
Abstract
Autophagy, which is one of the most important ways to maintain cell homeostasis plays an important regulatory role in cell survival and death. Currently, it is agreed that autophagy promotes or inhibits cell death depending on the internal and external environment and cell type. On the one hand, under normal nutritional conditions autophagy regulates cell survival by energy sensing through the main energy sensing cascade kinases. On the other hand, autophagy regulates the process of cell death. mTOR, Beclin 1, caspases, FLIPs, DAPK, and Tp53 play important regulatory roles in autophagy and apoptosis highlighting the crosstalk between the mechanisms underlying the two death modes. However, energy deficiency caused by PARP1 over-activation and DAPK-PKD pathway activation induces necrosis and autophagy, highlighting the interaction between the two pathways. In addition, autophagy regulates cell death through epigenetic regulation such as histone modification. More investigations on the relationship between autophagy and cell death is ongoing. In the future, there will be more challenges in the study of the relationship between autophagy and cell survival and death. As research increasingly focuses on cell death, the relationship between autophagy and existing and newly discovered cell death types is likely to become more complex. The elucidation of the regulatory role of autophagy in cell survival and death requires more research. Some research results are likely to provide hot topics for further investigations on diseases related to cell death disorders and an experimental basis for the targeted regulation of autophagy for specific treatment of diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Autophagy, which includes macroautophagy, microautophagy, and chaperone-mediated autophagy is an evolutionarily conserved catabolic process used to deliver damaged organelles and misfolded proteins to the lysosome for degradation. Macroautophagy (hereafter referred to as autophagy in this chapter), which is the most prevalent form of autophagy, plays a critical role in cellular homeostasis. Under starvation conditions cells can reuse metabolic precursors through autophagy to survive. Autophagy also promotes cell survival by maintaining the cell energy balance through the degradation of aged organelles and protein aggregates. Although early studies have suggested that autophagy is a mechanism responsible for cell survival, accumulated recent findings have confirmed that autophagy can also regulate the process of cell death including programmed cell death (such as apoptosis, necrosis, etc.), the inflammatory response, and acquired immune processes. Different mediators (such as different proteins in mitochondria, the endoplasmic reticulum, Golgi apparatus, etc.) and different pathways responsible for autophagic degradation regulate the physiological process of cells such as the occurrence and development of diseases. Currently, the causal relationship between autophagy and many regulatory or nonregulatory cell death patterns remains unclear. Autophagy can be associated with necrosis-like cell death induced by caspase inhibition; autophagy and apoptosis can occur simultaneously or inversely depending on the experimental conditions and the signaling pathways shared by both; and autophagy may regulate some other types of cell death such as necrosis. Recent studies have found that autophagy can inhibit the inflammatory response including inflammasome-dependent caspase-1 activation and proinflammatory body cytokine maturation; autophagy still participates in regulating caspase-1-dependent inflammatory cell death (pyroptosis). Due to the intricate relationship between autophagy and cell death, regulating the autophagy process in cell death disorder-related diseases has become a hot research field.
2 Autophagy and Cell Survival
As the “cell guard”, autophagy can enable cells to survive in harsh environments until the environment improves. From yeast to mammals autophagy has evolved from an ancient self-protection function in response to external stimuli. In addition, autophagy is associated with many diseases such as heart diseases, neurodegenerative diseases, and tumors. These diseases are related to changes in the cellular sensitivity to death caused by autophagy disorders. Although great progress has been made in exploring the mechanism and regulation of autophagy pathways, many problems still need to be elucidated. The relationship between the function of autophagy in metabolism and the process of cell life, the effect of autophagy flow on the metabolic status and cell death sensitivity, the metabolic pathway, metabolic and energy sensing, metabolic substrate supply, and metabolic transformation mechanism all require further investigation. In this section, we mainly discuss how autophagy regulates cell survival by sensing the energy environmental conditions and providing metabolic substrates, how autophagy regulates cell death through its effects on apoptotic and necrotic pathways, and how autophagic histone codes regulate cell death. By explaining the regulatory role of autophagy in cell survival and death we can understand the role of autophagy in the maintenance of cell homeostasis and regulation of the threshold of cell death and survival.
2.1 Pro-survival Function of Autophagy Under Stress
Autophagy plays an important role in the survival of cells, tissues, and whole organisms. When mammals were born the lack of nutrients caused by the sudden interruption of the placental nutrition supply activates autophagy to maintain sufficient amino acid metabolic pools for energy metabolism before the nutrition supply is guaranteed by breastfeeding. Another classic example in which autophagy promotes survival is the metabolic process of the liver when mammals have no food. The rapid activation of autophagy occurs in the liver in fasting rats which can be delayed by treatment with large doses of Cycloheximide (also known as actinomycin, a protein synthesis inhibitor). The number of autophagosomes in the starved liver is very small because the clearance rate of autophagosomes is higher than their formation rate. Studies investigating autophagy have observed the recruitment of LC3-labeled autophagosomes in some organs by transgenic techniques. The results indicate that nutritional deficiency could induce systemic autophagy. This finding demonstrates that tissues other than the liver, such as muscle tissue can provide metabolic substrates for the whole body mainly the brain and red blood cells to maintain life during fasting. However, in humans, starvation for more than three days can lead to a decline in autophagy; simultaneously, ketone replaces glucose as the major energy source to avoid the inadequate synthesis of key proteins in cells (see Chap. 16, Sect. 16.2).
Autophagy can also help maintain redox homeostasis. Under oxidative stress, reactive oxygen species (ROS) are the main intracellular signal transducers of autophagy. Nutrition deficiency could induce the production of reactive oxygen species and an imbalance in the thiol redox state which are important mediators of autophagy. ROS and reactive nitrogen species (RNS) irreversibly oxidize DNA and other cell macromolecules resulting in damage to the biological system and cell death. It has been reported that under oxidative stress autophagy is mainly triggered by the p62/Keap1/Nrf2 pathway which eliminates all irreversible oxidized biological molecules (proteins, DNA and lipids), plays an important role in the antioxidant and DNA damage repair system and promotes cell survival (Ichimura et al. 2013).
Misfolded proteins are toxic to the cells and the accumulation of some toxic proteins leads to neurodegenerative disorders. It is generally believed that the toxicity of misfolded proteins is due to the exposure of their hydrophobic surfaces which leads to their interaction with other normal proteins and interferes with key interactions between these proteins. To minimize the toxicity caused by misfolded proteins, the protein quantity control system in cells monitors protein folding in real time and removes misfolded components from the cytoplasm in a timely manner. Molecular chaperones can recognize and conceal the hydrophobic surfaces of misfolded monomers and transport these proteins to the ubiquitin-proteasome system or chaperone-mediated autophagy. To remove soluble misfolded proteins the autophagy-lysosome system is used to degrade these proteasome-resistant toxic proteins. In addition, microtubule-dependent transport systems can isolate soluble polymers or aggregates of these misfolded proteins into inclusion bodies. These systems are regulated by stress-induced transcription factors, cochaperone factors, and other cofactors thereby effectively eliminating misfolded proteins. Therefore, autophagy can be used as a supplement for the proteasome degradation system. Specifically, for example, mice with autophagic disorder exhibit neurodegenerative symptoms in the brain. Recently, the following two proteins have been found to associate polyubiquitination with autophagy: SQSTM1 (also known as sequestosome 1, p62) (Zaffagnini et al. 2018) and NBR1 (a neighbor of BRCA1 gene 1, BRCA1 gene 1 adjacent gene 1). These two proteins can serve as a bridge between ubiquitin which is a degradation marker molecule and LC3 which is a key member of autophagy. SQSTM1 and NBR1 can recognize soluble protein aggregates and guide their degradation through the autophagy-lysosome system. Another type of molecular chaperone-mediated autophagy (CMA) can directly lead cytoplasmic proteins to the lysosome for degradation thereby skipping the step of autophagosome entry. In the CMA pathway, the specific motifs (KFERQ and related sequences) of target proteins are first recognized by the molecular chaperone HSC70 to form complexes with HSP40, Hip, and Hop. Thirty percent of cytoplasmic proteins contain KFERQ homologous sequences suggesting that these proteins are likely to be degraded by CMA. Substrate proteins are transported into the lysosome by molecular chaperone complexes with the help of lysosome-associated membrane protein type 2A (LAMP2A). For example, mutations in the encoding gene of α-synuclein, which is associated with Parkinson’s disease can block its own degradation process; meanwhile, CMA plays an important role in the degradation of α-synuclein. Recent studies have found that CMA can degrade myocyte enhancer factor 2D (MEF2D), which is a transcription factor necessary for the survival of nerve cells. Although the inhibition of CMA can increase the content of MEF2D in the cytoplasm and whole-cell, the content of MEF2D in the nucleus and its DNA binding ability decrease simultaneously. The overexpression of α-synuclein decreases the transcriptional activity of MEF2D, leading to enhanced cell death. Therefore, CMA plays an important role in the quality control and cell survival of MEF2D and associates the degradation of α-synuclein with the activity of MEF2D, thus playing an important role in the pathological process of Parkinson’s disease. Therefore, autophagy plays an important role in the protein quantity control system that protects cells from toxic damage caused by protein misfolding and promotes cell survival (see Chaps. 18 and 20).
2.2 Autophagy-Related Genes and Cell Survival
Organisms or cells that lack key autophagy genes are more likely to die suggesting that autophagy is essential for cell survival. For example, mice lacking BECN1 die during the early stage of embryonic development while mice lacking Atg5 die within one day of birth unless forced breastfeeding is initiated immediately suggesting that autophagy is a key way to overcome hunger. In addition, the deletion of Atg5 or Atg7 in mouse nerve cells can lead to age-related neurodegeneration and shortens the life span of mice. A disruption in the Beclin 1-BCL2 interaction could significantly increase the level of autophagy, inhibit premature aging, and significantly improve the life span in mice (Fernandez et al. 2018). Autophagy genes are essential for maintaining cell homeostasis and cell survival when cells are unable to absorb nutrients such as growth factors from the environment. Autophagy also promotes cell survival through other mechanisms such as by removing damaged organelles and degrading pathogens and large protein aggregates that cannot be degraded through the ubiquitin-proteasome system. Therefore, in aging, infection, neurodegenerative diseases, and tumors autophagy is closely related to the maintenance of cell survival (see Chaps. 22 and 24).
2.3 Autophagy-Mediated Homeostasis for Cell Survival
Autophagy plays an important role in maintaining cell homeostasis. Under a normal nutrient supply autophagy regulates cell survival mainly through energy sensing. Recently, it has been found that the mechanism of the autophagy and metabolism interaction presents a complicated dynamic feedback loop that regulates the energy state of cells to prevent cell death. The process and activity of autophagy can be directly or indirectly affected by the metabolism or energy state inside and outside the cell. Autophagy performs the following three main functions in cell metabolic networks in response to external stimuli: first, autophagy synthetically senses the energy state of cells; second, autophagy generates many metabolic substrates as feedback by adjusting the flow of autophagy; and third, autophagy balances ATP consumption and mitochondrial restoration activity through the autophagy process to ensure cell survival.
The three main energy sensing pathways link the cell energy levels to the autophagy process to guarantee an adequate ATP supply, higher productivity, and cell restoration capacity. Therefore, some downstream amplification effects can regulate the stability of cell metabolism more effectively, which, in turn, could improve cell survival. The main energy sensing cascade kinases in cells include protein kinase A (PKA), AMPK, and mTOR. These three signal transduction pathways are also closely related to the regulation of autophagy.
2.3.1 mTOR Complex
The mTOR pathway can regulate protein synthesis and degradation, and mTOR can sense the intracellular ATP concentration. In yeast, the interaction between Atg1 and Atg13 initiated by the hyperphosphorylation of Atg13 is mediated by rapamycin receptor complex 1 (TorC1) and triggers the formation of autophagosomes. The interaction between Atg1 and Atg13 seems to be very stable because it cannot be altered by nutrient conditions or mTOR inhibition. In mammals, mTORC1 phosphorylates ATG1 homologous protein ULK1 which forms a complex with ATG13 and RB1CC1/FIP200/ATG101 to inhibit the initiation of phagophores. TOR/mTOR is a central regulatory pathway that connects the autophagic activity of cells to the energy condition thus enhancing ATP generation efficiency under energy-deficient conditions. For example, ATG13 could be highly phosphorylated by TOR leading to autophagy activation. TOR activity depends on its high sensitivity to available nitrogen sources, such as L-glutamine or L-asparagine. The sensitivity of mTOR to amino acids is based on the outflow of L-glutamine, the inflow of L-leucine and the flow of other essential amino acids in the bidirectional transport system. L-leucine is one of the most effective amino acids activating mTOR signaling. Therefore, mTOR participates in the regulation of the metabolism efficiency of amino acids by a negative feedback loop. During C. elegans embryogenesis, mTORC1 can regulate the liquid–liquid phase separation (LLPS) of PGL-1/-3 to help embryos adapt to developmental stresses through autophagic degradation (Zhang et al. 2018). However, the more specific mechanisms by which mTOR regulates autophagy have not been fully elucidated (see Chaps. 3 and 16).
2.3.2 AMPK
AMPK is a sensor of the cell energy level and is necessary for the regulation of autophagy because a mutation in AMPK or the inhibition of AMPK could block autophagy. AMPK can be activated by excessive ADP and AMP or the lack of ATP due to glucose starvation. AMPK can regulate autophagy by affecting the following two key signal transduction complexes: inhibition of mTOR or direct phosphorylation of ULK1. Therefore, AMPK acts as a “fail-safe” mechanism by regulating autophagy activity through two different molecular pathways regulating the induction of autophagy and providing ATP-producing metabolites. Furthermore, AMPK plays a key role in remodeling whole-cell metabolism and improving the metabolic ability of autophagy induction (Mihaylova and Shaw 2011). For example, by closing the ATP-dependent metabolic process, AMPK can promote the retention of ATP. This process may be important when oxygen levels are low and the yields of ATP per mole of oxygen need to be increased. Therefore, AMPK improves ATP levels by the following dual actions: first, AMPK improves the ATP levels available to cells by providing metabolites that can be used as ATP synthetic substrates and second, AMPK reduces ATP consumption and minimizes ATP loss by closing the ATP consumption metabolic process (see Chaps. 4 and 16).
2.3.3 PKA
In HeLa cells treated with amino acids and serum starvation PKA could mediate the phosphorylation of DNM1L/DRP1, thus inhibiting cell proliferation, breaking the division/fusion balance, and promoting cell development (Chang and Blackstone 2007). This process results in a larger mitochondrial network containing more elongated mitochondria which leads to higher ATP synthesis efficiency. This higher energy production rate is due to the increased mitochondrial ATP synthesis and higher mitochondrial ridge density and quantity. If a cellular energy state with sufficient residual oxygen and metabolite supply can be maintained the growing energy demand could be met. In this case, ATP is usually retained because of the mitophagy caused by depolarization, and damaged mitochondria are reduced. In addition, the available ATP in the cytoplasm can be more effectively utilized because nonelongated mitochondria consume more ATP in the cytoplasm to maintain their membrane potential leading to the consumption of the ATP storage and accelerating the initiation of cell death.
In addition to the above three forms it has also been mentioned that autophagy could regulate cell survival under pressure by regulating the removal of damaged organelles and misfolded proteins to reduce functional mitochondrial stress and the risk of toxic protein aggregates. In conclusion, these results suggest that changes in the autophagic flow can effectively regulate metabolic efficiency, thus regulating cell survival.
3 Autophagy and Cell Death
It is generally believed that cell death is an irreversible process leading to the termination of cell life. Recent studies have shown that there are reversible cell death procedures under certain conditions but the regulatory mechanism is still unclear. The current consensus regarding cell death is that cell membrane permeability cannot be reversibly restored and that macromolecules in cells are completely fragmented; it is conceivable that under such conditions cells will surely die. However, cell death often occurs in normal tissues, which is necessary for maintaining the normal function and morphology of life. According to the latest recommendations of the Nomenclature Committee on Cell Death (NCCD) regarding the classification of cell death, cell death can be divided into the following two categories: accidental cell death (ACD) and regulated cell death (RCD). ACD mainly refers to the rapid collapse of the cell structure caused by extreme physical, chemical or mechanical stimulation leading to cell death in an uncontrollable way. RCD can be considered a part of normal physiological development or an adaptive response of cells to intracellular and extracellular environmental changes. According to the specific biochemical mechanisms of cell death these death patterns can be subdivided into different subtypes. From a biochemical perspective apoptosis can be defined as a caspase-dependent form of RCD. According to the origin of the death signal apoptosis can be divided into internal induced apoptosis, external induced apoptosis, and anoikis (Fig. 29.1). Necroptosis, which is a key subprogram of death programs is considered a caspase-independent form of cell death. In addition, many different types of RCDs can be identified according to the different substances (short peptides, metal ions, etc.) on which cell death depends. Therefore, the classification of cell death is always in the process of continuous improvement. The factors leading to cell death are intricate. There are also intersections among the mechanisms of different types of cell death. Under stress conditions, different types of cells under different experimental conditions may also have various characteristics of cell death (Galluzzi et al. 2018). It is believed that cell death is a type of specific behavior depending on the cell and environment to some extent. Due to the complexity of cell death classification the most classical classification method was chosen in this chapter and cell death was classified into the following three categories: type I cell death (apoptosis), type II cell death (autophagic cell death), and type III cell death (necrosis) (Table 29.1).

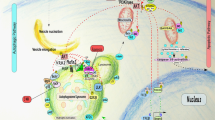
Crosstalk between autophagy and apoptosis. A Intrinsic apoptosis pathway. Many proteins, lipids, and metabolites can induce mitochondrial damage further triggering mitochondrial outer membrane permeabilization (MOMP) which leads to the release of catabolic hydrolases such as AIF (apoptosis-inducing factor) or Endo G (Endonuclease G), and caspase activators (such as cytochrome c and SMAC (second mitochondria-derived activator of caspase)) into the cytosol. These changes usually indicate that the internal trigger of apoptosis is irreversible. Selective autophagy clearance of damaged mitochondria (mitophagy) can limit the release of pro-apoptotic factors thereby delaying the initiation of apoptosis. B Extrinsic apoptosis pathway. The extrinsic apoptotic pathway is triggered by the binding of a trimer ligand to a so-called death receptor. A key event in this pathway is the activation of caspase 8. Autophagy selectively removes active caspase 8 and delays the onset of externally triggered apoptosis after death receptor activation. Since caspase 8-induced apoptosis depends on the cleavage and activation of BID (BH3-interacting domain death agonist), BID is a possible MOMP-inducer and mitophagy may inhibit the lethal signaling of death receptor transduction. C Anoikis. Autophagy can selectively eliminate overactivated SRC induced by disrupted FAK (focal adhesion kinase) signal transduction thereby delaying anoikis due to cell detachment from the extracellular matrix. D, E Role of autophagy proteins in apoptosis. Although autophagosomes and ATG proteins usually constitute cytoprotective mechanisms they may also be involved in lethal signal transduction processes under certain special conditions. D(a) Autophagosomes can serve as a platform for caspase 8 activation although the detailed mechanism of this process has not been elucidated (marked with a question mark in the figure). D(b) In Drosophila melanogaster, the clearance of anti-apoptotic proteins, such as Bruce (BIR-containing ubiquitin-conjugating enzyme; an orthologue of the mammalian inhibitor of apoptosis proteins (IAPs)) by selective autophagy can promote the induction of apoptosis. E(a) Specific pro-apoptotic stimulation signals such as C6 ceramide or tunicamycin can induce an interaction between ATG12 and the anti-apoptotic BCL-2 or MCL1 (myeloid cell leukemia sequence 1) proteins thereby inhibiting their functions to enhance apoptosis. E(b) ATG7 promotes lysosomal membrane permeabilization after lysosomal damage thereby promoting the activation of the apoptotic pathway
In the first section, it was mentioned that autophagy was originally considered a means of maintaining homeostasis and resisting environmental stresses, including nutritional deficiency, energy shortage, endoplasmic reticulum (ER) stress, reactive oxygen species, and hypoxia. Then, the phenotype and biochemical markers of autophagy were frequently found in dead cells and the relationship between autophagy and cell death became a new research hotspot. However, autophagy plays a causal role in a small number of cell deaths. Moreover, many studies claiming that autophagy causes cell death are insufficient. The concept of “autophagic cell death” was proposed in the 1970s as programmed cell death, which was initially described as caspase-dependent necrotizing cell death that is usually accompanied by the accumulation of autophagosomes in cells. This concept sometimes misleads people to believe that autophagy plays a role in the mechanism of cell death. In fact, the function of death mediation is not a part of this definition. Currently, “autophagy-mediated cell death” can be understood according to the following three different meanings: (1) autophagy-associated cell death, which is the original meaning of this concept (2) autophagy-mediated cell death, which suggests that cell death such as apoptosis is triggered by autophagy, and (3) a unique mechanism of cell death independent of apoptosis or necrosis. Of the three explanations the explanatory specificity of (2) is stronger than that of (1), which can be proved by observing whether cell death is blocked when autophagy is inhibited by biological or chemical methods. Explanation (3) is more specific because even if the inhibition of autophagy blocks cell death subsequent evidence is needed to prove that the mechanism of cell death is not apoptosis or necrosis. Therefore, some scholars believe that if the proof requirements of the interpretation of (3) cannot be met, it is better to replace the concept of “autophagy-related cell death” with “autophagy-mediated cell death”. According to the above principles it is recommended that autophagy-dependent cell death be used instead of autophagy-mediated cell death because while autophagy has been shown to be a precondition of cell death, it cannot be proven that autophagy mediates the cells to switch to the death state. The term “phagocyte death” is controversial but still widely accepted; thus, this term is still used in the follow-up of this book and most of the literature.
Since the exact relationship between autophagy and cell death has not been confirmed many studies have focused on clarifying the relationship between autophagy and the cell death process. Recent studies have also confirmed that crosslinks exist between signaling pathways regulating autophagy and cell death processes including apoptosis and necrosis. This section focuses on the relationship between autophagy and cell death and the regulation of the death process.
3.1 Autophagy and Apoptosis
Apoptosis is a cell-regulated mode of death requiring protease and nuclease that plays a synergistic role in the whole plasma membrane. The morphological characteristics of apoptosis include DNA breakage, vesicles in the plasma membrane, cell condensation, and eventually cell decomposition into apoptotic bodies surrounded by membrane, which are eventually removed by cell phagocytosis. The typical biochemical characteristics of apoptotic cells include mitochondrial dysfunction, respiratory chain inhibition, the loss of mitochondrial internal membrane potential, increased mitochondrial membrane permeability, and exposure of phosphatidylserine on the outer side of apoptotic bodies. Apoptosis plays an important role in maintaining tissue homeostasis and development under physiological conditions. In addition, apoptosis may play an important role in the process of some diseases. Many signaling pathways can activate apoptosis. The “internally initiated” (mitochondrial-dependent) apoptotic pathway is the main cause of apoptotic initiation and is usually triggered by harmful extracellular stimuli. This pathway depends on a series of chain reactions regulated by BCL-2 family proteins. The externally activated apoptotic pathways need to be triggered by receptor–ligand interactions and some downstream signaling pathways are shared with the internally initiated apoptotic pathways (Fig. 29.1).
Autophagy and apoptosis are strictly regulated biological processes in cells that play important roles in development, tissue homeostasis, and diseases. Similar stimulus signals could activate these two pathways and the interaction of some signaling molecules in these two pathways indicates that crosstalk exists between these two mechanisms. For example, studies have shown that both apoptosis and autophagy can be activated by metabolic stress. The lack of growth factors, nutrition, and energy metabolism could activate the LKB1-AMPK pathway in mammalian cells, improve the stability of the cyclin-dependent kinase inhibitor p27kip1, and promote cell survival by inducing autophagy. Conversely, silencing p27kip1 under the same conditions activates apoptosis. In addition, autophagy could be used as an adaptive response to ER stress. Notably, a disturbance in the ER calcium balance or function of ER can simultaneously increase autophagy and apoptotic cell death. In different tissues, autophagy plays different roles in resisting ER stress and promoting survival. In colorectal and prostate cancer cells, ER stress-induced autophagy plays an important role in clearing aggregates of ubiquitinated proteins thus protecting cells from death. However, in normal human colon cells and nontransformed mouse embryonic fibroblasts autophagy cannot alleviate endoplasmic reticulum stress but tends to promote endoplasmic reticulum stress-induced apoptosis. Studies have increasingly shown that autophagy and apoptosis could cooccur and antagonize or promote each other resulting in different effects on cell fate. Recent studies have also found that many pathways mediating autophagy can interact with apoptotic complexes laying the foundation for a profound understanding of the regulatory networks of the two pathways.
3.1.1 Crosstalk Between Autophagy and Apoptosis
(1) Autophagy inhibits apoptosis
Studies using different experimental systems suggest that the role of autophagy in cell death depends on the environment and cell type. Autophagy can delay the initiation of apoptotic processes including starvation, DNA damage, and hemodynamic stress-induced apoptosis. Fasting for one day leads to autophagy in the liver of rats but if starvation lasts for several days liver cells undergo apoptosis. The lack of growth factor IL-3 in hematopoietic cell lines first activates autophagy and eventually leads to apoptosis. This finding indicates that ATG genes can promote survival in cells via a complete apoptotic mechanism. SQSTM1-labeled protein aggregates have protective effects on cell death because SQSTM1 mutants lacking the domain of ubiquitin ligase activity inhibit the formation of SQSTM1-positive aggregates and promote cell death.
However, inhibition at an early stage of autophagy enhances the sensitivity of cells to stress-induced apoptosis. In rectal cancer cells, the inhibition of autophagy by 3-MA due to p38 inhibition induces autophagic cell death and apoptosis. These findings suggest that autophagy plays a protective role in promoting survival against harmful stimuli allowing cells to die only after sustained stimulation. In fact, the removal of death signal stimuli can reduce the number and volume of autophagic vesicles in colon cancer cells and the cells could re-enter the cell cycle. Studies using genetic techniques have also shown that autophagic proteins are directly involved in the balance between cell survival and cell death. The inhibition of autophagy induced by nutritional deficiency via the silencing of autophagy genes in HeLa cells leads to apoptosis which can be delayed by apoptotic inhibitors. When autophagy is inhibited during autophagy nucleation fasting cells accelerate cell death through classical apoptotic processes. Similarly, HeLa cells lacking the ubiquitin-like genes required for autophagosome formation are more sensitive to starvation-induced apoptosis. In vitro experiments have confirmed that ATG gene knockdown induces cell death. AMBRA1-deficient embryos exhibit neurodevelopmental deficits associated with autophagy disorders and excessive apoptosis confirming the regulatory role of autophagy-related genes in cell death. In contrast, cell death with apoptotic and autophagic characteristics occurs if autophagy is blocked at the late fusion stage by drugs or LAMP2 gene silencing. Therefore, the observed accumulation of autophagic vesicles does not necessarily imply an increase in autophagic activity because autophagy is inhibited under these conditions. When autophagy is induced through different pathways and the autophagy process is inhibited during the early or late stages, apoptosis occurs due to the inability to adapt to stress. However, in malignant glioma cells 3-MA can inhibit apoptosis induced by alkylating reagents, while Bafilomycin A1 enhances the sensitivity of cells to apoptosis induced by alkylating reagents indicating that the inhibition of autophagy at different stages can lead to different effects on apoptosis.
Autophagy reportedly inhibits cell apoptosis mainly through the mechanism of mitophagy. Mitochondria constitute the “battlefield” on which survival and death signals struggle to determine whether endogenous apoptotic pathways marked by mitochondrial outer membrane permeability (MOMP) are activated. In fact, hundreds of different factors including proteins, lipids, and metabolites affect the function and physiological integrity of the mitochondrial membrane. During cell death, MOMP causes the release of hydrolytic enzymes (such as apoptosis-inducing factor (AIF) and endonuclease G) in catabolism and caspase activating factors (such as cytochrome c and secondary mitochondria-derived activator of caspase, SMAC, also known as DIABLO) from mitochondria leading to the disappearance of mitochondrial transmembrane potential (ΔΨm) and finally, biological energy could be severely lost. These changes are irreversible in endogenous apoptotic pathways (Fig. 29.1A). Damaged mitochondria are particularly prone to activating apoptotic processes; thus, removing substances released due to mitochondrial damage via autophagy by increasing the threshold of apoptotic induction.
How does autophagy specifically degrade damaged mitochondria? A decrease in ΔΨm usually indicates the loss of respiratory chain function or an increase in endometrial permeability resulting in the ubiquitination of the Lys63 of the protein anchored on the outer membrane. These outer membrane proteins include voltage-dependent anion-selective channel 1 (VDAC1), mitochondrial fusion protein 1 (mitofusin 1, MFN1), and MFN2 which connect organelles required by mitophagy. This pathway is feasible because the disappearance of ΔΨm shuts down a protein enzyme that destroys PTEN-induced putative kinase 1 (PINK1) leading to PINK1 aggregation on the surface of damaged mitochondria. Then, PINK1 can recruit and phosphorylate PARKIN (Parkinson’s disease protein) which is an E3 ubiquitin ligase that ubiquitizes the substrate of the mitochondrial outer membrane, thus marking out organelles that need to be degraded by autophagy. In addition to the disappearance of ΔΨm, mitochondrial fragmentation is necessary for mitophagy which constitutes a fail-safe mechanism to prevent the unnecessary degradation of functional mitochondria. Mitochondria in starving cells undergo a loss of ΔΨm but do not break up suggesting that they do not undergo mitophagy. In contrast, the open mitochondria in permeability transition pores (PTPs), which are pores composed of many proteins and can permeate the inner membrane of mitochondria leading to a decrease in ΔΨm and the fragmentation of mitochondria; then, these mitochondria could be cleared by autophagy.
Autophagy also plays a role in inhibiting unnecessary cell death in the pancreas. Pancreatic acinar cells contain many inactive enzyme precursors (zymogens) such as trypsinogen (progenitors of trypsin) which exist as isolated particles but may also leak into the cytoplasm and cause cell death. In a cerulein-induced pancreatitis model, the overexpression of vacuole membrane protein 1 (VMP1) by a transgene could promote the clearance of microparticles containing abnormally activated trypsin. The overexpression of VMP1 can alleviate some enzymatic, histopathological and naked eye indicators of pancreatitis, and blocking the autophagic flow by reducing the expression of VMP1 can aggravate pancreatitis induced by ethanol and bacterial lipopolysaccharide. Therefore, VMP1 can inhibit cell death by promoting the removal of microsomes through autophagy.
Autophagy can also alleviate apoptotic cell death by selectively reducing the abundance of pro-apoptotic proteins in the cytoplasm (Fig. 29.1B). In fact, autophagy can specifically select ubiquitinated proteins as target proteins. Ubiquitination modification enables these proteins to interact with autophagic receptors that belong to a series of adaptor proteins including SQSTM1 which can bind both ubiquitinated substrates and LC3. For example, colorectal cancer cells lacking BAX can resist cell death induced by TNF-related apoptosis-inducing ligand (TRAIL) but this resistance disappears when autophagy is suppressed because autophagy can mediate the selective scavenging of active caspase 8. Similarly, the inhibition of autophagy by a specific Atg7 knockout can enhance the activity of caspase 8 in a TNF-induced mouse hepatocyte apoptosis model which may be due to the inability to effectively eliminate caspase 8 by autophagy. In addition, autophagy can specifically degrade the proto-oncogene tyrosine kinase SRC thus avoiding anoikis induced by blocking the signal transduction of focal adhesion kinase (FAK) (Fig. 29.1C). Under these conditions SRC binds the E3 ubiquitin protein ligase CBL, which contains an LC3 binding region that can be used as a molecular junction to localize SRC on LC3-positive autophagosomes and thus be degraded. In addition, autophagy can inhibit the production of reactive oxygen species (ROS) and cell death induced by SQSTM1 by degrading SQSTM1.
(2) Autophagy promotes apoptosis
Some experimental results suggest that the inhibition of autophagy can block the apoptotic process; thus, autophagy can induce apoptosis. When 3-MA was used to inhibit autophagy in the breast, prostate, colorectal cancer cells, and glioma cells apoptosis could not occur in these cells. Resveratrol-induced autophagy can be switched into the apoptotic process which can be blocked by the inhibition of autophagy. Recent studies have confirmed that autophagy plays a role in upstreaming of apoptosis in CD4+ T lymphocytes that are not infected by HIV because apoptosis can be inhibited by the gene silencing of BECN1 and ATG7 indicating that autophagy can induce apoptosis under certain conditions. In Drosophila tissues, the overexpression of Atg1 induces autophagy and inhibits Tor signal transduction and cell growth while high levels of autophagy lead to apoptosis.
There are many examples of apoptotic activation via the induction of autophagic cell death. Proteins that play key roles in autophagy may also play roles in apoptotic signal transduction. What is the mechanism by which autophagy promotes the induction of apoptosis? Autophagosome formation rather than degradation is thought to be associated with the activation of caspase 8 after SKI-I (a pan-sphingosine kinase inhibitor) and bortezomib (also known as Velcade, a proteasome signaling pathway inhibitor) treatment (Fig. 29.1D(a)). Under these conditions caspase 8 can form a complex with FADD (FAS-associated death domain) and ATG5, colocalize with ATG5, LC3, and SQSTM1 finally become activated in an ATG5, FADD and, SQSTM1dependent way. In SKI-I- or bortexamib-treated cells, inhibition at the early stage of autophagy (through ATG3 or ATG5 knockout) decreases the activation of caspase 8 and caspase 3 effector factors while inhibition at the late stage of autophagy (through bafilomycin A1 treatment) enhances caspase-dependent cell death. These data suggest that the autophagosome formation process rather than the whole autophagy process may provide favorable conditions for the activation of caspase 8. However, caspase 8 is usually not activated when autophagy is induced and the activation of caspase 8 can be confirmed by a TNF test in vivo in mouse models following the liver-specific knockout of Atg7, which functions in the early stage of autophagy. The factors determining caspase 8 activations upon autophagosome formation are unclear.
Autophagy may also promote apoptosis by removing endogenous inhibitors of apoptosis. In Drosophila melanogaster, autophagy is mainly regulated by the interaction between caspases and inhibitors of apoptotic proteins (IAPs). One IAP called Bruce (BIR containing ubiquitin-conjugating enzyme, ubiquitin-conjugating enzyme containing BIR) can be degraded by autophagy which explains why inhibiting autophagy genetically (by targeting mutations in Atg1, Atg13 or vps34) can prevent oocyte developmental apoptosis at later stages of oogenesis (Fig. 29.1D(b)). Recently, autophagy has been reported to promote palmitic acid-induced apoptosis by degrading Caveolin-1, which is a key protein that promotes astrocyte survival (Chen et al. 2018).
To date, many studies investigating the genetic manipulation of autophagy have indicated that autophagy is closely related to the promotion of apoptosis in selective toxicity models. Recent studies have found that exposure to cigarette smoke extract (CSE) can activate the exogenous apoptotic pathway and cause death in human lung epithelial cells. CSE-induced cell death involves the activation of a Fas-dependent death-inducing signaling complex (DISC) and downstream activation of caspases (8, 9, 3). When exposed to CSE, autophagy formation and transformation from LC3B-I to LC3B-II occur simultaneously in human lung epithelial cells. The silencing of the autophagic protein Beclin 1 or LC3 can inhibit apoptosis induced by CSE exposure in vitro indicating that the increase in the autophagy level is closely related to epithelial cell death. Subsequent studies have found that LC3B may function as an exogenous apoptotic regulator in this model. LC3B can form a complex with Fas which is the key component of DISC in the form of lipid raft protein caveolin-1-dependent. Exposure to CSE can lead to a rapid dissociation between LC3B and Fas which is related to the activation of apoptotic signal transduction. In conclusion, these results from RNA interference silencing experiments suggest that LC3B can promote apoptosis in a CSE-induced toxicity specific model, but the function of autophagic activity in promoting cell death in this model is still unclear. Notably, CSE-induced autophagy may differ from starvation-induced autophagy because the former requires the formation of complexes by members of different pathways which may alter the function of autophagy. Therefore, the concept of “toxic autophagy” may include this altered function depending not only on whether the degree of the CSE-induced activation of autophagy is at the physiological level or excessive but also on the nature of the exogenous substrates (such as intricate symbiotic organisms in vivo including tar or virus fragments) and their interaction with autophagosomes.
There are also many examples of simultaneous autophagy and apoptosis; for example, Tp53-dependent autophagy requires the upregulation of damage-regulated autophagy modulator (DRAM) which is consistent with the upregulation of apoptosis. TNFα can induce autophagy in embryonic trophoblast cells which leads to the activation of endogenous apoptotic pathways. In this model, the silencing of Atg5 inhibits TNFα-dependent apoptotic caspase activation. Moreover, Atg5 knockout protects mouse embryonic fibroblasts from death-promoting stimuli. However, the authors attributed this effect to a chaperone-dependent autophagic compensatory activation rather than autophagy inhibition. These studies also propose the following important conclusion, i.e., under any conditions, genetically silencing certain autophagy-related factors cannot explain whether autophagy has a cytoprotective function because the silencing of any gene expression may affect or trigger other signal transduction pathways unrelated to autophagy or some compensation mechanisms such as other forms of autophagy.
It has also been found that many ATG proteins do not depend on autophagy to participate in lethal signaling. ATG12 is thought to play a role in activating caspases through the mitochondrial pathway because its absence can reduce caspase activation induced by treatment with C6 ceramide, epipodophyllotoxin glucopyranoside, Taxol, and chlamydiamycin (Fig. 29.1E(a)). This ATG12 activity requires the BH3 domain rather than its autophagic activity to mediate its interaction with BCL-2 and MCL1. Similarly, in mouse hepatoma Hepa-1c1c7 cells, Atg7 (not Atg5) promotes autophagy induction after lysosomal injury induced by lysosomal membrane permeation which is similar to MOMP and both can induce apoptosis (Fig. 29.1E(b)). These examples illustrate the mechanism by which a single ATG protein promotes lethal signal transduction.
However, whether autophagy can be used as a mechanism of death when cells have complete apoptotic mechanisms remains unclear. The recovery of beclin-1 expression in breast cancer cells with a single allele knockout of BECN1 results in enhanced autophagy. In human cervical cancer cells treated with IFNα, ATG5 can promote autophagic cell death by interacting with FADD. Correspondingly, cells may die from apoptosis unless they are stimulated by other sufficiently strong death mechanisms. Autophagy is more likely to serve as a survival-promoting mechanism than a death-promoting mechanism. At least in cells with complete apoptotic mechanisms the characteristics of autophagy can be used as a resort to maintain survival during the process of death.
3.1.2 mTOR
Mammalian target of rapamycin (mTOR) can sense and balance signals from nutrition, growth factors, energy, and stress thus plays an important regulatory role in early development, growth, and adult aging. Low insulin and insulin-like growth factor-1 (IGF1) signaling, nutrient or energy deficiencies, and stress signals can be converted into the downstream regulation of TOR activity. The inhibition of TOR activity can lead to a decrease in the rate of gene translation and thus activate autophagy. Under starvation, TOR is rapidly suppressed to activate autophagy. Interestingly, recent studies have found that mTOR signaling is suppressed at the beginning of autophagy but reactivated when starvation persists. As a result, reactivated mTOR can alleviate the autophagic process by producing prelysosomal tubules and prelysosomal vesicles (also known as autophagic lysosome reformation, ALR) from the autophagic lysosome, which can develop into functional lysosomes thus restoring lysosome balance (Yu et al. 2010). This negative feedback regulation ensures the termination of autophagy under conditions of sufficient nutrients thus avoiding cell death caused by the excessive accumulation of vesicles in the cytoplasm.
mTOR reportedly performs distinct functions in apoptosis depending on the cell environment and different downstream targets such as Tp53, BAD, and BCL-2 proteins. Recently, the following two new mTOR-acting proteins have been identified: the proline-rich AKT substrates (PRAS40) and Q6MZQ0/FLJ14213/CAE45978 proteins; these proteins have been suggested to play a role in apoptotic regulation thereby controlling the balance between cell growth and death. Recent studies have found that MCL1 which is an anti-apoptotic protein BCL-2 homolog can act as a stress sensor to coordinate the control of autophagy and apoptosis. The final result is determined by the interaction between BAX and Beclin 1 activation downstream of MCL1 degradation. Consistent with the possibility of the simultaneous regulation of autophagy and apoptosis by TOR, the inhibition of mTOR leads to inadequate nutritional supply causing the degradation of MCL1. While it is widely accepted that TOR can regulate various anabolic and catabolic processes in a positive or negative way further studies are required to understand the regulation mechanism of TOR in autophagy and apoptosis.
3.1.3 Beclin 1
BECN1 is a mammalian ortholog of yeast Atg6. Its expression products form core complexes with VPS34 and VPS15 which can induce autophagy and play a key role in autophagosome formation (Fig. 29.2). Beclin 1 is widely expressed in many tissues in humans and mice. Similarly, Bec-1 is a homologous gene of mammalian BECN1 which is expressed in all remodeling tissues during dauer larval formation (a stage of developmental arrest). Bec-1 is critical during embryonic development and is necessary for normal larval development and the whole adult growth process. Moreover, the loss of BECN1 leads to the death of mouse embryos, and Becn 1+/−mice are more likely to suffer from tumors. These phenomena are closely related to autophagy deficiency but there is no defect in apoptotic cell death. It has been confirmed that the interaction between autophagy and apoptosis is partially mediated by the functional and structural interaction between Beclin 1 and BCL-2/BCL-XL. The Beclin 1/BCL-2 interaction is very evolutionarily conservative. In C. elegans, Bec-1 forms two complexes with the Bcl-2 homologs Ced-9 and Let-512/Vps34 which have different functions. The absence of the lipid product PtdIns 3-phosphate of LET-512/VPS34 in Bec-1-deficient larvae confirms that Bec-1 is essential for let-512/Vps34 which plays a key role in autophagy membrane trafficking, and endocytosis. In addition, the inactivation of Bec-1 induces apoptosis, which can be confirmed by the increase in the number of apoptotic cells in animal germ cells and body tissues. In conclusion, these studies confirm that Bec-1 plays a key regulatory role in both autophagy and apoptosis. Recently, the structural basis of the Beclin 1/BCL-2/BCL-XL interaction has been elucidated (Pattingre et al. 2005). Beclin 1 contains a BCL-2 homology (BH) 3 region which is responsible for the interaction with the BCL-2/BCL-XL protein. The BH3 domain interacts with the BH3 receptor to inhibit anti-apoptotic proteins, such as BCL-2 and BCL-XL, or activate members of the apoptotic BCL-2 family such as BAX and BAK. Interestingly, mutations in the BH3 domain of Beclin 1 or the BH3 receptor domain of BCL-XL block the interaction between Beclin 1 and BCL-XL thus eliminating the inhibition of BCL-XL-mediated autophagy. Notably, BCL-2 is specifically located in the endoplasmic reticulum rather than mitochondria which can effectively inhibit starvation-induced autophagy in yeast, mammalian cells and the heart muscle in transgenic mice coexpressing cardiac BCL-2 and the fluorescent autophagy marker GFP-LC3. The depletion or deletion of BH3-only protein BAD by siRNA has been shown to reduce starvation-induced autophagy, while the overexpression of BAD or the addition of BH3 analogues induce autophagy in human cells. Similarly, a defect of the BH3-only protein Egl-1 in C. elegans alleviates starvation-induced autophagy while a gain-of-function mutation in Egl-1 can activate autophagy. In conclusion, these studies suggest that BH3 domain-containing proteins or BH3 analogues can be used not only as inducers of cell death but also as regulators of autophagy. Interestingly, although the binding of BCL-2 to Beclin 1 weakens the ability of Beclin 1 to activate autophagy and Beclin 1 contains only one BH3 motif similar to apoptotic proteins, Beclin 1 cannot regulate the anti-apoptotic ability of BCL-2 to induce apoptosis. The interaction between Beclin 1 and Bcl-2 could not regulate apoptosis in autophagy-deficient mouse embryonic fibroblasts (MEFs), arguing against the protective effect of Beclin 1-mediated autophagy in cells.

Mechanism of the complex interplay between apoptosis and autophagy. Apoptosis can be stimulated by external receptor-dependent signaling or internal mitochondria-mediated signal transduction. The extrinsic pathway is initiated by the binding of a death receptor to its cognate ligands such as FASL, TRAIL or TNF. This results in the binding of FADD (FAS-associated death domain protein) to the death receptor and subsequent activation of caspase 8. Activated caspase 8 can directly cleave and activate caspase 7 and caspase 3 to promote apoptosis. The intrinsic pathway is regulated by a BH3 domain-containing protein that is activated by different types of cellular stress, such as DNA damage or endoplasmic reticulum stress which then activates BAX/BAK located in the mitochondrial outer membrane (MOM) and induces mitochondrial outer membrane permeabilization (MOMP). MOMP leads to the release of different apoptosis mediated factors such as cytochrome c, which activate caspase 9. In turn, caspase 9 cleaves and activates caspase 3 and caspase 7 which induce apoptotic cell death. Both pathways converge on the point of caspase 3 activations. Autophagosome formation requires beclin 1 with members of the polyprotein (PI3K) complex such as VPS34, VPS15, etc. The crosstalk between the autophagy and apoptotic pathways is at least partially mediated by the structural and functional interactions between beclin 1 and the anti-apoptotic proteins BCL-2 and BCL-XL. Diverse apoptotic stimuli (internal or external) can result in the caspase-mediated cleavage of beclin 1. Ultimately, beclin 1 loses its ability to induce autophagy. Instead, the C-terminal fragment translocates to the mitochondria, rendering the cell sensitive to apoptotic signals. In contrast to the phenomenon in which beclin 1 and ATG5 lose their ability to induce autophagy after cleavage, caspase cleavage of ATG4D can enhance its ability to promote autophagy. Tp53 plays a key role in both the apoptosis and autophagy pathways. At the transcriptional level Tp53 upregulates BAX, PUMA, and BID or reduces the expression of BCL-2, which inhibits BAX. In addition to apoptosis Tp53 is capable of inducing autophagy through the inhibition of TOR and the transcriptional activation of DRAM. Interestingly, the inhibition of autophagy by cytosol Tp53 suggests that an extremely complex relationship exists between Tp53 and the autophagy/survival pathways. DAPK belongs to a class of Ca2+/CaM-regulated Ser/Thr kinases and is closely related to apoptosis and autophagic cell death. See text for details
3.1.4 Caspases
Recent studies have provided new clues further elucidating the molecular mechanism of the crosstalk between autophagy and apoptosis. If the growth factor interleukin 3 (IL3) is depleted from mouse hematopoietic cell lines autophagy is induced as a mechanism of cell survival. If the deprivation is sustained, apoptotic cell death occurs. Apoptosis caused by growth factor deficiency is closely related to caspase-mediated cleavage in Beclin 1 and PI3K. This deficiency impairs the function of Beclin 1 in autophagy. In addition, the degradation of Beclin 1 and PI3K does not depend on the cell type, and the initiation of apoptosis is either endogenous (through the release of mitochondrial death-promoting factors) or exogenous (death receptor-dependent). Importantly, in IL3-dependent mouse pre B cell Ba/F3, the C-terminal fragment of Beclin 1 produced by caspase-mediated degradation is located in mitochondria rendering cells sensitive to apoptosis likely due to the release of pro-apoptotic factors (Fig. 29.2). The apoptotic protein BAX can inhibit autophagy by enhancing the caspase-mediated cleavage of Beclin 1 at D149; Beclin 1 and BCL-XL which cannot be degraded by caspase and can rescue BAX-induced autophagy (Luo and Rubinsztein 2010). These two phenomena indicate that apoptosis can inhibit autophagy. Further, evidence supporting the association between autophagy and apoptosis is that other autophagic proteins can also be caspase-induced apoptotic degradation substrates. Caspase 3 cleaves human ATG4D producing serine protease which, in turn, degrades the C-terminal of newly synthesized ATG8 (called LC3 in mammals) (Fig. 29.2). Atg4D cleavage by caspase results in increased priming and delipidation activities against yeast Atg8 homologous protein γ–aminobutyric acid receptor-associated protein-like 1 (GABARAP-L1), while silencing the expression of Atg4D inhibits autophagy and renders cells sensitive to starvation and staurosporine-induced cell death. This phenomenon confirms that caspase can activate autophagy-mediated by Atg4D to promote the survival of starved cells. Interestingly, the overexpression of caspase cleaved ATG4D induces apoptosis in human cells which requires the rapid recruitment of ATG4D to mitochondria. ATG5 is essential for the formation of autophagosomes also enhances cell sensitivity to apoptotic stimuli upon cleavage by caspase. Truncated ATG5 is transferred to mitochondria to regulate the mitochondrial apoptotic pathway. Notably, the caspase-mediated cleavage of ATG5 and Beclin 1 can switch autophagy to apoptosis and the truncated products produced by ATG4D cleavage can enhance autophagy activity (Fig. 29.2). These phenomena strongly suggest that autophagy is closely related to apoptosis.
3.1.5 FLIPs
Based on the discovery of the crosstalk between the autophagic and apoptotic mechanisms, recent studies have found that the anti-apoptotic protein Flice inhibitory protein (FLIP) can function as a new negative regulator of autophagy. Inhibitors of apoptosis such as the cellular and viral orthologs of the FADD-like interleukin-1 beta-converting enzyme (FLICE)-like inhibitory proteins (c-FLIP and v-FLIP, respectively), are triggered by known death receptors in the TNF/NGF (tumor necrosis/nerve growth factor) family. Strong evidence suggests that FLIPs compete with LC3 to bind ATG3 which is an E2-like enzyme in the normal environment. As a result, FLIPs inhibited ATG3-mediated autophagosome extension, thus decreasing the autophagy levels. However, under stress conditions FLIPs allow the ATG3-LC3 interaction to induce autophagy (Lee et al. 2009). In conclusion, FLIPs can not only act as an anti-apoptotic factor but also function as an autophagy inhibitor due to inhibitory binding to ATG3.
3.1.6 DAPK
DAPK is a serine/threonine kinase regulated by Ca2+/CaM (calmodulin) that can regulate cell death induced by various death signals. DAPK inhibits tumors and its expression is inhibited by DNA methylation in most tumors. DAPK is thought to be associated with apoptosis and autophagic cell death. DAPK is activated under endoplasmic reticulum stress, which, in turn, induces apoptotic and autophagic cell death (Gozuacik et al. 2008). In addition, Dapk knockout can protect against ER stress in vitro in isolated fibroblasts and in vivo in a mouse kidney toxicity model. Under these two experimental conditions, both apoptosis and autophagy were inhibited confirming the view that DAPK could integrate signals from both apoptosis and autophagy pathways to induce cell death under ER stress. The regulatory function of DAPK on autophagy has been confirmed in mammalian cells and C. elegans whereby reducing the expression of the DAPK homologous gene Dapk-1 in the worms by mutation or RNAi silencing reduced the occurrence of starvation-induced autophagy in the pharyngeal muscle cells. However, the mechanism through which DAPK promotes autophagy remains unclear. A recent study investigating this issue found that Beclin 1 can be used as a target for the downstream regulation of DAPK (Fig. 29.2). DAPK mediates Thr119 phosphorylation on the BH3 domain of Beclin 1 and promotes the dissociation of Beclin 1 from its inhibitor BCL-2 family members thereby activating Beclin 1-induced autophagy.
3.1.7 Tp53
As mentioned in the previous chapters, Tp53 can be activated by a wide range of stress signals in cells such as DNA damage, hypoxia or abnormal oncogene expression induced cell cycle checkpoints, DNA repair, cell death, and apoptosis. The regulation of apoptosis by Tp53 through endogenous and exogenous pathways has been well studied. Tp53 functions to integrate pressure signals and induce apoptosis by activating the transcription of BAX, PUMA, and BID which are members of the BCL-2 family with multiple domains or reducing the expression of BCL-2 which inhibits BAX (Fig. 29.2). Notably, Tp53-induced apoptosis usually depends on the environment. For example, Tp53 can induce apoptosis in fibroblasts transformed by an oncogene under BAX inactivation but Tp53 has no obvious ability to mediate apoptosis in normal thymocytes. In addition to controlling the transcription of apoptotic members in the BCL-2 protein family, Tp53 can transactivate the core members of the apoptotic mechanism such as the gene encoding caspase 9 costimulator APAF1. In addition, Tp53 can regulate caspase 6 effector expression through transcription regulation. Recently, it has been reported that Atg7 can interact with Tp53 in Cnot3-depleted mouse cardiomyocytes and regulate the activity of Tp53 to adjust the expression of cell death-promoting genes (Yamaguchi et al. 2018). Although Tp53 can also activate gene transcription which plays a role in exogenous apoptotic pathways, it mainly functions in apoptosis through endogenous mechanisms. Surprisingly, Tp53 has the function of not only transcriptional activation but also transcriptional inhibition which may be involved in the regulation of apoptosis. Although most studies have focused on the apoptotic regulatory activity of Tp53 many recent reports have also highlighted the functional relationship between Tp53 and autophagy. Tp53 can inhibit mTOR by activating AMP-dependent protein kinase (AMPK) or trigger autophagy by activating DRAM which is a gene encoding a lysosomal protein with an autophagy induction function (Fig. 29.2). Under genomic instability conditions, Tp53 induces autophagy through DRAM, which will eventually lead to apoptotic cell death. Therefore, DRAM seems to be a key component in the apoptotic and autophagic pathway network regulated by Tp53. Interestingly, new studies have found that Tp53 has a novel function in the cytoplasm in addition to inducing apoptosis and inhibiting autophagy. The knockout or inhibition of Tp53 expression by drugs or interference can induce autophagy in human, mouse, and nematode cells. The enhancement of autophagy can promote survival in Tp53 inactivated cancer cells under hypoxia or nutrition deficient conditions indicating that the inhibition of autophagy induced by Tp53 is a self-protection mechanism in cells. Notably, Tp53 in the cytoplasm but not the nucleus can inhibit the intense autophagy observed in Tp53-inactivated cells suggesting that Tp53 can play a role in the regulation of autophagy by forming complexes. Valid evidence confirms that AMPK is activated in Tp53−/− cells while rapamycin target protein (mTOR) which is a nutrient-sensing kinase is inhibited. Because AMPK and mTOR play key roles in autophagy regulation the inhibition of Tp53 is likely to regulate autophagy through AMPK/mTOR dependent pathways. Overall, these findings suggest that Tp53 depends on different subcellular localization to regulate autophagy. Thus, Tp53 associates autophagy with apoptosis in an intricate and environment-dependent manner and coordinates these processes to restore metabolic balance in cells and organisms (see Chap. 6 for details).
3.1.8 Mitophagy
Mitoptosis is a process of mitochondrial suicide that is usually caused by outer membrane permeabilization (MOMP) of mitochondria and subsequent potential loss. Recent studies have shown that after BAX/BAK mediated MOMP an intermembrane space (IMS) protein called DDP/TIMM8a is released into the cytoplasm where it binds the dynamin-like GTP enzyme DRP1. This interaction activates DRP1-mediated mitochondrial fission and subsequent mitoptosis. Mitochondrial dysfunction and ROS production are the main factors inducing mitoptosis. Interestingly, growing evidence suggests that programmed mitochondrial damage leads to autophagy. In fact, recent studies have suggested that dysfunctional mitochondria can be removed by the formation of autophagosomes during mitophagy or the formation of mitoptotic bodies which are eventually released into the extracellular space through atypical exocytosis processes (Jangamreddy and Los 2012). Recently, it has been reported that the inhibition of proteasome function by the proteasome inhibitor MG132 can activate BAX/BAK-dependent mitoptosis in epithelial cancer cells. However, further clarification of the molecular mechanism of mitoptosis is needed to clarify its significance in pathophysiology (see Chap. 19).
3.2 Autophagy and Necrosis
Necrosis is a type of cell death caused by severe, accidental or nonphysiological damage which is usually closely related to cell lysis caused by membrane destruction and subsequently the cell contents leak into the extracellular space which can lead to local inflammation and the destruction of surrounding tissues. In some specific cases, swelling or carcinogenesis of cells may precede necrosis. Although necrosis and apoptosis greatly differ in morphological characteristics the two processes are not mutually exclusive. Apoptosis and necrosis can respond in a dose-dependent manner to injury stimuli caused by various reagent treatments. Many reagents that cause apoptosis at low doses can eventually cause necrosis at relatively high doses. Many intracellular events determine the balance between apoptosis and necrosis. Changes in cell energy such as the ATP level can represent one of the factors influencing cell fate. Because ATP is necessary for some specific steps in caspase activation a rapid reduction in ATP levels in cells usually leads to necrotic cell death.
The interplay between autophagy and necrosis is more complex. These two processes can be activated simultaneously or sequentially and they can lead to the same or opposite results. The ability of autophagy to inhibit various forms of necrotic cell death which is usually caused by blocking apoptosis or inhibiting necrotic cell death is considered one of the most important pro-survival functions of autophagy.
3.2.1 Crosstalk Between Autophagic Death and Necrosis
Some experiments in cancer cells suggest the possibility of crosstalk between the mechanisms of autophagy and necrosis. In the process of the metabolic stress response autophagy can resist tumor necrosis and inflammation thus performing a protective function in cells. Although autophagy buffers against metabolic stress the damage caused by apoptosis and autophagy can promote cell death caused by necrosis both in vivo and in vitro. Although the triggers of necrosis in cancer cells are unclear an inadequate ATP content which may affect the integrity of the plasma membrane is likely to play a role leading to serious metabolic disorders and cell lysis. It can be inferred that the need for ATP rapidly decreases in the process of cell necrosis. In contrast, autophagy integrates the signals of the metabolic feedback system to help produce enough ATP to maintain cell survival. Natural polyamine spermidine can enhance autophagy and inhibit the destruction of the plasma membrane integrity and can cause the release of high mobility group B1 (HMGB1), which is a biomarker of necrosis.
However, recent studies have confirmed that Necrostatin-1 (Nec-1), which is a specific necroptosis inhibitor cannot only inhibit the process of nerve cell necrosis in nerve cells but also inhibit autophagy. These phenomena suggest that autophagy may be caused by necroptosis and there is a possibility that autophagy can be induced by increasing cell stress during cell death. Necrosis can be triggered when the cell metabolism and integrity are disrupted by nonphysiological factors. The cascade activation of programmed self-destruction includes cathepsin activation and lysosome breakdown. Therefore, autophagy and necrosis are closely interrelated because they share cytotoxic events. Some studies have confirmed the possibility of autophagy transforming into necrosis. For example, it was recently found that when hypoxia-induced autophagy reaches an irreversible point necrotic cell death can be triggered in a BNIP3-dependent manner. In protists of Dictyostelium, the inactivation of Atg1 inhibits vacuolation rather than cell death transforming the cell death caused by developing autophagy into a very similar death mode that is not identical to that caused by mammalian cell necrosis. Interestingly, in L929 mouse cellulosome cell lines TNFα can induce necrosis through the well-known autophagy and apoptosis-inducing factor FADD.
Similar to the relationship between autophagy and apoptosis evidence suggests that autophagy can promote, inhibit, or be unrelated to necrosis. For example, treatment with rapamycin combined with glucocorticoid dexamethasone causes autophagy-dependent cell death with necrotic apoptotic characteristics in acute lymphoblastic leukemia cells suggesting that autophagy can promote necroptosis in this particular system. Recent reports have shown that Atg9a drives necrosis in an autophagy independent manner during developmental bone formation in mice (Imagawa et al. 2016). However, most reports have the following opposite conclusion that autophagy can inhibit necroptosis in many cells such as L929 cells, lymphocytes or human cancer cells treated with TNFα, antigen stimulation or starvation. Currently, additional studies investigating the relationship between autophagy and necroptosis are ongoing. In addition, autophagy can be used as a means of cell survival against PARP-mediated cell necrosis. Many studies have shown that ROS, DNA destructive agents or ionizing radiation can lead to the following events including PARP activation, ATP consumption, AMPK activation, mTOR inhibition, and autophagy induction. More importantly, this induced autophagy can protect cells against cell death caused by PARP activation caused by DNA damage which is a way of cell survival.
3.2.2 RIPK1 and RIPK3
Necrosis has been previously described as death caused by extreme physical and chemical stress. However, the widely accepted theory is that specific genes can regulate necrosis which is a process known as necroptosis. Receptor-interacting serine/threonine protein kinase 1 (RIPK1), which is also known as kinase receptor-interacting protein 1 (RIP1), and RIPK3 are key signaling molecules in the process of necroptosis. Some studies suggest that the treatment of L929 cells with the general caspase inhibitor zVAD leads to autophagy and cell death and that this process requires the involvement of RIPK1 suggesting that autophagy is also involved in the process of necroptosis. In many models autophagy has been found to regulate necroptosis. In epithelial cells the inhibition of autophagy can rescue necroptosis induced by palmitic acid. Recently, it has been reported that Map3k7-deleted mouse prostate cells are more sensitive to TRAIL (TNF-related apoptotic inducing ligand)induced cell death and that this necroptosis is mainly induced by the assembly of the necrosome in association with the autophagy machinery which is mediated by the p62/SQSTM1 recruitment of RIPK1 (Goodall et al. 2016).
The replication of coxsackievirus in the intestinal epithelium reportedly uses autophagy regulated by RIPK3 to aid in the assembly of its replication mechanism while inhibiting the initiation of necrosis. The knockdown of RipK3 in Atg7-deficient mouse pancreatic cells enhances the process of necroptosis due to defects in autophagy. Photodynamic therapy based on 5-aminolevulinic acid can render the human glioblastoma cell line LN-18 sensitive to RIPK3-dependent cell death, and this process can be reversed by the activation of autophagy. These studies suggest that RIPK1 and RIPK3 associate autophagy and necrosis either synergistically or antagonistically to regulate the process of cell death. However, further research is needed to clarify the complex interactions between the two processes and their molecular mechanism. These experimental phenomena illustrate the functional correlation between autophagy and necroptosis. However, further studies are needed to elucidate the molecular mechanisms underlying the complex interplay between these two processes.
3.2.3 PARP
PARP1 belongs to a family of nuclear enzymes that can regulate DNA repair, transcription regulation, chromatin modification, and genome stability through poly-ADP ribosylation. The over-activation of PARP1 leads to an inadequate ATP supply, which can induce necrotic cell death and inhibit energy-dependent cell apoptosis. Interestingly, PARP1 activation may be involved in known signal transduction pathways that promote autophagy. As mentioned above AMPK can be used as a biosensor of intracellular energy and is activated in the absence of ATP. AMPK can promote autophagy through the inhibition of the mTOR signaling pathway (Fig. 29.3) or activation of the ULK1 complex (Egan et al. 2011). The activation of PARP1 in response to DNA damage can lead to ATP deficiency, AMPK activation, mTOR inhibition, and autophagy induction. DNA damage-induced autophagy protects cells against necrotic cell death induced by PARP1 activation and may serve as a mechanism for cell survival. These findings suggest that the activation of PARP1 can elicit the following opposing functions: ATP deficiency induces necrosis while autophagy is triggered simultaneously to protect cells from death. Ultimately, whether a cell survives or dies depends on the balance between autophagy and necrosis. In this setting, autophagy induction, which promotes survival, becomes the final means of survival for cells before they die under extreme stress.

Modulation of cell death mode by caspase activity. PARP-1 mediates oxidative stress-induced autophagy via the LKB1-AMPK-mTOR signaling pathway and acts as a cellular survival mechanism against ROS-induced necrosis
3.2.4 DAPK-PKD
Protein Kinase D1 (PKD) is a type of serine/threonine protein kinase that involves in many cellular biological processes including cell proliferation, migration, and death and this kinase can be activated by oxidative stress. The accumulation of ROS (reactive oxygen species) eventually leads to cell damage and the activation of various cellular responses including autophagy, apoptosis, and necrosis. The activation of autophagy promotes cell survival by removing damaged proteins and organelles to maintain cell homeostasis. Recently, PKD has been reportedly used as a new regulator of autophagy under oxidative stress. PKD can promote autophagy formation by phosphorylating VPS34. DAPK can also respond to oxidative stress, release Beclin 1 from BCL-XL through phosphorylation, and activate PKD by phosphorylation. Interestingly, PKD plays a role downstream of DAPK and both are required for the induction of autophagy under oxidative stress (Eisenberg-Lerner and Kimchi 2012). Therefore, DAPK can coordinate the induction of autophagy under stress through the following two different mechanisms: first through the phosphorylation of Beclin 1 and second through the phosphorylation of PKD resulting in activated PKD phosphorylating VPS34 to activate autophagy.
3.3 Autophagy and Other Forms of Cell Death
As research progressed increasing forms of cell death have been identified. Autophagy, as a means of maintaining cell homeostasis and fighting against death is bound to interact with these newly discovered forms of death. For example, entosis can be induced by AMPK under glucose starvation. AMPK is also a positive regulator of autophagy. TM9SF4 plays an important role in the phagocytosis of human malignant tumor cells and is also involved in the regulation of autophagy induced by nutritional deficiency (Sun et al. 2018). These phenomena suggest that autophagy and entosis are coregulated. Recent studies have shown that autophagy can help muscle stem cells prevent senescence and thus maintain stemness (Garcia-Prat et al. 2016). In erastin-treated mouse embryonic fibroblasts autophagy can also increase labile iron and ROS by degrading ferritin leading to ferroptosis. In addition, in acrolein-treated human umbilical vein endothelial cells (HUVECs) the inhibition of autophagy by 3-MA aggravates the process of pyroptosis while rapamycin which is an autophagy inducer alleviates pyroptosis. In the non-alcoholic steatohepatitis (NASH) model established by treatment with As2O3, the inhibition of autophagy alleviates the process of pyroptosis. Based on the above observations the relationship between autophagy and pyroptosis is very complex and more in-depth research is needed to clarify their relationship.
3.4 Epigenetic Regulation of Autophagy and Cell Death
The concept of a “histone code” was originally proposed by Allis and Turner who summarized the effect of histone modification on the chromosome structure and the corresponding regulation of nuclear function. According to this hypothesis, histones with specific covalent modifications affect the structure of chromatin and consequently affect the transcription process. This hypothesis was further developed, i.e., the post-translational modification of histones has been proposed to be regulated by specific chromatin-related functions and processes (Tan et al. 2011).
Histone acetylation and deacetylation are known to be in a dynamic balance in the nucleus accurately regulating gene transcription and expression. The process of adding acetyl groups to histone lysine residues under the action of acetyltransferases is a mechanism by which cells control gene expression, protein activity or physiological processes. Histone acetylation occurs mostly in specific lysine residues in the N-terminal alkaline amino acid-rich region of core histone, i.e., the acetyl group of acetyl coenzyme A is transferred to –NH3+ of lysine to neutralize a positive charge. Histone acetylation is determined by histone acetyltransferase (HATs) and histone deacetylase (HDACs).
Initially, the post-translational modification of histones during autophagy has not attracted much attention. Until recently, nuclear regulation was not considered to exist in autophagy because it was reported that autophagic vesicles could still accumulate in cells without nuclei under the action of autophagy inducers. However, recently, contrary experimental evidence suggested that the nucleus plays a key role in autophagy. The transcription factors TFEB, ZKSACN3, and TFE3 have also been shown to be closely related to autophagy regulation (Pan et al. 2017). In fact, autophagy seems to involve transcriptional and epigenetic processes, including the epigenetic processes of post-translational modification of histones. The demethylation of H3K9 in histone modification participates in the early stage of autophagy, and the deacetylation of H4K16 participates in the late stage of autophagy, ensuring an adequate level of autophagy required for survival. Notably, this modification of autophagy is also significantly negatively correlated with apoptotic histone markers, suggesting that autophagic histone codes can promote cell survival (Fullgrabe et al. 2014). Moreover, this histone modification may regulate the transcription of autophagy-related genes by regulating the affinity of the autophagy-related transcription factors TFEB, ZKSCAN3, and TFE3 to corresponding DNA sequences thus regulating the autophagy process (see Chap. 11).
Unsurprisingly, the regulation of histone modification is significantly related to apoptosis, autophagy, and cancer progression. Apoptosis, autophagy, and the dysregulation of histone modification are closely related to tumorigenesis. Undoubtedly, the survival related histone code represents only the tip of the iceberg of the regulatory mechanism of autophagic histone modification. Since the concept of survival related histone code has only been born the concept of death-related histone code is still in infancy. Furthermore, more in-depth research is needed to elucidate how these histone post-translational modifications participate in autophagy regulation processes (transcription, silencing, stagnation, flow control of autophagy, etc.).
4 Autophagic Cell Death and Diseases
Recently, experimental evidence has increasingly confirmed that autophagy plays a key regulatory role in different aspects of human health. These studies have reported both the positive and negative effects of autophagy depending on the type and progression of the disease. Generally, autophagy plays a certain role in bacterial and viral infection diseases caused by protein aggregation, lysosomal storage diseases, tumors, and other diseases autophagic cell death participates in the above processes.
During pathogen infection autophagy locates bacteria in the cytoplasm (Streptococcus pyogenes), immature autophagosomes (Mycobacterium tuberculosis) or damaged autophagosome-like vesicles (Salmonella typhi). During Salmonella typhimurium infection of macrophages (rather than epithelial cells), bacteria can induce autophagy-dependent host cell death rather than lysosome degradation. This process is likely the host’s self-protection mechanism thus avoiding bacterial growth and limiting further bacterial infection of nearby cells. In the process of virus infection host cells secrete interferon (IFN) which triggers cellular antiviral mechanisms to restrict virus replication. IFN has been shown to upregulate autophagy. Type I IFN such as IFN-α can induce the activation of PKR. PKR is a type of eIF2α kinase that can inhibit protein synthesis and restrict viral replication. PKR signal transduction can promote autophagy induction, etc. Type II IFN and IFN-γ can also induce autophagy or autophagic cell death and cause the autophagic degradation of intracellular mycobacterium.
The correlation between autophagy and cancer has already been reported as early as 20 years ago and some possible molecular mechanisms have recently been widely investigated. Although autophagy has been reported to have both positive and negative effects on tumorigenesis it plays context-dependent roles in cancer. Due to this finding the therapeutic targeting of autophagy in cancer is sometimes considered controversial. In fact, autophagic cell death mainly plays a role when apoptosis is defective which can lead to unregulated cell growth. In this case, cytotoxic stimuli can induce autophagic cell death to kill cancer cells mainly through the inhibition of the mTOR pathway or the activation of the type I PI3K-kinase/Akt pathway or type III PI3K-kinase/beclin 1 pathway. In this case, the self-killing function of autophagy provides a possible way to inhibit the process of cancer. In addition, necrosis can occur if cells can effectively resist apoptosis or nutritional deficiencies are very severe. It has recently been reported that autophagy can promote tumor growth through circulating arginine (Poillet-Perez et al. 2018). Therefore, understanding the relationship among nutritional deficiency, autophagy, and cell death through apoptosis and necrosis is critical for inhibiting the metabolism of cancer cells through new metabolic therapy strategies.
Studies have increasingly confirmed that intricate relationships exist among the three main cell death pathways, i.e., apoptosis, necrosis, and autophagy. Autophagy plays a dual role in cell survival and death. Therefore, autophagy can be used as a mechanism to balance cell survival and death. With the development of cell imaging technology and drug inhibitors with higher specificity the understanding of the process of cell death has become increasingly profound. Numerous cell death subtypes have been identified because of the more detailed definition of different cell types responding to different death stimuli. One such example is the study of the treatment of mouse embryonic stem cell death by etoposide (ETO) which is an inhibitor of DNA topoisomerase II that produces DNA double-strand breaks. This type of cell death is caspase-independent and necrosis independent but is partially dependent on lysosomal protease activity. Moreover, autophagy is not considered to play a role in the process of cell death, suggesting that etoposide induces a new form of programmed cell death in this cell in which Endo G (an endonuclease) participates in DNA breakage. The inhibition of Tp53 transcriptional activity or the role of Tp53 in mitochondria by pifithrinα which is a Tp53 inhibitor, can significantly rescue cell death. The author of this report named this type of cell death in mouse embryonic stem cells after Charon, the ferryman of the dead in Greek mythology, a unique programmed death process called “charontosis”.
Abbreviations
- ACD:
-
Accidental cell death
- AIF:
-
Apoptosis-inducing factor
- AMPK:
-
Adenosine 5′-monophosphate (AMP)-activated protein kinase
- Bruce:
-
BIR-containing ubiquitin-conjugating enzyme
- CMA:
-
Chaperone-mediated autophagy
- CSE:
-
Cigarette smoke extract
- DISC:
-
Fas-dependent death-inducing signaling complex
- DRAM:
-
Damage-regulated autophagy modulator
- FADD:
-
FAS-associated death domain
- FAK:
-
Focal adhesion kinase
- FLICE:
-
FADD-like interleukin-1 beta-converting enzyme
- FLIP:
-
Flice inhibitory protein
- GABARAP-L1:
-
Γ-aminobutyric acid receptor-associated protein-like 1
- HAT:
-
Histone acetyltransferase
- HDAC:
-
Histone deacetylase
- HMGB1:
-
High mobility group B1
- IAPs:
-
Inhibitor of apoptosis proteins
- IFN:
-
Interferon
- IL3:
-
Interleukin 3
- IMS:
-
Intermembrane space
- LAMP2A:
-
Lysosome-associated membrane protein type 2A
- MEF2D:
-
Myocyte enhancer factor 2D
- MEFs:
-
Mouse embryonic fibroblasts
- MFN1:
-
Mitofusin 1
- MOMP:
-
Mitochondrial outer membrane permeabilization
- mTOR:
-
Mammalian target of rapamycin
- NBR1:
-
The neighbor of BRCA1 gene 1
- NCCD:
-
Nomenclature Committee on Cell Death
- PARKIN:
-
Parkinson’s disease protein
- PINK1:
-
PTEN-induced putative kinase 1
- PKA:
-
Protein kinase A
- PKD:
-
Protein kinase D1
- PTP:
-
Permeability transition pore
- RCD:
-
Regulated cell death
- RIP1:
-
Kinases receptor-interacting protein 1
- ROS:
-
Reactive oxygen species
- SMAC:
-
Second mitochondria-derived activator of caspase
- SQSTM1:
-
Sequestosome 1, p62
- TNF:
-
Tumour necrosis factor
- TNF/NGF:
-
Tumor necrosis factor/nerve growth factor
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- VDAC1:
-
Voltage-dependent anion-selective channel 1
- VMP1:
-
Vacuole membrane protein
References
Chang CR, Blackstone C (2007) Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem 282(30):21583–21587
Chen Z, Nie SD, Qu ML et al (2018) The autophagic degradation of Cav-1 contributes to PA-induced apoptosis and inflammation of astrocytes. Cell Death Dis 9(7):771
Egan DF, Shackelford DB, Mihaylova MM et al (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331(6016):456–461
Eisenberg-Lerner A, Kimchi A (2012) PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell Death Differ 19(5):788–797
Fernandez AF, Sebti S, Wei YJ et al (2018) Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558(7708):136–140
Fullgrabe J, Heldring N, Hermanson O et al (2014) Cracking the survival code autophagy-related histone modifications. Autophagy 10(4):556–561
Galluzzi L, Vitale I, Aaronson SA et al (2018) Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25(3):486–541
Garcia-Prat L, Martinez-Vicente M, Perdiguero E et al (2016) Autophagy maintains stemness by preventing senescence. Nature 529(7584):37–42
Goodall ML, Fitzwalter BE, Zahedi S et al (2016) The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell 37(4):337–349
Gozuacik D, Bialik S, Raveh T et al (2008) DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ 15(12):1875–1886
Ichimura Y, Waguri S, Sou Y et al (2013) Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 51(5):618–631
Imagawa Y, Saitoh T, Tsujimoto Y (2016) Vital staining for cell death identifies Atg9a-dependent necrosis in developmental bone formation in mouse. Nat Commun 7:13391
Jangamreddy JR, Los MJ (2012) Mitoptosis, a novel mitochondrial death mechanism leading predominantly to activation of autophagy. Hepat Mon 12(8):e6159
Lee JS, Li QL, Lee JY et al (2009) FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol 11(11):1355–1362
Luo S, Rubinsztein DC (2010) Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ 17(2):268–277
Mihaylova MM, Shaw RJ (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 13(9):1016–1023
Pan HH, Yan Y, Liu CY et al (2017) The role of ZKSCAN3 in the transcriptional regulation of autophagy. Autophagy 13(7):1235–1238
Pattingre S, Tassa A, Qu XP et al (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122(6):927–939
Poillet-Perez L, Xie XQ, Zhan L et al (2018) Autophagy maintains tumour growth through circulating arginine. Nature 563(7732):569–573
Sun L, Meng ZY, Zhu YF et al (2018) TM9SF4 is a novel factor promoting autophagic flux under amino acid starvation. Cell Death Differ 25(2):368–379
Tan MJ, Luo H, Lee S et al (2011) Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146(6):1015–1027
Yamaguchi T, Suzuki T, Sato T et al (2018) The CCR4-NOT deadenylase complex controls Atg7-dependent cell death and heart function. Sci Signal 11(516):eaan3638
Yu L, McPhee CK, Zheng LX et al (2010) Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465(7300):942–946
Zaffagnini G, Savova A, Danieli A et al (2018) p62 filaments capture and present ubiquitinated cargos for autophagy. EMBO J 37(5):e98308
Zhang GM, Wang Z, Du Z et al (2018) mTOR regulates phase separation of PGL granules to modulate their autophagic degradation. Cell 174(6):1492–1506
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Science Press and Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Yan, X., Zhou, R., Ma, Z. (2019). Autophagy—Cell Survival and Death. In: Qin, ZH. (eds) Autophagy: Biology and Diseases. Advances in Experimental Medicine and Biology, vol 1206. Springer, Singapore. https://doi.org/10.1007/978-981-15-0602-4_29
Download citation
DOI: https://doi.org/10.1007/978-981-15-0602-4_29
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-0601-7
Online ISBN: 978-981-15-0602-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)