
Abstract
Circular RNAs (circRNAs) are abundant in the brain and are often expressed in complex spatiotemporal patterns that coincide with distinct developmental transitions. This suggests that circRNAs play a significant role in the central nervous system. This book chapter will review research progress into the function of circRNAs during neuronal development. The major themes to be discussed are the enrichment of circRNAs in the synapse and their possible contributions to synaptopathologies, in addition to the findings that neural circRNAs accumulate with age and appear beneficial for neuronal repair. Although more research is needed, some of the possible functions of circRNAs with in the brain are already beginning to come to light.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
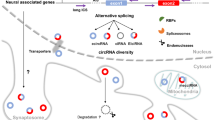
High-throughput sequencing (RNA-seq) has greatly expanded our understanding of circRNA biology. The first half of this book chapter will review insights gained from the vast amounts of RNA-seq data, including the general properties for circRNA expression in the brain and their complex spatiotemporal expression patterns. The second half will focus on two potential functions for circRNAs in the brain. The first in synaptic learning, as evidenced by the enrichment (and activity-dependent transcription) of circRNAs in the synapse and their deregulation in some synaptopathologies. Already, a convincing circRNA-associated competing endogenous RNA network has been identified in Alzheimer’s disease. The second in the neuroprotection of the ageing brain, on the basis that neuronal circRNAs accumulate with age, are spatiotemporally deregulated with age, and are differentially expressed during the recovery period after stroke or brain injury.
Although many unanswered questions still remain, this book chapter will summarise the current understanding of circRNA function in neuronal development and will put forward two potential biological roles for circRNAs in the brain.
2 The Expression of Circular RNAs in the Brain
2.1 Circular RNAs Are Lowly But Diversely Expressed in the Brain
Circular RNAs are expressed at very low levels in all tissues. The same can be said for the brain. In mouse brain, less than 0.1% of RNA-seq reads from circular junctions can be mapped back to the reference genome [1]. In comparison, 44% of all protein-coding genes RNA-seq reads from the mouse brain are mappable [1]. The majority of the distinct circRNA species that make up this small population of RNAs also appear to be expressed at low levels. For example, 41,027 out of 65,731 circRNA candidates are supported by fewer than 10 RNA-seq reads in the human brain [2]. Similarly, in mouse brain, 10,081 out of 15,849 circRNA candidates are supported by fewer than 10 RNA-seq reads [2]. Further contributing to their low abundance is that only a small percentage of genes are able to synthesise circRNAs. This is also true for the brain, where 21% of expressed genes produce circRNAs in the adult mouse brain [1], 15.8% in the foetal pig cortex [3], and 13% in the human brain [2]. However, the percentage of circRNA-hosting genes in the brain is greater than in other tissues like the heart, liver, and lung, where less than 10% of expressed genes synthesise circRNAs [3]. Therefore, in the brain, a small number of expressed genes are able to synthesise few but a diverse set of circRNAs. This is not unlike in other tissues, although the relative abundance of circRNAs in the brain is much greater.
The brain also stands apart from other tissues in its ability to synthesise the greatest number of distinct circRNAs. This is consistent across species. For example, the most comprehensive analysis of circRNA expression across tissues and developmental staging to date, which involved mining 10 billion RNA-seq reads from 103 fly sample libraries, identified that 90–95% of all circRNAs are expressed in the head [4]. In adult mouse, the brain expresses by far the greatest number of distinct circRNAs: an average of 5925 circRNA candidates (from an average total reads of 19,479,587) compared the second highest tissue, the testis, which expresses an average of 3018 circRNA candidates (out of an average total reads of 20,081,654) [1]. Similarly in the human foetus, the brain produces the greatest variety of circRNAs out of any of the other 14 tissues analysed [5]. Furthermore, one study identified 65,731 distinct circRNAs in the human brain alone [2].
Two factors may help to explain why the brain has a unique capacity to synthesise such a diverse set of circRNA species. One is that the population of circRNA-synthesising genes in the brain, on average, synthesise multiple distinct circRNAs. For example, circRNA-host genes of the adult mouse brain are able to produce an average of 2.4 distinct circRNAs, compared to other tissues like the heart, liver, and lungs, which produce an average of 1.2–1.5 circRNAs per circRNA-host gene [1]. In the human brain, this appears to be even greater, where the average circRNA-producing host genes synthesise 6.4 distinct circRNAs, or a median of three circRNAs per host gene [2]. The second factor is that many circRNA-hosting genes are exclusively expressed in the brain. For example, 225 circRNA-producing genes are exclusively expressed in the brain, relative to 140 in the testis, and fewer than 20 in the heart, liver, and lungs [1]. A similar trend has been reported in adult rat, where 60 circRNA-producing genes are exclusively expressed in the brain, ~35 in the testis, and fewer than 10 in the heart, liver, and lung [1].
Given that circRNA expression is dependent on host gene transcription, it is not unexpected that different tissues should express different subsets of circRNAs. For example, the gene ontologies of linear transcripts derived from the liver are enriched in liver-specific processes like lipoprotein metabolism and extracellular exosomes, while in the brain, the gene ontologies are enriched in brain-specific processes such as protein phosphorylation, postsynaptic density, and protein kinase activity [6]. Likewise, the host genes of tissue-specific circRNAs should also be enriched in pathways specific to that tissue, and indeed this has been shown. For instance, the host genes of brain-specific circRNAs are enriched in pathways specific to neuron development, differentiation, and synaptic transmission, while the host genes of liver-specific circRNAs are enriched in ion transport, proton transport, and caton transport [5]. Therefore, it can be concluded that the brain likely expresses the greatest number of circRNAs out of any tissue simply because of the transcriptional properties of the linear circRNA-synthesising host genes.
2.2 Circular RNA–Hosting Genes May Be Enriched in Long Neuronal Genes
Another point of difference between the linear transcripts of the brain and other tissues is that brain-specific genes tend to have much longer introns. This is particularly obvious when studying topoisomerases, enzymes that catalyse the winding and unwinding of supercoiled DNA strands during transcription or cell replication. Understandably, DNA topoisomerases are particularly important for long genes. This is exemplified by the strong negative correlation between the length of long genes (>67 kb) and their expression levels when DNA topoisomerases are inhibited [7]. Topoisomerases have recently been shown to regulate synaptic genes [7] and to maintain normal synaptic functions [8]. More specifically, the inhibition of topoisomerases at excitatory synapses reduces the number of synapses, while inhibition at inhibitory synapses interferes with the membrane trafficking of GABAA receptor subunits. Therefore, neuronal genes, particularly those involved in synaptic processes, tend to be long (and therefore dependent on DNA topoisomerases).
Circular RNAs tend to be bracketed by longer introns. The flanking introns of circRNAs in human forebrain neurons are on average five times longer than randomly selected introns [9]. Furthermore, a comprehensive analysis of 10 billion RNA-seq reads in a fly identified that the median length of a fly intron is 96 bps, the median length of introns longer than 200 bps is 1009 bp, while the median lengths of introns upstream and downstream of circRNAs are 4662 and 2962 bps, respectively [4]. Moreover, splice sites that are involved in the biogenesis of two or more circRNA isoforms tend to be flanked by even longer introns than splice sites that drive a single circRNA isoform [3]. Given that circRNA-synthesising genes are flanked by much longer introns, neuronal genes (particularly those related to the synapse) may have greater circRNA-synthesising capabilities simply because of their increased length.
2.3 Circular RNAs Are Actively Regulated in the Brain
Although circRNAs are dependent on their host gene for the initiating of transcription, hundreds of neuronal circRNAs are expressed several times higher than their host genes [1,2,3, 10]. In cases where circRNA-host genes are equivalently expressed in the brain and other tissues, the numbers of circRNAs produced from the brain host genes are significantly higher [1]. This suggests that preferentially expressed circRNAs may have biological roles independent from their linear host genes. CircRNAs are not only more relatively abundantly and diversely expressed in the brain, they are also differentially expressed in the different regions and cell types of the brain. For example, the adult mouse cortex and hippocampus share 4030 out of 6231 circRNA candidates; however, 2201 circRNAs are differentially expressed [11]. Some specific examples of circRNAs with region- or cell-specific expression profiles are circRims2 and circDym, which are expressed at greater than 50% in the mouse adult cerebellum versus the striatum, prefrontal cortex, olfactory bulb, midbrain, and hippocampus [2]. Another example is circPlxnd1, which is predominantly expressed in the prefrontal cortex (<60%) versus the other aforementioned brain regions [2].
Many circRNA isoforms, which are derived from the same host gene splice acceptor or donor sites, also display divergent expression profiles in the brain [3]. An intriguing example in human cells are the circStau2a (containing exons 2–5) and circStau2b (containing exons 2–3) isoforms, which display inverse expression patterns. circStau2b is highly expressed in the adult brain relative to almost all other tissues, while the longer circStau2a isoform is highly expressed in the adult lung and relatively lowly expressed elsewhere including the brain [2]. The divergent expression profiles of circRNAs in the brain suggests that cis-regulatory elements and brain-specific trans-acting factors may regulate these processes.
There are many possibilities as to why some circRNAs are preferentially upregulated in the brain. First, the brain may be enriched for neuron-specific splicing factors and/or RNA-binding proteins that regulate circRNA biogenesis. The divergent expression profiles and levels of circRNA isoforms from identical splice acceptor or donor sites in particular support this. Second, as already discussed, circularised exons require longer introns, and neuronal genes tend to fulfil this requirement. Third, circRNAs have a half-life almost five times longer than their host transcripts [10], and in quiescent and postmitotic tissues like neurons, this allows for circRNA to accumulate. An accumulation of circRNAs relative to their linear isoforms has already been documented in the ageing fly brain [4]; conversely, circRNAs tend to be reduced in cancers [12]. Finally, highly polarised cells, such as neurons, must regulate multiple cellular functions and translate different combinations of proteins within their different cellular compartments, and this is often mediated by RNA-dependent mechanisms, not unlike circRNAs. One exciting possibility is that circRNAs may form an additional layer of localised posttranscriptional regulation.
Circular RNAs are also temporally regulated in the brain. Differentiating human primary cortical neurons upregulate 1926 and downregulate 797 (out of 5265) circRNA candidates [2]. Similarly, differentiating mouse embryonic carcinoma cells upregulate 1116 and downregulate 238 (out of 2735) circRNAs [2]. Furthermore, metabolic tagging of differentiating human forebrain neuron progenitor cells revealed that 785 (out of 11,185) circRNA candidates are upregulated over 26 days of in vitro differentiation (27068474). The temporal expression patterns of some circRNAs in the brain appear highly coordinated and complex. For example, the 200 most highly expressed circRNAs of the foetal pig cortex can be clustered into seven categories based on their temporal expression patterns. For instance, 29 circRNAs are expressed highly in the early half of gestation, 11 circRNAs in the late half of gestation, and 130 at a specific developmental stage during mid-gestation [3]. The tight temporal regulation of circRNAs in the brain, which coincides with distinct developmental transitions, strongly supports a biological function.
3 Circular RNAs Appear Important at the Synapse
3.1 Circular RNAs Are Greatly Enriched in the Synapse
Given that the expression of a particular circRNA is dependent on the transcription of its host gene (although, as already discussed, the relative abundance of circular and linear host transcripts can be regulated divergently), valuable insights can be gained from analysing the molecular function and biological process ontologies of host genes. Intriguing, the synapse is consistently one of the most significantly enriched ontologies for circRNA-associated genes from the brain. This is true for the set of circRNAs that are consistently upregulated in mouse hippocampal cells during postnatal development [1] and for the set of circRNAs most highly elevated with age in the fly brain [4]. Further to this, circRNAs are highly expressed in synaptic subcellular fractions. The majority of circRNAs in the adult mouse brain (1117) are enriched in the synaptoneurosomes, relative to the corresponding cytoplasmic (709) and whole brain (847) fractions [2]. Similarly, in the mouse and rat hippocampus, circRNAs are enriched in synaptoneurosomes and/or synaptic neuropils (which exhibit robust synaptic plasticity), compared to corresponding cell body or whole hippocampal homogenates [1]. The enrichment of circRNAs in synaptic compartments has also been validated visually using high-resolution in situ hybridisation, where at least eight circRNAs derived from synapse-related host genes were located throughout the dendritic arbours of the mouse hippocampus [1].
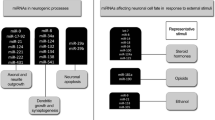
Evidence is also beginning to suggest that circRNAs may play a direct role in homeostatic scaling. Homeostatic adaptation, or scaling, is the ability of neurons to maintain excitability while the brain is adjusting to environmental change. It involves a cell-wide increase or decrease in postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor in excitatory synapses; this scales all synapses by the same multiplicative factor—to become stronger or weaker—while maintaining their relative strengths. Bicuculline, a gamma-aminobutyric acid (GABA)A-receptor antagonist, can also be used to induce homeostatic plasticity. Recently, 37 circRNAs (versus 7 host genes) were transcribed in response to bicuculline-induced homeostatic plasticity, while 5 circRNAs (versus 3 host genes) were downregulated [1]. These circRNAs are examples of synaptic activity-dependent transcription, which suggests a role in neural plasticity. The most dramatically upregulated transcripts were circHomer1_a and its host gene, homer homolog 1 (Drosophila) (Homer1) [1].
Homer1 can be translated into three different protein products at the postsynaptic density: Homer1a, Homer1b, and Homer1c. Homer1b/c is constitutively expressed, while Homer1a is transiently upregulated during increases to network activity, such as those created by long-term bicuculline treatment [13]. Under neutral network conditions, Homer1b/c interacts with group I metabotropic glutamate receptor (mGluR) at the postsynaptic density; however, when Homer1a is expressed, it interferes with native interactions between mGluR and Homer1b/c [14]. This leads to a cell-wide reorganisation of the postsynaptic density, which activates group I mGluR signalling that in turn initiates homeostatic adaptation [13]. During homeostatic adaptation,circHomer1_a is also synthesised. The synthesis of circHomer1_a requires the same splice sites as those for Homer1b/c transcripts [1]. Therefore, circHomer1_a and Homer1b/c transcripts cannot be mutually expressed. Circular RNAs are thought to regulate translation by competing with the canonical splicing of the host gene [15]. This may be such an example. That is, the biogenesis of circHomer1_a may be actively and purposefully competing with the transcription of Homer1b/c transcripts during homeostatic adaptation, with the goal of reducing competition between Homer1a and Homer1b/c mRNA synthesis. Further experimental validation is required.
3.2 Circular RNAs May Be Linked to Neurodegenerative Synaptopathies
Alzheimer’s disease (AD) is the most common form of progressive dementia in the ageing brain. The pathological features of AD are intracellular tau-containing neurofibrillary tangles and extracellular amyloid-β plaques, which accumulate in vulnerable regions of the brain such as the cortex and hippocampus. Alzheimer’s disease is thought to be a synaptic pathology. Some evidence for this are that subtle alterations in the synaptic efficacy of the hippocampus occur in AD patients prior to the detection of neurofibrillary tangles and amyloid-β plaques [16]. Also, patients within 2–4 years of the clinical onset of AD have reduced numbers of spines per neuron in layers II–III (38%) and V (30%) of the temporal cortex and in layer V (30%) of the frontal cortex [17]. Furthermore, cognitive deficits associated with AD are more strongly correlated to neocortical synapse loss compared to the number of plaques and tangles [18].
Recently, circRNA dysfunction was identified in a sporadic mouse model of AD [19]. More specifically, 94 and 141 circRNAs were, respectively, up- and downregulated (out of 34,096) in the adult brain relative to controls. Based on this, in combination with RNA-seq reads on deregulated miRNAs and linear mRNAs, a circRNA-associated-competing endogenous RNA network was built. This RNA regulatory network did not take into account the number and density of miRNA-binding seed sequences and therefore likely contains a large proportion of false-positive pairings. Nonetheless, the network identified two interactions formally linked to AD involving the genes deiodinase, iodothyronine, type II (Dio2), and high-mobility group box 2 (HMGB2). Dio2, which activates myelination [20] and is reduced in AD [21], putatively associated with miR-122-5p and five deregulated circRNAs. HMGB2, which activates pathways involved in amyloid-β plaque clearance [22], was paired with let-7 g-3p and deregulated 3 circRNAs. These circRNAs may be competitively modulating the activity of miR-122-5p and let-7 g-3p, to affect the expression of Dio2 and HMGB2, in a process commonly described as ‘miRNA sponging’ [23]. This study did not identify the most convincing circRNA-mRNA pairing involved in human AD, as would be expected: ciRS-7 and ubiquitin-conjugating enzyme E2A, RAD6 homolog (S. cerevisiae) (UBE2A).
The circRNA ciRS-7 is produced from the antisense of the cerebellar degeneration-related protein 1 (CDR1 as) gene. ciRS-7 contains 74 tandem seed matches to miR-7, and 63 of these are conserved from annelids to humans [24]. Unsurprisingly, ciRS-7 is a potent negative regulator of miR-7. For example, based on RNA-seq data, a single HEK293 cell is estimated to contain ~1400 ciRS-7 molecules which can sequester up to 20,000 miR-7 molecules [24]. In patients with sporadic AD, ciRS-7 is downregulated by 5.4-fold in the hippocampal CA1 region [25, 26] and is significantly reduced by more than fivefold in the superior temporal lobe neocortex (Brodmann area 22). A decrease of ciRS-7 in hippocampal CA1 and Brodmann area 22 would enable the excess accumulation of miR-7 in these regions. Indeed, miR-7 is upregulated in the brain of AD patients by an average of threefold [26]. Excesses of miR-7 would in turn repress UBE2A, which is essential for the proteolytic clearance of amyloid-β peptides in Alzheimer’s disease. Indeed, UBE2A is also downregulated by 3.7-fold in the hippocampal CA1 region and by 2.8-fold in Brodmann area 22 [26]. Thus, a deficiency in ciRS-7 ‘miRNA sponging’ would enable miR-7 to potentially and efficiently downregulate target genes essential for the clearance of amyloid-β plaques. In a similar fashion, ciRS-7 could be protective against Parkinson’s disease by preventing miR-7 from silencing epidermal growth factor receptor (EGFR), alpha-synuclein (SNCA), and insulin receptor substrate 2 (IRS2) [27].
The fervent sponging capacity of ciRS-7 is not, however, a general feature of circRNAs. The majority of circRNAs expressed in the brain have comparable numbers of miRNA-binding sites as linear mRNAs and therefore would not make for strong ‘sponges’ [1, 23, 28]. Also, organisms completely devoid of RNA interference, such as the yeast Saccharomyces cerevisiae [29], clearly produce circRNAs [30]. Hence, the function of the majority of circRNAs must extend beyond being competitive-binding moderators of the RNA interference pathway.
4 Circular RNAs Appear Important for the Ageing Brain
4.1 Circular RNAs Accumulate in the Ageing Brain
Circular RNAs accumulate in the ageing brain. This is substantiated by the most comprehensive survey of circRNA expression to date that mined 103 fly tissues from various developmental stages [4]. More specifically, the total circRNA levels were found to increase across fly embryo development and dramatically increase in the adult head relative to earlier time points from the head or all other adult tissues [4]. Similarly in the mouse, the global levels of circRNAs—measured by transcript per kilobase million—significantly increase from 1 to 22 months in the cortex and hippocampus, but not in the heart [11]. CircRNAs not only accumulate in the brain with time, but a subset is differentially upregulated with age independently from host genes. In a fly, 262 (out of 2513) circRNA candidates are significantly upregulated by more than twofold in 20-day-old heads compared to 1-day-old heads [4]. Similarly in the ageing mouse hippocampus, 250 (out of 5528) circRNA candidates are significantly upregulated at 22 months compared to 1 month, and in the ageing cortex, 258 (out of 4733) circRNA candidates are significantly upregulated at 22 months compared to 1 month [11]. In the mouse cortex, the functional ontologies of host genes synthesising age-upregulated circRNAs are enriched for synapse assembly, synapse organisation, neurotransmitter secretion, and neurotransmitter transport [11]. Alternatively, in the mouse hippocampus, the host genes of age-upregulated circRNAs are enriched for protein and chromatin modifications [11].
Given that linear RNA expression does not change with age in the mouse brain [11] and that age-upregulated circRNAs were largely independent to the expression level of the host gene [4, 11], the mechanisms that drive circRNA age-accumulation are not host-dependent. One mechanism may be that circRNAs are especially stable in the quiescent and postmitotic cells of the ageing brain, which allow a greater proportion of circRNAs to accumulate over time. Another mechanism may be related to the phenomenon that more than one-third of genes expressed in the ageing human brain undergo changes to alternative splicing that encourage back-splicing [31]. The next steps would be to determine whether the accumulation of circRNAs is innocuous or serves a protective or detrimental function to the brain. Recent findings that circRNAs are deregulated following stroke provide insights into the potential role of age-upregulated circRNAs.
4.2 Circular RNAs Are Linked to Neural Repair
Stroke is the third leading cause of death in the United States. It is an acute neurological event that leads to the death of neural tissues. The majority of strokes result from vascular occlusions, called ischemic strokes. For some ischemic stroke patients, the restoration of blood flow exacerbates the initial injury, producing a so-called cerebral reperfusion injury. The specific role of circRNAs during ‘cerebral reperfusion injury’ has been investigated by three different laboratories, using either the intraluminal middle cerebral artery occlusion model in the mouse [32, 33] or oxygen-glucose deprivation and then reoxygenation in cell culture [34]. The most comprehensive analyses to date identified 283 deregulated (out of 1064) circRNA candidates over a 6-, 12-, and/or 24-h time course, with 239 significantly altered at 6 h [32]. This great peak of circRNA deregulation at 6 h, but not at 12 and 24 h, is suggestive of complex temporal regulatory processes taking place. Another study profiled circRNAs at 48 h after artery occlusion and identified over a thousand deregulated circRNAs [33]. Only modest changes in the expression of 15 circRNAs were found at 24 h in the cell culture model [34]. These three studies lack consistencies in the particular circRNAs involved. For example, none of the circRNAs altered post-stroke were shared between the cell culture [34] and intraluminal middle cerebral artery occlusion model [32] at 24 h. Also, there was a far greater number of circRNAs deregulated at 48 h [33] compared to before 24 h [32], perhaps highlighting differences in the models used and the brain regions assayed.
The host genes of stroke-responsive circRNAs across these studies were, however, enriched for repair processes. For example, at 48 h post-stroke, host genes were most significantly enriched for the cell survival and proliferation pathways of Rap1 and Hippo signalling [33]. Similarly, the host genes of all stroke-responsive circRNAs over the entire 24-h time course were most significantly enriched for mitogen-activated kinase signalling, cell cycle and actin cytoskeletal regulators, and focal adhesions molecules that are related to cell growth, proliferation, and death [32]. In a rat model of traumatic injury to the hippocampus, the host genes of injury-responsive circRNAs were most significantly enriched for neurogenesis, neuronal differentiation and development, and in cellular components related to the synapse (5 out of the top 10) [35]. Therefore, circRNAs that are deregulated following brain injury are tightly associated with neuronal repair processes. This tentatively implies that age-upregulated circRNAs may serve as a biological function during recovery in the injured brain, although much more research is required.
References
You X, Vlatkovic I, Babic A et al (2015) Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci 18(4):603–610
Rybak-Wolf A, Stottmeister C, Glazar P et al (2015) Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. gs. Mol Cell 58(5):870–885
Veno MT, Hansen TB, Veno ST et al (2015) Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol 16:245
Westholm JO, Miura P, Olson S et al (2014) Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep 9(5):1966–1980
Xia S, Feng J, Lei L et al (2017) Comprehensive characterization of tissue-specific circular RNAs in the human and mouse genomes. Brief Bioinform 18(6):984–992
Li L, Zheng YC, Kayani MUR et al (2017) Comprehensive analysis of circRNA expression profiles in humans by RAISE. Int J Oncol 51(6):1625–1638
King IF, Yandava CN, Mabb AM et al (2013) Topoisomerases facilitate transcription of long genes linked to autism. Nature 501(7465):58–62
Mabb AM, Kullmann PH, Twomey MA et al (2014) Topoisomerase 1 inhibition reversibly impairs synaptic function. Proc Natl Acad Sci U S A 111(48):17290–17295
Zhang XO, Wang HB, Zhang Y et al (2014) Complementary sequence-mediated exon circularization. Cell 159(1):134–147
Jeck WR, Sorrentino JA, Wang K et al (2013) Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19(2):141–157
Gruner H, Cortes-Lopez M, Cooper DA et al (2016) CircRNA accumulation in the aging mouse brain. Sci Rep 6:38907
Bachmayr-Heyda A, Reiner AT, Auer K et al (2015) Correlation of circular RNA abundance with proliferation—exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep 5:8057
Hu JH, Park JM, Park S et al (2010) Homeostatic scaling requires group I mGluR activation mediated by Homer1a. Neuron 68(6):1128–1142
Tu JC, Xiao B, Yuan JP et al (1998) Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron 21 (4):717–726; Brakeman PR, Lanahan AA, O’Brien R et al (1997) Homer: a protein that selectively binds metabotropic glutamate receptors. Nature 386 (6622):284–288
Ashwal-Fluss R, Meyer M, Pamudurti NR et al (2014) circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 56(1):55–66
Selkoe DJ (2002) Alzheimer’s disease is a synaptic failure. Science 298(5594):789–791
Davies CA, Mann DM, Sumpter PQ et al (1987) A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J Neurol Sci 78(2):151–164
Terry RD, Masliah E, Salmon DP et al (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30(4):572–580
Zhang S, Zhu D, Li H et al (2017) Characterization of circRNA-associated-ceRNA networks in a senescence-accelerated mouse prone 8 brain. Mol Ther 25(9):2053–2061
Calza L, Fernandez M, Giuliani A et al (2002) Thyroid hormone activates oligodendrocyte precursors and increases a myelin-forming protein and NGF content in the spinal cord during experimental allergic encephalomyelitis. Proc Natl Acad Sci U S A 99(5):3258–3263
Humphries CE, Kohli MA, Nathanson L et al (2015) Integrated whole transcriptome and DNA methylation analysis identifies gene networks specific to late-onset Alzheimer’s disease. J Alzheimers Dis 44(3):977–987
Yamanaka Y, Faghihi MA, Magistri M et al (2015) Antisense RNA controls LRP1 sense transcript expression through interaction with a chromatin-associated protein, HMGB2. Cell Rep 11(6):967–976
Guo JU, Agarwal V, Guo H et al (2014) Expanded identification and characterization of mammalian circular RNAs. Genome Biol 15(7):409
Memczak S, Jens M, Elefsinioti A et al (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495(7441):333–338
Lukiw WJ (2013) Circular RNA (circRNA) in Alzheimer’s disease (AD). Front Genet 4:307
Zhao Y, Alexandrov PN, Jaber V et al (2016) Deficiency in the ubiquitin conjugating enzyme UBE2A in Alzheimer’s disease (AD) is linked to deficits in a natural circular miRNA-7 sponge (circRNA; ciRS-7). Genes (Basel) 7(12):116
Hansen TB, Jensen TI, Clausen BH et al (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495 (7441):384–388; Junn E, Lee KW, Jeong BS et al (2009) Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A 106 (31):13052–13057
Guo J (2014) Transcription: the epicenter of gene expression. J Zhejiang Univ Sci B 15(5):409–411
Drinnenberg IA, Fink GR, Bartel DP (2011) Compatibility with killer explains the rise of RNAi-deficient fungi. Science 333(6049):1592
Wang PL, Bao Y, Yee MC et al (2014) Circular RNA is expressed across the eukaryotic tree of life. PLoS One 9(6):e90859
Mazin P, Xiong J, Liu X et al (2013) Widespread splicing changes in human brain development and aging. Mol Syst Biol 9:633; Harries LW, Hernandez D, Henley W et al (2011) Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing. Aging Cell 10 (5):868–878
Mehta SL, Pandi G, Vemuganti R (2017) Circular RNA expression profiles alter significantly in mouse brain after transient focal ischemia. Stroke 48(9):2541–2548
Liu C, Zhang C, Yang J et al (2017) Screening circular RNA expression patterns following focal cerebral ischemia in mice. Oncotarget 8(49):86535–86547
Lin SP, Ye S, Long Y et al (2016) Circular RNA expression alterations are involved in OGD/R-induced neuron injury. Biochem Biophys Res Commun 471(1):52–56
Xie B, Wang Y, Lin Y et al (2018) Circular RNA expression profiles alter significantly after traumatic brain injury in rats. J Neurotrauma. https://doi.org/10.1089/neu.2017.5468
Acknowledgement
I would like to thank Professor Brandon J. Wainwright for encouraging me to pursue noncoding RNAs as a field of research.
Competing Financial Interests
The author declares no competing financial interests.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Constantin, L. (2018). Circular RNAs and Neuronal Development. In: Xiao, J. (eds) Circular RNAs. Advances in Experimental Medicine and Biology, vol 1087. Springer, Singapore. https://doi.org/10.1007/978-981-13-1426-1_16
Download citation
DOI: https://doi.org/10.1007/978-981-13-1426-1_16
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-1425-4
Online ISBN: 978-981-13-1426-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)