key factors in the pathogenesis of the disease [137]. It is well known that IgE expression in nasal polyps is high in patients with CRS, regardless of systemic IgE levels [138], suggesting that, unlike allergic rhinitis, inflammation in CRS is not allergen specific. Therefore, in addition to Th2 cells, ILC2s may contribute to the pathogenesis of ECRS through type 2 cytokines in response to IL-33, TSLP, and IL-25. The discovery of ILC2s is expected to accelerate our understanding of CRS pathogenesis and lead to the development of new therapies.
6.4.3 Nasal Allergy and ILC1s, ILC3s
ILC1s, including NK cells, suppress ILC2 function through the production of IFNγ. ILC1s in allergic rhinitis patients produce less IFNγ upon in vitro stimulation than that produced in nonallergic rhinitis patients [139], suggesting that ILC2s may be activated in these patients. In fact, in a study of patients who responded to allergen immunotherapy (AIT), a treatment for allergic rhinitis showed a reduced response of ILC2s to allergen stimulation compared to ILC2s in healthy subjects [140]. In addition, the ratio of ILC2 to ILC1 in patients who responded to AIT was similar to that in healthy controls, suggesting that ILC1s suppress allergic rhinitis by inhibiting the function of ILC2s. The frequency of ILC3s, along with ILC2s, significantly increased only during the grass pollen season in grass-allergic patients, while the frequency of ILC1s did not change. Therefore, ILC3s may have a different function from ILC1s in this condition, but the role of ILC3s in allergic rhinitis requires further analysis. Although ILC2s selectively accumulate in polyps in CRSwNPs, the increased production of IFNγ and IL-17A in the nasal mucosa in CRSsNP supports the possibility that ILC3s may have a different function than ILC1s under these conditions. Although there are reports that increased ILC1s and ILC3s are observed in the nasal mucosa in CRSsNP pathologies, there is no significant difference in the frequency of ILC1s and ILC3s in the nasal mucosa compared to differences in CRSwNPs or healthy individuals, although there is an increasing trend [128]. Therefore, the involvement of ILC1s and ILC3s in the pathogenesis of the disease is still unclear and requires further analysis.
6.5 Food Allergy, Including Anaphylaxis, and ILC2s
Food allergy (FA) is an allergic disease that is increasing worldwide, affecting one in ten people in developed countries. FA is defined as an antigen-specific biological response resulting from exposure to orally ingested food or food-derived components. Most of the symptoms of FA depend on the production of antigen-specific IgE, while anaphylaxis, caused by IgE-mediated mast cell degranulation, is a serious reaction that can lead to death. Various forms of food immunotherapy including oral, sublingual, and epicutaneous delivery routes have been used for the treatment of FA; clinical trials of anti-IgE therapy have also been initiated. Although anti-IgE therapy has not yet been approved for the treatment of FA, the combination of anti-IgE therapy and oral immunotherapy (OIT) is expected to be useful in peanut, milk, and multiple FAs, because anti-IgE treatment decreases the adverse events during OIT and shortens the treatment duration [141, 142].
Various types of ILCs reside in the intestine and contribute to homeostasis. In particular, the intestinal lamina propria (LP) has a high frequency of ILCs compared to that in the intestinal epithelium (IE). In mice, ILCs account for approximately 2.5% of all lymphocytes in the small intestinal LP fraction and 1.8% in the large intestinal LP fraction, which is more than 20 times higher than that in the IE fraction [143]. In the LP fraction of the small intestine, ILC3s are the most abundant, accounting for approximately 60% of all ILCs, while ILC2s and ILC1/NK cells account for approximately 20% each. On the other hand, in the LP fraction of the large intestine, ILC2s are the most abundant, accounting for approximately 50% of all ILCs, while ILC1/NK cells and ILC3s account for approximately 30% and 20%, respectively, which is opposite to the composition of the small intestine. Since FA is a type 2 immune response similar to other allergies, ILC2s among ILCs are thought to be involved in the pathogenesis of FA.
In studies of FA, models in which OVA/alum or peanuts/cholera toxin (PN/CT) are administered orally or peritoneally to mice to induce antigen-specific IgE production are often used. In these FA models, subsequent oral administration of OVA or peanuts to mice can induce type 2 inflammation, gastrointestinal symptoms such as diarrhea, and anaphylactic symptoms such as body temperature decrease through mast cell degranulation. The cytokines involved in the activation of ILC2s, such as IL-25, IL-33, and TSLP, are elevated, and the number of intestinal ILC2s is increased in an IL-33-dependent manner in the FA model mice. Consistent with these data, eosinophil infiltration, IgE production, and anaphylactic symptoms are reduced in IL-33 receptor-deficient mice, suggesting that ILC2s are involved in the pathogenesis of FA [144]. It has been reported that in the OVA/alum-induced FA model, IL-25 production is enhanced and elicits IL-13 production from ILC2s by direct stimulation or indirect activation via IL-25 receptor-positive Th2 cells, resulting in mastocytosis and diarrhea symptoms [145]. Furthermore, in the PN/CT-induced FA model using IL4RαF709 mice, in which the unrestrained form of the IL-4Rα chain lacking the immunoreceptor tyrosine-based inhibitory motif (ITIM) motif is knocked out, ILC2s contribute to the disruption of immune tolerance by suppressing antigen-specific Tregs via IL-4 production and further enhancing IgE reactivity of mast cells via IL-4/IL-13 production, leading to the exacerbation of anaphylaxis [146, 147]. Interestingly, disruption of the skin barrier induces the activation of intestinal ILC2s via IL-33, which enhances mastocytosis and anaphylaxis in OVA-induced FA models [148]. This study indicates that improper activation of ILC2s is involved in the development of allergic marches. On the other hand, Chu et al. reported that ILC2s contribute to type 2 inflammation in the abdominal cavity induced by FA, but not to IgE production, gastrointestinal symptoms, and anaphylaxis, based on the result of ILC2 depletion using Thy1-neutralizing antibody in a PN/CT-induced FA model [149]. These differences may be due to the varying FA models and mouse strains used, or due to the effects of the gut microbiota. Although ILC2s are abundant in the intestinal tracts and can be involved in the pathogenesis of FA as a source of type 2 cytokines, FA studies focusing on ILC2s have not been widely reported, especially in humans, compared to other allergic diseases such as asthma and CRS. Further research in both humans and mice will help to understand the role of ILC2s in the development of FA.
6.6 Allergic Conjunctivitis and ILC2s
Allergic conjunctivitis is an allergic inflammatory disease caused by foreign substances in the eyes, such as pollen, house dust, and contact lenses. The main symptoms of allergic conjunctivitis are itching, redness, and increased tear production, which are thought to be caused by the acquired immune system triggered by foreign substances. However, the involvement of the innate immune system in allergic conjunctivitis has recently attracted attention because of the elevated levels of epithelial cell-derived type 2 initiating cytokines such as IL-33 and TSLP [150, 151]. The mouse line hK14mIL33Tg is a keratin 14-driven transgenic mouse, which overexpresses IL-33 in an epithelial cell-specific manner and spontaneously develops allergic dermatitis and conjunctivitis. In these mice, ILC2s in the conjunctiva were increased by more than 20 times compared to ILC2s in naïve mice, and ILC2-derived IL-5 and IL-13 production was enhanced [152]. It was also reported that ILC2s increased with eosinophils in conjunctivitis induced by papain contact lenses [153]. Interestingly, a similar level of inflammation was induced in Rag2-/- mice lacking the acquired immune system, suggesting that ILC2-mediated allergic inflammation plays an essential role in the pathogenesis of allergic conjunctivitis. It is expected that understanding the mechanism by which ILC2s contribute to the maintenance of homeostasis and the induction of allergic inflammation in the conjunctiva, a barrier tissue in contact with the outside world similar to the skin, nasal mucosa, and bronchi would aid in the development of novel therapeutic approaches.
6.7 Conclusion
The word “allergy” is a concept, and the actual diseases, such as asthma, atopic dermatitis, and food allergy, are differentiated according to the organ in which the allergy occurs and are treated by the respective clinical departments. As a result, patients are referred to the Department of Respiratory Medicine for asthma, the Department of Dermatology for atopic dermatitis, and the Department of Gastroenterology for FA, with limited coordination between doctors in each department. On the other hand, in the field of basic research, the existence of ILCs has been recently revealed, and the role of ILC2s in allergic diseases has been rapidly elucidated. As a result, allergies, regardless of the organ, can now be roughly divided into antigen-specific mediated by Th2 cells and antigen-nonspecific mediated by ILC2s, mainly IL-33. Therefore, future research, drug development, and medical treatment for allergies must emphasize antigen and organ specificity to comprehensively understand the diseases. In the face of the explosive increase in allergies worldwide, it is necessary to establish methods that accurately determine whether each patient’s allergy is dependent on Th2 cells or ILC2s, and to identify targets that can completely suppress each of these cell types.
References
Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. 2011;12(11):1045–54.
Yudanin NA, Schmitz F, Flamar AL, Thome JJC, Tait Wojno E, Moeller JB, et al. Spatial and temporal mapping of human innate lymphoid cells reveals elements of tissue specificity. Immunity. 2019;50(2):505–19.
Marquardt N, Kekalainen E, Chen P, Kvedaraite E, Wilson JN, Ivarsson MA, et al. Human lung natural killer cells are predominantly comprised of highly differentiated hypofunctional CD69(−)CD56(dim) cells. J Allergy Clin Immunol. 2017;139(4):1321–30.
Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol. 2016;17(6):626–35.
Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med. 2014;20(1):54–61.
Mazzurana L, Czarnewski P, Jonsson V, Wigge L, Ringnér M, Williams TC, et al. Tissue-specific transcriptional imprinting and heterogeneity in human innate lymphoid cells revealed by full-length single-cell RNA-sequencing. Cell Res. 2021;31(5):554–68.
Klein Wolterink RG, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y, et al. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur J Immunol. 2012;42(5):1106–16.
Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage- CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. 2012;188(3):1503–13.
Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. 2012;36(3):451–63.
Arae K, Ikutani M, Horiguchi K, Yamaguchi S, Okada Y, Sugiyama H, et al. Interleukin-33 and thymic stromal lymphopoietin, but not interleukin-25, are crucial for development of airway eosinophilia induced by chitin. Sci Rep. 2021;11(1):5913.
Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, et al. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J Allergy Clin Immunol. 2012;129(1):216–27.
Han M, Rajput C, Hong JY, Lei J, Hinde JL, Wu Q, et al. The innate cytokines IL-25, IL-33, and TSLP cooperate in the induction of type 2 innate lymphoid cell expansion and mucous metaplasia in rhinovirus-infected immature mice. J Immunol. 2017;199(4):1308–18.
Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12(7):631–8.
Kouzaki H, Iijima K, Kobayashi T, O'Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J Immunol. 2011;186(7):4375–87.
Cayrol C, Duval A, Schmitt P, Roga S, Camus M, Stella A, et al. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat Immunol. 2018;19(4):375–85.
Furusawa J, Moro K, Motomura Y, Okamoto K, Zhu J, Takayanagi H, et al. Critical role of p38 and GATA3 in natural helper cell function. J Immunol. 2013;191(4):1818–26.
Kabata H, Moro K, Koyasu S. The group 2 innate lymphoid cell (ILC2) regulatory network and its underlying mechanisms. Immunol Rev. 2018;286(1):37–52.
Hammad H, Lambrecht BN. The basic immunology of asthma. Cell. 2021;184(6):1469–85.
Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. 2015;16(1):45–56.
Torgerson DG, Ampleford EJ, Chiu GY, Gauderman WJ, Gignoux CR, Graves PE, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat Genet. 2011;43(9):887–92.
Prefontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, et al. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol. 2009;183(8):5094–103.
Christianson CA, Goplen NP, Zafar I, Irvin C, Good JT Jr, Rollins DR, et al. Persistence of asthma requires multiple feedback circuits involving type 2 innate lymphoid cells and IL-33. J Allergy Clin Immunol. 2015;136(1):59–68.
Prefontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, Chakir J, et al. Increased IL-33 expression by epithelial cells in bronchial asthma. J Allergy Clin Immunol. 2010;125(3):752–4.
Smith SG, Chen R, Kjarsgaard M, Huang C, Oliveria JP, O'Byrne PM, et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol. 2016;137(1):75–86.
Bartemes KR, Kephart GM, Fox SJ, Kita H. Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J Allergy Clin Immunol. 2014;134(3):671–8.
Al-Sajee D, Sehmi R, Hawke TJ, El-Gammal A, Howie KJ, Watson RM, et al. Expression of IL-33 and TSLP and their receptors in asthmatic airways after inhaled allergen challenge. Am J Respir Crit Care Med. 2018;198(6):805–7.
Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med. 2014;190(12):1373–82.
Winkler C, Hochdorfer T, Israelsson E, Hasselberg A, Cavallin A, Thorn K, et al. Activation of group 2 innate lymphoid cells after allergen challenge in asthmatic patients. J Allergy Clin Immunol. 2019;144(1):61–9.
Mazzurana L, Czarnewski P, Jonsson V, Wigge L, Ringner M, Williams TC, et al. Tissue-specific transcriptional imprinting and heterogeneity in human innate lymphoid cells revealed by full-length single-cell RNA-sequencing. Cell Res. 2021;31(5):554–68.
Moro K, Kabata H, Tanabe M, Koga S, Takeno N, Mochizuki M, et al. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat Immunol. 2016;17(1):76–86.
Liu S, Verma M, Michalec L, Liu W, Sripada A, Rollins D, et al. Steroid resistance of airway type 2 innate lymphoid cells from patients with severe asthma: the role of thymic stromal lymphopoietin. J Allergy Clin Immunol. 2018;141(1):257–68 e6.
Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2010;463(7280):540–4.
Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12(11):1055–62.
Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science. 2016;351(6279):1329–33.
Barlow JL, Peel S, Fox J, Panova V, Hardman CS, Camelo A, et al. IL-33 is more potent than IL-25 in provoking IL-13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J Allergy Clin Immunol. 2013;132(4):933–41.
Kabata H, Moro K, Fukunaga K, Suzuki Y, Miyata J, Masaki K, et al. Thymic stromal lymphopoietin induces corticosteroid resistance in natural helper cells during airway inflammation. Nat Commun. 2013;4:2675.
Shikotra A, Choy DF, Ohri CM, Doran E, Butler C, Hargadon B, et al. Increased expression of immunoreactive thymic stromal lymphopoietin in patients with severe asthma. J Allergy Clin Immunol. 2012;129(1):104–11.
Motomura Y, Morita H, Moro K, Nakae S, Artis D, Endo TA, et al. Basophil-derived interleukin-4 controls the function of natural helper cells, a member of ILC2s, in lung inflammation. Immunity. 2014;40(5):758–71.
Yu X, Pappu R, Ramirez-Carrozzi V, Ota N, Caplazi P, Zhang J, et al. TNF superfamily member TL1A elicits type 2 innate lymphoid cells at mucosal barriers. Mucosal Immunol. 2014;7(3):730–40.
Meylan F, Hawley ET, Barron L, Barlow JL, Penumetcha P, Pelletier M, et al. The TNF-family cytokine TL1A promotes allergic immunopathology through group 2 innate lymphoid cells. Mucosal Immunol. 2014;7(4):958–68.
Machida K, Aw M, Salter BMA, Ju X, Mukherjee M, Gauvreau GM, et al. The role of the TL1A/DR3 axis in the activation of group 2 innate lymphoid cells in subjects with eosinophilic asthma. Am J Respir Crit Care Med. 2020;202(8):1105–14.
Nagashima H, Okuyama Y, Fujita T, Takeda T, Motomura Y, Moro K, et al. GITR cosignal in ILC2s controls allergic lung inflammation. J Allergy Clin Immunol. 2018;141(5):1939–43.
Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J Allergy Clin Immunol. 2013;132(1):205–13.
Xue L, Salimi M, Panse I, Mjosberg JM, McKenzie AN, Spits H, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol. 2014;133(4):1184–94.
Lund SJ, Portillo A, Cavagnero K, Baum RE, Naji LH, Badrani JH, et al. Leukotriene C4 potentiates IL-33-induced group 2 innate lymphoid cell activation and lung inflammation. J Immunol. 2017;199(3):1096–104.
Wojno ED, Monticelli LA, Tran SV, Alenghat T, Osborne LC, Thome JJ, et al. The prostaglandin D(2) receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal Immunol. 2015;8(6):1313–23.
Salimi M, Stoger L, Liu W, Go S, Pavord I, Klenerman P, et al. Cysteinyl leukotriene E4 activates human group 2 innate lymphoid cells and enhances the effect of prostaglandin D2 and epithelial cytokines. J Allergy Clin Immunol. 2017;140(4):1090–100.
von Moltke J, O'Leary CE, Barrett NA, Kanaoka Y, Austen KF, Locksley RM. Leukotrienes provide an NFAT-dependent signal that synergizes with IL-33 to activate ILC2s. J Exp Med. 2017;214(1):27–37.
White AA, Stevenson DD. Aspirin-exacerbated respiratory disease. N Engl J Med. 2018;379(11):1060–70.
Eastman JJ, Cavagnero KJ, Deconde AS, Kim AS, Karta MR, Broide DH, et al. Group 2 innate lymphoid cells are recruited to the nasal mucosa in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2017;140(1):101–8.
Duerr CU, McCarthy CD, Mindt BC, Rubio M, Meli AP, Pothlichet J, et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol. 2016;17(1):65–75.
Molofsky AB, Van Gool F, Liang HE, Van Dyken SJ, Nussbaum JC, Lee J, et al. Interleukin-33 and interferon-gamma counter-regulate group 2 innate lymphoid cell activation during immune perturbation. Immunity. 2015;43(1):161–74.
McHedlidze T, Kindermann M, Neves AT, Voehringer D, Neurath MF, Wirtz S. IL-27 suppresses type 2 immune responses in vivo via direct effects on group 2 innate lymphoid cells. Mucosal Immunol. 2016;9(6):1384–94.
Okuzumi S, Miyata J, Kabata H, Mochimaru T, Kagawa S, Masaki K, et al. TLR7 agonist suppresses ILC2-mediated inflammation via IL-27-producing interstitial macrophages. Am J Respir Cell Mol Biol. 2021;65(3):309–18.
Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201(6):937–47.
Djukanovic R, Harrison T, Johnston SL, Gabbay F, Wark P, Thomson NC, et al. The effect of inhaled IFN-beta on worsening of asthma symptoms caused by viral infections. A randomized trial. Am J Respir Crit Care Med. 2014;190(2):145–54.
Psallidas I, Backer V, Kuna P, Palmer R, Necander S, Aurell M, et al. A phase 2a, double-blind, placebo-controlled randomized trial of inhaled TLR9 agonist AZD1419 in asthma. Am J Respir Crit Care Med. 2021;203(3):296–306.
Maric J, Ravindran A, Mazzurana L, Bjorklund AK, Van Acker A, Rao A, et al. Prostaglandin E2 suppresses human group 2 innate lymphoid cell function. J Allergy Clin Immunol. 2018;141(5):1761–73.
Zhou Y, Wang W, Zhao C, Wang Y, Wu H, Sun X, et al. Prostaglandin E2 inhibits group 2 innate lymphoid cell activation and allergic airway inflammation through E-prostanoid 4-cyclic adenosine monophosphate signaling. Front Immunol. 2018;9:501.
Zhou W, Toki S, Zhang J, Goleniewksa K, Newcomb DC, Cephus JY, et al. Prostaglandin I2 signaling and inhibition of group 2 innate lymphoid cell responses. Am J Respir Crit Care Med. 2016;193(1):31–42.
Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, et al. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med. 2013;5(174):174ra26.
Ohne Y, Silver JS, Thompson-Snipes L, Collet MA, Blanck JP, Cantarel BL, et al. IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat Immunol. 2016;17(6):646–55.
Lim AI, Menegatti S, Bustamante J, Le Bourhis L, Allez M, Rogge L, et al. IL-12 drives functional plasticity of human group 2 innate lymphoid cells. J Exp Med. 2016;213(4):569–83.
Morita H, Kubo T, Ruckert B, Ravindran A, Soyka MB, Rinaldi AO, et al. Induction of human regulatory innate lymphoid cells from group 2 innate lymphoid cells by retinoic acid. J Allergy Clin Immunol. 2019;143(6):2190–201.
Kabata H, Artis D. Neuro-immune crosstalk and allergic inflammation. J Clin Investig. 2019;129(4):1475–82.
Sui P, Wiesner DL, Xu J, Zhang Y, Lee J, Van Dyken S, et al. Pulmonary neuroendocrine cells amplify allergic asthma responses. Science. 2018;360(6393):8546.
Nagashima H, Mahlakoiv T, Shih HY, Davis FP, Meylan F, Huang Y, et al. Neuropeptide CGRP limits group 2 innate lymphoid cell responses and constrains type 2 inflammation. Immunity. 2019;51(4):682–95.
Klose CSN, Mahlakoiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature. 2017;549(7671):282–6.
Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature. 2017;549(7672):351–6.
Cardoso V, Chesne J, Ribeiro H, Garcia-Cassani B, Carvalho T, Bouchery T, et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature. 2017;549(7671):277–81.
Talbot S, Abdulnour RE, Burkett PR, Lee S, Cronin SJ, Pascal MA, et al. Silencing nociceptor neurons reduces allergic airway inflammation. Neuron. 2015;87(2):341–54.
Galle-Treger L, Suzuki Y, Patel N, Sankaranarayanan I, Aron JL, Maazi H, et al. Nicotinic acetylcholine receptor agonist attenuates ILC2-dependent airway hyperreactivity. Nat Commun. 2016;7:13202.
Moriyama S, Brestoff JR, Flamar AL, Moeller JB, Klose CSN, Rankin LC, et al. β2-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science. 2018;359(6379):1056–61.
Martinez-Gonzalez I, Matha L, Steer CA, Ghaedi M, Poon GF, Takei F. Allergen-experienced group 2 innate lymphoid cells acquire memory-like properties and enhance allergic lung inflammation. Immunity. 2016;45(1):198–208.
Verma M, Michalec L, Sripada A, McKay J, Sirohi K, Verma D, et al. The molecular and epigenetic mechanisms of innate lymphoid cell (ILC) memory and its relevance for asthma. J Exp Med. 2021;218(7):e20201354.
Gold MJ, Antignano F, Halim TY, Hirota JA, Blanchet MR, Zaph C, et al. Group 2 innate lymphoid cells facilitate sensitization to local, but not systemic, TH2-inducing allergen exposures. J Allergy Clin Immunol. 2014;133(4):1142–8.
Mirchandani AS, Besnard AG, Yip E, Scott C, Bain CC, Cerovic V, et al. Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J Immunol. 2014;192(5):2442–8.
Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity. 2014;41(2):283–95.
Drake LY, Iijima K, Kita H. Group 2 innate lymphoid cells and CD4+ T cells cooperate to mediate type 2 immune response in mice. Allergy. 2014;69(10):1300–7.
Halim TYF, Rana BMJ, Walker JA, Kerscher B, Knolle MD, Jolin HE, et al. Tissue-restricted adaptive type 2 immunity is orchestrated by expression of the costimulatory molecule OX40L on group 2 innate lymphoid cells. Immunity. 2018;48(6):1195–207.
Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity. 2014;40(3):425–35.
Sugita K, Steer CA, Martinez-Gonzalez I, Altunbulakli C, Morita H, Castro-Giner F, et al. Type 2 innate lymphoid cells disrupt bronchial epithelial barrier integrity by targeting tight junctions through IL-13 in asthmatic patients. J Allergy Clin Immunol. 2018;141(1):300–10.
Yu QN, Guo YB, Li X, Li CL, Tan WP, Fan XL, et al. ILC2 frequency and activity are inhibited by glucocorticoid treatment via STAT pathway in patients with asthma. Allergy. 2018;73(9):1860–70.
Kabata H, Flamar AL, Mahlakoiv T, Moriyama S, Rodewald HR, Ziegler SF, et al. Targeted deletion of the TSLP receptor reveals cellular mechanisms that promote type 2 airway inflammation. Mucosal Immunol. 2020;13(4):626–36.
Gauvreau GM, O'Byrne PM, Boulet LP, Wang Y, Cockcroft D, Bigler J, et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. N Engl J Med. 2014;370(22):2102–10.
Corren J, Parnes JR, Wang L, Mo M, Roseti SL, Griffiths JM, et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med. 2017;377(10):936–46.
Menzies-Gow A, Corren J, Bourdin A, Chupp G, Israel E, Wechsler ME, et al. Tezepelumab in adults and adolescents with severe, uncontrolled asthma. N Engl J Med. 2021;384(19):1800–9.
Porsbjerg CM, Sverrild A, Lloyd CM, Menzies-Gow AN, Bel EH. Anti-alarmins in asthma: targeting the airway epithelium with next-generation biologics. Eur Respir J. 2020;56(5):2000260.
Patel G, Pan J, Ye L, Shen X, Rosloff D, D'Souza SS, et al. Blockade of IL-4Rα inhibits group 2 innate lymphoid cell responses in asthma patients. Clin Exp Allergy. 2020;50(2):267–70.
Kobayashi T, Voisin B, Kim DY, Kennedy EA, Jo JH, Shih HY, et al. Homeostatic control of sebaceous glands by innate lymphoid cells regulates commensal bacteria equilibrium. Cell. 2019;176(5):982–97.
Dutton EE, Gajdasik DW, Willis C, Fiancette R, Bishop EL, Camelo A, et al. Peripheral lymph nodes contain migratory and resident innate lymphoid cell populations. Sci Immunol. 2019;4(35):8082.
Paternoster L, Standl M, Waage J, Baurecht H, Hotze M, Strachan DP, et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet. 2015;47(12):1449–56.
Tamari M, Hirota T. Genome-wide association studies of atopic dermatitis. J Dermatol. 2014;41(3):213–20.
Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, et al. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. 2013;5(170):170ra16.
Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med. 2013;210(13):2939–50.
Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, et al. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat Immunol. 2013;14(6):564–73.
Imai Y, Yasuda K, Sakaguchi Y, Haneda T, Mizutani H, Yoshimoto T, et al. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc Natl Acad Sci USA. 2013;110(34):13921–6.
Imai Y, Yasuda K, Nagai M, Kusakabe M, Kubo M, Nakanishi K, et al. IL-33-induced atopic dermatitis-like inflammation in mice is mediated by group 2 innate lymphoid cells in concert with basophils. J Investig Dermatol. 2019;139(10):2185–94.
Leyva-Castillo JM, Galand C, Mashiko S, Bissonnette R, McGurk A, Ziegler SF, et al. ILC2 activation by keratinocyte-derived IL-25 drives IL-13 production at sites of allergic skin inflammation. J Allergy Clin Immunol. 2020;145(6):1606–14.
Beck LA, Thaçi D, Hamilton JD, Graham NM, Bieber T, Rocklin R, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371(2):130–9.
Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375(24):2335–48.
Honda T, Egawa G, Grabbe S, Kabashima K. Update of immune events in the murine contact hypersensitivity model: toward the understanding of allergic contact dermatitis. J Investig Dermatol. 2013;133(2):303–15.
O'Leary JG, Goodarzi M, Drayton DL, von Andrian UH. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat Immunol. 2006;7(5):507–16.
Peng H, Jiang X, Chen Y, Sojka DK, Wei H, Gao X, et al. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J Clin Investig. 2013;123(4):1444–56.
Wang X, Peng H, Cong J, Wang X, Lian Z, Wei H, et al. Memory formation and long-term maintenance of IL-7Rα(+) ILC1s via a lymph node-liver axis. Nat Commun. 2018;9(1):4854.
Carbone T, Nasorri F, Pennino D, Eyerich K, Foerster S, Cifaldi L, et al. CD56highCD16-CD62L-NK cells accumulate in allergic contact dermatitis and contribute to the expression of allergic responses. J Immunol. 2010;184(2):1102–10.
Xu G, Zhang L, Wang DY, Xu R, Liu Z, Han DM, et al. Opposing roles of IL-17A and IL-25 in the regulation of TSLP production in human nasal epithelial cells. Allergy. 2010;65(5):581–9.
Asaka D, Yoshikawa M, Nakayama T, Yoshimura T, Moriyama H, Otori N. Elevated levels of interleukin-33 in the nasal secretions of patients with allergic rhinitis. Int Arch Allergy Immunol. 2012;158(Suppl 1):47–50.
Sakashita M, Yoshimoto T, Hirota T, Harada M, Okubo K, Osawa Y, et al. Association of serum interleukin-33 level and the interleukin-33 genetic variant with Japanese cedar pollinosis. Clin Exp Allergy. 2008;38(12):1875–81.
Haenuki Y, Matsushita K, Futatsugi-Yumikura S, Ishii KJ, Kawagoe T, Imoto Y, et al. A critical role of IL-33 in experimental allergic rhinitis. J Allergy Clin Immunol. 2012;130(1):184–94.
Nakanishi W, Yamaguchi S, Matsuda A, Suzukawa M, Shibui A, Nambu A, et al. IL-33, but not IL-25, is crucial for the development of house dust mite antigen-induced allergic rhinitis. PLoS One. 2013;8(10):e78099.
Ramasamy A, Curjuric I, Coin LJ, Kumar A, McArdle WL, Imboden M, et al. A genome-wide meta-analysis of genetic variants associated with allergic rhinitis and grass sensitization and their interaction with birth order. J Allergy Clin Immunol. 2011;128(5):996–1005.
Andiappan AK, Wang de Y, Anantharaman R, Suri BK, Lee BT, Rotzschke O, et al. Replication of genome-wide association study loci for allergic rhinitis and house dust mite sensitization in an Asian population of ethnic Chinese in Singapore. J Allergy Clin Immunol. 2013;131(5):1431–3.
Birben E, Sahiner UM, Karaaslan C, Yavuz TS, Cosgun E, Kalayci O, et al. The genetic variants of thymic stromal lymphopoietin protein in children with asthma and allergic rhinitis. Int Arch Allergy Immunol. 2014;163(3):185–92.
Nilsson D, Henmyr V, Hallden C, Sall T, Kull I, Wickman M, et al. Replication of genome-wide associations with allergic sensitization and allergic rhinitis. Allergy. 2014;69(11):1506–14.
Sun Q, Liu Y, Zhang S, Liu K, Zhu X, Liu J, et al. Thymic stromal lymphopoietin polymorphisms and allergic rhinitis risk: a systematic review and meta-analysis with 6351 cases and 11,472 controls. Int J Clin Exp Med. 2015;8(9):15752–8.
Zhu DD, Zhu XW, Jiang XD, Dong Z. Thymic stromal lymphopoietin expression is increased in nasal epithelial cells of patients with mugwort pollen sensitive-seasonal allergic rhinitis. Chin Med J. 2009;122(19):2303–7.
Mou Z, Xia J, Tan Y, Wang X, Zhang Y, Zhou B, et al. Overexpression of thymic stromal lymphopoietin in allergic rhinitis. Acta Otolaryngol. 2009;129(3):297–301.
Li Z, Wang H, Liu L. Interleukin-25 enhances allergic inflammation through p38MAPK and NF-kappaB pathways in mouse models of allergic rhinitis. Iran J Allergy Asthma Immunol. 2014;13(6):412–9.
Zhong H, Fan XL, Yu QN, Qin ZL, Chen D, Xu R, et al. Increased innate type 2 immune response in house dust mite-allergic patients with allergic rhinitis. Clin Immunol. 2017;183:293–9.
Sun R, Yang Y, Huo Q, Gu Z, Wei P, Tang X. Increased expression of type 2 innate lymphoid cells in pediatric patients with allergic rhinitis. Exp Ther Med. 2020;19(1):735–40.
Fan D, Wang X, Wang M, Wang Y, Zhang L, Li Y, et al. Allergen-dependent differences in ILC2s frequencies in patients with allergic rhinitis. Allergy Asthma Immunol Res. 2016;8(3):216–22.
Shaw JL, Fakhri S, Citardi MJ, Porter PC, Corry DB, Kheradmand F, et al. IL-33-responsive innate lymphoid cells are an important source of IL-13 in chronic rhinosinusitis with nasal polyps. Am J Respir Crit Care Med. 2013;188(4):432–9.
Payne SC, Early SB, Huyett P, Han JK, Borish L, Steinke JW. Evidence for distinct histologic profile of nasal polyps with and without eosinophilia. Laryngoscope. 2011;121(10):2262–7.
Miljkovic D, Bassiouni A, Cooksley C, Ou J, Hauben E, Wormald PJ, et al. Association between group 2 innate lymphoid cells enrichment, nasal polyps and allergy in chronic rhinosinusitis. Allergy. 2014;69(9):1154–61.
Ho J, Bailey M, Zaunders J, Mrad N, Sacks R, Sewell W, et al. Group 2 innate lymphoid cells (ILC2s) are increased in chronic rhinosinusitis with nasal polyps or eosinophilia. Clin Exp Allergy. 2015;45(2):394–403.
Tojima I, Kouzaki H, Shimizu S, Ogawa T, Arikata M, Kita H, et al. Group 2 innate lymphoid cells are increased in nasal polyps in patients with eosinophilic chronic rhinosinusitis. Clin Immunol. 2016;170:1–8.
Poposki JA, Klingler AI, Tan BK, Soroosh P, Banie H, Lewis G, et al. Group 2 innate lymphoid cells are elevated and activated in chronic rhinosinusitis with nasal polyps. Immun Inflamm Dis. 2017;5(3):233–43.
Baba S, Kondo K, Kanaya K, Suzukawa K, Ushio M, Urata S, et al. Expression of IL-33 and its receptor ST2 in chronic rhinosinusitis with nasal polyps. Laryngoscope. 2014;124(4):E115–22.
Reh DD, Wang Y, Ramanathan M Jr, Lane AP. Treatment-recalcitrant chronic rhinosinusitis with polyps is associated with altered epithelial cell expression of interleukin-33. Am J Rhinol Allergy. 2010;24(2):105–9.
Kimura S, Pawankar R, Mori S, Nonaka M, Masuno S, Yagi T, et al. Increased expression and role of thymic stromal lymphopoietin in nasal polyposis. Allergy Asthma Immunol Res. 2011;3(3):186–93.
Mjosberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, et al. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity. 2012;37(4):649–59.
Liu T, Li TL, Zhao F, Xie C, Liu AM, Chen X, et al. Role of thymic stromal lymphopoietin in the pathogenesis of nasal polyposis. Am J Med Sci. 2011;341(1):40–7.
Ouyang Y, Fan E, Li Y, Wang X, Zhang L. Clinical characteristics and expression of thymic stromal lymphopoietin in eosinophilic and non-eosinophilic chronic rhinosinusitis. ORL J Otorhinolaryngol Relat Spec. 2013;75(1):37–45.
Nakayama T, Hirota T, Asaka D, Sakashita M, Ninomiya T, Morikawa T, et al. A genetic variant near TSLP is associated with chronic rhinosinusitis with nasal polyps and aspirin-exacerbated respiratory disease in Japanese populations. Allergol Int. 2020;69(1):138–40.
Park SK, Jin YD, Park YK, Yeon SH, Xu J, Han RN, et al. IL-25-induced activation of nasal fibroblast and its association with the remodeling of chronic rhinosinusitis with nasal polyposis. PLoS One. 2017;12(8):e0181806.
Bachert C, Han JK, Desrosiers M, Hellings PW, Amin N, Lee SE, et al. Efficacy and safety of dupilumab in patients with severe chronic rhinosinusitis with nasal polyps (LIBERTY NP SINUS-24 and LIBERTY NP SINUS-52): results from two multicentre, randomised, double-blind, placebo-controlled, parallel-group phase 3 trials. Lancet. 2019;394(10209):1638–50.
Gevaert P, Holtappels G, Johansson SG, Cuvelier C, Cauwenberge P, Bachert C. Organization of secondary lymphoid tissue and local IgE formation to Staphylococcus aureus enterotoxins in nasal polyp tissue. Allergy. 2005;60(1):71–9.
Lombardi V, Beuraud C, Neukirch C, Moussu H, Morizur L, Horiot S, et al. Circulating innate lymphoid cells are differentially regulated in allergic and nonallergic subjects. J Allergy Clin Immunol. 2016;138(1):305–8.
Mitthamsiri W, Pradubpongsa P, Sangasapaviliya A, Boonpiyathad T. Decreased CRTH2 expression and response to allergen re-stimulation on innate lymphoid cells in patients with allergen-specific immunotherapy. Allergy Asthma Immunol Res. 2018;10(6):662–74.
Burks AW, Sampson HA, Plaut M, Lack G, Akdis CA. Treatment for food allergy. J Allergy Clin Immunol. 2018;141(1):1–9.
Sampath V, Sindher SB, Alvarez Pinzon AM, Nadeau KC. Can food allergy be cured? What are the future prospects? Allergy. 2019;75(6):1316–26.
Kim CH, Hashimoto-Hill S, Kim M. Migration and tissue tropism of innate lymphoid cells. Trends Immunol. 2016;37(1):68–79.
Chu DK, Llop-Guevara A, Walker TD, Flader K, Goncharova S, Boudreau JE, et al. IL-33, but not thymic stromal lymphopoietin or IL-25, is central to mite and peanut allergic sensitization. J Allergy Clin Immunol. 2013;131(1):187–200.
Lee J-B, Chen C-Y, Liu B, Mugge L, Angkasekwinai P, Facchinetti V, et al. IL-25 and CD4+ TH2 cells enhance type 2 innate lymphoid cell-derived IL-13 production, which promotes IgE-mediated experimental food allergy. J Allergy Clin Immunol. 2016;137(4):1216–25.
Burton OT, Medina Tamayo J, Stranks AJ, Miller S, Koleoglou KJ, Weinberg EO, et al. IgE promotes type 2 innate lymphoid cells in murine food allergy. Clin Exp Allergy. 2018;48(3):288–96.
Noval Rivas M, Burton OT, Oettgen HC, Chatila T. IL-4 production by group 2 innate lymphoid cells promotes food allergy by blocking regulatory T-cell function. J Allergy Clin Immunol. 2016;138(3):801–11.
Leyva-Castillo J-M, Galand C, Kam C, Burton O, Gurish M, Musser MA, et al. Mechanical skin injury promotes food anaphylaxis by driving intestinal mast cell expansion. Immunity. 2019;50(5):1262–75.
Chu DK, Mohammed-Ali Z, Jiménez-Saiz R, Walker TD, Goncharova S, Llop-Guevara A, et al. T helper cell IL-4 drives intestinal Th2 priming to oral peanut antigen, under the control of OX40L and independent of innate-like lymphocytes. Mucosal Immunol. 2014;7(6):1395–404.
Matsuda A, Ebihara N, Yokoi N, Kawasaki S, Tanioka H, Inatomi T, et al. Functional role of thymic stromal lymphopoietin in chronic allergic keratoconjunctivitis. Investig Opthalmol Visual Sci. 2010;51(1):151.
Matsuda A, Okayama Y, Terai N, Yokoi N, Ebihara N, Tanioka H, et al. The role of interleukin-33 in chronic allergic conjunctivitis. Investig Opthalmol Vis Sci. 2009;50(10):4646.
Imai Y, Hosotani Y, Ishikawa H, Yasuda K, Nagai M, Jitsukawa O, et al. Expression of IL-33 in ocular surface epithelium induces atopic keratoconjunctivitis with activation of group 2 innate lymphoid cells in mice. Sci Rep. 2017;7(1):10053.
Sugita J, Asada Y, Ishida W, Iwamoto S, Sudo K, Suto H, et al. Contributions of interleukin-33 and TSLP in a papain-soaked contact lens-induced mouse conjunctival inflammation model. Immun Inflamm Dis. 2017;5(4):515–25.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Kabata, H., Motomura, Y., Kiniwa, T., Kobayashi, T., Moro, K. (2022). ILCs and Allergy. In: Sun, XH. (eds) Innate Lymphoid Cells. Advances in Experimental Medicine and Biology, vol 1365. Springer, Singapore. https://doi.org/10.1007/978-981-16-8387-9_6
Download citation
DOI: https://doi.org/10.1007/978-981-16-8387-9_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-8386-2
Online ISBN: 978-981-16-8387-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)