
Abstract
Thermal and noxious stimuli are detected by specialized nerve endings, which transform the stimuli into electrical signals and transmit the signals into central nervous system to facilitate the perception of temperature and pain. Several members within the transient receptor potential (TRP) channel family serve as the sensors for temperature and noxious stimuli and are involved in the development of pathological pain, especially inflammatory pain. Various inflammatory mediators can sensitize and modulate the activation threshold of TRP channels and result in the development of inflammatory pain behaviors. A brief review of the role of TRP channels in nociception and the modulatory mechanisms of TRP channels by inflammatory mediators, focusing on TRPV1, TRPA1, and TRPM2, will be presented. Recent advances in the development of therapeutic strategies targeting against TRP channels will also be reviewed.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
2.1 Introduction
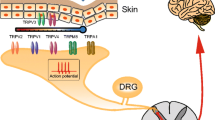
“Pain,” the word that comes from Greek goddess of revenge, Poine, describes an unpleasant experience that is elicited by noxious stimuli. Such experience often serves as a warning flag and reminds an individual of avoiding or eliminating the encountered threats. Pain can be divided into three categories including nociceptive pain, inflammatory pain, and neuropathic pain [66]. Nociceptive pain is generated by specialized nerve endings (nociceptors) with a relatively high activation threshold compared with those responsible for the sensation of light, sound, smell, and taste. Nociceptors serve as sensors for strong mechanical stimuli, chemical irritants, and noxious thermal stimuli, and only stimuli that are potentially capable of causing tissue injuries can reach the threshold to activate nociceptive nerve terminals and generate actions potentials, which are then transmitted and perceived as pain signals [21]. Transformation of these stimuli into electrical signals and transduction of the action potentials involve the participation of multiple receptors and channels located on free nerve endings and synaptic terminals. As a group of multimodal cation-permeable channels that depolarize the cells, TRP channels are involved in various aspects of physiological function, including nociception [70].
TRP channels were first discovered in Drosophila melanogaster and viewed as mutant structures exhibiting only a transient receptor potential (TRP) rather than normal sustained potential in response to light [16]. Later on, mammalian homologs of TRP channels were discovered [89], which then opens the opportunity for further investigation on the functional roles of TRP channels. To date, 28 mammalian TRP channels have been identified and are divided into 6 subfamilies according to their sequence homology: canonical or classic (TRPC), vanilloid (TRPV), melastatin (TRPM), polycystin (TRPP), mucolipin (TRPML), and ankyrin (TRPA) [77]. Among them, several types of TRP channels have been found to be involved in the generation of pain, including TRPV1 [13], TRPV2 [93], TRPV3 [58], TRPV4 [80], TRPA1 [60, 91], TRPM2 [30], and TRPM8 [27]. Each channel has its unique characteristics and contributes to the generation of various pain behaviors including heat hyperalgesia, mechanical hyperalgesia, cold allodynia, and inflammatory hyperalgesia.
As for the roles of TRP channels in inflammatory pain, inflammatory mediators can sensitize or alter the threshold of TRP channels, leading to pain behaviors including thermal hyperalgesia, mechanical allodynia, and spontaneous pain [41]. In this review, we will focus on the molecular mechanisms of TRP channel modulation in the generation of nociception and the development of inflammatory pain, focusing on TRPV1, TRPA1, and TRPM2.
2.2 TRPV1
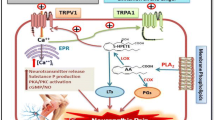
Among the members within the TRP channel family, TRPV1 is the one that has been most thoroughly investigated, and its pivotal role in sensing noxious stimuli and generating pain in primary afferent nociceptors has also been demonstrated across different studies [41]. Since the successful cloning of TRPV1 in 1997, follow-up studies have identified TRPV1 as a cation-permeable channel which is responsive to thermal stimuli in the range of noxious heat (over 43 °C) [82] and changes in pH [19]. As a polymodal channel, TRPV1 can also be activated by vanilloids (e.g., capsaicin from chili peppers or anandamide from inflammation process) [72], vanillotoxins [73], and protons [3]. The finding of TRPV1 being expressed almost exclusively in C-fibers indicates its role as a sensor for noxious stimuli [46]. Furthermore, the activity of TRPV1 can be enhanced by a variety of inflammatory mediators, including bradykinin, ATP, and nerve growth factor (NGF), through second messenger-signaling pathways such as phospholipase C (PLC) and protein kinase A (PKA) [56]. The sensitization and activation of TRPV1 in peripheral nociceptors lead to the transmission of the noxious signals to the central nervous system and, hence, the production of unpleasant and painful sensation warning the body of potentially harmful threat [41, 66].
The success in the application of cryo-microscopy to understand the molecular structure of TRPV1 has enabled us to gain deeper understanding in the gating mechanism of TRP channels [12]. TRPV1 is composed of four identical protein subunits assembled into a functional and cation-permeable channel [12]. Each subunit contains six transmembrane segments, a loop constructing pore helix between segment five and six, and intracellular N- and C-termini with two restriction points in the pore helix defined as the selectivity filter and the lower gate [12, 28]. During inactive state, both the selectivity filter and the lower gate are constricted, and the pathway for ion conduction is blocked. An intracellularly located hydrophobic pocket, the so-called vanilloid pocket, is composed of the external surface of the S3–S4 helices, S4–S5 linkers, and S6 helix [12, 32]. It allows small vanilloid molecules, such as resiniferatoxin (RTX) and capsaicin, to cross the plasma membrane to bind and allosterically modulate the pore, more precisely, expanding the lower gate of the pore domain. As for the extracellular outer pore region, the binding of chemicals, such as double-knot toxin (DkTx), or stimulation with thermal stimuli cause substantial conformational changes of TRPV1, resulting in marked change in the relative position of the pore helix in the outer pore region. The change of the relative position of the pore helix may also break down the potential hydrogen bonding formed between the amino acids on the chains within the outer pore region in resting state resulting in the widening of the selectivity filter. It is rather remarkable that the upper and lower gates could be allosterically coupled and regulate the activation of the channel. Such synergy between different levels of gates could contribute to the coordination of disparate physiologic signals [12].
Under physiological condition, TRPV1 has been shown to be co-expressed with PKCβII in a subset of sensory neurons. In these neurons, TRPV1 binds directly to PKCβII, which in return markedly enhances the responses of TRPV1 by phosphorylating TRPV1 at T705 [52]. The differences in the basal phosphorylation of TRPV1 at T705 may explain the differences in the threshold of TRPV1-expressing neurons to heat stimuli [45]. In addition, TRPV1-PKCβII complex-containing neurons have been suggested to represent a subset of hypersensitive nociceptive neurons [95]. Not only does TRPV1 play a pivotal role in generating proportionate pain under physiological condition, it also contributes to the generation of action potentials during inflammation, leading to pathological pain behaviors such as thermal hyperalgesia, spontaneous pain, and mechanical allodynia [7, 13, 56]. In response to tissue damages, numerous inflammatory mediators such as eicosanoids (e.g., prostaglandin E2), neuropeptides (e.g., substance P and bradykinin), excitatory amino acids (e.g., glutamate), leukotrienes, and cytokines (e.g., TNF-α, IL-6and INF-γ) [90] are released, and the inflammatory mediators lower the mechanical and thermal thresholds of the exposed sensory neurons, a process called “sensitization” [41]. Besides acting as a downstream target of proalgesic factors, TRPV1 itself can also trigger the secretion of neuropeptides, including substance P and calcitonin gene-related peptide (CGRP) upon activation [39, 91]. The neuropeptides secreted then bind to specific receptors expressed on the surrounding cells, such as lymphocytes, dendritic cells, mast cells, and macrophages, which then trigger a series of reactions involved in immune responses [4]. This process is known as neurogenic inflammation, and the involvement of TRPV1 in the development of inflammatory pain is clearly demonstrated by the significantly reduced thermal hyperalgesia in TRPV1 knockout mice after tissue injury [13, 18].
The mechanisms responsible for the exaggerated response of TRPV1 during inflammatory state are associated with protein kinases [8], which modulate the activities of proteins through phosphorylation. Phosphorylation of TRPV1 can be facilitated by inflammatory mediators through multiple protein kinases including cyclic AMP-dependent protein kinase (PKA) and protein kinase C (PKC), phosphatidylinositol-3 kinase (PI3K), Ca2+/calmodulin-dependent kinase II (CaMKII), and extracellular signal-regulated protein kinase/mitogen-activated protein kinase (ERK/MAPK) [56]. Receptors of several inflammatory mediators, including prostaglandin receptors (e.g., prostaglandin E2 receptor 2, prostaglandin E2 receptor 4, I prostanoid receptor, and prostaglandin D2 receptor 1), 5-hydroxytryptamine(5-HT) receptors and endothelin ETA receptors, are G protein-coupled receptors (GPCRs) which are coupled to the Gs type of Gα subunit [54]. When these inflammatory mediators bind to GPCRs, adenylyl cyclases are activated and cause increase of intracellularly cAMP level and full activation of PKA [78].
Another mechanism modulating the function of TRPV1 is through the PLC/PKC pathway. Receptors for inflammatory mediators such as histamine H1, bradykinin B2, protease-activated receptor-2 (PAR2), prostaglandin E2 receptor 1, substance P, neurokinin 1 (NK1), and purinergic P2Y are also GPCRs. Instead of being coupled to Gs type of Gα subunit, they are coupled to Gαq type of Gα subunit which then initiate the activation of phospholipase C (PLC). The activation of PLC leads to the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) and results in the production of two second messengers: 1,4,5-triphosphate (IP3) and diacylglycerol (DAG) [25]. Some studies showed results suggesting that PIP2 causes inactivation or desensitization of TRPV1, and the hydrolysis of PIP2 by PLC results in the activation of TRPV1 [25, 69]. However, there are conflicting results showing that direct application of PIP2 causes TRPV1 activation and absence of PIP2 results inTRPV1 inactivation [71]. Another study showed that TRPV1 can be fully functional in the absence of PIP2, suggesting that PIP2 contributes to the sensitization of TRPV1 by disinhibiting the channel [11]. In addition, PIP2 was shown to activate TRPV1 even in the absence of capsaicin, though the current intensity was much smaller than that elicited by capsaicin. The result suggests that PIP2 is a positive regulator of TRPV1 and the difference of amplitude caused by the presence of capsaicin may indicate that PIP2 serves as a cofactor rather than pure agonist of TRPV1 [44]. The lack of conclusion for the role of PIP2 on the regulation of TRPV1 indicates the sophisticated modulation of TRPV1. Meanwhile, DAG stimulates PKC and subsequently leads to the activation of TRPV1 [25, 35]. However, one study showed that 1-oleoyl-2-acetyl-sn-glycerol (OAG), an analog of DAG, causes TRPV1 activation in rat dorsal root ganglion neurons in the presence of chelerythrine, a PKC inhibitor, suggesting that DAG is a direct endogenous ligand of TRPV1 and the TRPV1 activation induced by OAG is independent of PKC. In addition, the study also showed that the binding site of DAG is similar to that of capsaicin, though the effect of DAG on TRPV1 is much smaller than that of capsaicin [92]. All these results indicate the involvement of Gαq-PLC pathway in the modulation of TRPV1 by inflammatory mediators.
The TRPV1 modulatory mechanisms mentioned above are basically at posttranslational level, which includes phosphorylation and proteolysis of protein subunits, resulting in the change of the activity of TRPV1. However, inflammatory mediators have a more comprehensive effect on TRPV1. For example, NGF, a neurotrophic factor that binds to tropomyosin receptor kinase A (trkA) [74], modulate TRPV1 through several different aspects, including level of transcription, translation, and posttranslation, and such interactions are also suggested to be responsible for the development of thermal hyperalgesia during inflammation [51]. One study showed an increased level of NGF and higher percentage of neurons expressing TRPV1 following inflammation induced by intraplantar injection of Freund’s complete adjuvant, and inhibiting the effect of NGF with anti-NGF was shown to prevent the increased TRPV1 expression within trk-A positive neurons and lessen the thermal hyperalgesia induced by inflammation [1]. These results indicate the mechanisms modulating TRPV1 at transcription or translation level.
2.3 TRPA1
As a member of TRP channel family, TRPA1 and TRPV1 share some common features, including the similarity in the structures consisting of six transmembrane domains with intracellular N- and C-termini and ion permeability to cation nonselectively. Within vertebrate TRP channel family, TRPA1 is characterized by a long N-terminus with multiple ankyrin repeats along with three critical cysteine residues. These three cysteine residues are located in the linker region connecting the ankyrin-rich domain to the transmembrane domain and are involved in the channel activation by electrophiles [34, 41, 67]. In addition, TRPV1 and TRPA1 are co-expressed in a specific subgroup of dorsal root ganglion neurons that is responsible for the detection and transduction of noxious stimuli [23]. Interestingly, TRPA1 was shown recently, to work together with TRPV1 and TRPM3 for acute noxious heat sensing in mice [84].
Like TRPV1, TRPA1 is also a polymodal receptor and has been shown to be activated by numerous natural pungent chemicals, including allyl isothiocyanate (AITC), cinnamaldehyde, and allicin, which are all electrophiles [85]. One intriguing question is the mechanism by which these structurally diverse electrophiles serve as specific agonists for TRPA1 activation and modulation. Instead of structural specificity, the activation of TRPA1 by the pungent chemicals depends on the covalent modification of cysteine residues on the N-terminal of the channel. The covalent modification causes conformational change in protein structure and modulates the channel permeability [34, 57].
Furthermore, presence of calcium ions may also play a pivotal role in TRPA1 modulation. TRPA1 currents evoked by some agonists, such as AITC and cinnamaldehyde, were shown to be potentiated in the presence of extracellular Ca2+ [40, 63]. Meanwhile, TRPA1 activation by icilin requires the presence of calcium, and adding BAPTA (an intracellular calcium chelator) to the pipette solution significantly reduces icilin-evoked currents. This indicates that intracellular calcium serves as a co-agonist of TRPA1 [20, 87]. Modulation of intracellular calcium on TRPA1 through direct binding to an EF-hand-like motif within intracellular N-terminus has been suggested by several studies [20, 96]. However, some other studies suggest that the activation might be contributed by Ca2+-binding protein calmodulin [31]. Both suggestions have been demonstrated in genetic deletion model with both positive and negative results [20, 31, 87]. These divergent results implie that the underlying mechanism is complicated and further revaluation is needed.
Besides pungent chemicals, TRPA1 can also be activated by environmental irritants, such as acrolein [5], formalin [60], and metabolic by-products of chemotherapeutic agents. The activation of the TRPA1-expressing C-fibers in the respiratory tract and bladder was proposed to be associated with the development of airway and urinary tract symptoms, as evidenced by the findings showing that TRPA1 agonists evoke coughing in both guinea pig and human volunteers and TRPA1 antagonists attenuate symptoms of cyclophosphamide-induced hemorrhagic cystitis [9, 61].
Apart from the activation by the chemicals mentioned above, TRPA1 is activated or potentiated during inflammatory state as well. During tissue injury, the reactive oxygen species generated cause superoxidation of membrane phospholipids and result in the production of 4-hydroxy-2-nonenal (HNE), which then causes activation of TRPA1 [2, 83]. The inhibition of the pain-related behaviors elicited by 4-HNE injection with TRPA1 antagonists and the absence of the pain-related behaviors in TRPA1-deficient mice demonstrate the importance of TRPA1 in mediating the effect of 4-HNE in the development inflammatory pain [2, 83]. In addition, the binding of bradykinin to bradykinin receptor B2 causes activation of PLC and PKA, and results in the enhancement of the TRPA1 current activated by AITC or cinnamaldehyde [86]. All the results above indicate the involvement of TRPA1 in the induction of acute pain and hyperalgesia during inflammation.
2.4 TRPM2
Interactions between neurons and immune cells contribute substantially to the initiation of pathological pain, in which neurogenic inflammation and generation of reactive oxygen species and reactive nitrogen species (ROS/RNS) are of fundamental importance. TRPM2 is a member within TRP channel family that plays a crucial role in serving as the downstream target of ROS/RNS [42] and can be activated by micromolar levels of H2O2 and agents producing ROS/RNS [29, 88]. TRPM2 is a cation channel characterized by a nudix hydrolase (NUDT9) homology region in the intracellular C-terminus, which was suggested to be responsible for channel activation and modulation by intracellular adenosine diphosphate ribose (ADPR) [48, 68]. In addition, cADPR and NAADP have synergistic effect with ADPR, as evidenced by the finding showing that the EC50 for cADPR and NAADP decrease significantly from 44 to 3 μM and from 95 to 1 μM, respectively, in the presence of subthreshold levels of ADPR (100 nM) [49]. However, whether they bind directly to the Nudix box motif as ADPR or to distinct synergetic sites or had been converted to ADPR beforehand remains unclear. ADPR can also be produced extracellularly. Extracellular NAD+ can be catalyzed into ADPR, cADPR, and NAADP with the enzymatic activity of nicotinamide adenine dinucleotide nucleosidase, such as CD38 [64] and CD157 [38] that are extensively expressed on hematopoietic and non-hematopoietic cells [55, 77]. However, as the binding site of ADPR for the activation of TRPM2 seems to be located inside the plasma membrane, whether and how these extracellularly formed ADPR crosses the membrane and activates TRPM2 channels is not entirely known. Nevertheless, extracellular ADPR has been reported to modulate the activity of TRPM2 indirectly from the extracellular area through activation of P2Y receptors [50] and PLC [36]. In either pathways, the activation of P2Y receptors and PLC results in an increase in intracellular calcium concentration and leads to the enhancement of TRPM2 channel sensitivity toward ADPR. Intracellular Ca2+ has also been shown to serve as a coactivator of TRPM2, and a minimum of 30 nM intracellular calcium concentration is required to cause partial TRPM2 activation with ADPR in the absence of extracellular Ca2+ [17, 76]. In addition to the metabolites and ROS/RNS mentioned above, TRPM2 can also be activated by thermal stimuli with an activation threshold at temperature above 35 °C [81]. Meanwhile, the temperature threshold for TRPM2 activation has also been shown to be lowered in the presence of H2O2, a phenomenon termed “sensitization” [43].
TRPM2 is ubiquitously expressed among various tissues (e.g., central nervous system, peripheral nervous system, bone marrow, and heart) and in different cell types (e.g., pancreatic β-cells, endothelial cells, microglial cells, neurons, and immune cells) [33, 37, 47, 65]. Importantly, the expression of TRPM2 in the phagocytic lineages (e.g., neutrophils and monocytes/macrophages) of immune cells enables the cells to respond to signals of ROS/RNS [49, 94].
The TRPM2 expressed in sensory neurons in dorsal root ganglion plays an important role in thermosensation and nociception. In the experiment investigating the effect of TRPM2 on thermal preference, wild-type male mice showed preference for a 33 °C plate over a 38 °C plate, while TRPM2 knockout male mice showed no such preference. The results indicate that the genetic deletion of TRPM2 causes a remarkable behavioral change in the thermal preference [79]. In addition to the crucial role of TRPM2 in the warmth sensation, TRPM2 has also been shown to be involved in the pathogenesis of various chronic pain in several studies. When tested with von Frey filament test for mechanical sensitivity and Hargreaves and hot plate test at 52 °C and 55 °C for noxious heat sensitivity, wild-type and TRPM2 knockout mice showed no difference in their basal sensitivity. However, TRPM2 knockout mice showed attenuated nocifensive responses when injected intraplantarly with formalin. In carrageenan-induced inflammatory pain and sciatic nerve injury-induced neuropathic pain models, in which the expression of TRPM2 mRNA in the inflamed paw and the area around the injured sciatic nerve were found to be increased, mechanical allodynia and thermal hyperalgesia were attenuated in TRPM2 knockout mice [30]. In addition, the mechanical allodynia in the monosodium iodoacetate-induced osteoarthritis pain model, the mechanical allodynia in paclitaxel-induced peripheral neuropathy and streptozotocin-induced painful diabetic neuropathy models have all been shown to be significantly attenuated in TRPM2 knockout mice [75]. In addition, econazole, a TRPM2 inhibitor, was shown to reduce the visceromotor response to noxious colorectal distention in rats in both baseline condition and trinitrobenzene sulfonic acid-induced colitis model. Furthermore, TRPM2 knockout mice showed significantly reduced visceral hypersensitivity induced by trinitrobenzene sulfonic acid. The results mentioned above all demonstrate the crucial role of the TRPM2 expressed in both sensory neurons and immune cells for the development of various types of pain.
2.5 TRP Channel and Analgesic Drug Development
TRP channels have attracted much attention for the development of analgesic agents. Huge effort has been attempted in the development of antagonists of TRPV1 to treat inflammatory pain and cancer-related pain since the results demonstrating the attenuation of thermal hyperalgesia in inflammatory pain model in TRPV1 knockout mice [13, 18]. A TRPV1 antagonist, AMG 9810, was shown to be effective at preventing capsaicin-induced eye wiping and reversed thermal and mechanical hyperalgesia induced by intraplantar injection of complete Freund’s adjuvant [26]. One study evaluated the effects of a TRPV1 antagonists, SB-705498, in humans and showed that SB-705498 reduced the area of capsaicin-evoked flare, increased the heat pain threshold on non-sensitized skin, and increased heat pain tolerance at the site of UVB-evoked inflammation [15]. The results all demonstrate the great potentials of TRPV1 as a therapeutic target for treating chronic pain. However, most previous TRPV1 antagonist programs have now been put on hold, due to the unwanted on-target side effects. One side effect was the development of marked hyperthermia after TRPV1 blockade, which caused the early termination of phase I clinical trials with AMG 517 for dental pain in humans [15]. Furthermore, TRPV1 antagonists also elevate noxious heat sensation threshold and cause higher risk of burn injuries in individuals receiving TRPV1 antagonists. Several TRPV1 antagonists (e.g., MK-2295 [62], SB-705498 [15] and JNJ-39439335 [59]) have been reported to have such adverse effect in human studies. Although direct blockade of TRPV1 causes the adverse effects mentioned above, an alternative strategy of developing therapeutic agents disrupting the sensitization of TRPV1 is showing promising effect. By disrupting the interactions between TRPV1 and AKAP79, the sensitization of TRPV1 under pathological conditions can be inhibited without changing the normal physiological function of TRPV1 [10]. An effective cell permeable peptide capable of preventing TRPV1-AKAP79 interaction was shown to be analgesic in three mouse models of inflammatory hyperalgesia without causing hyperthermia or decreased sensitivity to noxious heat [24]. The approach demonstrates the potentials for developing therapeutic agents targeting against TRPV1.
Meanwhile, TRPA1 antagonists also show some promising results in treating pathological pain. When applying TRPA1 selective antagonists, attenuation in mechanical hypersensitivity was shown in animal inflammatory and neuropathic pain models [14, 22]. Adverse effect regarding body temperature regulation (such as hyperthermia) is not common after TRPA1 antagonist application. However, another concern that has been brought up is whether TRPA1 antagonism will compromise the ability to elicit protective actions against harmful hazards, such as coughing, sneezing, and generation of nociception to eliminate foreign irritants. These protective responses were reported to be absent in TRPA1 knockout mice [6]. Whether TRPA1 antagonists will cause the loss of such protective reflexes will be challenges for the development of therapeutic agents targeting against TRPA1. Furthermore, TRPM2 channel has been proposed to be a therapeutic target for a wide variety of oxidative stress-related diseases including cardiovascular and cerebrovascular diseases. However, effects of therapeutic strategies targeting against TRPM2 selectively have not been reported, and future efforts are needed for the development of such therapeutic agents for clinical use [53].
2.6 Conclusions
We have gained much deeper understanding in the functions of TRP channels in nociception and chronic pain in the last two decades. However, the modulatory mechanisms of TRP channels are still not entirely known, which can have enormous effects on the chronic pain state. However, it is also due to this sophisticated and complex design that provides us the chance to develop therapeutic strategies for pain relief without affecting the physiological functions of TRP channels. Meanwhile, analgesic agents targeting against TRP channels without side effects are still under development. Hopefully, in the future, medication targeting against TRP channels and related pathways will be brought into clinical use with fewer side effects to fight against refractory pain and other associated disorders.
References
Amaya F, Shimosato G, Nagano M, Ueda M, Hashimoto S, Tanaka Y, Suzuki H, Tanaka M (2004) NGF and GDNF differentially regulate TRPV1 expression that contributes to development of inflammatory thermal hyperalgesia. Eur J Neurosci 20(9):2303–2310. https://doi.org/10.1111/j.1460-9568.2004.03701.x
Andersson DA, Gentry C, Moss S, Bevan S (2008) Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci Off J Soc Neurosci 28(10):2485–2494. https://doi.org/10.1523/JNEUROSCI.5369-07.2008
Aneiros E, Cao L, Papakosta M, Stevens EB, Phillips S, Grimm C (2011) The biophysical and molecular basis of TRPV1 proton gating. EMBO J 30(6):994–1002. https://doi.org/10.1038/emboj.2011.19
Assas BM, Pennock JI, Miyan JA (2014) Calcitonin gene-related peptide is a key neurotransmitter in the neuro-immune axis. Front Neurosci 8:23. https://doi.org/10.3389/fnins.2014.00023
Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D (2006) TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124(6):1269–1282. https://doi.org/10.1016/j.cell.2006.02.023
Bessac BF, Sivula M, von Hehn CA, Escalera J, Cohn L, Jordt SE (2008) TRPA1 is a major oxidant sensor in murine airway sensory neurons. J Clin Invest 118(5):1899–1910. https://doi.org/10.1172/jci34192
Bhave G, Gereau RW (2004) Posttranslational mechanisms of peripheral sensitization. J Neurobiol 61(1):88–106. https://doi.org/10.1002/neu.20083
Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW (2002) cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron 35(4):721–731
Birrell MA, Belvisi MG, Grace M, Sadofsky L, Faruqi S, Hele DJ, Maher SA, Freund-Michel V, Morice AH (2009) TRPA1 agonists evoke coughing in guinea pig and human volunteers. Am J Respir Crit Care Med 180(11):1042–1047. https://doi.org/10.1164/rccm.200905-0665OC
Btesh J, Fischer MJ, Stott K, McNaughton PA (2013) Mapping the binding site of TRPV1 on AKAP79: implications for inflammatory hyperalgesia. J Neurosci Off J Soc Neurosci 33(21):9184–9193. https://doi.org/10.1523/jneurosci.4991-12.2013
Cao E, Cordero-Morales JF, Liu B, Qin F, Julius D (2013a) TRPV1 channels are intrinsically heat sensitive and negatively regulated by phosphoinositide lipids. Neuron 77(4):667–679. https://doi.org/10.1016/j.neuron.2012.12.016
Cao E, Liao M, Cheng Y, Julius D (2013b) TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504(7478):113–118. https://doi.org/10.1038/nature12823
Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D (2000) Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science (New York, NY) 288(5464):306–313
Chen J, Joshi SK, DiDomenico S, Perner RJ, Mikusa JP, Gauvin DM, Segreti JA, Han P, Zhang XF, Niforatos W, Bianchi BR, Baker SJ, Zhong C, Simler GH, McDonald HA, Schmidt RG, McGaraughty SP, Chu KL, Faltynek CR, Kort ME, Reilly RM, Kym PR (2011) Selective blockade of TRPA1 channel attenuates pathological pain without altering noxious cold sensation or body temperature regulation. Pain 152(5):1165–1172. https://doi.org/10.1016/j.pain.2011.01.049
Chizh BA, O’Donnell MB, Napolitano A, Wang J, Brooke AC, Aylott MC, Bullman JN, Gray EJ, Lai RY, Williams PM, Appleby JM (2007) The effects of the TRPV1 antagonist SB-705498 on TRPV1 receptor-mediated activity and inflammatory hyperalgesia in humans. Pain 132(1–2):132–141. https://doi.org/10.1016/j.pain.2007.06.006
Cosens DJ, Manning A (1969) Abnormal electroretinogram from a Drosophila mutant. Nature 224(5216):285–287
Csanady L, Torocsik B (2009) Four Ca2+ ions activate TRPM2 channels by binding in deep crevices near the pore but intracellularly of the gate. J Gen Physiol 133(2):189–203. https://doi.org/10.1085/jgp.200810109
Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA (2000) Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405(6783):183–187. https://doi.org/10.1038/35012076
Dhaka A, Uzzell V, Dubin A, Mathur J, Petrus M, Bandell M, Patapoutian A (2009) TRPV1 senses both acidic and basic pH. J Neurosci Off J Soc Neurosci 29(1):153–158. https://doi.org/10.1523/JNEUROSCI.4901-08.2009
Doerner JF, Gisselmann G, Hatt H, Wetzel CH (2007) Transient receptor potential channel A1 is directly gated by calcium ions. J Biol Chem 282(18):13180–13189. https://doi.org/10.1074/jbc.M607849200
Dubin AE, Patapoutian A (2010) Nociceptors: the sensors of the pain pathway. J Clin Invest 120(11):3760–3772. https://doi.org/10.1172/JCI42843
Eid SR, Crown ED, Moore EL, Liang HA, Choong KC, Dima S, Henze DA, Kane SA, Urban MO (2008) HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol Pain 4:48. https://doi.org/10.1186/1744-8069-4-48
Fernandes ES, Fernandes MA, Keeble JE (2012) The functions of TRPA1 and TRPV1: moving away from sensory nerves. Br J Pharmacol 166(2):510–521. https://doi.org/10.1111/j.1476-5381.2012.01851.x
Fischer MJ, Btesh J, McNaughton PA (2013) Disrupting sensitization of transient receptor potential vanilloid subtype 1 inhibits inflammatory hyperalgesia. J Neurosci Off J Soc Neurosci 33(17):7407–7414. https://doi.org/10.1523/jneurosci.3721-12.2013
Gamper N, Shapiro MS (2007) Regulation of ion transport proteins by membrane phosphoinositides. Nat Rev Neurosci 8(12):921–934. https://doi.org/10.1038/nrn2257
Gavva NR, Tamir R, Qu Y, Klionsky L, Zhang TJ, Immke D, Wang J, Zhu D, Vanderah TW, Porreca F, Doherty EM, Norman MH, Wild KD, Bannon AW, Louis JC, Treanor JJ (2005) AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther 313(1):474–484. https://doi.org/10.1124/jpet.104.079855
Gentry C, Stoakley N, Andersson DA, Bevan S (2010) The roles of iPLA2, TRPM8 and TRPA1 in chemically induced cold hypersensitivity. Mol Pain 6:4. https://doi.org/10.1186/1744-8069-6-4
Geron M, Hazan A, Priel A (2017) Animal toxins providing insights into TRPV1 activation mechanism. Toxins 9(10):326. https://doi.org/10.3390/toxins9100326
Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K, Mori Y (2002) LTRPC2 Ca2+−permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell 9(1):163–173
Haraguchi K, Kawamoto A, Isami K, Maeda S, Kusano A, Asakura K, Shirakawa H, Mori Y, Nakagawa T, Kaneko S (2012) TRPM2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J Neurosci Off J Soc Neurosci 32(11):3931–3941. https://doi.org/10.1523/jneurosci.4703-11.2012
Hasan R, Leeson-Payne AT, Jaggar JH, Zhang X (2017) Calmodulin is responsible for Ca(2+)-dependent regulation of TRPA1 channels. Sci Rep 7:45098. https://doi.org/10.1038/srep45098
Hazan A, Kumar R, Matzner H, Priel A (2015) The pain receptor TRPV1 displays agonist-dependent activation stoichiometry. Sci Rep 5:12278. https://doi.org/10.1038/srep12278
Hecquet CM, Ahmmed GU, Vogel SM, Malik AB (2008) Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ Res 102(3):347–355. https://doi.org/10.1161/CIRCRESAHA.107.160176
Hinman A, Chuang HH, Bautista DM, Julius D (2006) TRP channel activation by reversible covalent modification. Proc Natl Acad Sci U S A 103(51):19564–19568. https://doi.org/10.1073/pnas.0609598103
Huang KP (1989) The mechanism of protein kinase C activation. Trends Neurosci 12(11):425–432
Ishii M, Shimizu S, Hagiwara T, Wajima T, Miyazaki A, Mori Y, Kiuchi Y (2006a) Extracellular-added ADP-ribose increases intracellular free Ca2+ concentration through Ca2+ release from stores, but not through TRPM2-mediated Ca2+ entry, in rat beta-cell line RIN-5F. J Pharmacol Sci 101(2):174–178
Ishii M, Shimizu S, Hara Y, Hagiwara T, Miyazaki A, Mori Y, Kiuchi Y (2006b) Intracellular-produced hydroxyl radical mediates H2O2-induced Ca2+ influx and cell death in rat beta-cell line RIN-5F. Cell Calcium 39(6):487–494. https://doi.org/10.1016/j.ceca.2006.01.013
Itoh M, Ishihara K, Tomizawa H, Tanaka H, Kobune Y, Ishikawa J, Kaisho T, Hirano T (1994) Molecular cloning of murine BST-1 having homology with CD38 and Aplysia ADP-ribosyl cyclase. Biochem Biophys Res Commun 203(2):1309–1317. https://doi.org/10.1006/bbrc.1994.2325
Jardin I, Lopez JJ, Diez R, Sanchez-Collado J, Cantonero C, Albarran L, Woodard GE, Redondo PC, Salido GM, Smani T, Rosado JA (2017) TRPs in pain sensation. Front Physiol 8:392. https://doi.org/10.3389/fphys.2017.00392
Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D (2004) Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427(6971):260–265. https://doi.org/10.1038/nature02282
Julius D (2013) TRP channels and pain. Annu Rev Cell Dev Biol 29:355–384. https://doi.org/10.1146/annurev-cellbio-101011-155833
Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E, Minami T, Takada Y, Kume T, Katsuki H, Mori Y, Akaike A (2006) A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J Pharmacol Sci 101(1):66–76
Kashio M, Sokabe T, Shintaku K, Uematsu T, Fukuta N, Kobayashi N, Mori Y, Tominaga M (2012) Redox signal-mediated sensitization of transient receptor potential melastatin 2 (TRPM2) to temperature affects macrophage functions. Proc Natl Acad Sci U S A 109(17):6745–6750. https://doi.org/10.1073/pnas.1114193109
Kim AY, Tang Z, Liu Q, Patel KN, Maag D, Geng Y, Dong X (2008) Pirt, a phosphoinositide-binding protein, functions as a regulatory subunit of TRPV1. Cell 133(3):475–485. https://doi.org/10.1016/j.cell.2008.02.053
Kirschstein T, Busselberg D, Treede RD (1997) Coexpression of heat-evoked and capsaicin-evoked inward currents in acutely dissociated rat dorsal root ganglion neurons. Neurosci Lett 231(1):33–36
Kobayashi K, Fukuoka T, Obata K, Yamanaka H, Dai Y, Tokunaga A, Noguchi K (2005) Distinct expression of TRPM8, TRPA1, and TRPV1 mRNAs in rat primary afferent neurons with adelta/c-fibers and colocalization with trk receptors. J Comp Neurol 493(4):596–606. https://doi.org/10.1002/cne.20794
Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, Schultz G, Harteneck C (2004) Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol 286(1):C129–C137. https://doi.org/10.1152/ajpcell.00331.2003
Kuhn FJ, Luckhoff A (2004) Sites of the NUDT9-H domain critical for ADP-ribose activation of the cation channel TRPM2. J Biol Chem 279(45):46431–46437. https://doi.org/10.1074/jbc.M407263200
Lange I, Penner R, Fleig A, Beck A (2008) Synergistic regulation of endogenous TRPM2 channels by adenine dinucleotides in primary human neutrophils. Cell Calcium 44(6):604–615. https://doi.org/10.1016/j.ceca.2008.05.001
Lange I, Yamamoto S, Partida-Sanchez S, Mori Y, Fleig A, Penner R (2009) TRPM2 functions as a lysosomal Ca2+−release channel in beta cells. Sci Signal 2(71):ra23. https://doi.org/10.1126/scisignal.2000278
Lewin GR, Rueff A, Mendell LM (1994) Peripheral and central mechanisms of NGF-induced hyperalgesia. Eur J Neurosci 6(12):1903–1912
Li L, Hasan R, Zhang X (2014) The basal thermal sensitivity of the TRPV1 Ion channel is determined by PKCbetaII. J Neurosci 34(24):8246–8258. https://doi.org/10.1523/jneurosci.0278-14.2014
Lima WG, Marques-Oliveira GH, da Silva TM, Chaves VE (2017) Role of calcitonin gene-related peptide in energy metabolism. Endocrine 58(1):3–13. https://doi.org/10.1007/s12020-017-1404-4
Linley JE, Rose K, Ooi L, Gamper N (2010) Understanding inflammatory pain: ion channels contributing to acute and chronic nociception. Pflugers Arch 459(5):657–669. https://doi.org/10.1007/s00424-010-0784-6
Lund FE, Cockayne DA, Randall TD, Solvason N, Schuber F, Howard MC (1998) CD38: a new paradigm in lymphocyte activation and signal transduction. Immunol Rev 161:79–93
Ma W, Quirion R (2007) Inflammatory mediators modulating the transient receptor potential vanilloid 1 receptor: therapeutic targets to treat inflammatory and neuropathic pain. Expert Opin Ther Targets 11(3):307–320. https://doi.org/10.1517/14728222.11.3.307
Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, Patapoutian A (2007) Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 445(7127):541–545. https://doi.org/10.1038/nature05544
Mandadi S, Sokabe T, Shibasaki K, Katanosaka K, Mizuno A, Moqrich A, Patapoutian A, Fukumi-Tominaga T, Mizumura K, Tominaga M (2009) TRPV3 in keratinocytes transmits temperature information to sensory neurons via ATP. Pflugers Arch 458(6):1093–1102. https://doi.org/10.1007/s00424-009-0703-x
Manitpisitkul P, Mayorga A, Shalayda K, De Meulder M, Romano G, Jun C, Moyer JA (2015) Safety, tolerability and pharmacokinetic and pharmacodynamic learnings from a double-blind, randomized, placebo-controlled, sequential group first-in-human study of the TRPV1 antagonist, JNJ-38893777, in healthy men. Clin Drug Investig 35(6):353–363. https://doi.org/10.1007/s40261-015-0285-7
McNamara CR, Mandel-Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M, Hayward NJ, Chong JA, Julius D, Moran MM, Fanger CM (2007) TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci U S A 104(33):13525–13530. https://doi.org/10.1073/pnas.0705924104
Meotti FC, Forner S, Lima-Garcia JF, Viana AF, Calixto JB (2013) Antagonism of the transient receptor potential ankyrin 1 (TRPA1) attenuates hyperalgesia and urinary bladder overactivity in cyclophosphamide-induced haemorrhagic cystitis. Chem Biol Interact 203(2):440–447. https://doi.org/10.1016/j.cbi.2013.03.008
Moran MM, Szallasi A (2017) Targeting nociceptive transient receptor potential channels to treat chronic pain: current state of the field. Br J Pharmacol. https://doi.org/10.1111/bph.14044
Nagata K, Duggan A, Kumar G, Garcia-Anoveros J (2005) Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J Neurosci Off J Soc Neurosci 25(16):4052–4061. https://doi.org/10.1523/jneurosci.0013-05.2005
Nishina H, Inageda K, Takahashi K, Hoshino S, Ikeda K, Katada T (1994) Cell surface antigen CD38 identified as ecto-enzyme of NAD glycohydrolase has hyaluronate-binding activity. Biochem Biophys Res Commun 203(2):1318–1323. https://doi.org/10.1006/bbrc.1994.2326
Olah ME, Jackson MF, Li H, Perez Y, Sun HS, Kiyonaka S, Mori Y, Tymianski M, MacDonald JF (2009) Ca2+−dependent induction of TRPM2 currents in hippocampal neurons. J Physiol 587(Pt 5):965–979. https://doi.org/10.1113/jphysiol.2008.162289
Patapoutian A, Tate S, Woolf CJ (2009) Transient receptor potential channels: targeting pain at the source. Nat Rev Drug Discov 8(1):55–68. https://doi.org/10.1038/nrd2757
Paulsen CE, Armache J-P, Gao Y, Cheng Y, Julius D (2015) Structure of the TRPA1 ion channel suggests regulatory mechanisms. Biophys J 110:26a
Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP, Scharenberg AM (2001) ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 411(6837):595–599. https://doi.org/10.1038/35079100
Prescott ED, Julius D (2003) A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science (New York, NY) 300(5623):1284–1288. https://doi.org/10.1126/science.1083646
Ramsey IS, Delling M, Clapham DE (2006) An introduction to TRP channels. Annu Rev Physiol 68:619–647. https://doi.org/10.1146/annurev.physiol.68.040204.100431
Rohacs T (2015) Phosphoinositide regulation of TRPV1 revisited. Pflugers Arch 467(9):1851–1869. https://doi.org/10.1007/s00424-015-1695-3
Ross RA (2003) Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol 140(5):790–801. https://doi.org/10.1038/sj.bjp.0705467
Siemens J, Zhou S, Piskorowski R, Nikai T, Lumpkin EA, Basbaum AI, King D, Julius D (2006) Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature 444(7116):208–212. https://doi.org/10.1038/nature05285
Silos-Santiago I, Molliver DC, Ozaki S, Smeyne RJ, Fagan AM, Barbacid M, Snider WD (1995) Non-TrkA-expressing small DRG neurons are lost in TrkA deficient mice. J Neurosci Off J Soc Neurosci 15(9):5929–5942
So K, Haraguchi K, Asakura K, Isami K, Sakimoto S, Shirakawa H, Mori Y, Nakagawa T, Kaneko S (2015) Involvement of TRPM2 in a wide range of inflammatory and neuropathic pain mouse models. J Pharmacol Sci 127(3):237–243. https://doi.org/10.1016/j.jphs.2014.10.003
Starkus J, Beck A, Fleig A, Penner R (2007) Regulation of TRPM2 by extra- and intracellular calcium. J Gen Physiol 130(4):427–440. https://doi.org/10.1085/jgp.200709836
Sumoza-Toledo A, Penner R (2011) TRPM2: a multifunctional ion channel for calcium signalling. J Physiol 589(Pt 7):1515–1525. https://doi.org/10.1113/jphysiol.2010.201855
Sun L, Ye RD (2012) Role of G protein-coupled receptors in inflammation. Acta Pharmacol Sin 33(3):342–350. https://doi.org/10.1038/aps.2011.200
Tan CH, McNaughton PA (2016) The TRPM2 ion channel is required for sensitivity to warmth. Nature 536(7617):460–463. https://doi.org/10.1038/nature19074
Todaka H, Taniguchi J, Satoh J, Mizuno A, Suzuki M (2004) Warm temperature-sensitive transient receptor potential vanilloid 4 (TRPV4) plays an essential role in thermal hyperalgesia. J Biol Chem 279(34):35133–35138. https://doi.org/10.1074/jbc.M406260200
Togashi K, Hara Y, Tominaga T, Higashi T, Konishi Y, Mori Y, Tominaga M (2006) TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J 25(9):1804–1815. https://doi.org/10.1038/sj.emboj.7601083
Tominaga M, Caterina MJ (2004) Thermosensation and pain. J Neurobiol 61(1):3–12. https://doi.org/10.1002/neu.20079
Trevisani M, Siemens J, Materazzi S, Bautista DM, Nassini R, Campi B, Imamachi N, Andre E, Patacchini R, Cottrell GS, Gatti R, Basbaum AI, Bunnett NW, Julius D, Geppetti P (2007) 4-hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci U S A 104(33):13519–13524. https://doi.org/10.1073/pnas.0705923104
Vandewauw I, De Clercq K, Mulier M, Held K, Pinto S, Van Ranst N, Segal A, Voet T, Vennekens R, Zimmermann K, Vriens J, Voets T (2018) A TRP channel trio mediates acute noxious heat sensing. Nature. https://doi.org/10.1038/nature26137
Vriens J, Nilius B, Vennekens R (2008) Herbal compounds and toxins modulating TRP channels. Curr Neuropharmacol 6(1):79–96. https://doi.org/10.2174/157015908783769644
Wang S, Dai Y, Fukuoka T, Yamanaka H, Kobayashi K, Obata K, Cui X, Tominaga M, Noguchi K (2008a) Phospholipase C and protein kinase A mediate bradykinin sensitization of TRPA1: a molecular mechanism of inflammatory pain. Brain J Neurol 131(Pt 5):1241–1251. https://doi.org/10.1093/brain/awn060
Wang YY, Chang RB, Waters HN, McKemy DD, Liman ER (2008b) The nociceptor ion channel TRPA1 is potentiated and inactivated by permeating calcium ions. J Biol Chem 283(47):32691–32703. https://doi.org/10.1074/jbc.M803568200
Wehage E, Eisfeld J, Heiner I, Jungling E, Zitt C, Luckhoff A (2002) Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J Biol Chem 277(26):23150–23156. https://doi.org/10.1074/jbc.M112096200
Wes PD, Chevesich J, Jeromin A, Rosenberg C, Stetten G, Montell C (1995) TRPC1, a human homolog of a Drosophila store-operated channel. Proc Natl Acad Sci U S A 92(21):9652–9656
Wesseldijk F (2008) Inflammatory soup mediators of inflammation in CRPS
Wick EC, Hoge SG, Grahn SW, Kim E, Divino LA, Grady EF, Bunnett NW, Kirkwood KS (2006) Transient receptor potential vanilloid 1, calcitonin gene-related peptide, and substance P mediate nociception in acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 290(5):G959–G969. https://doi.org/10.1152/ajpgi.00154.2005
Woo DH, Jung SJ, Zhu MH, Park CK, Kim YH, Oh SB, Lee CJ (2008) Direct activation of transient receptor potential vanilloid 1(TRPV1) by diacylglycerol (DAG). Mol Pain 4:42. https://doi.org/10.1186/1744-8069-4-42
Woodbury CJ, Zwick M, Wang S, Lawson JJ, Caterina MJ, Koltzenburg M, Albers KM, Koerber HR, Davis BM (2004) Nociceptors lacking TRPV1 and TRPV2 have normal heat responses. J Neurosci Off J Soc Neurosci 24(28):6410–6415. https://doi.org/10.1523/jneurosci.1421-04.2004
Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, Kaneko S, Lange I, Fleig A, Penner R, Nishi M, Takeshima H, Mori Y (2008) TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med 14(7):738–747. https://doi.org/10.1038/nm1758
Zhang X (2015) Molecular sensors and modulators of thermoreception. Channels (Austin) 9(2):73–81. https://doi.org/10.1080/19336950.2015.1025186
Zurborg S, Yurgionas B, Jira JA, Caspani O, Heppenstall PA (2007) Direct activation of the ion channel TRPA1 by Ca2+. Nat Neurosci 10(3):277–279. https://doi.org/10.1038/nn1843
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Hung, CY., Tan, CH. (2018). TRP Channels in Nociception and Pathological Pain. In: Shyu, BC., Tominaga, M. (eds) Advances in Pain Research: Mechanisms and Modulation of Chronic Pain. Advances in Experimental Medicine and Biology, vol 1099. Springer, Singapore. https://doi.org/10.1007/978-981-13-1756-9_2
Download citation
DOI: https://doi.org/10.1007/978-981-13-1756-9_2
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-1755-2
Online ISBN: 978-981-13-1756-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)