
Abstract
The green revolution has increased the quantum of major cereals (wheat and rice), which otherwise would have been substantially low across the developing countries; a significant contribution that one can never undervalue. However, focused monoculture of either crop was realized particularly with respect to loss of agro-biodiversity and sustenance of nutri-rich minor crops resulting in poor food grain diversity. Albeit, these crops provide sufficient calories, they do not make a complete diet resulting in malnutrition of over 2000 million people worldwide. Millets are versatile grains valued for their exceptional nutritional profile. Being the reservoir of essential micronutrients and trace elements they are often termed as nature’s nutraceutical basket. Furthermore, their climate resilient nature and adaptation to low input agriculture makes them “harbingers for evergreen revolution.” The latest advancements in genomics and automate phenotyping techniques for searching genes and metabolites, coupled with high-throughput transformation processes have opened new avenues for product development in millets. Furthermore, various computational biology platforms help us in analyzing big molecular data of crop plants to identify valuable genes hidden in them. In addition, molecular breeding platforms may be utilized to speed up the introgression of value-added genes in high welding and widely adapted genetic backgrounds. This chapter focuses on searching for values in the natural millet gene pool of millets for developing climate smart crops with value-added traits.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
10.1 Introduction
In nature, plants are a treasure trove of interesting and useful substances because they must collect everything from the place on earth where they are rooted and when challenged they cannot escape; thus, despite these constraints, they have created the most impressive panoply of products to survive in ever-changing environments. Major cereals comprise a major part of the diets of people, but they lack the essential micronutrients resulting in micronutrient malnutrition across the world. Among the cereal crops, millets, which are having the status of neglected and minor crops, are quite important from the point of nutritional and food security.
Millets are the best-kept secrets of our ancestors. Millets were important staple food in human history, especially for poor people of Asia and Africa’s semi-arid tropics. As per an old Kannada (south Indian language) saying, it is said that “The rice eater is weightless like a bird; the one who eats Jowar (sorghum) is strong like a wolf: one who eats ragi (finger millet) remains ‘nirogi’ (free from illness) throughout his life.” Sorghum and pearl millet share the major cultivated area under millets and share 95% of total millet output across the world (Yadav and Rai 2013; Nedumaran et al. 2014). Among the six minor millets, foxtail millet is cultivated as a food crop in semi-arid tropics of Asia and Africa and as a forage crop in Europe, North America, Australia, and North Africa (Austin 2006). Finger millet is grown as a major food crop in Central and East Africa and Asia (Vijayakumari et al. 2003). Millets are hardy in nature, thrive well in drylands under low soil fertility, and humid environments. Some of them have a short life cycle as from seed planting to harvest it takes only 65–70 days. When properly stored and preserved, whole millets can be used as seed for 5 years or more. Therefore, improvement in millet production and value addition through harnessing the genes available in natural gene pool is one of the sustainable approach for achieving food and nutritional security.
10.2 Minor Millets: Neglected Crop Goes Mainstream
Kids, teenagers, pregnant women, and lactating mothers are most vulnerable parts of the population suffering from malnutrition (Arlappa et al. 2010). For instance, the national family health survey of India revealed that approximately 46% of the children under 5 years of age are severely underweight, 38% are stunted, and 19% are wasted (Kanjilal et al. 2010). The overdependence on modern monocultural agricultural systems of major crops (rice, wheat, and potato) for dietary requirements have resulted in the loss of diversity of many nutritionally rich plant species from agricultural production systems. One such group of highly promising crop plant is that of minor millets. Once regarded as major player in household food security, they have been replaced by rice and wheat in the last few decades. However, in the context of the ongoing weather extremes and rising insecurities for food supplies, there has been a revived interest in cultivation and consumption of minor millets (Ravi et al. 2010).
10.3 Natural Gene Pool of Millets and Its Ex Situ Conservation
Mainstream and wild gene pool of millets harbor genes for agronomic and nutritional traits. Modest efforts have been made for collection and conservation of millet genetic resources across the world (Table 10.1). International Crop Research Institute for Semi-arid Tropics (ICRISAT) gene bank is the global repository for millets with a total of 24.7% accessions belonging to millets (Upadhyaya et al. 2015). The Indian germplasm collection of millets is the second largest collection in the world (Table 10.1). The millet diversity spots are well represented in these collections. However, major share belongs to the landraces and genetic stocks of the cultivated gene pool. Therefore, systematic and exhaustive efforts are required for enriching the collections with wild and unadapted gene pool of millets.
10.4 Searching Nutraceuticals Properties Hidden in Millets
10.4.1 Harnessing Nutraceutical Potential of the Natural Gene Pool
Diverse germplasm collections of millets assembled in gene banks made a modest impact on cultivar development (Upadhyaya et al. 2015). These large collections need rigorous phenotyping for nutraceutical traits in diverse environmental conditions for identification of genes and molecular mechanism governing the traits. However, extremely large size of these collections is the major hindrance in large-scale characterization for searching valuable genes. Concept of core and minicore collection to retain the total genetic diversity within a fraction of the entire germplasm collection with 95% certainty has been proposed for the enhanced utilization of germplasm in breeding programs (Brown 1989; Upadhyaya et al. 2015). Core and minicore collections with drastically reduced size, capturing maximum diversity, and minimum repetitiveness have been developed in millets and served as an ideal gene pool in extracting genes regulating nutraceutical traits.
Promising trait-specific accessions containing high concentration of micronutrients (Fe, Zn, and Ca) and protein in grains have been identified through rigorous multi-locational evaluation of core and minicore collections in sorghum), finger millet (Upadhyaya et al. 2011a), and foxtail millet (Upadhyaya et al. 2011b). In addition to conventional core and minicore subsets, global composite germplasm collections (GCGC), and genotyping-based reference sets have also been developed in millets (Upadhyaya et al. 2015). These collections comprised of wild and weedy relatives, advanced breeding lines, and accessions selected from phenotypic and genotypic evaluation of core and minicore collections.
Genome-wide analysis of the core and diversity panels and its integration with physiochemical characterization can be used to select parental combinations that can maximize genetic polymorphism for traits required for biofortification. Additionally, characterization of these collections through high fidelity next-generation molecular markers will facilitate analysis of population structure and association genetics to identify the genomic regions (QTLs) governing nutraceutical traits in millets.
10.4.2 Genomic Resources for Exploring Nutraceutical Potential of Millets
Deciphering molecular mechanism controlling nutraceutical traits is essential for Designing strategies for crop biofortification program. However, dissection of the nutritional quality traits through conventional methods is cumbersome, owing to their complex genetic control and high G × E interaction. In recent years, phenomenal development of high-throughput genomic tools facilitated comprehensive genome analysis of nutritional traits in coarse cereals (Singh et al. 2016). Application of such tools has helped in the identification and characterization of genes for genetic biofortification of crops (Velu et al. 2016). Despite well documented genetic variation for grain physiochemical properties in millets, little information is available with respect to genetics and genomics of these traits. Well assembled reference genome sequence is one of the most important requirements for gene identification and trait improvement through molecular breeding. Among the millets, genome sequences are available for sorghum (Paterson et al. 2009), pearl millet (Varshney et al. 2010), foxtail millet (Zhang et al. 2012), and finger millet (Hittalmani et al. 2017). Utilizing the genome sequencing information, a large set of simple sequence repeat (SSR) markers and a physical map has been developed in foxtail millet (Muthamilarasan et al. 2014). Thus, genome sequencing of these four species will provide a thorough insight into the gene structure and genetic network system governing the nutrient trait pathways in millets. However, no genome sequencing information is available for other lesser explored millets. In the absence of reference genome sequence, mutagenesis-based high-throughput genomic approaches such as TILLING and EcoTILLING may be exploited for the identification of genetic determinants governing nutraceutical properties in these neglected small millets. Among the millets, exploitation of genomic resources has just begun for characterizing QTLs, genes, and gene families governing valuable traits (Mudge et al. 2016). For instance, genome-wide association analysis (GWAS) followed by candidate gene-based approach identified genomic regions for ten grain quality traits in community resource sorghum diversity panel comprising 300 lines (Sukumaran et al. 2012). Likewise, QTLs and candidate genes controlling grain iron and zinc content in pearl millet were identified (Anuradha et al. 2017) in GWAS diversity panel. There have been no reports on application of GWAS analysis for genomic regions deciphering nutritional traits in other millets until recent times, though GWAS for agro-morphological traits and stress tolerance has been conducted in foxtail millet and finger millet (Jia et al. 2013).
In addition to QTLs, some specific genes or mechanisms associated with nutritional traits have been identified in millets. In sorghum, GWAS followed by candidate gene identification revealed that polymorphism in five genes (Sh2, Bt, Sssl, Ae1, and Wx) regulating starch biosynthesis and one gene governing protein content (O2) was associated with grain quality traits such as grain hardiness, endosperm texture, and protein content. In another study, polymorphism in coding regions of two alleles of Tannin-1 gene controlling tannin biosynthesis in sorghum grains revealed that tan-1a has a 1-bp deletion and the tan-1b has a 10-bp insertion in the coding region (Wu et al. 2012). The polymorphism observed in these genes can be exploited for the development of SNPs to improve protein digestibility of sorghum grain. Haplotype trait association analysis revealed considerable allelic variation in starch metabolism gene pullulanase (SbPUL) of sorghum and demonstrated that low-frequency haplotype of SbPUL gene enhances the starch digestibility, a critical factor for poor acceptability of sorghum grain (Gilding et al. 2013).
In recent years, transcriptome sequencing has emerged as a cost-effective and high-throughput approach to characterize the genes regulating nutraceutical properties in millets. Recently, finger millet seed transcriptome has emerged as a model to decipher the complex mechanism involved in higher calcium accumulation in grains (Kumar et al. 2014; Kumar et al. 2015a, b; Singh et al. 2014; Mirza et al. 2014, Table 10.2). Singh et al. (2014) generated a unique transcriptome assembly based on Illumina sequencing platform comprising 120, 130 Transcript Assembly Contigs (TACs) through RNA-seq analysis of two finger millet genotypes with contrasting grain calcium content. Analysis of TACs identified 82 calcium sensors and transporter genes classified into eight gene families, namely one calmodulin gene (CaM), one two-pore channel (TPC) protein, four calcium interacting protein kinases (CIPKs), and two calcium dependent protein kinases (CDPKs). These interesting findings in finger millet provide valuable functional markers for improving calcium content in crop breeding programs and encourage development of comprehensive transcriptome assemblies in all millets for functional characterization of genes and gene families governing nutritional traits.
Studies using transcriptomics to understand grain quality traits have been limited in other millets. Nevertheless, studies analyzed transcriptomes of various tissues to provide molecular understanding of other essential traits. For instance, several informative differentially expressed sequence tags (ESTs) during salinity and moisture stress have been generated through cDNA libraries (Lata et al. 2010) and suppression subtraction hybridization (SSH) (Puranik et al. 2011) in foxtail millet. However, major disadvantage of these traditional transcriptomes is that it is expensive, time-consuming, and labor intensive. Keeping this in view, whole transcriptome of foxtail millet through RNA-seq analysis of various tissues based on Illumina GA II platform have been generated (Zhang et al. 2012). In sorghum, a large transcriptome database “Morokoshi” comprising high-quality ESTs and TACs generated from cDNA libraries and RNA-seq analysis of various tissues and 23 previous studies have been developed (Makita et al. 2014). The foxtail and sorghum transcriptome data generated in these recent studies would enable identification of functional markers and candidate genes associated with different traits of interest including nutraceutical properties. Therefore, efforts need to be directed towards the development of transcriptome databases in other neglected millets for getting insight into the molecular mechanisms of nutraceutical traits and ultimately help to develop biofortified millet cultivars.
RNAi-induced gene silencing has been well exploited in deciphering the molecular mechanism underlying poor digestibility of sorghum protein and stable transformants producing grains with improved digestibility were developed (Grootboom et al. 2014). This approach could potentially be used in many other millets as well mainly for silencing the genes governing anti-nutritional factors reducing digestibility and bioavailability of various nutritional compounds including starch, protein, and micronutrients. Availability of an efficient in vitro regeneration protocol is a prerequisite for utilizing RNAi-mediated gene silencing. Therefore, immediate attention need to be paid towards developing efficient regeneration and transformation protocols in many of the small millets for harnessing the potential of functional gene validation through RNAi approach.
Proteomics involves large-scale study of the complete set of proteins and detailed analysis of their expression, structure, and function. In millets, proteomics pipelines can play a driving role in deciphering molecular events involved in nutrient trait pathways by characterizing bioactive proteins and peptides to address the question of nutritional bio-efficacy through proteome mapping, comparative proteomics, and protein–protein interactions. Furthermore, integration of experimental biology and computational modeling with proteomics will greatly facilitate characterization of versatile properties of proteins governing nutraceutical properties. So far, proteomics studies for nutritional traits in millets have been carried out only in finger millet (Kumar et al. 2014, 2016a, b) and sorghum (Cremer et al. 2014). Two calcium-binding proteins, calmodulin (Kumar et al. 2014) and calreticulin (Kumar et al. 2016a, b) responsible for higher calcium accumulation during grain filling stage in finger millet were identified through peptide mass fingerprinting. In sorghum, seed proteome analysis of different sorghum genotypes revealed significant genetic variation in concentration of β, γ, and δ kafirin proteins (Cremer et al. 2014). Further proteome investigation revealed that many of these proteins were involved in formation of kafirin–starch complex, which is a major reason for poor digestibility of sorghum protein. Allelic variation for these proteins may be utilized for improving protein digestibility of sorghum cultivars. Comparative proteomics of small millets and coarse cereals could be exploited for extracting the common functional and regulatory proteins involved in nutrition biosynthesis pathways. Although lesser information is available for nutraceutical traits currently, proteomics enriched datasets for the minor millets will be enhanced with the increasing availability of millet reference genome sequences (Hittalmani et al. 2017).
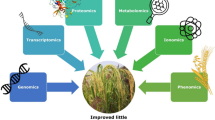
In recent years, metabolomics has emerged as a potential tool for identification and quantification of essential metabolites produced in specific tissues at a particular developmental stage (Kumar et al. 2016a, b). In addition to structural and functional properties of metabolites, metabolomics can also be used to analyze the metabolism pattern of nutrients in the gastrointestinal tract and its effect on growth and health of the organism (Fig. 10.1).

An overview of an integrative genomics and breeding approach for achieving higher genetic gain in millets biofortification program
Almost all the analytical techniques utilized for metabolomics profiling are based on the principle of mass spectroscopy (MS) and nuclear magnetic resonance (Pandey et al. 2016). Metabolomics profiling of finger millet grain through the combination of analytical techniques such as high performance liquid chromatography (HPLC), electrospray ionization mass spectrometry (ESI-MS), and NMR revealed that a wide range of secondary metabolites (polyphenols) are associated with its high nutritive value (Chandrasekara and Shahidi 2010; Banerjee et al. 2012). The integration of this metabolic information with transcriptome datasets and reverse genetics approaches will facilitate the development of biomarkers for effective metabolomics profiling of large breeding populations for deciphering the novel metabolic QTLs in millets.
10.4.3 Bioinformatics and Computational Biology Tools
The increasing availability of big molecular data about the gene pool of crop plants resulting from enhancements in the precision of molecular biology results and in systems biology-based integrated simulation technologies provides us a unique opportunity to analyze and integrate these data through bioinformatics for novel discovery (Kumar et al. 2015a, b; Pathak et al. 2017). Data-driven research has made data integration strategies which are crucial for the advancements and novel insight. Technically, data integration methods have been developed and utilized in both corporate and academic sectors. When it arrives to biological research, there are different types of interpretations and levels of data integration ranging from genomics to proteomics. Data integration and annotation is necessary to ensure the reproducibility of the experimental findings through systems biology and bioinformatics (Lapatas et al. 2015; Kumar et al. 2015a, b).
10.4.3.1 Integration and Visualization of Big Molecular Data
In the post genomic era, big data has really become the most popular term. It refers to the huge quantity of structured, semi-structured, and unstructured data (Rodríguez-Mazahua et al. 2016). These data are generated through high-throughput platforms by using molecular sample of organisms obtained via specialized molecular biology protocol so far it is said to be big molecular data in the molecular biology and biotechnology domain. During the last 10–12 years, technical advances in the field of plant molecular biology and biotechnology have made the opportunity to produce enormous amounts of data, heretofore it was impossible to generate such type of data in minimum time and cost. Advances in DNA sequencing technology have shortened the time for the gathering of the raw sequencing data from years to days, and the costs have also reduced 1000-fold from millions to few thousands of dollars. Besides, a similar reduction was also seen in the amount of required material for generation of such data (Frelinger 2015).
In recent years rapid developments in the field of computational systems biology and bioinformatics have made tremendous contributions to decoding the intricacy of such big data. The application of computational methods in the analysis of big molecular data for detection of important genes/proteins, which are directly linked to human health and nutrition have gained a lot of attention. From that aspect, these computational approaches are very crucial for novel insight (Demir 2015). Therefore, harnessing the potential of such computational approaches for integration and analysis of the gene pool data has significant opportunity to identify and visualize the key genes and alleles that can be further utilized to enhance the performance of major crop species for ensuring food and nutritional security (Henry 2011).
10.4.3.2 Data Mining for Computational Systems Biology and Bioinformatics
In recent years, rapid developments in omics platforms have generated big molecular data about crop plants. Decoding of these data for identification of novel genes/proteins which are essential for biofortification program can be utilized for agri-food nutrition and health requires sophisticated computational analyses (Kumar et al. 2015a, b). Due to availability of huge molecular data in public domain, the development of data mining tools and techniques to solve the complexity of biological problems recently received a lot of attention. Data Mining is the finding of new interesting knowledge from big data. It is also known as Knowledge Discovery in Databases. It requires next-generation technologies and the motivation to investigate the hidden knowledge that resides in the data. It has several tasks to uncover the complexity of big molecular data for extraction of meaningful informations such as classification, estimation, prediction, association rule, clustering, and visualization (Raza 2012). It was suggested that data mining approaches should be applied to overcome the drawbacks of traditional methods. Accordingly, future work should be focussed on the development of integrated databases and user-friendly software for routine use to analyze the natural gene pool data of crop plants (Kumar et al. 2015a, b). This is challenging because gene pools are highly complex and several things are defined poorly. Nonetheless, data mining should facilitate researchers to build earlier and critical decisions in the searching of useful information that can further be utilized in development of the functional food for human health and nutrition (Lee et al. 2008).
Data-driven research has made data integration strategies which are crucial for the advancements and novel insight. Technically, data integration methods have been developed and utilized in both corporate and academic sectors. When it arrives to biological research, there are different types of interpretations and levels of data integration ranging from genomics to proteomics. Data integration and annotation is necessary to ensure the reproducibility of the experimental findings through computational systems biology and bioinformatics (Lapatas et al. 2015; Kumar et al. 2015a, b).
It is mainly based on three approaches called as top-down approach, intermediate approach, and bottom-up approach. In case of top-down, big molecular data of natural gene pool of plants generated through wet-lab experimentation were used to develop integrative models that can be used to predict dynamic behavior of systems at specific scale. This is also known as integrative systems biology. Whereas, bottom-up approach deals with the molecular mechanism and function of cell components via formulating the hypothesis based on perturbation results that can be further utilized in validation through experimental approaches. To determine the intricacy of systems at different levels such as intercommunicating system of systems that cell components, interaction of many molecules in any pathway, and altered states to build profiling data that can be used for filling the gaps between top-down and bottom-up approaches. Intermediate approaches are utilized for the integration of various biomolecules in a controlled way at molecular to systems level that can assess the complexity of biological systems for filling the unknown information (Pathak et al. 2013, 2017; Kumar et al. 2015a, b; Gupta and Misra 2016). Therefore, systems biology and bioinformatics have immense potential to decode the natural gene pool of crops for agri-food nutrition and health.
10.4.3.3 Data Mining Approaches in Gene Discovery
Converting the biological data repository into an informative electronic format is the key function of system biology tools widely used in genomics studies. These large genomic data repositories help in understanding the sequences and expression pattern of the genes regulating complex biological possesses such as biosynthesis and accumulation of nutrients (Zweiger 1999; Kumar et al. 2015a, b). Classification, estimation, prediction, clustering, description, and visualization are the common data mining approaches used in defining big genomic data sets. Based on these available information multiple algorithms are developed for each task. All of these algorithms attempt to fit either a descriptive or predictive model to the data (Dunham 2002). A descriptive model deciphers patterns or relationships in data, whereas a predictive model makes a prediction about data based on already available examples (Sindhu and Sindhu 2017). The key tasks well suited for mining of meaningful new patterns from the big data that information will be further utilized for sustainable agriculture.
10.5 Searching of Genes Responsible for Production of Nutritionally and Health-Related Molecules in Millets with Higher Contents for Biofortification
Various epidemiological reports have suggested a reverse connection between the daily intake of polyphenols-rich diet like millets and threat of human sickness (Scalbert et al. 2005; Arts and Hollman 2005). Millets are a family of cereals, very rich source of nutritionally and health-related molecules like phenolic acids, carotenoids, and various minerals in comparison to wheat and rice (Saleh et al. 2013; Hegde et al. 2005). These small molecules act as therapeutic agents in severe diseases such as diabetes, cancer, and cardiac disorders. These small molecules are synthesized in various pathways as secondary metabolites with numerous metabolic roles in plants. Apart from these poly-phenolic molecules some millet proteins also have the capability to work against chronic human diseases.
Diabetes mellitus is a chronic metabolic disease recognized by hyperglycemia either due to insufficient insulin production or secretion. It was very well known that all millets have some phytochemical which is present in the outer membrane of their seeds have antioxidant properties, and help in lowering glycemic response (Hegde et al. 2005, Table 10.2). Quercetin, a particular polyphenol extract from finger millet seed coats showed effective inhibition against aldolase enzyme (Shobana et al. 2009) and may act as potential alternatives to clinically tested artificially synthesized AR inhibitors (Lee and Kim 2001). Quercetin lowers the hyperglycemic condition by various pathways one is by modulating insulin-dependent pathways such as akt, another one by stimulating GLUT4 translocation which is associated with AMPK activation (Coskun et al. 2005; Stewart et al. 2009; Eid et al. 2015). In another study, proso millet protein concentrate (PMP) upon feeding revamped the glycemic response in severe type-2 diabetic mice by enhancing the plasma levels of adiponectin, HDL cholesterol, and insulin. Some previous studies have shown that intake of calcium and magnesium minerals in daily diet can reduce the effect of type-2 diabetes. Enrichment of finger millet with these calcium, magnesium, iron, and potassium could be directly correlated with its role in reducing the type-2 diabetes risk. Perhaps the presence of some anti-nutritional molecules in millets especially in finger millet like tannins, phenolics, and phytates may also reduce the glucose concentration in cells due to reduction in starch digestibility and absorption. Oxidation stress to cells or ROS generation is an interdependent process which is responsible for regulating several pathological conditions and aging (Chandrasekara and shahidi 2011c). In millet grains, insoluble bound forms of phenolics are present in the majority of free form of phenolics, and main contributor as an antioxidant as well as anti-aging. But apart from the above statement, both free and bound phenolic form present in proso millet were shown to possess very good antioxidation property (Zhang et al. 2014). Use of flavonoids, including quercetin protects cell damage from oxidative stress by reducing the activity of induced nitric oxide synthase (Shoskes 1998).
Millets have varieties of phytochemicals that may protect human population from different types of cancers by suppressing cellular oxidation. Millet’s phenolics, tannins, and phytates are known to possess a variety of anticarcinogenic properties (Graf and Eaton 1990; Chandrasekara and Shahidi 2011a, b). For example, ferulic acid, a major bound phenolic acid in millets is known to have anticarcionogenic effect on tongue, colon, and breast cancer cells (Mori et al. 1999; Kawabata et al. 2000; Choi and Park 2015). Similarly, extracts of edible part of proso millet, rich in ferulic acid and chlorogenic acid inhibited the growth of human cancer cell lines, providing a better supplement as an anticancerous agent (Zhang et al. 2014). Phenolics of finger and pearl millet have shown anticancerous activity in HepG2 hepatic cancer cell lines (Singh et al. 2015). Studies conducted in Africa and China reported lower incidence of oesophageal cancer on consumption of sorghum and millet (van Rensburg 1981; Chen et al. 1993). Not only the phenolics and phytates but also the protein of millets is known to possess anticancerous properties. Recently, novel 35 kDa protein homologus to peroxidize extracted from foxtail millet seeds is reported to inhibit colon cancer.
In recent years, various reports have shown a strong relationship between uptake of polyphenols rich diet with decrease risk of coronary heart diseases (Renaud and de Lorgeril 1992; Nardini et al. 2007). Atherosclerosis, major cause of myocardial infarction or cardiac death is caused by oxidation of LDL followed by deposition in narrow arteries (Aviram et al. 2000; Vita 2005). Polyphenols play anti-coronary diseases by function as anti LDL oxidation, antioxidant, anti-platelet, anti-inflammatory as well as by increasing HDL, and improving endothelial function 6 g/kg of pearl millet bran have showed similar antihyperlipidemic effect to with 0.6 mg/kg Simvastatin, a synthetic cholesterol reducing drug in albino rats through stimulating fecal bile acid secretion and by lowering all lipid profile. In one study, streptozotocin-induced diabetes rats showed lower lipid levels after treatment with Nigerian finger millet seed coat matter, suggested high phenolic content in finger millet seed coat. Biosynthesis of all these secondary metabolites initiate by formation of phenylalanine with the help of intermediate products of cellular metabolism through shikimate pathway which further proceed to phenylpropanoid pathway and flavonoid branch pathway.
Expression of genes involved in biosynthesis pathway regulated by different transcription factors, which influenced the generation of specific secondary metabolite in the particular plant in an environment conditions. Myeloblastosis viral oncogene homolog (MYB) represents a large class of transcription factors which is the main regulator of synthesis of plant phenolics. 1R-MYB, 2R-MYB, and 3R-MYB of foxtail millet have been characterized as phenylpropanoid pathway regulator (Muthamilarasan et al. 2014). Expression level of flavonoid biosynthesis genes encoding enzymes were affected in overexpressed soyabean R2R3-MYB in transgenic Arabidopsis plants. In Arabidopsis thaliana, AtDOF4;2 have shown environmental and tissue-specific regulation on phenylpropanoid metabolism (Skirycz et al. 2007). Searching and identification of genes involved in the biosynthesis, degradation pathways, and in regulation of these metabolites and proteins in millets through various biological approaches will help us to create a plant with desired metabolites as use of nutraceuticals and as a therapeutic.
10.6 Genomic Selection
In recent years, genomic selection (GS) has emerged as a robust tool for enhancing genetic gain of complex traits with low heritability. In GS, potential of a genotype for a trait of interest is estimated based on its genomic estimated breeding value (GEBV). Based on the GEBVs, derived from numerous genome-wide distributed SNPs, training population (TP) and candidate population (CP) are formed for deducing genetics of the trait. GS has been successfully utilized for deducing genetics and enhancing genetic gain for grain micronutrient content (Velu et al. 2016). So far, genomic selection has not been tested in millets. However, with the emerging genome sequence information in many of the minor millets, it will be possible to use GS for enhancing genetic gains for nutritional traits in the biofortification program.
10.7 Genome Editing
In recent years, genome editing technology opened exciting new avenues for trait improvement. Unlike MAS, genome editing inserts desirable alleles or QTLs into a promising genotype to obtain desired phenotypes without following a lengthy backcross cycle. Different genome editing methods modify plant genomes through different sequence-specific nucleases (SSNs) that can be engineered to target genomic sites. The traditional nuclease-based gene editing approaches requiring complex gene constructs are now replaced with more robust and easy to handle CRISPR/Cas9 mediated gene editing. To date, genome editing has not been used in any of the millets. However, advances in establishing regeneration and transformation protocols present an opportunity to address the major production constraints of millets through genome editing technology in near future. One further application of CRISPR/Cas9 that is likely to expand in the future is the molecular stacking of genes governing nutraceutical properties and potential medical compounds in millets.
10.8 Metabolic Pathway Engineering
The metabolic engineering of plants with desirable quality traits involves both conventional plant breeding and biotechnology techniques. Metabolic engineering involves redirecting cellular behavior by modifying the cell’s enzymatic, transport, and regulatory functions. In recent years, metabolic engineering has revealed the molecular mechanism of import pathways and key regulatory genes have been cloned. With the rapid advances in omics, bioinformatics, and system biology tools it is possible to identify and clone the valuable genes governing important nutrient pathways in millets through metabolic engineering.
10.9 Conclusion
The emerging tools for next-generation sequencing favors rapid millet genome sequencing. The various omics platforms have great potential in enhancing the use of millets to cope up with malnutrition by micronutrients and foster millets as model systems for sustainable crop biofortification. Identifying rate-limiting synthesis steps may provide objectives for the genetic manipulation of important nutrient accumulation pathways to enhance the production of nutraceutical compounds. Targeted gene expression and gene knockdown through genome editing may help to minimize or remove anti-nutritional factors. However, millet value enhancement is constrained by poor knowledge of regulatory mechanism of complex metabolic pathways. With the advancements in “omics” and “informatics” millet researchers have the ability to search valuable genes in the gene pool and simultaneously define their role in value addition and biofortification. To accomplish this, interdisciplinary endeavor ranging from plant science to human physiology and clinical science are needed.
References
Anuradha N, Satyavathi CT, Bharadwaj C, Nepolean T, Sankar SM, Singh SP, Srivastava RK (2017) Deciphering genomic regions for high grain iron and zinc content using association mapping in pearl millet. Front Plant Sci 8:412
Arlappa N, Laxmaiah A, Balakrishna N, Brahmam GNV (2010) Consumption pattern of pulses, vegetables and nutrients among rural population in India. Afr J Food Sci 4(10):668–675
Arts ICW, Hollman PCH (2005) Polyphenols and disease risk in epidemiologic studies. Am J Clin Nutr 81:317–325
Austin D (2006) Fox-tail millets (Setaria: Poaceae): abandoned food in two hemispheres. Econ Bot 60:143–158
Aviram M, Dornfeld L, Rosenblat M, Volkova N, Kaplan M, Coleman R, Hayek T, Presser D, Fuhrman B (2000) Pomegranate juice consumption reduces oxidative stress, atherogenic modifications to LDL, and platelet aggregation: studies in humans and in atherosclerotic apolipoprotein E-deficient mice. Am J Clin Nutr 71:1062–1076
Banerjee S, Sanjay KR, Chethan S, Malleshi NG (2012) Finger millet (Eleusine coracana) polyphenols: investigation of their antioxidant capacity and antimicrobial activity. Afr J Food Sci 6:362–374
Brown AHD (1989) Core collections: a practical approach to genetic resources management. Genome 31(2):818–824
Chandrasekara A, Shahidi F (2010) Content of insoluble bound phenolics in millets and their contribution to antioxidant capacity. J Agric Food Chem 58:6706–6714. https://doi.org/10.1021/jf100868b
Chandrasekara A, Shahidi F (2011a) Antiproliferative potential and DNA scission inhibitory activity of phenolics from whole millet grains. J Funct Foods 3:159–170
Chandrasekara A, Shahidi F (2011b) Inhibitory activities of soluble and bound millet seed phenolics on free radicals and reactive oxygen species. J Agric Food Chem 59:428–436
Chandrasekara A, Shahidi F (2011c) Determination of antioxidant activity in free and hydrolysed fractions of millet grains and characterization of their phenolic profiles by HPLC-DAD-ESI-MSn. J Funct Foods 3:144–148
Chen F, Cole P, Mi Z, Zing LY (1993) Corn and wheat-flour consumption and mortality from esophageal cancer in Shanxi, China. Int J Cancer 53:902–906
Choi YE, Park E (2015) Ferulic acid in combination with PARP inhibitor sensitizes breast cancer cells as chemotherapeutic strategy. Biochem Biophys Res Commun 458:520–524
Coskun O, Kanter M, Korkmaz A, Oter S (2005) Quercetin, a flavonoid antioxidant, prevents and protects streptozotocin-induced oxidative stress and beta-cell damage in rat pancreas. Pharmacol Res 51:117–123
Cremer JE, Bean SR, Tilley MM, Ioerger BP, Ohm JB, Kaufman RC, Godwin ID (2014) Grain sorghum proteomics: integrated approach toward characterization of endosperm storage proteins in kafirin allelic variants. J Agric Food Chem 62(40):9819–9831
Demir A (2015) Possible effect of biotechnology on plant gene pools in Turkey. Biotechnol Biotechnol Equip 29(1):1–9
Dunham MH (2002) Data mining: introductory and advanced topics. Prentice Hall, Upper Saddle River, NJ
Eid HM, Nachar A, Thong F, Sweeney G, Haddad PS (2015) The molecular basis of the antidiabetic action of quercetin in cultured skeletal muscle cells and hepatocytes. Pharmacogn Mag 11:74–81
Frelinger JA (2015) Big data, big opportunities, and big challenges. J Invest Dermatol Symp Proc 17(2):33–35
Gilding EK, Frere CH, Cruickshank A, Rada AK, Prentis PJ, Mudge AM, Godwin ID (2013) Allelic variation at a single gene increases food value in a drought-tolerant staple cereal. Nat Commun 4:1483
Graf E, Eaton JW (1990) Antioxidant functions of phytic acid. Free Radic Biol Med 8:61–69
Grootboom AW, Mkhonza NL, Mbambo Z, O’Kennedy MM, Da Silva LS, Taylor J, Mehlo L (2014) Co-suppression of synthesis of major α-kafirin sub-class together with γ-kafirin-1 and γ-kafirin-2 required for substantially improved protein digestibility in transgenic sorghum. Plant Cell Rep 33(3):521–537
Gupta MK, Misra K (2016) A holistic approach for integration of biological systems and usage in drug discovery. Netw Model Anal Health Informatics Bioinformatics 5(1):4
Hegde PS, Rajasekaran NS, Chandra TS (2005) Effects of the antioxidant properties of millet species on oxidative stress and glycemic status in alloxan induced rats. Nutr Res 25:1109–1120
Henry RJ (2011) Next-generation sequencing for understanding and accelerating crop domestication. Brief Funct Genomics 11(1):51–56
Hittalmani S, Mahesh HB, Shirke MD, Biradar H, Uday G, Aruna YR, Mohanrao A (2017) Genome and transcriptome sequence of finger millet (Eleusine coracana (L) Gaertn) provides insights into drought tolerance and nutraceutical properties. BMC Genomics 18(1):465
Jia G, Huang X, Zhi H, Zhao Y, Zhao Q, Li W, Zhu C (2013) A haplotype map of genomic variations and genome-wide association studies of agronomic traits in foxtail millet (Setaria italica). Nat Genet 45(8):957
Joshi DC, Meena RP, Chandora R (2020) Genetic resources: collection, characterization, conservation and documentation. In: Singh M, Sood S (eds) Millets and pseudocereals genetic resources and breeding advancements. Wood Head Publishing, Cambridge, UK, pp 19–28
Kanjilal B, Mazumdar PG, Mukherjee M, Rahman MH (2010) Nutritional status of children in India: household socio-economic condition as the contextual determinant. Int J Equity Health 9(1):19
Kawabata K, Yamamoto T, Hara A, Shimizu M, Yamada Y, Matsunaga K, Tanaka T, Mori H (2000) Modifying effects of ferulic acid on azoxymethane-induced colon carcinogenesis in F344 rats. Cancer Lett 157:15–21
Kumar A, Mirza N, Charan T, Sharma N, Gaur VS (2014) Isolation, characterization and immunolocalization of a seed dominant CaM from finger millet (Eleusine coracana L Gartn) for studying its functional role in differential accumulation of calcium in developing grains. Appl Biochem Biotechnol 172:2955–2973
Kumar A, Gaur VS, Goel A, Gupta AK (2015a) De novo assembly and characterization of developing spikes transcriptome of finger millet (Eleusine coracana): a minor crop having nutraceutical properties. Plant Mol Biol Report 33(4):905–922
Kumar A, Pathak RK, Gupta SM, Gaur VS, Pandey D (2015b) Systems biology for smart crops and agricultural innovation: filling the gaps between genotype and phenotype for complex traits linked with robust agricultural productivity and sustainability. Omics 19(10):581–601
Kumar A, Metwal M, Kaur S, Gupta AK, Puranik S, Singh S, Yadav R (2016a) Nutraceutical value of finger millet [Eleusine coracana (L) Gaertn], and their improvement using omics approaches. Front Plant Sci 7:934
Kumar A, Sharma D, Tiwari A, Jaiswal JP, Singh NK, Sood S (2016b) Genotyping-by-sequencing analysis for determining population structure of finger millet germplasm of diverse origins. Plant Genome 9. https://doi.org/10.3835/plantgenome2015070058
Lapatas V, Stefanidakis M, Jimenez RC, Via A, Schneider MV (2015) Data integration in biological research: an overview. J Biol Res-Thessalon 22(1):9–9
Lata C, Sahu PP, Prasad M (2010) Comparative transcriptome analysis of differentially expressed genes in foxtail millet (Setaria italica L) during dehydration stress. Biochem Biophys Res Commun 393(4):720–727
Lee MS, Kim MK (2001) Rat intestinal α-glucosidase and lens aldose reductase inhibitory activities of grain extracts. Food Sci Biotechnol 10:172–177
Lee JK, Williams PD, Cheon S (2008) Data mining in genomics. Clin Lab Med 28(1):145–166
Makita Y, Shimada S, Kawashima M, Kondou-Kuriyama T, Toyoda T, Matsui M (2014) MOROKOSHI: transcriptome database in Sorghum bicolor. Plant Cell Physiol 56(1):e6
Mirza N, Taj G, Arora S, Kumar A (2014) Transcriptional expression analysis of genes involved in regulation of calcium translocation and storage in finger millet [Eleusine coracana (L) Gartn]. Gene 550:171–179. https://doi.org/10.1016/jgene201408005
Mori H, Kawabata K, Yoshimi N, Tanaka T, Murakami T, Okada T, Murai H (1999) Chemopreventive effects of ferulic acid on oral and rice germ on large bowel carcinogenesis. Anticancer Res 19:3775–3778
Mudge SR, Campbell BC, Mustapha NB, Godwin ID (2016) Genomic approaches for improving grain quality of Sorghum. In: The Sorghum genome. Springer, Cham, pp 189–205
Muthamilarasan M, Prasad M (2015) Advances in Setaria genomics for genetic improvement of cereals and bioenergy grasses. Theor Appl Genet 128:1–14
Muthamilarasan M, Khandelwal R, Yadav CB, Bonthala VS, Khan Y, Prasad M (2014) Identification and molecular characterization of MYB transcription factor superfamily in C-4 model plant foxtail millet (Setaria italica L). PLoS One 9:e109920
Nardini M, Natella F, Scaccini C (2007) Role of dietary polyphenols in platelet aggregation a review of the supplementation studies. Platelets 18:224–224
Nedumaran S, Bantilan MCS, Gupta SK, Irshad A, Davis JS (2014) Potential welfare benefit of millets improvement research at ICRISAT: multi country-economic surplus model approach. Socioeconomics Discussion Paper Series Number 15. ICRISAT, Hyderabad
Pandey MK, Roorkiwal M, Singh VK, Ramalingam A, Kudapa H, Thudi M, Varshney RK (2016) Emerging genomic tools for legume breeding: current status and future prospects. Front Plant Sci 7:455
Patel GS, Dubey M, Chandel G (2015) Characterization of metal homeostasis related rice gene orthologs in nutri-rich minor millets. Int J Plant Anim Environ Sci 5(4):14–23
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Schmutz J (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457(7229):551
Pathak RK, Taj G, Pandey D, Arora S, Kumar A (2013) Modeling of the MAPK machinery activation in response to various abiotic and biotic stresses in plants by a system biology approach. Bioinformation 9(9):443
Pathak RK, Baunthiyal M, Pandey N, Pandey D, Kumar A (2017) Modeling of the jasmonate signaling pathway in Arabidopsis thaliana with respect to pathophysiology of Alternaria blight in Brassica. Sci Rep 7:16790
Puranik S, Jha S, Srivastava PS, Sreenivasulu N, Prasad M (2011) Comparative transcriptome analysis of contrasting foxtail millet cultivars in response to short-term salinity stress. J Plant Physiol 168:280–287
Ravi SB, Hrideek TK, Kumar AK, Prabhakaran TR, Mal B, Padulosi S (2010) Mobilizing neglected and underutilized crops to strengthen food security and alleviate poverty in India. Indian J Plant Genet Resour 23(1):110–116
Raza K (2012) Application of data mining in bioinformatics. arXiv preprint arXiv:12051125
Renaud S, de Lorgeril M (1992) Wine, alcohol, platelets, and the French paradox for coronary heart disease. Lancet 339:1523–1526
Rodríguez-Mazahua L, Rodríguez-Enríquez CA, Sánchez-Cervantes JL, Cervantes J, García-Alcaraz JL, Alor-Hernández G (2016) A general perspective of Big Data: applications, tools, challenges and trends. J Supercomput 72(8):3073–3113
Saleh AS, Zhang Q, Chen J, Shen Q (2013) Millet grains: nutritional quality, processing, and potential health benefits. Compr Rev Food Sci 12(3):281–295
Scalbert A, Manach C, Morand C, Remesy C (2005) Dietary polyphenols and the prevention of diseases. Crit Rev Food Sci Nutr 45:287–306
Shobana S, Sreerama YN, Malleshi NG (2009) Composition and enzyme inhibitory properties of finger millet (Eleusine coracana L) seed coat phenolics: mode of inhibition of a-glucosidase and pancreatic amylase. Food Chem 115:1268–1273
Shoskes DA (1998) Effect of bioflavonoids quercetin and curcumin on ischemic renal injury: a new class of renoprotective agents. Transplantation 66:147–152
Sindhu S, Sindhu D (2017) Data mining and gene expression analysis in bioinformatics. Int J Comput Sci Mob Comput 6(5):72–83
Singh UM, Chandra M, Shankhdhar SC, Kumar A (2014) Transcriptome wide identification and validation of calcium sensor gene family in developing spikes of finger millet genotypes for elucidating their role in grain calcium accumulation. PLoS One. https://doi.org/10.1371/0103963
Singh N, Meenu G, Sekhar A, Abraham J (2015) Evaluation of antimicrobial and anti-cancer properties of finger millet (Eleusine coracana) and pearl millet (Pennisetum glaucum) extracts. Pharma Innov J 3:82–86
Singh AK, Singh R, Subramani R, Kumar R, Wankhede DP (2016) Molecular approaches to understand nutritional potential of coarse cereals. Curr Genomics 17(3):177–192
Skirycz A, Jozefczuk S, Stobiecki M, Muth D, Zanor MI, Witt I, Roeber BM (2007) Transcription factor AtDOF4;2 affects phenylpropanoid metabolism in Arabidopsis thaliana. New Phytol 175:425–438
Stewart LK, Wang Z, Ribnicky D, Soileau JL, Cefalu WT, Gettys TW (2009) Failure of dietary quercetin to alter the temporal progression of insulin resistance among tissues of C57BL/6 J mice during the development of diet-induced obesity. Diabetologia 52:514–523
Sukumaran S, Xiang W, Bean SR, Pedersen JF, Kresovich S, Tuinstra MR, Yu J (2012) Association mapping for grain quality in a diverse sorghum collection. Plant Genome 5(3):126–135
Upadhyaya HD, Ramesh S, Sharma S, Singh SK, Varshney RK, Sarma NDRK, Ravishankar CR, Narasimhudu Y, Reddy VG, Sahrawat KL et al (2011a) Genetic diversity for grain nutrients contents in a core collection of finger millet (Eleusine coracana (L) Gaertn) germplasm. Field Crops Res 121:42–52. https://doi.org/10.1016/jfcr201011017
Upadhyaya HD, Ravishankar CR, Narasimhudu Y, Sarma NDRK, Singh SK, Varshney SK, Nadaf HL (2011b) Identification of trait-specific germplasm and developing a mini core collection for efficient use of foxtail millet genetic resources in crop improvement. Field Crop Res 124(3):459–467
Upadhyaya HD, Sharma S, Dwivedi SL, Singh SK (2014) Sorghum genetic resources: conservation and diversity assessment for enhanced utilization in Sorghum improvement. In: Genetics, genomics and breeding of Sorghum, Genetics, genomics and breeding of crop plants. CRC Press (Taylor & Francis), Boca Raton, FL, pp 28–55
Upadhyaya HD, Wang YH, Sastry DV, Dwivedi SL, Prasad PV, Burrell AM, Klein PE (2015) Association mapping of germinability and seedling vigor in sorghum under controlled low-temperature conditions. Genome 59(2):137–145
Van Rensburg SJ (1981) Epidemiological and dietary evidence for a specific nutritional disposition to esophageal cancer. J Natl Cancer Inst 67:243–241
Varshney RK, Glaszmann JC, Leung H, Ribaut JM (2010) More genomic resources for less-studied crops. Trends Biotechnol 28(9):452–460
Velu G, Crossa J, Singh RP, Hao Y, Dreisigacker S, Perez-Rodriguez P, Tiwari C (2016) Genomic prediction for grain zinc and iron concentrations in spring wheat. Theor Appl Genet 129(8):1595–1605
Vijayakumari J, Mushtari Begum J, Begum S, Gokavi S (2003) Sensory attributes of ethnic foods from finger millet (Eleusine coracana). In: Proc of the National seminar on Processing and utilization of millet for nutrition security: recent trends in millet processing and utilization. CCSHAV, Hisar, pp 7–12
Vinoth A, Ravindhran R (2017) Biofortification in millets: a sustainable approach for nutritional security. Front Plant Sci 8:29
Vita JA (2005) Polyphenols and cardiovascular disease: effects on endothelial and platelet function. Am J Clin Nutr 81:292–297
Wu Y, Li X, Xiang W, Zhu C, Lin Z, Wu Y, Li J et al (2012) Presence of tannins in sorghum grains is conditioned by different natural alleles of Tannin1. Proc Natl Acad Sci U S A 109:10281–10286. https://doi.org/10.1073/pnas1201700109
Yadav OP, Rai KN (2013) Genetic improvement of pearl millet in India. Agric Res 2:275–292
Yadav OP, Upadhyaya HD, Reddy KN, Jukanti AK, Pandey S, Tyagi RK (2017) Genetic resources of pearl millet: status and utilization. Indian J Plant Genet Res 30:31–47
Zhang G, Liu X, Quan Z, Cheng S, Xu X, Pan S, Tao Y (2012) Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat Biotechnol 30(6):549
Zhang L, Liu R, Niu W (2014) Phytochemical and antiproliferative activity of proso millet. PLoS One 9(8):e104058. https://doi.org/10.1371/journalpone0104058
Zweiger G (1999) Knowledge discovery in gene-expression-microarray data: mining the information output of the genome. Trends Biotechnol 17(11):429–436
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Kumar, A., Sharma, D., Pathak, R.K., Tomar, R.S.S., Agrawal, A., Tripathi, M.K. (2021). Science-Led Innovation for Searching and Creating Values in Natural Gene Pool of Millets for Agri-Food Nutrition and Health. In: Kumar, A., Tripathi, M.K., Joshi, D., Kumar, V. (eds) Millets and Millet Technology. Springer, Singapore. https://doi.org/10.1007/978-981-16-0676-2_10
Download citation
DOI: https://doi.org/10.1007/978-981-16-0676-2_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-0675-5
Online ISBN: 978-981-16-0676-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)