
Abstract
With an increasingly ageing population that is expected to double by 2050 in the U.S., it is paramount that we further understand the neurological changes that occur during ageing. This is relevant not only in the context of “pathological” ageing, where the development of many neurodegenerative disorders is typically a feature of only the older population (and indeed, age is the primary risk factor for many conditions such as Alzheimer’s disease), but also for what is considered to be “normal” or “healthy” ageing. Specifically, a significant proportion of the older population are affected by “age-related cognitive decline” (ARCD), which is both independent of dementia and has an incidence 70% higher than dementia alone. However, whilst it is reported that there are pathogenic and phenotypic overlaps between healthy and pathological ageing, it is clear that there is a need to identify the pathways and understand the mechanisms that contribute to this loss of cognitive function with normal ageing, particularly in light of the increasing life expectancy of the global population. Importantly, there is an increasing body of evidence implicating zinc homeostasis as a key player in learning and memory and also potentially ARCD. Further research will ultimately contribute to the development of targeted therapeutics that will promote successful brain ageing. In this chapter we will explore the notion of ARCD, with a perspective on potential key neurochemical pathways that can be targeted for future intervention.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
By the year 2100, the population is estimated to increase by nearly 3.6 billion people worldwide (United Nations 2017). This growth is largely due to previous advances in modern medicine, improved hygiene and the implementation of antibiotics and pesticides which have resulted in an increase in life expectancy, especially in developed countries (Lunenfeld 2008). That being said, developing countries are experiencing the highest growth in the ageing population. For instance, the elderly population in Singapore and Malaysia is expected to increase by 372% and 277% respectively by the year 2030 (Kinsella and Velkoff 2002). In contrast, France and the United Kingdom are predicted to see a 56% and 55% increase in the ageing population by the same year (Kinsella and Velkoff 2002). The elderly population is, therefore, growing at a faster rate than ever before. These demographic differences can be explained by the increase in birth and mortality rates. Indeed, birth rates remain much higher in developing countries (3.3 births per woman in Malaysia in 2000) as compared to developed countries (1.7 births per woman in United Kingdom in 2000) (Kinsella and Velkoff 2002). Whereas developed countries have seen a large decline in mortality rates (39.3% of individuals in Japan aged 65 years or more will be older than 80 by 2030), so have developing countries (27.3% of individuals in Argentina aged 65 years or more will be older than 80 by 2030) (Kinsella and Velkoff 2002). Furthermore, it is estimated that 40% of the population aged 60 years or more is affected by ARCD (VanGuilder and Freeman 2011). Age-related cognitive decline significantly hampers quality of life and these individuals therefore require specialized care. Unfortunately, developing countries with low economic power and less resources will experience more difficulty in taking care of the elderly as compared to developed countries (Lunenfeld 2008). For these reasons, a better understanding of the neurochemical basis underlying the ageing process will aid us in determining the specific biochemical pathways that contribute to cognitive decline, and thus will be critical for the development of targeted therapeutics in the pursuit of promoting healthy ageing.
What is Ageing and What Causes It?
Ageing, which is a normal and complex biological process that remains poorly understood, is characterized by a steady decline in various physiological functions that result in both physical and cognitive impairment (Deary et al. 2009). Physiologically, a number of biological changes occur to impact upon normal daily function. For instance, body composition is altered with age, such that lean muscle mass declines while fat mass increases (Bigby 2004). Because the amount of collagen decreases with age, flexibility of ligaments, tendons, muscles and joints decline and this affects muscle function over time. This reduction in muscle tone is directly linked to reduced lung capacity and therefore reduced oxygen consumption (Bigby 2004). Ageing also causes thinning of arterial walls and increases fat deposition within arteries. These cardiovascular problems, along with reduced lung capacity, render physical activity a difficult task. It is therefore no surprise that the elderly are susceptible to a range of medical complications over time. Moreover, the skin undergoes changes in pigmentation, water content and elasticity via a decrease in elastin and collagen content which presents as wrinkles on the face and body (Bigby 2004). Hair loss, which is perhaps one of the most well-known physical consequence of ageing, is dependent upon genetics and involves thinning and de-pigmentation of hair follicles perceived as greying of the hair. In addition, bone density decreases with age and this is threefold greater in women than men (Bigby 2004). Finally, vision and hearing loss are debilitating consequences of ageing to many individuals as well as decreased sensitivity to touch, smell and taste over time (Bigby 2004). It is therefore suggested that ageing is, for the most part, caused by the deterioration of these core biological functions (Deary et al. 2009).
There are, however, several factors that are likely to impact upon the rate and magnitude of the ageing process which varies greatly between individuals (Deary et al. 2009), such as changes in diet and lifestyle, inflammation, genetics and the presence of confounding illness. Individuals with co-morbidities such as cardiovascular disease, for example, display increased ageing rates compared to healthy individuals (VanGuilder and Freeman 2011). Interestingly, diets high in antioxidants have been shown to improve cognitive function during ageing (Robertson et al. 2013). Furthermore, age-related chronic illness and stress may lead to inflammation initiating the release of pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and interleukin-6 (IL-6), which only exacerbate the inflammatory response (Robertson et al. 2013). Finally, testosterone, which decreases with age, is suggested to have neuroprotective properties through the enhancement of hippocampal synaptic plasticity as well as limiting the aggregation of proteins involved in neurodegenerative diseases, namely amyloid beta (Maggio et al. 2012). Hence, it is suggested that amelioration of these factors that can be relatively easily controlled, such as diet for example, might promote “healthy” ageing and this is discussed in further detail in a subsequent section.
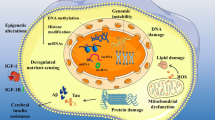
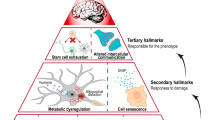
López-Otín et al. (2013) have proposed nine probable hallmarks contributing to the ageing process and these include genomic instability, mitochondrial dysfunction, telomere attrition, cellular senescence, stem cell exhaustion and altered intercellular communication, epigenetics, proteostasis and nutrient sensing. Indeed, as individuals age, their genes become increasingly susceptible to damage from endogenous and exogenous sources. Errors in DNA replication, point mutations, reactive oxygen species (ROS) generation and alterations in DNA methylation all contribute to accelerated ageing (Moskalev et al. 2013). Evidence for this is supported by studies in transgenic mice that overexpress the BubR1 gene (a gene involved in chromosomal separation during mitosis), such that these mice exhibit an increased life expectancy and reduced chances of developing cancer (Baker et al. 2013). As such, ageing is favored by DNA damage and suppressed by DNA repair (Kirkwood 2005). DNA damage as a result of ageing occurs in a fairly random fashion, whereas the telomeres at the end of chromosomes are specifically vulnerable to damage with increasing age (López-Otín et al. 2013). That is, telomeres bind to shelterin, a protein complex that prevents repair processes from restoring damaged DNA thereby causing persistent damage at telomeres with increasing age (Palm and de Lange 2008). Experimental studies in mice have demonstrated that telomere attrition causes a decrease in lifespan (Armanios et al. 2009), and conversely that lifespan can be prolonged by telomere activation (López-Otín et al. 2013). As we age, ATP generation becomes less efficient causing mitochondrial dysfunction and increased ROS generation which ultimately leads to widespread cellular damage. Furthermore, adequate proteostasis is essential for the normal functioning of the proteome where misfolded proteins are repaired (Powers et al. 2009). During ageing, proteins are misfolded and/or aggregated and this can lead to a variety of neurodegenerative pathologies (Bishop et al. 2010). In addition, nutrient sensing, in which glucose is sensed by insulin and insulin-like growth factor 1 (IGF-1) is dysregulated in ageing such that genetic alterations of these factors or even dietary restriction has been shown to increase lifespan (Fontana et al. 2010). Finally, ageing has been linked with decreased stem cell function and altered intercellular communication ultimately leading to global cellular senescence (López-Otín et al. 2013). As such, the following sections will more specifically interrogate the influence of ageing on the brain.
The Impact of Ageing on the Brain
Ageing is the biggest risk factor for the development of neurodegenerative diseases including Alzheimer’s disease (AD) and other disorders such as Parkinson’s disease and Huntington’s disease. Whilst such diseases will precipitate degenerative changes in the brain, their evolution and progression may be impacted by normal age-related changes in various pathways. These concepts on the intersection between ageing and neurodegenerative disease have been reviewed extensively in the past (Celsis 2000; Harada et al. 2013; Hulette et al. 1998) and are not the focus of this current chapter. Rather, we will explore the biochemical and anatomical changes that occur across the lifespan that may contribute to ARCD.
During normal ageing, various neuroanatomical regions are affected but these structural changes are not identical in all brain regions. In early adulthood, grey matter volume but more specifically the prefrontal cortex, decreases in size (Harada et al. 2013). In fact, the whole brain volume, as estimated in longitudinal studies, decreases in size by 0.2–0.5% every year (Fjell and Walhovd 2010). That being said, white matter changes during ageing are more prominent than that of grey matter changes (Harada et al. 2013). While there is a steady increase in structural grey matter alterations from childhood to adulthood, the white matter undergoes significant alterations in adulthood where this plateaus and resumes in old age (Madden et al. 2008). For example, diffusion tensor magnetic resonance images (DT-MRI) comparing white matter integrity between young and old individuals show greater anisotropic diffusion of water molecules and therefore greater white matter density in young individuals as compared to older individuals (Head et al. 2004). Depending on where these structural alterations occur in the white matter tracts, these will have varying functional outcomes. The largest volumetric changes recorded include the putamen, accumbens, thalamus and frontal and temporal cortices (Fjell and Walhovd 2010). Interestingly, the ventricular system has been shown to increase in size during ageing. Clinical data from Rogalski et al. (2012) demonstrates reductions in parahippocampal white matter volumes in healthy aged participants. They suggest that this plays a role in age-related memory impairment as the parahippocampal tract is involved in the relay of sensory information between the hippocampus and the entorhinal cortex. Indeed, axon myelination greatly reduces with increasing age therefore impeding upon axonal signal transduction (Fjell and Walhovd 2010). Similarly, Madden et al. (2008) suggest that decreased integrity of a portion of the corpus callosum during ageing leads to reduced perceptual speed and memory retrieval. Furthermore, functionality of posterior regions of the brain also decline with age, particularly the ventral visual cortex that exhibits less neural selectivity to inputs from the visual system (Park et al. 2004).
It is clear, therefore, that there are widespread changes throughout various regions of the brain across age. The hippocampus, however, is an area of particular interest in ageing and cognition as it governs spatial learning and memory; processes that are intimately linked to cognitive decline (VanGuilder and Freeman 2011). Hippocampal atrophy is one of the defining anatomical alterations that occur as a result of ageing, and is likely due to a reduction in synaptic density and neuronal size (Harada et al. 2013). The hippocampus is made up of several structures namely the cornu ammonis areas CA1-4 which together form what is known as the “hippocampus proper” and along with the dentate gyrus and the subiculum are collectively known as the “hippocampal formation”. During normal ageing, neuronal loss has been demonstrated in the dentate gyrus and the subiculum but not in the hippocampus proper (Jagust 2013). Some have suggested that the hippocampus may serve in the discrimination of “healthy” and “pathological” ageing. Indeed, studies have shown that neuronal loss in the dentate gyrus and subiculum of the hippocampus are due to age-related changes only, whereas neuronal loss in CA1 and the entorhinal cortex are implicated in neurodegenerative diseases and particularly in Alzheimer’s disease (Jagust 2013). In contrast to this view, others have suggested that older adults display decreased volumes of the dentate gyrus and the CA3 region of the hippocampus (Shing et al. 2011) while others have found smaller volumes of CA1 in memory impaired individuals (Yassa et al. 2010). Therefore, while there is a current disagreement in the field as to which hippocampal regions are affected or spared during ARCD, this may be due to the fact that few studies have investigated the link between volumetric changes in hippocampal sub-regions and subsequent changes in functional endpoints – a notion discussed in greater detail in the following section. Nonetheless, these morphometric changes in the ageing brain may only be partly attributed to neuronal loss, rather, it seems that reductions in spine density and decreased neuron sizes and number of synapses more likely contribute to the volumetric alterations observed during normal ageing (Fjell and Walhovd 2010). During ageing, neurons undergo drastic morphological changes via a decrease in synaptic branching and the number and length of dendritic spines (Harada et al. 2013) and this has been demonstrated in humans, rats and non-human primates (Morisson and Baxter 2012). Interestingly, increased neuronal activity in the prefrontal cortex has been reported during normal ageing and it is suggested that this functions to compensate for the overall dampened activity in other neuroanatomical regions (Reuter-Lorenz 2002). This compensatory mechanism is thought to be evidence of the brain’s adaptation to ageing (Ballesteros et al. 2009). Indeed, during memory tasks, the activation of the brain’s memory networks becomes bilateral perhaps due to the decreased activation in other areas specifically involved in memory (Peters 2006). Furthermore, functional reorganization of dendritic synapses via dendritic sprouting has been suggested to compensate for cell death during ageing (Peters 2006).
Other pathways contributing to the ageing process include reduced vascular density, elevations in inflammation and oxidative stress, compromised neurotransmitter performance such as GABA (Rozycka and Liguz-Lecznar 2017) and the dysregulation of a number of key hippocampal proteins namely the glutamate receptors AMPA (alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) and NMDA (N-methyl-D-aspartate) and other pre and post-synaptic proteins involved in learning and memory processes (VanGuilder and Freeman 2011). A number of these processes, which will be discussed in further detail in the next section, play key roles in the development of ARCD.
The Impact of Ageing on Cognition
Cognition involves complex information processing, planning and reasoning (Buckner and Fridland 2017). During ageing, specific cognitive domains decline such as reasoning, memory and processing speed (Ballesteros et al. 2009). While some of these domains decline throughout the lifespan, some decline only during late life whilst other cognitive domains are relatively preserved (Hedden and Gabrieli 2004). For example, a consequence of ARCD that is widely agreed upon in the field is a decrease in episodic memory which occurs at around 70 years of age (Celsis 2000; Salthouse 2011). In an MRI study consisting of 50 elderly healthy participants, Kramer et al. (2007) reported that decreased grey matter and hippocampal volumes were associated with impaired episodic memory. Furthermore, processing speed and working memory have also been reported to decrease linearly across the lifespan (Hedden and Gabrieli 2004). For example, Meijer et al. (2009) showed that middle-aged adults (50–60 years old) displayed longer reaction times to a word processing task than did younger adults (25–35 years old), demonstrating that this cognitive domain is already susceptible to decline in mid-life. The decline in this cognitive domain can pose an important threat to society as the elderly are at higher risk of car accidents due to their reduced processing speed, executive function and visual processing (Harada et al. 2013). In contrast, short-term memory and tasks involving world knowledge show little decline until late life (Hedden and Gabrieli 2004). In addition, emotional processing is a cognitive function that has been reported to be well preserved into old age (Hedden and Gabrieli 2004). For instance, Mather (2006) reports that older individuals remember positive memories more in comparison to younger adults who recall negative information more, suggesting that age-related changes in processing emotional information dictates which emotional memories are remembered. Moreover, there have been inconsistent reports regarding verbal ability during normal ageing. Longitudinal data, for example, have suggested that verbal ability declines with age particularly after 60 years of age (Hedden and Gabrieli 2004). In contrast, Ballesteros et al. (2009) suggest that verbal ability and general knowledge do not deteriorate with age but instead show improvements, perhaps owing to the compensatory process discussed above. Indeed, Meunier et al. (2014) reported that age-related reductions in grey matter density in language areas lead to functional reorganization of the network via an increase in functional connectivity which results in the recruitment of new areas closely located to core language regions. Overall, these data highlight that ARCD has the potential to negatively impact upon the daily life of those affected and over time can greatly limit the breadth of activities one can perform.
It is important to note that the rate and degree of cognitive decline varies widely across individuals and there are various reasons to explain this. In the same way that there is heterogeneity in the onset and progression of disease, there is heterogeneity in the lifestyle of each individual which can ultimately influence the degree and susceptibility to ARCD development. For example, individuals may have inherent differences in plasticity based on differing life experiences or even due to differing levels of physical activity throughout their lives (Hedden and Gabrieli 2004). Based on their genetic background and epigenetic influences throughout the lifespan, certain individuals are more susceptible to cognitive decline. Education level has also been implicated, with highly educated individuals being less susceptible to cognitive impairment such as memory loss and language ability (Celsis 2000).
Cognition is intimately linked to synaptic function and neuronal activity (VanGuilder and Freeman 2011). During ageing, neuronal synapses at the hippocampus become dysregulated which may be due to morphological alterations of the synapse itself, impaired neurotransmitter signaling or changes in protein and/or gene expression. Decreased synaptic density has been demonstrated in the dentate gyrus and the CA3 region of the hippocampus but not in the CA1 region, as evidenced in aged rats exhibiting a loss of axospinous synapses in these regions (Hara and Morrison 2014). Similarly, in a study of aged female monkeys, there was a reduction in the number of presynaptic boutons and thus, fewer connections made in multiple synapses of the dentate gyrus and this reduction in synaptic strength was correlated to ARCD (Hara et al. 2011). Interestingly, the loss of excitatory synapses is correlated with cognitive disability in aged monkeys, even though the amount of both excitatory and inhibitory synaptic loss during ageing is similar (Petralia et al. 2014). Morisson and Baxter (2012) report that during ageing, the hippocampus loses the large perforated mushroom synapses that modulate learning and memory while the prefrontal cortex loses thin spines, which they suggest causes a decrease in cognitive performance. Interestingly, old rats in which protein kinase C is activated (a protein believed to modulate synaptic growth), demonstrate an increase of mushroom-like spines which improves spatial memory (Petralia et al. 2014). Moreover, synaptic release of neurotransmitters modulates memory, executive function, hormone release and motor function amongst other things and is therefore essential for normal functioning of the central nervous system (Azpurua and Eaton 2015). During ageing, both excitatory and inhibitory neurotransmitters play a role in the destabilization of the synaptic machinery which significantly contributes to cognitive decline. Indeed, changes in the activity of excitatory glutamatergic synapses during ageing, which are heavily implicated in learning and memory, are evidenced via a number of anatomical and chemical mechanisms. Firstly, key hippocampal proteins are decreased in aged mice namely the glutamate receptors AMPA and NMDA, the presynaptic protein SNAP25 and postsynaptic protein PSD95 (Adlard et al. 2010; Canas et al. 2009). In addition, several markers of synaptic plasticity such as CaMKII (calmodulin-dependent kinase II) and synaptophysin have also been shown to decrease in aged mice (Adlard et al. 2014; VanGuilder and Freeman 2011). Smith et al. (2000) reported a decrease in CA3 hippocampal synaptic terminals in aged rats that correlated with the observed deficits in spatial learning, from which they concluded that synaptic loss within the hippocampus is implicated in ARCD. On the other hand, inhibitory neurons function to control overall excitability of neural circuits, thereby maintaining homeostatic balance in the brain (Rozycka and Liguz-Lecznar 2017). During ageing, this control is impaired and therefore contributes to cognitive disability in old age. As such, neuroplasticity therefore functions to overcome these alterations in order to promote homeostatic balance of excitatory and inhibitory activity within the synapse (Rozycka and Liguz-Lecznar 2017). Indeed, impaired GABA signaling has been well characterized in the ageing synapse. In the hippocampus of aged rats, Stanley et al. (2012) observed a decrease in inhibitory dendritic input and reduced GABA release in the entorhinal cortex. Additionally, it has been demonstrated via patch-clamp electrophysiology that the ageing hippocampus exhibits a decrease in inhibitory postsynaptic potential frequency and in the frequency and amplitude of currents mediated by GABA (McQuail et al. 2015). Together, these data suggest that the disruption in synaptic integrity and neurotransmission greatly contributes to ARCD (Petralia et al. 2014). Furthermore, these synaptic alterations further contribute to cognitive decline by prohibiting the induction of long-term potentiation (LTP; one of the primary cellular mechanisms underlying learning and memory) and encouraging long-term depression (LTD; which opposes LTP) (Barnes et al. 2000). For instance, Barnes et al. (2000) found that there was an increased threshold for LTP induction in cognitively impaired old rats compared to middle-aged and young controls, suggesting that reduced NMDA signaling at the synapse causes a decrease in neuroplasticity in aged mice. Over time, this decrease in synaptic health and plasticity results in impaired learning and memory abilities (Celsis 2000). These cognitive deficits are substantiated via a decreased ability of aged mice to perform on the Morris Water maze, a well-established spatial memory task (Adlard et al. 2014). On the other hand, we can genetically modify mice to increase specific proteins that are normally decreased with age in order to enhance learning and memory via increases in LTP. An example of this are mice where the voltage-dependent Ca2+ channel β subunit is knocked out, causing them to exhibit an increase in NMDA receptor expression in the hippocampus and therefore an increase in LTP (Lee and Silva 2009). This was evidenced by increased performance on a range of learning and memory tasks such as novel-object recognition and contextual fear conditioning (Lee and Silva 2009). Another example of this are transgenic mice where increasing CREB levels, a transcription factor regulated by cAMP-PKA/phosphatase signaling, leads to increased memory (Lee and Silva 2009). Indeed, increasing CREB levels is suggested to increase the expression of proteins required for memory and neuroplasticity (Lee and Silva 2009).
Finally, the dysregulation of neuronal synapses observed during ageing are also influenced by age-related changes in gene expression, otherwise known as epigenetics. Ageing individuals undergo changes in histone methylation, acetylation and DNA demethylation which have been reported to modulate the observed age-related changes in synapse loss and structural changes (Azpurua and Eaton 2015). The expression of certain gene categories changes with increasing age. For example, the gene categories involved in the stress response, inflammatory response, metal ion homeostasis, myelin-related proteins and glial genes are all increased during ageing in humans (Bishop et al. 2010). In contrast, those gene categories involved in neural plasticity, synaptic function and inhibitory interneuron function decrease in ageing humans (Bishop et al. 2010). Guan et al. (2009) overexpressed histone deacetylase 2 (HDAC2) in wild-type mice and demonstrated a decrease in the number of synapses, dendritic spine density but also synaptic plasticity and memory formation in the CA1 pyramidal neurons of the hippocampus. Moreover, memory formation has been shown to transiently increase acetylation of histone H3, and the administration of HDAC inhibitors in mice was demonstrated to increase LTP induction (Levenson et al. 2004). Similarly, Peleg et al. (2010) showed that aged mice exhibit changes in acetylation of histone H4 lysine 12 (H4K12) during learning which prevents the expression of specific hippocampal genes that are required for memory consolidation. They further demonstrate that expression of these memory-consolidating genes can be repaired via reestablishment of H4K12 acetylation and that this improves cognitive ability.
How Can We Intervene to Prevent Cognitive Failure with Age?
It is clear, then, that there are a variety of pathways (genetic, environmental, behavioural etc) that can lead to cognitive decline, many of which are no doubt additive in nature. As such, there is a large number of different pharmacological and behavioural approaches that have been used in an attempt to enhance cognitive function with ageing. For instance, increased physical activity, social involvement and taking part in intellectually demanding activities such as computer use, reading and music have shown to increase cognitive performance (Harada et al. 2013). Indeed, physical activity induces proteins that improve cognitive function, learning and memory (Chen et al. 2007) with, for example, profound effects shown on critical proteins such as brain-derived neurotrophic factor (BDNF) in both health and disease (Adlard and Cotman 2004, Adlard et al. 2005a, b). Indeed, BDNF levels were shown to significantly increase following 4 weeks of exercise in rats who also showed improved performance on the Morris water maze, a well characterized learning task (Adlard et al. 2004). Alternatively, cognitive training has been shown to increase individuals’ performance in day to day activities. The advantage to this is that cognitive training is a method accessible to most of the population as it can be delivered within a clinical setting or at home on video (Harada et al. 2013). These behavioural methods of either preserving, or enhancing, cognition with age may also be augmented through the use of targeted therapeutics. In this regard, there have been innumerable compounds tested for their ability to enhance cognition (Froestl et al. 2012, 2013a, b), largely via actions on discrete proteins or signaling cascades, or on other auxiliary pathways that may either directly or indirectly influence cognition (such as through the use of growth factors to enhance neuronal health and function with the hope of a downstream impact on gross function).
With the number of possibilities open to testing, the use of techniques such as gene profiling in the hippocampus during aging, for example, has enabled the more targeted study of specific pathways that impact upon cognitive function (VanGuilder and Freeman 2011). These advancements in the field have allowed for the identification of specific molecules and pathways that are dysfunctional or altered during ageing and which may represent tractable therapeutic targets.
Finally, whilst efforts over many decades have gone into targeting specific hippocampal proteins that are dysregulated in ageing, recent evidence suggests that there may be a crucial link between zinc levels in the brain and cognitive function. Specifically, the decline in cognitive performance observed during ageing has been linked to the dysregulation of zinc homeostasis (Hancock et al. 2014; Mocchegiani et al. 2006).
Is Zinc Dyshomeostasis a Common Pathway for Age-Related Cognitive Decline?
Zinc is essential for the normal functioning of the brain and there is an increasing body of evidence to suggest that zinc plays an important role in ARCD. The levels of zinc in the brain are tightly controlled as it is essential for various cellular and molecular processes, enzymatic activity and transcription factors (Levenson and Tassabehji 2007). In fact, the concentration of zinc in the brain surpasses that of the body by ten-fold (Portbury and Adlard 2017). While the majority of zinc is bound to proteins or amino acids, around 10% of zinc remains as chelatable, or free, zinc (Levenson and Tassabehji 2007). Under normal conditions, chelatable zinc is located within vesicles of presynaptic glutamatergic neurons and therefore plays a crucial role in modulating neuroplasticity and cognition (Besser et al. 2009). Approximately 300 μM of zinc is released with glutamate into the synapse upon depolarization (Assaf and Chung 1984; Frederickson et al. 2005). Zinc acts via a range of transporters, ion channels (such as voltage-gated Ca2+ channels), synaptic receptors namely the AMPA and NMDA receptors and post-synaptic factors such as TrkB for example (Besser et al. 2009), in order to mediate synaptic communication and neuroplasticity (Paoletti et al. 2009).
Under healthy conditions, zinc levels are maintained by a variety of zinc transporters and other transporter proteins that balance its export/import into discrete cellular compartments (in fact, when these transporters are decreased or dysfunctional, significant pathologies can result) (Kambe et al. 2015). Of particular relevance here is the zinc transporter-3 protein (ZnT3), which was shown to be responsible for loading zinc into pre-synaptic vesicles in the hippocampus (Cole et al. 1999, Linkous et al. 2008, Palmiter et al. 1996). For example, Saito et al. (2000) demonstrated that reduced ZnT3 expression causes a decrease in synaptic zinc levels in the hippocampal mossy fiber pathway. This decreased synaptic zinc leads to excitotoxicity due to excessive glutamate release ultimately impairing learning and memory. Furthermore, studies in mice exhibiting accelerated ageing present with brain atrophy and impaired learning and memory functions probably due to reduced zinc levels at the synapse (Saito et al. 2000). Indeed, studies have shown that the onset of age-related cognitive decline is influenced by ZnT3 mutations (Rocha et al. 2014). Impaired memory has also been observed in ZnT3 knock out mice and there is a reduction in ZnT3 protein levels in the cortex of aged mice and humans (in both health and disease) (Adlard et al. 2010).
Furthermore, metallothioneins (MTs) which are molecules that bind zinc and other metals, have been shown to increase with age. Because plasma levels of zinc decrease with age, this suggests that MTs retain zinc intracellularly thereby causing an overall decrease in extracellular zinc (Mocchegiani et al. 2006). However, although both vesicular and synaptic zinc levels have been demonstrated to reduce with increasing age, the overall amount of zinc is unchanged in ageing humans and rodents (Takahashi et al. 2001).
It is becoming increasingly apparent that zinc dyshomeostasis may play an important role in ARCD. As such, several pharmacological strategies that target zinc have been assessed for their therapeutic efficacy in both cognitive ageing and a range of neurodegenerative diseases over the last decade. An example of this is a recent study that showed that PBT2 (Prana Biotechnology), an 8-hydroxy quinolone that functions as a cellular chaperone for zinc, works by redistributing zinc within the brain and increasing the amount of total zinc within the hippocampus of aged mice (Adlard et al. 2014). This lead to an improvement in cognitive performance, neurogenesis and increased synaptic plasticity that was underscored by changes in a suite of relevant synaptic proteins (Adlard et al. 2014). A related compound, clioquinol, was similarly shown to restore cognition in ZnT3 KO mice (Adlard et al. 2015). These studies corroborate the idea that redistribution of zinc within different compartments of the brain, and therefore re-establishment of zinc ion homeostasis may prove to be a valuable avenue for preventing age-related cognitive decline.
Zinc dyshomeostasis has also been shown to have a number of implications for other age-related and non-ageing diseases. For example, zinc dyshomeostasis has been identified as a key player in the onset and progression of AD. In fact, PBT2 treatment of transgenic mice exhibiting an AD-like phenotype has previously been shown to improve cognitive function and to reduce disease pathology (Adlard et al. 2008). Further, zinc has also been implicated in the cognitive failure associated with non-ageing disorders such as schizophrenia, depression, attention-deficit hyperactivity-disorder (ADHD) and malnutrition in children. Research has shown that malnutrition in young infants may lead to zinc deficiency and that dietary zinc supplements may have favorable effects on cognitive performance (Black 2003). Zinc deficiency has also been linked to schizophrenia, ADHD and depression. In depression, when low plasma zinc levels are restored, the depressive symptoms are reduced (Levenson and Tassabehji 2007). Similarly, Scarr et al. (2016) increased the expression of zinc transporter SLC39A12 (which was shown to be higher in schizophrenia patients in comparison to controls) in CHO cells and found an increased uptake of zinc in these cells, suggesting that zinc homeostasis may be altered in schizophrenia due to dysregulation of the SLC39A12 zinc transporter. Finally, ADHD is caused by the dysfunction of the dopamine transporter which has been shown to have a high affinity binding site for zinc (Lepping and Huber 2010). As zinc binds to the dopamine receptor, this causes inhibition of dopamine re-uptake. There has been cumulative evidence to suggest that zinc deficiency is involved in ADHD such that various groups have reported that children with ADHD have decreased plasma zinc levels and that this zinc deficiency is positively correlated with more severe ADHD symptoms (Arnold et al. 2005). Taken together, these data highlight the impact that zinc dyshomeostasis can have on cognitive decline and demonstrate its potential as a therapeutic target in the prevention of age-related cognitive decline.
Summary and Conclusion
Currently, there is no treatment available for age-related cognitive decline. This is due to the fact that our current knowledge on normal ageing is still relatively limited, perhaps because there is more urgency in understanding the pathology of neurodegenerative diseases. However, future research into the biological implications of ageing will help reveal what aberrant processes contribute to cognitive decline in both normal and pathological ageing. As outlined in this chapter, one avenue under investigation is zinc, which may represent a tractable therapeutic target for a number of disorders where a failure in zinc ion homeostasis is implicated either more generally in the pathogenesis of disease or specifically in the cognitive deficits associated with the condition. While the research is ongoing, immediate efforts should work to maximize and preserve cognitive function in order to improve the quality of life of those affected.
References
Adlard P, Cotman C (2004) Voluntary exercise protects against stress-induced decreases in brain-derived neurotrophic factor protein expression. Neuroscience 124:985–992
Adlard PA, Perreau VM, Engesser-Cesar C, Cotman CW (2004) The timecourse of induction of brain-derived neurotrophic factor mRNA and protein in the rat hippocampus following voluntary exercise. Neurosci Lett 363:43–48
Adlard PA, Perreau VM, Cotman CW (2005a) The exercise-induced expression of BDNF within the hippocampus varies across life-span. Neurobiol Aging 26:511–520
Adlard PA, Perreau VM, Pop V, Cotman CW (2005b) Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease. J Neurosci 25:4217–4221
Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E et al (2008) Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Aβ. Neuron 59:43–55
Adlard PA, Parncutt JM, Finkelstein DI, Bush AI (2010) Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J Neurosci 30:1631–1636
Adlard PA, Sedjahtera A, Gunawan L, Bray L, Hare D et al (2014) A novel approach to rapidly prevent age-related cognitive decline. Aging Cell 13:351–359
Adlard PA, Parncutt J, Lal V, James S, Hare D et al (2015) Metal chaperones prevent zinc-mediated cognitive decline. Neurobiol Dis 81:196–202
Armanios M, Alder JK, Parry EM, Karim B, Strong MA, Greider CW (2009) Short telomeres are sufficient to cause the degenerative defects associated with aging. Am J Hum Genet 85:823–832
Arnold LE, Bozzolo H, Hollway J, Cook A, DiSilvestro RA et al (2005) Serum zinc correlates with parent-and teacher-rated inattention in children with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol 15:628–636
Assaf S, Chung S-H (1984) Release of endogenous Zn2+ from brain tissue during activity. Nature 308:734
Azpurua J, Eaton BA (2015) Neuronal epigenetics and the aging synapse. Front Cell Neurosci 9:208
Baker DJ, Dawlaty MM, Wijshake T, Jeganathan KB, Malureanu L et al (2013) Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat Cell Biol 15:96
Ballesteros S, Nilsson L-G, Lemaire P (2009) Ageing, cognition, and neuroscience: an introduction. Eur J Cogn Psychol 21:161–175
Barnes C, Rao G, Houston F (2000) LTP induction threshold change in old rats at the perforant path–granule cell synapse. Neurobiol Aging 21:613–620
Besser L, Chorin E, Sekler I, Silverman WF, Atkin S et al (2009) Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J Neurosci 29:2890–2901
Bigby C (2004) Ageing with a lifelong disability: a guide to practice, program, and policy issues for human services professionals. Jessica Kingsley, London
Bishop NA, Lu T, Yankner BA (2010) Neural mechanisms of ageing and cognitive decline. Nature 464:529
Black MM (2003) The evidence linking zinc deficiency with children’s cognitive and motor functioning, 2. J Nutr 133:1473S–1476S
Buckner C, Fridland E (2017) What is cognition? angsty monism, permissive pluralism (s), and the future of cognitive science. Springer, Heidelberg
Canas PM, Duarte JM, Rodrigues RJ, Köfalvi A, Cunha RA (2009) Modification upon aging of the density of presynaptic modulation systems in the hippocampus. Neurobiol Aging 30:1877–1884
Celsis P (2000) Age-related cognitive decline, mild cognitive impairment or preclinical Alzheimer’s disease? Ann Med 32:6–14
Chen WQ, Viidik A, Skalicky M, Höger H, Lubec G (2007) Hippocampal signaling cascades are modulated in voluntary and treadmill exercise rats. Electrophoresis 28:4392–4400
Cole TB, Wenzel HJ, Kafer KE, Schwartzkroin PA, Palmiter RD (1999) Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc Natl Acad Sci 96:1716–1721
Deary IJ, Corley J, Gow AJ, Harris SE, Houlihan LM et al (2009) Age-associated cognitive decline. Br Med Bull 92:135–152
Fjell AM, Walhovd KB (2010) Structural brain changes in aging: courses, causes and cognitive consequences. Rev Neurosci 21:187–222
Fontana L, Partridge L, Longo VD (2010) Extending healthy life span—from yeast to humans. Science 328:321–326
Frederickson CJ, Koh J-Y, Bush AI (2005) The neurobiology of zinc in health and disease. Nat Rev Neurosci 6:449
Froestl W, Muhs A, Pfeifer A (2012) Cognitive enhancers (nootropics). Part 1: drugs interacting with receptors. J Alzheimers Dis 32:793–887
Froestl W, Muhs A, Pfeifer A (2013a) Cognitive enhancers (nootropics). Part 2: Drugs interacting with enzymes. J Alzheimers Dis 33:547–658
Froestl W, Pfeifer A, Muhs A (2013b) Cognitive enhancers (nootropics). Part 3: drugs interacting with targets other than receptors or enzymes. disease-modifying drugs. J Alzheimers Dis 34:1–114
Guan J-S, Haggarty SJ, Giacometti E, Dannenberg J-H, Joseph N et al (2009) HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459:55
Hancock SM, Finkelstein DI, Adlard PA (2014) Glia and zinc in ageing and Alzheimer’s disease: a mechanism for cognitive decline? Front Aging Neurosci 6:137
Hara Y, Morrison JH (2014) Synaptic correlates of aging and cognitive decline. In: The synapse: structure and function. Elsevier Inc, Amsterdam, pp 301–342
Hara Y, Park CS, Janssen WG, Punsoni M, Rapp PR, Morrison JH (2011) Synaptic characteristics of dentate gyrus axonal. boutons and their relationships with aging, menopause, and memory in female rhesus monkeys. J Neurosci 31:7737–7744
Harada CN, Love MCN, Triebel KL (2013) Normal cognitive aging. Clin Geriatr Med 29:737–752
Head D, Buckner RL, Shimony JS, Williams LE, Akbudak E et al (2004) Differential vulnerability of anterior white matter in nondemented aging with minimal acceleration in dementia of the Alzheimer type: evidence from diffusion tensor imaging. Cereb Cortex 14:410–423
Hedden T, Gabrieli JD (2004) Insights into the ageing mind: a view from cognitive neuroscience. Nat Rev Neurosci 5:87
Hulette CM, Welsh-Bohmer KA, Murray MG, Saunders AM, Mash DC, McIntyre LM (1998) Neuropathological and neuropsychological changes in “normal” aging: evidence for preclinical Alzheimer disease in cognitively normal individuals. J Neuropathol Exp Neurol 57:1168–1174
Jagust W (2013) Vulnerable neural. systems and the borderland of brain aging and neurodegeneration. Neuron 77:219–234
Kambe T, Tsuji T, Hashimoto A, Itsumura N (2015) The physiological, biochemical, and molecular roles of zinc transporters in zinc homeostasis and metabolism. Physiol Rev 95:749–784
Kinsella K, Velkoff VA (2002) The demographics of aging. Aging Clin Exp Res 14:159–169
Kirkwood TB (2005) Understanding the odd science of aging. Cell 120:437–447
Kramer JH, Mungas D, Reed BR, Wetzel ME, Burnett MM et al (2007) Longitudinal MRI and cognitive change in healthy elderly. Neuropsychology 21:412
Lee Y-S, Silva AJ (2009) The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci 10:126
Lepping P, Huber M (2010) Role of zinc in the pathogenesis of attention-deficit hyperactivity disorder. CNS Drugs 24:721–728
Levenson C, Tassabehji N (2007) Role and regulation of copper and zinc transport proteins in the central nervous system. In: Handbook of neurochemistry and molecular neurobiology. Springer, New York, pp 257–284
Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD (2004) Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem 279:40545–40559
Linkous DH, Flinn JM, Koh JY, Lanzirotti A, Bertsch PM et al (2008) Evidence that the ZNT3 protein controls the total amount of elemental zinc in synaptic vesicles. J Histochem Cytochem 56:3–6
López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153:1194–1217
Lunenfeld B (2008) An Aging World – demographics and challenges. Gynecol Endocrinol 24:1–3
Madden DJ, Spaniol J, Costello MC, Bucur B, White LE et al (2008) Cerebral white matter integrity mediates adult age differences in cognitive performance. J Cogn Neurosci 21:289–302
Maggio M, Dall’Aglio E, Lauretani F, Cattabiani C, Ceresini G et al (2012) The hormonal pathway to cognitive impairment in older men. J Nutr Health Aging 16:40–54
Mather M (2006) Why memories may become more positive as people age. Blackwell Publishing, Malden
McQuail JA, Frazier CJ, Bizon JL (2015) Molecular aspects of age-related cognitive decline: the role of GABA signaling. Trends Mol Med 21:450–460
Meijer WA, de Groot RH, van Gerven PW, van Boxtel MP, Jolles J (2009) Level of processing and reaction time in young and middle-aged adults and the effect of education. Eur J Cogn Psychol 21:216–234
Meunier D, Stamatakis EA, Tyler LK (2014) Age-related functional reorganization, structural changes, and preserved cognition. Neurobiol Aging 35:42–54
Mocchegiani E, Costarelli L, Giacconi R, Cipriano C, Muti E et al (2006) Zinc homeostasis in aging: two elusive faces of the same “metal”. Rejuvenation Res 9:351–354
Morisson JH, Baxter MG (2012) The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci 13:240
Moskalev AA, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Budovsky A et al (2013) The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res Rev 12:661–684
Palm W, de Lange T (2008) How shelterin protects mammalian telomeres. Annu Rev Genet 42:301–334
Palmiter RD, Cole TB, Quaife CJ, Findley SD (1996) ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc Natl Acad Sci 93:14934–14939
Paoletti P, Vergnano A, Barbour B, Casado M (2009) Zinc at glutamatergic synapses. Neuroscience 158:126–136
Park DC, Polk TA, Park R, Minear M, Savage A, Smith MR (2004) Aging reduces neural specialization in ventral visual cortex. Proc Natl Acad Sci U S A 101:13091–13095
Peleg S, Sananbenesi F, Zovoilis A, Burkhardt S, Bahari-Javan S et al (2010) Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 328:753–756
Peters R (2006) Ageing and the brain. Postgrad Med J 82:84–88
Petralia RS, Mattson MP, Yao PJ (2014) Communication breakdown: the impact of ageing on synapse structure. Ageing Res Rev 14:31–42
Plassman BL, Langa KM, Fisher GG, Heeringa SG, Weir DR et al (2007) Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology 29:125–132
Plassman BL, Langa KM, Fisher GG, Heeringa SG, Weir DR et al (2008) Prevalence of cognitive impairment without dementia in the United States. Ann Intern Med 148:427–434
Portbury SD, Adlard PA (2017) Zinc signal in brain diseases. Int J Mol Sci 18:2506
Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE (2009) Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 78:959–991
Reuter-Lorenz PA (2002) New visions of the aging mind and brain. Trends Cogn Sci 6:394–400
Robertson DA, Savva GM, Kenny RA (2013) Frailty and cognitive impairment—a review of the evidence and causal mechanisms. Ageing Res Rev 12:840–851
Rocha TJ, Blehm CJ, Bamberg DP, Fonseca TLR, Tisser LA et al (2014) The effects of interactions between selenium and zinc serum concentration and SEP15 and SLC30A3 gene polymorphisms on memory scores in a population of mature and elderly adults. Genes Nutr 9:377
Rogalski E, Stebbins G, Barnes C, Murphy C, Stoub T et al (2012) Age-related changes in parahippocampal white matter integrity: a diffusion tensor imaging study. Neuropsychologia 50:1759–1765
Rozycka A, Liguz-Lecznar M (2017) The space where aging acts: focus on the GABAergic synapse. Aging Cell 16(4):634–643
Saito T, Takahashi K, Nakagawa N, Hosokawa T, Kurasaki M et al (2000) Deficiencies of hippocampal Zn and ZnT3 accelerate brain aging of Rat. Biochem Biophys Res Commun 279:505–511
Salthouse TA (2011) Neuroanatomical substrates of age-related cognitive decline. Psychol Bull 137:753
Scarr E, Udawela M, Greenough MA, Neo J, Seo MS et al (2016) Increased cortical expression of the zinc transporter SLC39A12 suggests a breakdown in zinc cellular homeostasis as part of the pathophysiology of schizophrenia. NPJ Schizophrenia 2:16002
Shing YL, Rodrigue KM, Kennedy KM, Fandakova Y, Bodammer N et al (2011) Hippocampal subfield volumes: age, vascular risk, and correlation with associative memory. Front Aging Neurosci 3:2
Smith TD, Adams MM, Gallagher M, Morrison JH, Rapp PR (2000) Circuit-specific alterations in hippocampal synaptophysin immunoreactivity predict spatial learning impairment in aged rats. J Neurosci 20:6587–6593
Stanley EM, Fadel JR, Mott DD (2012) Interneuron loss reduces dendritic inhibition and GABA release in hippocampus of aged rats. Neurobiol Aging 33:431. e1–31. e13
Takahashi S, Takahashi I, Sato H, Kubota Y, Yoshida S, Muramatsu Y (2001) Age-related changes in the concentrations of major and trace elements in the brain of rats and mice. Biol Trace Elem Res 80:145–158
United Nations, Department of Economic and Social Affairs, Population Division (2017) World population prospects: the 2017 revision, key findings and advance tables. Working Paper No. ESA/P/WP/248
VanGuilder H, Freeman W (2011) The hippocampal neuroproteome with aging and cognitive decline: past progress and future directions. Front Aging Neurosci 3:8
Vincent GK, Velkoff VA (2010) The next four decades: the older population in the United States: 2010 to 2050. US Department of Commerce, Economics and Statistics Administration, US Census Bureau
Yassa MA, Stark SM, Bakker A, Albert MS, Gallagher M, Stark CE (2010) High-resolution structural and functional MRI of hippocampal CA3 and dentate gyrus in patients with amnestic mild cognitive impairment. NeuroImage 51:1242–1252
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Juan, S.M.A., Adlard, P.A. (2019). Ageing and Cognition. In: Harris, J., Korolchuk, V. (eds) Biochemistry and Cell Biology of Ageing: Part II Clinical Science. Subcellular Biochemistry, vol 91. Springer, Singapore. https://doi.org/10.1007/978-981-13-3681-2_5
Download citation
DOI: https://doi.org/10.1007/978-981-13-3681-2_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-3680-5
Online ISBN: 978-981-13-3681-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)