
Abstract
The primarily pathogenesis of IPF, an incurable respiratory disease is believed to over-repair to lung injury. The development of new drugs for IPF has increased the necessity of identifying biomarkers for predicting clinical behavior and the selection of the appropriate treatment strategy for individual patient.
We and another group found that periostin, a matricellular protein expressed specifically in areas of ongoing fibrotic lesions, such as fibroblastic foci in lung tissues from human IPF or murine bleomycin-induced lung injury models. Murine bleomycin-induced lung injury was improved by the constant suppression of periostin expression and treatment with neutralizing anti-periostin antibodies at the fibroproliferative phase. Moreover, total periostin can predict both short-term declines of pulmonary function and overall survival in IPF patients. Our group also established a new enzyme-linked immunosorbent assay (ELISA) kit that is more specific for IPF compared with the conventional kit. This new periostin ELISA kit specifically detects monomeric form, whereas the conventional kit detects both monomeric and oligomeric forms. The monomeric periostin levels can be used to predict pulmonary function decline and to distinguish IPF patients from healthy controls.
In conclusion, periostin may play an important role in fibrogenesis and could be a potential biomarker for predicting disease progression and therapeutic effect in IPF patients.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Monomeric periostin
- Matricellular protein
- Idiopathic pulmonary fibrosis
- Fibroblast
- Myofibroblast
- Biomarker
- Interleukin-13
- Interleukin-4
- TGF-β
1 Idiopathic Pulmonary Fibrosis Is a Chronic Respiratory Disease with Unknown Etiology and a Grave Prognosis
Interstitial lung disease (ILD) is one of respiratory disorders with significant morbidity and mortality [1]. Patients with ILD present mainly with chronic progressive exertional dyspnea, a restrictive pulmonary dysfunction , and a radiological diffuse lung infiltration and/or fibrotic change. ILDs are classified into (1) idiopathic interstitial pneumonias (IIPs) with unknown etiology and (2) secondary ILD with known and heterogeneous etiology [2]. The most common underlying disease of secondary ILD is a connective tissue disease, such as rheumatoid arthritis, scleroderma , or polymyositis/dermatomyositis [3].
Idiopathic pulmonary fibrosis (IPF), a pathologically usual interstitial pneumonia (UIP), is the most common and incurable form of IIPs [4, 5]. Estimates of the IPF prevalence per 100,000 people were reported as ranging from 2 to 43 cases worldwide [4]. Patients with IPF have a grave prognosis with a median survival of 2–5 years [2, 4, 5] and the major causes of death being acute exacerbation (AE), gradual respiratory failure, and cardiovascular disease [6]. AE is a complication of IPF that shows rapid and lethal respiratory failure at a frequency of 8.5–14.2% per year during the clinical course of this disease [4, 7,8,9].
2 Understanding of Pathogenesis and Development of Molecular Biomarkers for IPF
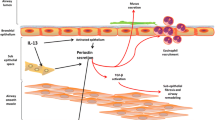
Although the pathogenesis of IPF has not been fully elucidated, the primarily mechanism is believed to attribute to aberrant wound healing responses to repetitive lung injury that targets alveolar epithelial cells (AECs). The death and apoptosis of AECs trigger abundant fibroblast recruitment, proliferation , and activation, as well as extracellular matrix (ECM) protein secretion. Moreover, fibroblasts frequently differentiate into myofibroblasts that express α-smooth muscle actin (α-SMA) in IPF. Myofibroblasts have the highly activated contractile ability, and they secrete ECM proteins with characteristics that are intermediate between fibroblasts and smooth muscle cells [10,11,12]. Small aggregates of proliferating myofibroblasts and fibroblasts, termed fibroblastic foci (FF), play an important role in ongoing fibrogenesis and have been reported to be relevant for predicting the prognosis of IPF patients [13, 14].
The over-repairing environment in IPF is activated by innate and adaptive immune responses involving the activation of type 2 T helper (Th2) cytokines, M2-like macrophages, and/or growth factors. TGF-β1 is an important growth factor in the fibrotic process of IPF; it promotes myofibroblast differentiation as well as an anti-apoptotic phenotype in fibroblasts and myofibroblasts [10,11,12, 15]. An inappropriate shift in the Th1/Th2 cytokine balance, favoring the Th2 profile, can contribute to lung fibrosis in IPF [10]. A previous report revealed that the expressions of interleukin (IL)-4 and IL-13 receptors in the lungs were increased in IPF patients compared with patients having other IIP subtypes [16]. Lee et al. reported a model for lung fibrosis in which the overexpression of IL-13 in CC10-IL-13 transgenic mice caused lung fibrosis by selectively stimulating and activating TGF-β [17].
Recently, an evolving understanding of IPF pathogenesis has contributed to the development of two therapeutic drugs, pirfenidone and nintedanib, for which New Drug Applications had been approved by the U.S. Food and Drug Administration [18, 19]. Moreover, ongoing or recently completed clinical trials of new drug candidates targeting lung over-repair have been reported. Human monoclonal antibodies targeting connective tissue growth factor (CTGF), a matricellular protein (FG-3019), a selective autotaxin inhibitor that reduces plasma concentrations of lysophosphatidic acid (GLPG1690), and pentraxin 2 that inhibits M2-like macrophage differentiation (PRM-151) were all found to slow the disease progression of IPF in phase 2 randomized, double-blind, placebo-controlled trials [20,21,22]. On the other hands, these paradigm shifts in the available therapeutic strategies for IPF treatment have raised the new problem of selecting an appropriate treatment type and intervention time for individual patient.
The natural course of IPF has been described as a progressive decline in pulmonary function until eventual death [4]. The short-term progression measured via the decline of pulmonary function from baseline values has been reported as a promising prognostic predictor. A decline of 10% for forced vital capacity (FVC) and that of 15% for diffusing capacity of the lung for carbon monoxide (DLCO) over 6 months were associated with the survival of IPF patients in some previous reports [23, 24]. It should be noted that there is heterogeneity in the clinical behavior of individual IPF patients, including gradual or accelerated progression or AE development [2, 4, 25]. There is no established predictor that can accurately determine the clinical behavior or survival of IPF patients. Some epithelial or macrophage-related proteins, such as CC-chemokine ligand-18 (CCL-18), matrix metalloproteinase-7 (MMP-7), Krebs von den Lungen-6 (KL-6), surfactant protein D (SP-D), and SP-A, were reported as biomarkers for declining pulmonary function or those for survival [2, 26,27,28,29,30,31]. Molecular biomarkers may allow the application of “precision medicine” by aiding in the selection of a suitable treatment for individual IPF patients, such as an anti-fibrotic drug, lung transplantation, or palliative therapy [2, 25, 26, 32].
3 Periostin, a Matricellular Protein, Is Involved in the Pathogenesis of IPF
Periostin is an ECM protein belonging to the fasciclin family that acts as a matricellular protein modulating cell–matrix interactions via the αvβ1, αvβ3, or αvβ5 integrin receptor [33]. Periostin is secreted from fibroblasts, epithelial cells, and endothelial cells via stimulation by IL-4 , IL-13 , TGF-β , angiotensin II, CTGF, bone morphogenetic protein 2, mechanical stretch, and cancer-derived factors [33]. Periostin contributes to tissue development and wound healing by stabilizing collagen cross-linking and fibrotic disease progression [34]. Takayama et al. suggested that periostin secreted from lung fibroblasts is involved in subepithelial fibrosis via binding to other ECM proteins in a murine ovalbumin-induced allergic asthma model [35]. Periostin also contributes to the development of skin fibrosis in scleroderma [36]. Experiments in vitro showed that periostin can cooperate with TGF-β to promote the expression of messenger ribonucleic acid (mRNA) for α-SMA and procollagen type-I alpha 1 on dermal fibroblasts in a bleomycin (BLM)-induced murine scleroderma model. These mechanisms depend on the phosphatidylinositol 3-kinase/Akt (PI3K/Akt) pathway [37].
We and other groups attempted to clarify whether periostin is involved in the fibrotic mechanism in IPF [38,39,40,41,42,43,44]. We hypothesized that periostin was strongly expressed in fibroblasts, especially in FF areas, but not in regenerative alveolar epithelium , inflammatory cells, or areas showing established fibrosis with dense collagen depositions in IPF lungs (Fig. 9.1) [38]. In histochemical analyses of other subtypes of IIP, periostin was strongly expressed in fibrotic non-specific interstitial pneumonia (NSIP), whereas the periostin expression was weak in cellular NSIP and cryptogenic organizing pneumonia, as well as in normal lungs [38]. Uchida et al. also reported the high expression of periostin in lung fibrotic tissues, particularly in the α-SMA-positive myofibroblasts , from BLM-administered mice that are a representative murine model of IPF [39]. Naik et al. demonstrated that periostin localizes to FF areas as well as to subendothelial and subepithelial regions in lung tissue from IPF patients [40]. Some experiments in vitro revealed that lung fibroblasts and circulating fibrocytes are sources of periostin in the circulation of IPF patients [40, 41]. Together, these results suggest that the expression of periostin is localized specifically to areas of ongoing fibrotic lesions in IPF lungs .

Immunohistochemical (IHC) findings. (This figure was quoted from Ref. [38] and modified)
Expression of periostin in lungs of a 69-years-old female nonsmoker (a–c), a 64-years-old male with usual interstitial pneumonia (UIP) (d–f). The tissues were stained with haematoxylin and eosin (HE) 40×, HE 100× or periostin 100×, as shown
Uchida et al. used periostin-deficient mice in which periostin expression was constantly suppressed for clarifying the pathogenesis of IPF. Periostin-deficient mice administered with BLM exhibited less lung fibrosis and mortality compared with wild type mice that were treated similarly [39]. Naik et al. reported that treatment of wild type mice with neutralizing antibody against periostin (OC-20) on 10 and 15 days after BLM administration protected them from lung fibrosis and improved survival [40]. Therefore, periostin suppression at only the fibroproliferative phase of the disease was able to improve murine BLM-induced lung injury similarly to constant suppression.
Previous studies suggested that periostin induces type 1 collagen production from lung fibroblasts and/or circulating fibrocytes and promotes collagen deposition , mesenchymal cell proliferation , and wound closure in the lungs [40, 41]. Periostin and TGF-β upregulate the production of one another by fibroblasts and fibrocytes [41]. Ashley et al. demonstrated that periostin promotes CTGF production from fibrocyte and myofibroblast differentiation , leading to pulmonary fibrosis [41]. Periostin is thought to augment ECM protein deposition in IPF lungs via activating soluble factors, such as growth factors and other matricellular proteins, and mesenchymal cells. Nance et al. sequenced mRNA from the lung tissues of eight IPF patients and seven healthy controls, and their results suggested that the spliced-out exon 21 of periostin gene (POSTN) occurred more highly in IPF samples than in control samples [42].
Some drugs, particularly chemotherapeutic agents, often cause an acute and lethal subtype of ILD with the typical histopathological features of diffuse alveolar damage (DAD). We revealed that periostin was expressed not only in IPF lungs but also in human lung tissue with BLM- or gefinitib-induced DAD. Periostin staining was evident in the thickened alveolar walls adjacent to α-SMA-positive cells in the lung tissues with either drug-induced ILD [39]. During day 1–7 of BLM administration, mouse lungs showed acute lung injury-like features; histologically, the accumulation of inflammatory cells and upregulation of chemokines and pro-inflammatory cytokines were observed [45]. Uchida et al. found that the increase of chemokines, pro-inflammatory cytokines , and neutrophil and macrophage recruitment in the lung tissues or bronchoalveolar lavage fluids collected at day 7 of a murine BLM-induced lung injury model were impaired in periostin-deficient mice [39]. Therefore, periostin promotes the production of chemokines and pro-inflammatory cytokines, followed by the recruitment of neutrophils and macrophages, subsequently leading to lung injury. Periostin may be important in rapidly progressive ILD with acute onset or AE of IPF showing a histological DAD pattern. We will investigate these issues in future analyses .
4 Periostin Is a Potential Prognostic Biomarker of IPF
We first reported that periostin is a potential prognostic biomarker for predicting the short-term progression of IPF patients in a single-center study. In that work, baseline serum levels of periostin were positively correlated with a decline of vital capacity (VC) and DLCO over 6 months (VC: N = 26, r = −0.498, p < 0.01; DLCO: N = 21, r = −0.467, p < 0.05) [38]. Moreover, we attempted to clarify whether periostin is useful for predicting long-term survival by analyzing 29 IPF subjects who were observed for at least 2 years and for up to 5 years [43]. Log-rank tests revealed that a higher serum periostin level was a predictor of a shortened overall survival (OS) and time-to-events (TTE) defined as a complicating AE or a relative decline in VC of ≥10% from the baseline (OS: relative risk (RR) = 3.6, 95% confidence interval (CI) = 1.3–9.9, p < 0.01; TTE: RR = 6.0, 95%CI = 1.8–21.1, p < 0.01) [43]. In that study, the baseline periostin levels significantly correlated with an increase in the extent of honeycombing visible on high resolution computed tomography images during 6 months [43]. Naik et al. demonstrated that the baseline periostin level was able to predict progression-free survival within 48 weeks for IPF patients, as determined by the time until any of the following: death, complicating AE, lung transplant, or relative decline in FVC of ≥10% or in DLCO of ≥15% (N = 54, hazard ratio = 1.47, 95%CI = 1.03–2.10, p < 0.05) [40]. Thus, the serum level of periostin can predict short-time disease course and survival.
Unfortunately, periostin is not a specific biomarker for IPF because it is upregulated in various diseases other than IPF [36]. This fact may affect its accuracy as prognostic biomarker for IPF in patients who complicated other high-periostin diseases. Izuhara et al. and the Shino-Test established a new enzyme-linked immunosorbent assay (ELISA ) kit that is more specific for IPF compared with the conventional kit [44]. The new periostin ELISA kit specifically detects the monomeric form (SS20A × SS19D, the capture and detection antibody ), whereas the conventional kit detects both the monomeric and oligomeric forms (total periostin, SS18A × SS17B). We found that the index of total periostin/monomeric periostin was significantly lower in IPF patients (2.1, N = 40) than that in either patients with atopic dermatitis (14.2, N = 224), systemic sclerosis (11.7, N = 37), or bronchial asthma (7.3, N = 143), all of which are also high-periostin diseases [44]. These results suggest that a high ratio of monomeric periostin to total periostin is characteristic of IPF patients. Serum periostin mostly exists in the oligomeric form assembled by intramolecular disulfide bonds, with only small amounts existing in the monomeric form. The fact that monomeric periostin is predominantly upregulated compared with the level of total periostin may be explained by the aberrant redox status in IPF. We will investigate this issue in future work.
We conducted a multi-center analysis to examine the ability of monomeric periostin to serve as a prognostic biomarker of IPF. The changes in VC and DLCO were inversely associated with both the monomeric periostin level (VC: r = −0.492, p < 0.01; DLCO: r = −0.587, p < 0.001) and the total periostin level (VC: r = −0.428, p < 0.01; r = −0.460, p < 0.01) (Fig. 9.2) [44]. We also suggested that periostin could be a useful diagnostic biomarker of IPF. The receiver operating characteristic curve analyses for distinguishing IPF patients (n = 60) from healthy control donors (n = 137) revealed that monomeric periostin had the highest area under the curve (AUC, 0.958) among the investigated biomarkers (total periostin: 0.843, KL-6: 0.948, SP-D: 0.953, lactate dehydrogenase [LDH]: 0.898). When we set the cut-off values for monomeric periostin at 11.2 ng/mL and total periostin at 77 ng/mL, the sensitivities and specificities were respectively evaluated as 90.0% and 91.2% for monomeric periostin and at 73.3% and 79.6% for total periostin (Fig. 9.3). Therefore, monomeric periostin is a potential diagnostic and prognostic biomarker for IPF patients that is less influenced by complicatied high-periostin diseases.

Ability of each biomarker to predict the short-term progression of idiopathic pulmonary fibrosis. (This figure was quoted from Ref. [44])
Correlations between monomeric periostin , total periostin , Krebs von den Lungen-6 (KL-6), surfactant protein D (SP-D) or lactate dehydrogenase (LDH) and short-term change of vital capacity (VC, A) or diffusing capacity of the lung for carbon monoxide (DLCO, B) in idiopathic pulmonary fibrosis patients (N = 44 for VC and 39 for DLCO)

Abilities of each biomarker to diagnose as idiopathic pulmonary fibrosis. (This figure was quoted from Ref. [44])
A receiver operating characteristic curve analysis of each biomarker between idiopathic pulmonary fibrosis (IPF) patients and healthy donors. Monomeric periostin (red), total periostin (orange), Krebs von den Lungen-6 (KL-6), (black), surfactant protein D (SP-D) (green), and lactate dehydrogenase (LDH) (blue) between IPF patients (n = 60) and healthy donors (n = 137)
Neighbors et al. performed post-hoc analyses of two replication IPF cohorts from the clinical trials for pirfenidone (CAPACITY and ASCEND). That was a comprehensive study evaluating the properties of biomarkers useful for predicting FVC changes as well as the therapeutic effect as assessed by a difference in the FVC decline between the pirfenidone and placebo groups over 12 months [46]. Total periostin measured using the Elecsys® periostin assay (Roche Diagnostics, Penzberg, Germany) was included among the analyzed biomarkers. The baseline serum levels of periostin and many other biomarkers were able to predict the specified parameters in the CAPACITY cohort but not in the ASCEND cohort [46]. These findings raise the problem that the utility of a prognostic biomarker may be affected by the characteristics of the analyzed IPF population. Their study indicated that evaluations with a combination of CCL18, C-X-C motif chemokine ligand 14, and total periostin were better at predicting the prognosis and therapeutic effect compared with any single biomarker [46]. The evaluation of a combination of biomarkers associated with different pathologies of IPF may be useful for prognostic prediction. Periostin is also expected to be applicable as a biomarker for predicting the therapeutic effect and stratification of subjects in clinical trials on IPF patients.
5 Periostin as a Therapeutic Target
We and another group reported the improvement of murine BLM-induced lung injury via the constant suppression of periostin expression [39] or treatment with neutralizing anti-periostin antibodies at the fibroproliferative phase [40]. A previous report additionally suggested that osteopontin reduced murine BLM-induced lung injury via the αV integrin [47]. Periostin or the αV integrin may be a potential target in new treatments for IPF.
6 Conclusion
Periostin may play an important role in fibrogenesis and be applicable as a potential biomarker for predicting both the disease progression and therapeutic effect in IPF patients. The further study of periostin is expected to accelerate the development of new diagnostic and treatment strategies for IPF .
References
Ryerson CJ, Collard HR (2013) Update on the diagnosis and classification of ILD. Curr Opin Pulm Med 19:453–459
Travis WD, Costabel U, Hansell DM et al (2013) An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 188:733–748
Mathai SC, Danoff SK (2016) Management of interstitial lung disease associated with connective tissue disease. BMJ 352:h6819
Raghu G, Collard HR, Egan JJ et al (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183:788–824
Raghu G, Rochwerg B, Zhang Y et al (2011) An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med 192:e3–e19
Fernández Pérez ER, Daniels CE, Schroeder DR et al (2010) Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest 137:129–137
Collard HR, Moore BB, Flaherty KR et al (2007) Idiopathic pulmonary fibrosis clinical research network investigators. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 176:636–643
Song JW, Hong SB, Lim CM et al (2011) Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 37:356–363
Kim DS, Park JH, Park BK et al (2006) Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur Respir J 27:143–150
Strieter RM, Mehrad B (2009) New mechanisms of pulmonary fibrosis. Chest 136:1364–1370
Ahluwalia N, Shea BS, Tager AM (2014) New therapeutic targets in idiopathic pulmonary fibrosis. Aiming to rein in runaway wound-healing responses. Am J Respir Crit Care Med 190:867–878
Scotton CJ, Chambers RC (2007) Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest 132:1311–1321
King TE Jr, Schwarz MI, Brown K et al (2001) Idiopathic pulmonary fibrosis: relationship between histopathologic features and mortality. Am J Respir Crit Care Med 164:1025–1032
Nicholson AG, Fulford LG, Colby TV et al (2002) The relationship between individual histologic features and disease progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 166:173–177
Border WA, Noble NA (1994) Transforming growth factor beta in tissue fibrosis. N Engl J Med 331:1286–1292
Jakubzick C, Choi ES, Kunkel SL et al (2004) Augmented pulmonary IL-4 and IL-13 receptor subunit expression in idiopathic interstitial pneumonia. J Clin Pathol 57:477–486
Lee CG, Homer RJ, Zhu Z et al (2001) Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med 194:809–821
Richeldi L, du Bois RM, Raghu G et al (2014) Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370:2071–2082
King TE Jr, Bradford WZ, Castro-Bernardini S et al (2014) A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 370:2083–2092
Raghu G, Scholand MB, de Andrade J et al (2016) FG-3019 anti-connective tissue growth factor monoclonal antibody: results of an open-label clinical trial in idiopathic pulmonary fibrosis. Eur Respir J 47:1481–1491
Maher TM, van der Aar EM, Van de Steen O et al (2018) Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): a phase 2a randomised placebo-controlled trial. Lancet Respir Med 6:627–635
Raghu G, van den Blink B, Hamblin MJ et al (2018) Effect of recombinant human pentraxin 2 vs placebo on change in forced vital capacity in patients with idiopathic pulmonary fibrosis: a randomized clinical trial. JAMA 319:2299–2307
Latsi PI, du Bois RM, Nicholson AG, Colby TV, Bisirtzoglou D, Nikolakopoulou A, Veeraraghavan S, Hansel DM, Wells AU (2003) Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 168:531–537
Flaherty KR, Mumford JA, Murray S et al (2003) Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia. Am J Respir Crit Care Med 168:543–548
Ley B, Collard HR, King TE Jr (2011) Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 183:431–440
Ley B, Brown KK, Collard HR (2014) Molecular biomarkers in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 307:L681–L691
Prasse A, Probst C, Bargagli E et al (2009) Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 179:717–723
Rosas IO, Richards TJ, Konishi K et al (2008) MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med 5:e93
Yokoyama A, Kondo K, Nakajima M et al (2006) Prognostic value of circulating KL-6 in idiopathic pulmonary fibrosis. Respirology 11:164–168
Takahashi H, Fujishima T, Koba H et al (2000) Serum surfactant proteins a and D as prognostic factors in idiopathic pulmonary fibrosis and their relationship to disease extent. Am J Respir Crit Care Med 162:1109–1114
Ishikawa N, Hattori N, Yokoyama A et al (2012) Utility of KL-6/MUC1 in the clinical management of interstitial lung diseases. Respir Investig 50:3–13
Brownell R, Kaminski N, Woodruff PG et al (2016) Precision medicine: the new frontier in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 193:1213–1218
Conway SJ, Izuhara K, Kudo Y et al (2014) The role of periostin in tissue remodeling across health and disease. Cell Mol Life Sci 71:1279–1288
Izuhara K, Nunomura S, Nanri Y et al (2017) Periostin in inflammation and allergy. Cell Mol Life Sci 74:4293–4303
Takayama G, Arima K, Kanaji T et al (2006) Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J Allergy Clin Immunol 118:98–104
Yamaguchi Y, Ono J, Masuoka M et al (2013) Serum periostin levels are correlated with progressive skin sclerosis in patients with systemic sclerosis. Br J Dermatol 168:717–725
Yang L, Serada S, Fujimoto M et al (2012) Periostin facilitates skin sclerosis via PI3K/Akt dependent mechanism in a mouse model of scleroderma. PLoS One 7:e41994
Okamoto M, Hoshino T, Kitasato Y et al (2011) Periostin, a matrix protein, is a novel biomarker for idiopathic interstitial pneumonias. Eur Respir J 37:1119–1127
Uchida M, Shiraishi H, Ohta S et al (2012) Periostin, a matricellular protein, plays a role in the induction of chemokines in pulmonary fibrosis. Am J Respir Cell Mol Biol 46:677–686
Naik PK, Bozyk PD, Bentley JK et al (2012) Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 303:L1046–L1056
Ashley SL, Wilke CA, Kim KK et al (2017) Periostin regulates fibrocyte function to promote myofibroblast differentiation and lung fibrosis. Mucosal Immunol 10:341–351
Nance T, Smith KS, Anaya V et al (2014) Transcriptome analysis reveals differential splicing events in IPF lung tissue. PLoS One 9:e92111
Tajiri M, Okamoto M, Fujimoto K et al (2015) Serum level of periostin can predict long-term outcome of idiopathic pulmonary fibrosis. Respir Investig 53:73–81
Ohta S, Okamoto M, Fujimoto K et al (2017) The usefulness of monomeric periostin as a biomarker for idiopathic pulmonary fibrosis. PLoS One 12:e0174547
Moore BB, Hogaboam CM (2008) Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 294:L152–L160
Neighbors M, Cabanski CR, Ramalingam TR et al (2018) Prognostic and predictive biomarkers for patients with idiopathic pulmonary fibrosis treated with pirfenidone: post-hoc assessment of the CAPACITY and ASCEND trials. Lancet Respir Med 6:615–626
Takahashi F, Takahashi K, Okazaki T et al (2001) Role of osteopontin in the pathogenesis of bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 24:264–271
Acknowledgment
We thank the colleagues and collaborators as follows: Kiminori Fujimoto, Koichi Ohshima, Tomotaka Kawayama, Yasuhiko Kitasato, Yuki Sakazaki, Morihiro Tajiri (Kurume University), Koichiro Takahashi, Shinichiro Hayashi, Masaru Uchida, Hiroshi Shiraishi, Kazuto Taniguchi, Shoichi Suzuki, Atsushi Kawaguchi (Saga University), Shigeki Kohno, Noriho Sakamoto (Nagasaki University), Junichi Kadota (Oita University), Masayuki Hanaoka, Hiroshi Yamamoto (Shinshu University), Masao Ichiki (Kyushu Medical Center), Hisako Kushima, Hiroshi Ishii (Fukuoka University), Keiichi Akasaka (Nigata Universtity Medical and Dental Hospital), Hironori Sagara (Syowa University), Takeshi Johkoh (Kinki Central Hospital), Seiya Kato (Saiseikai Fukuoka General Hospital), Hisako Matsumoto (Kyoto University), Yukie Yamaguchi, Michiko Aihara (Yokohama City University), Ayami Kamei, Yoshinori Azuma (Shino-Test Corp.), Simon J. Conway (Indiana University).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Okamoto, M., Izuhara, K., Ohta, S., Ono, J., Hoshino, T. (2019). Ability of Periostin as a New Biomarker of Idiopathic Pulmonary Fibrosis. In: Kudo, A. (eds) Periostin. Advances in Experimental Medicine and Biology, vol 1132. Springer, Singapore. https://doi.org/10.1007/978-981-13-6657-4_9
Download citation
DOI: https://doi.org/10.1007/978-981-13-6657-4_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-6656-7
Online ISBN: 978-981-13-6657-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)