
Abstract
Circular RNAs (cirRNAs) are long, noncoding endogenous RNA molecules and covalently closed continuous loop without 5′–3′ polarity and polyadenylated tail which are largely concentrated in the nucleus. CirRNA regulates gene expression by modulating microRNAs and functions as potential biomarker. CirRNAs can translate in vivo to link between their expression and disease. They are resistant to RNA exonuclease and can convert to the linear RNA by microRNA which can then act as competitor to endogenous RNA. This chapter summarizes the evolutionary conservation and expression of cirRNAs, their identification, highlighting various computational approaches on cirRNA, and translation with a focus on the breakthroughs and the challenges in this new field.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
In 1976 Sanger and coworkers proposed that the viroids are single-stranded structures covalently bound to circular RNAs (cirRNAs). These are pathogenic to certain plants of higher class. It was primitively reported as a viroid, consisting of a covalently closed cirRNA molecule, and pathogenic to particular higher plants [1].
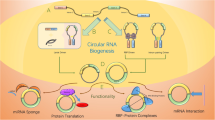
CirRNAs, a class of noncoding endogenous RNA, regulate gene expression in mammals at the transcriptional or posttranscriptional level by interacting with microRNAs [2,3,4,6]. For many years, cirRNAs were overlooked as rare isoforms that result from splicing artifacts or gene rearrangements [7]. These rediscovered RNA molecules mainly arise from exon circularization or intron circularization and covalently joined 3′ and 5′ ends of a single-stranded RNA molecule by backsplice events (an upstream splice acceptor is joined to a downstream splice donor), thus presenting as covalently closed continuous loops [1, 6,7,9].
CirRNAs are misinterpreted as splicing errors. Recently, cirRNAs have shown to be widespread and diverse in eukaryotic cells [10]. CirRNAs are relatively stable in the cytoplasm [5]. These are produced by a backsplicing process, wherein downstream exons are spliced to upstream exons in reverse order [2]. CirRNAs are more stable than linear RNA isoforms due to the lack of accessible ends, which resist exonucleases. However, the mechanism of cirRNA formation and their cellular function are still unclear. Relating to the function of cirRNA, it is hypothesized that these molecules are epigenetic microRNA sponges [7]. Human CDR1as/ciRS-7 are examples of functional exonic cirRNAs which have been experimentally validated to function as miRNA sponges and involve in gene expression regulation [10]. However, it is not clear whether all cirRNA molecules work as miRNA sponges or not [7]. The stable nature of cirRNAs makes these moieties intriguing candidates as functional molecules in circulating body fluid [11]. However, currently, there is no systematic approach available for identifying exonic cirRNAs in the human transcriptome [10].
Various challenges associated with the detection of cirRNA include exclusion of sequencing errors, unfair treatment between exonic cirRNAs, and other types of RNAs (e.g., trans-spliced RNAs and genetic rearrangements) on the basis of prejudice, adjustment of errors, in vitro artifacts, and the reconciliation of heterogeneous results [10].
CirRNAs are specific to certain diseases such as neuronal disorders and atherosclerosis [4, 5]. Our insensitivity about cirRNAs is due to an insufficiency of available sequencing data for cirRNA detection [12]. CirRNAs have great potential as clinical diagnostic markers and new therapeutic molecules for the disease therapy. Till today few reports have been published on cirRNAs due to low expression level. Originally these molecules were considered as by-products of alternative splicing and were named as a genetic accident or experimental errors [1].
2 Identification and Appropriate Validation of cirRNA
The cirRNA was firstly recognized in the early 1990s. The recognition on a large scale was not focused in the early stages because of its tedious traditional method of study and due to the lack of useful information. Therefore, the developed recent method of study and identification brings a very precise way to explore cirRNA. Moreover, a key element of cirRNA is out of-order arrangement of exons kenned as a backsplice (described beneath) is not one of a kind to cirRNAs. An early RNA-seq mapping algorithm filtered out such sequences. These issues have been tended to through the improvement of exonuclease-based enhancement approaches. Novel bioinformatic devices such as sequencing with longer reads and higher throughput and sequencing of ribosomal RNA (rRNA)-depleted RNA libraries (as opposed to poly(A)-advanced libraries) make easy to separate cirRNAs from other RNAs and also maintain its circularity [13].
The identification of cirRNAs is exceptionally valuable for understanding the regulatory mechanisms and for potential ramifications for remedial applications, for instance, working as miRNA sponges for oncogenic miRNAs. lncRNA is effectively recognized from other little ncRNA, such as miRNA, siRNA, and snoRNA, by utilizing straightforward property transcript size. However, for cirRNA identification from different lncRNAs, it has been nearly unrealistic to distinguish them just on simple features. cirRNA has shown some extraordinary succession attributes from different lncRNAs, for example, GT-AG match of sanctioned graft locales, combined Alu rehash, and backsplice [14]. Sequence features cumulating with machine learning are accounted to be puissant to prognosticate gene regulation, splicing sites, and chromatin 18. They promote sequence-based strategy possibly used to recognize cirRNA from different lncRNAs efficaciously [15].
Discovery of cirRNA articulation can be accomplished utilizing various techniques such as polymerase chain reaction (PCR) of the Northern blot, two-dimensional gel electrophoresis, gel trap electrophoresis, in situ hybridization, and RNase degradation assay [16].
2.1 Identification and Validation of cirRNA by PCR
PCR is the speediest and most effortless technique to distinguish the expression of cirRNAs. Primers are utilized as a part of PCR for recognition of protein-coding or noncoding RNAs. These are essentially planned and focused to permit enhancement of the primer-flanked nucleic acid region. The utilization of different oriented primer sets is fundamental for the recognition of cirRNA articulation utilizing PCR [17]. Sanger sequencing is the fundamental technique to identify various circular transcripts by semiquantitative or quantitative PCR. Sanger sequencing is also useful for further refinement of the PCR product to validate backsplice site. Backsplice sequence information can be generated from RNA sequencing data or publicly accessible sets of non-poly(A)-culled RNA sequencing data from the National Center for Biotechnology Information – Gene Expression Omnibus (NCBI – GEO) database. Primers to categorically detect cirRNAs by PCR should be planned divergently which can be straightforwardly achieved utilizing free online implements such as Primer3.
Identification of circular RNA by PCR is done in the following steps:
-
a.
Primer design : For the determination of chromosome position of the terminuses presaged to pair for backsplicing, we need to determine which exons/introns are to be included in the backsplicing utilizing the genome browser. To get a general summary of the required exons in cirRNA, an entire backsplice sequence is inserted in the BLAT implement by using https://genome.ucsc.edu/cgi-bin/hgBlat. The corresponding exon sequences are fetched to the respective gene and species using www.ensembl.org. The exon order is reversed, keeping 5′→3′ orientation of both exons. The sequence has to be pasted into the corresponding box using http://primer3.ut.ee/. The box is changed from a product size in the range of 70–150 bp and cull pick primers. Cull primer pair ascertains amplified region covering the backsplice site and controls the presaged primer tm to 60 °C. It is suggested that the primers should not overlap the backsplice site. The primer sequence is examined by UCSC in silico PCR implement to check the amplification in genome assembly and the UCSC-annotated genes; no presaged amplification is expected.
-
b.
Semiquantitative PCR : The accompanying convention is depicted for the enhancement of cirRNAs. For reference, articulation of the straight RNA of the quality of intrigue ought to be evaluated. Briefly, the buffer concentrate is defrosted, dNTP is mixed, and random hexamer-primed cDNA is kept on the ice. The quantity of responses is computed, and 1–2 extra responses are incorporated to make up for inevitable misfortune by pipetting out. PCR Master is mixed maintaining Taq Reaction Buffer (10×) 2.5 μL, 1 μL of Taq polymerase (1 U/μL), 0.5 μL of 10 mM dNTPs, forward and reverse primers (1 μL each), and 14 μL of RNase/DNase-free water. PCR Master Mix (20 μL) is distributed for each reaction, and 5 μL of random hexamer-primed cDNA (an RNA/cDNA equivalent of >10 ng per reaction is recommended) is added. Control PCR Master (15 μL) containing H2O and 5 μL of RNase-/DNase-free H2O is added and mixed. PCR is carried out at 95 °C for 2 min, 95 °C for 10 s, 60 °C for 20 s at 30–35 PCR cycles, and 72 °C for 15 s. The time and temperature are subject to the individual polymerase and the item estimate. PCR items are broken down by gel electrophoresis utilizing 2% agarose gels.
-
c.
The accompanying convention is portrayed utilizing the SYBR Green Master Mix for a standard 96-well qPCR : Heumuller and Boeckel described specificity of the PCR examine for cirRNA discovery utilizing semiquantitative PCR and gel electrophoresis preceding qPCR. Besides, the qPCR item ought to dependably be prepared by liquefying bend investigation and in any event once by consequent gel electrophoresis. Dissolve bend investigation is not vital when utilizing hydrolysis test-based qPCR. In this situation, a hydrolysis test ace blend is utilized rather than the SYBR Green Master Mix (SYBR-GMM) in the accompanying convention. SYBR-GMM is defrosted, and irregular hexamer cDNA is prepared on ice. The quantity of responses is figured out, and 1–2 extra responses are incorporated to make up for inevitable misfortune by pipetting. This is followed by planning qPCR Master Mix. The qPCR Master Mix contains 10 μL of SYBR-GMM, 3 μL of water, and 1 μL of the forward and switch 10 μM groundwork stock. To perform hydrolysis test-based qPCR, it is recommended to utilize 10 μL of hydrolysis test ace blend, 2 μL of water, 1 μL of the forward and turnaround 10 μM preliminary stock each, and 1 μL of the hydrolysis test. PCR ace blend (15 μL) is circulated for every response, and 5 μL irregular hexamer-prepared cDNA is incorporated. This is followed by the incorporation of H2O control comprising of 15 μL of the PCR ace blend and includes 5 μL H2O. It is ensured to liquefy bend for every groundwork. The information is broken down utilizing the 2-CT strategy or the 2-∆CT technique when a housekeeping quality (e.g., the mRNA of RPLP0) has been estimated.
-
d.
PCR items increasing the back-grafted area ought to be filtered utilizing phenol/chloroform/isoamyl liquor precipitation : In this way, Sanger sequencing (PCR sequencing) is utilized to approve the presence of the back-join site and to control the specificity of the differently oriented preliminaries utilized as a part of the PCR. An equivalent volume of phenol/chloroform/isoamyl liquor (25:24:1 (v/v/v)) is added to the PCR item in a 1.5 mL response tube mixed and centrifuged for 5 min at 12,000 × g at room temperature (25 °C). The tube is handled deliberately and abstained from irritating stage partition-exchange, the upper (watery) stage to another 1.5 mL response tube. It is recommended not to aggravate the lower (natural) stage. Tainting with the lower stage can bring about diminished extraction productivity. The tube containing the lower stage (phenol squander) is disposed of. The product was blended with 2.5 mL of ice-cold ethanol and mixed for 15 min at 4 °C to hasten the DNA. The supernatant is evacuated using a pipette. The pellet is dried on a warm obstruct with open cover at 37 °C for 0.5–2 min. The pellets are resuspended in 10 μL TE cushion. DNA sum ought to be resolved, and the test is sent to PCR sequencing utilizing the disparate forward and invert preliminary.
2.2 Identifying cirRNAs by RNA Fluorescence In Situ Hybridization (FISH)
FISH permits the representation of various RNA species inside the cell. This section contains an all-around appropriate technique to recognize cirRNA through a junction specific test. CirRNAs are set apart by making a beeline for a tail-ligated intersection that is not found in some other RNAs. Till today, this convention is very strong and delicate. Numerous tests marked by an alternate fluor can be taken into consideration to achieve synchronous identification of different targets [18]. RNA FISH depends on the straightforward idea of uncovering settled cells or tissues to short DNA oligonucleotides in adequately high fixations to enable blending with corresponding RNA molecules to frame stable DNA-RNA half-breeds. The tests comprise of a pooled set of ~32–48 DNA oligos of various arrangements, every 20 nucleotides in length and named with a solitary fluorophore at its 3′ end. The convention can recognize single RNA molecule with high specificity (a couple of false positives) and high affectability (a couple of false negatives) and does not require flag intensification steps, which tend to render single-atom identification approaches less quantitatively [19].
2.3 Northern Blot Analysis of cirRNAs
Northern smear hybridization makes the strategy for the decision to convincingly show round setup of putative cirRNAs. CirRNA identification can be proficient by short tests spreading over the round graft intersection or by longer tests covering as much as a whole circularized exon. This alternative winds up significantly if the specificity for roundabout isoform is not fundamental (for instance, if the straight structures do not enter the gel, if both direct and roundabout isoforms ought to be identified in parallel, or if there should be an occurrence of solely roundabout RNAs). Northern blots are thus basic part of any cirRNA portrayal, because of their incredible flexibility. To start with the decision of test districts (round or straight joint intersection or exonic areas) and identification standard (digoxigenin or 32P-named tests) decides the specificity for roundabout versus direct isoforms. The decision of gel network includes greater adaptability in northern smudge examine. Agarose gels are reasonable for cirRNAs from 0.2 kb up to a few kb. In agarose gels, round and straight RNAs of a similar size cannot be recognized by their running conduct. Actually, in denaturing polyacrylamide gels, direct RNA keeps running at the normal size, while cirRNAs have a lower evident versatility with respect to straight markers; this hindrance impact is upgraded by expanding acrylamide fixations [20]. Due to this impediment, cirRNAs up to 1 kb can be examined by polyacrylamide gel electrophoresis. Thus, at any rate for a far-reaching investigation of one or a couple of putative cirRNAs, not for a medium- to high-throughput screening endeavors, Northern blot tests give an extremely profitable and exceedingly useful approach.
2.4 Portrayal of cirRNA Concatemers
The model on cirRNA biogenesis suggests that the rearranged rehashes take part in base blending, accordingly situating the two splice sites in nearness. Wang and his colleagues outlined the embodiment of exon 2 from beta-globin (HBB) in the middle of modified components. As far as anyone is concerned, this exon is not creating cirRNA in its normal setting. However, when flanked with transformed rehashes, the exon produces one particular cirRNA, as well as a step of cirRNA-like items (cirRNA concatemers) [21]. The identification and profiling of cirRNA are normally done by cutting-edge sequencing (NGS) or by qRT-PCR [22]. The cirRNA in the first place contained exon rehashes or whether the monotony was presented by the RT chemical. Barrett et al. presented a blueprint of basic biochemical tests projected by northern smearing to ponder the idea of these cirRNAs and demonstrated that they are made out of exon rehashes (cirRNA concatemers). To recognize concatemers and interwoven cirRNAs (topologically bolted single exon cirRNAs), three particular examinations such as (1) RNase R absorption to approve the roundabout structure of the cirRNA species, (2) RNase H absorption to decide the structure of exons by crumbling the cirRNAs into their exon units, and (3) a soluble treatment to tenderly scratch the cirRNA into a relating direct RNA have been suggested [23].
3 Computational Approaches on cirRNA
The hereditary data streams of life, in which DNA and protein are considered as primary on-screen characters of cell life, retain RNA as basic part of protein synthesis. However, this perspective of the organic part of RNA experienced various challenges [24]. Computational approaches to deal with RNA tertiary structure expectation are based on the examination of RNA tertiary themes, and diagram hypothesis for RNA and RNA endeavors plan to enhance the in vitro test choice for aptamer outline. Thus, the examination of RNA basic correlation is important thought of root-mean-square deviation (RMSD) since the forecasts are not exact for RNA during this phase [25]. There are numerous different zones of advancement in RNA bioinformatics, for instance, auxiliary structure forecasts [26].
Current discoveries in the field of noncoding RNA are focused on cirRNA [27] which are produced by nonlinear backsplicing linked to a downstream splice donor and upstream splice acceptor. CirRNA is present in all eukaryotic clades, including insects, fungi, and plants, and it also exists in humans to establish several thousand different cirRNAs. In the immense majority, the function of cirRNA is not clear. A small subset of cirRNAs has been reported to act as steerers for miRNAs [28,28,, 29] or to bind and regulate protein function [30, 31]. The diversity of cirRNAs can be explicated based on the gene fraction and antisense strand of some genes and from intergenic regions [33,34,34]. The length of cirRNA ranges from 100 bp to 4 kb [35]. cirRNAs may hold multiple or single exon [36] and are present in different cell lines, tissues, and extracellular exosomes. Biogenesis of cirRNA is based on lariat-driven, intron-pairing-driven, and RNA-binding protein-driven circularization mechanisms [37, 38]. It has been proposed that cirRNA acts as microRNA sponges and regulates multiple gene expressions. The source quality, mode of exon creation, biogenesis, and capacity make them different than other RNAs. Comment-free recognition calculations can be utilized as a part of an extensive variety of living beings. However, it requires more careful systems to guarantee unwavering quality.
The majority of the location techniques are upgraded for their assigned aligners, and these can be additionally partitioned into joint mindful aligners and adaptable read mappers. Paired-end sequencing gives more data to diminish false positives for discovery techniques that receive sifting in light of paired-end mapping. In view of identification calculations, promising computational techniques have been developed for the downstream investigations of cirRNAs. However, new computational techniques to remake full length of cirRNAs and measure their demeanor are critically required. Late examinations have shown that cirRNAs are universal and have different capacities and systems of biogenesis. In such investigations, computational profiling of cirRNAs has been pervasively utilized as an irreplaceable strategy to give high-throughput ways to deal with identifying and breaking down of cirRNAs. In any case, without a general comprehension of the basic methodologies, these computational techniques may not be exactly chosen or utilized for a particular research reason, and a few misguided judgments may bring about predispositions in the examinations. Gao and Zhao reviewed the key advances and abridged trade-off of various systems, covering every single prominent calculation for cirRNA discovery and different downstream investigations [39].
The computational approach plays an important role in high-throughput RNA-seq data examination and in expression of cirRNA profiling. Till today about 11 computational approaches for cirRNA detection have been reported. CIRI, CIRCexplorer [40], and KNIFE [41] are more functional than other approaches. All the reported computational approaches have their own advantages and essential point sensitivity, precision, and computational cost (Tables 1.1 and 1.2). Downstream computational approaches are significant due to their primary detection results. These approaches are linked to the quantification and differential expression analysis (Table 1.3) [42, 43].
3.1 Detection of cirRNA Using Annotation, Genome Reference, and GT-AG Splicing Signals
Genomes are essential for algorithm sensing and can be used in the detection workflows. It mainly works for the direct alignment of sequencing reads against the standard genome. UROBORUS [44], CIRCexplorer [45], find circ [46], and CIRI [47] are the examples of detection algorithms. The circularity pathway of cirRNA is different from other categories of RNAs, and thus an evident feature can be captured from the circle junction alignment which is used as a backsplice junction (BSJ). By contrast, forward-spliced junction (FSJ) in mRNA, the developed sequencing reads that are collinearly aligned on the genome, interprets spanning BSJs are divided into segment and are aligned to the address/reference sequence in reverse order. Hence, detection algorithms in this category can be termed as split-alignment-based approaches.
For different algorithms, such as NCLscan [48] and KNIFE [49], the reference genome is combined with the corresponding genome annotation to build pseudo-sequence around putative BSJs in the first few steps (Fig. 1.1). Consequent steps are centered on the complete alignment of sequencing reads against such pseudo-sequences to identify BSJ reads. In addition to a BSJ pseudo-sequence database, KNIFE also constructed a FSJ sequence information according to the annotation to remove candidate reads with high-score alignment in both databases. In this category, detection algorithms may be termed pseudoreference-based approaches.

Pseudoreference-based approach for the cirRNA detection
The reference genome is combined with the corresponding genome annotation to build pseudo-sequence
The application of annotation is much useful. As an example, a comprehensive evaluation of RNA-seq aligners which actively addressed that annotation can help to increase the sensitivity for junction reorganization versus de novo detection [50].
3.2 Various Read Mappers or Specified Splice-Aware Aligner
BSJ reads such as split-alignment and pseudoreference approaches are identified with the help of alignment of transcriptomic read. Many algorithms, for example, KNIFE and CIRI, prefer read mappers which are generally applicable in reference-based RNA/DNA sequence studies. The simplest way is splice-aware aligners which were developed for RNA-seq reads across intron-sized gaps on genome references such as Novoalign, STAR [51], and TopHat [52]. Detection algorithms, such as CIRCexplorer and DCC [53], depend on this type of aligner. An evident advantage is that the aligners are optimized according to eukaryotic transcription which is easier than various read mappers. The flexibility of splice-aware aligners is less than the versatile mappers, which have developed both end-to-end and local alignment. In other condition, nearly all splice-aware aligners are based on versatile read mappers.
4 Translation of cirRNA
The introduction of nearly all cirRNAs from exons and their confinement in cytoplasm increase the probability of cirRNA translation [54]. Translation of several viral proteins in many organisms, including humans, depends on internal ribosome entry site (IRES) and cellular IRESs. However, their mechanism of action remains controversial [55]. Based on this theory, artificial cirRNAs with IRES have been translated [56, 57]. It has also been noticed that principal cirRNAs can be translated in vitro and in vivo [58]. Translation of cirRNAs in living human cells is based on rolling circle amplification mechanism. The elements including IRES are not required for the translation of cirRNA in eukaryotic translation system [59]. The cirRNAs have been endogenously translated and indirectly tested [59].
Binding of open pre-initiation complex containing small ribosome is the beginning step for canonical translation process in eukaryotes [59]. The interaction between the cap-binding protein (CBP) poly(A) regions leads to the circularization of mRNA competent for translation [60]. Small ribosomal subunits and mRNAs are further scanned by small ribosomal subunits for the start codon. After that the 60S ribosomal subunit is required. Also, the internal start codons can recirculate the ribosomes internally by an IRES-dependent mechanism.
4.1 Translation of cirMbl
Till date, the investigations are limited to the in vivo translation of endogenous cirRNAs. Ribo-cirRNAs have specific ribosome profiling and are denoted as cirRNAs. Ribosome profiling datasets of Drosophila have shown presence of cirRNA-specific junctions [61]. Expression of cirRNA in Drosophila S2 cells depends on the presence of intron-exon-intron minigenes. The minigenes have been reported to express V5-tagged proteins. V5-tagged proteins have been reported from the cells transfected with circCdiV5, circPde8V5, or circMblV5. This was not observed with the cells which are not transfected with minigenes of circHaspinV5 or circCamKIV5. The protein of desired size has been observed using antiMBL antibody. For cirMbl, cirRNA has been established as the main source of detected protein. Transgenic flies containing MBL-immunoreactive bands have been originated to express in vivo cirMbl minigene [11]. In ribosome footprinting (RFP) reads of fly heads about 122 ribo-cirRNAs have been identified. Protein domains have been identified in many ribo-cirRNA-encoded proteins. Protein expression of V5-tagged cirRNA minigenes was not affected by co-expression of 4E-BP that inhibits cap-dependent translation. circMbls are translated in a cap-independent manner, and the untranslated region (UTR) sequence of circMbl is capable of facilitating cirRNA translation [61].
4.2 Translation of circ-ZNF609
Circ-ZNF609 codon starts with a linear transcript and terminates at a stop codon. It contains 753-nt ORF [62]. A substantial percentage of circ-ZNF609 was indicated to sediment with heavy polysome. Treatment with puromycin disrupted active translation of ribosomes which shifted circ-ZNF609 to lighter polysomes. p-circ3XF containing 3XFLAG-coding sequence of stop codon has been tested to express circ-ZNF609 protein-coding ability. The study resulted in two flagged isoforms. On the other side, p-lin3XF containing circ-ZNF609 ORF was also produced and expressed same proteins more efficiently. The RNA amount was normalized in both circ3XF and p-lin3XF. The results suggested that the translation efficiency of p-circ3XF was lower than that of p-lin3XF. CRISPR/Cas9 has been utilized to introduce 3XFLAG-code in endogenous ZNF609 gene which translated circZNF609 from the chromosomal gene. Positive clone with alleles (1) holding expected flag and (2) holding a deletion that prevents circ-ZNF609 production has been reported. However, only clone carrying flagged allele can develop circ-ZNF609.
In an investigation the positive clone cell lysates were immunoprecipitated with anti-FLAG antibody and subject to mass spectrometry. One peptide mapping to the cir-ORF was observed in the positive clone cell, whereas several peptides were present in the protein from cells overexpressing p-circ3XF and p-lin3XF. This resulted in the formation of lower cir-RNAs from the chromosomal gene and lower translational capacity. The heat shock resulted to increased translation of circZNF609. In this process no cap structure was present in cirRNA. The study proposed the possibility of translation through sequences with internal ribosome entry site (IRES) activity [62].
5 m6 A Modification in cirRNA Translation
Yang and coworkers discovered sequences to induce translation of cirRNA [56]. RRACH fragment involves in the methylation of N6 position of adenosine (m6 A). m6 Higher peak density of m6 A has been observed in cirRNA when compared to mRNAs [63, 64] suggesting its role in mRNA translation [65, 66]. These findings support the hypothesis that m6 A plays an important role in cirRNA translation. To validate this hypothesis, a short fragment containing m6 A motif was introduced before the start codon in cirRNA reporter. The level of GFP protein output was assessed in all the transfected cells [56], and it was observed that cirRNA containing m6 A motif was translated expeditiously. The methylation of cirRNAs with m6 A motif is associated with RNA-immunoprecipitation (RNA-IP). Negative effect of m6 ademethylase FTO co-expression on the amount of immunoprecipitated RSV-containing cirRNA and GFP translation from cirRNA has been observed. eIF4G2, a noncanonical protein which recognizes IRES, also plays an important role in cirRNA translation and partakes in cap-independent translation and m6 A reader protein YTHDF3 [56, 67]. N6-methylation requires the translation of mRNA and leads to GFP protein translation from m6 A-containing cirRNA by heat shock stress [56, 65, 66].
6 Conclusions
It is evident that the number of cirRNAs with known functions is expanding during the last few years. However, the function of various cirRNAs remains unknown, and very little information is available about the control of backsplicing process of cirRNA generation. This could be due to the availability of limited and challengeable methods to detect and characterize cirRNAs. A detailed study of cirRNA biogenesis and in vivo research may allow testing for functional consequences of cirRNA expression and will provide novel insights into cellular development human disease.
Competing Financial Interests
The authors declare no competing financial interests.
References
Liu L, Wang J, Khanabdali R et al (2017) Circular RNAs: isolation, characterization and their potential role in diseases. RNA Biol 14(12):1715–1721
Li J, Yang J, Zhou P, et al (2015a) Circular RNAs in cancer: novel insights into origins, properties, functions and implications. Am J Cancer Res 5(2):472
Li P, Chen S, Chen H et al (2015b) Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin Chim Acta 444:132–136
Greene J, Baird AM, Brady L et al (2017) Circular RNAs: biogenesis, function and role in human diseases. Front Mol Biosci 4:38
Hansen TB, Kjems J, Damgaard CK (2013) Circular RNA and miR-7 in cancer. Cancer Res 73(18):5609–5612
Bachmayr-Heyda A, Reiner AT, Auer K et al (2015) Correlation of circular RNA abundance with proliferation–exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep 5:8057
Barrett SP, Salzman J (2016) Circular RNAs: analysis, expression and potential functions. Development 143(11):1838–1847
Lasda E, Parker R (2014) Circular RNAs: diversity of form and function. RNA 20(12):1829–1842
Chen I, Chen CY, Chuang TJ (2015) Biogenesis, identification, and function of exonic circular RNAs. Wiley Interdiscip Rev RNA 6(5):563–579
Bahn JH, Zhang Q, Li F et al (2015) The landscape of microRNA, Piwi-interacting RNA, and circular RNA in human saliva. Clin Chem 61(1):221–230
Gao Y, Wang J, Zhao F (2015) CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol 16(1):4
Jeck WR, Sharpless NE (2014) Detecting and characterizing circular RNAs. Nat Biotechnol 32(5):453
Jeck WR, Sorrentino JA, Wang K et al (2013) Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19(2):141–157
Xiong HY, Alipanahi B, Lee LJ et al (2015) The human splicing code reveals new insights into the genetic determinants of disease. Science 347(6218):1254806
Hansen TB (2018) Characterization of circular RNA concatemers. In: Circular RNAs. Humana Press, New York, pp 143–157
Hansen TB, Wiklund ED, Bramsen JB (2011) miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J 30(21):4414–4422
Gingeras TR (2009) Implications of chimaeric non-co-linear transcripts. Nature 461(7261):206
Itzkovitz S, Van Oudenaarden A (2011) Validating transcripts with probes and imaging technology. Nature Methods 8(4s):S12
Günzl A, Palfi Z, Bindereif A (2002) Analysis of RNA–protein complexes by oligonucleotide-targeted RNase H digestion. Methods 26(2):162–169
Tabak HF, Van der Horst G, Smit J (1988) Discrimination between RNA circles, interlocked RNA circles and lariats using two-dimensional polyacrylamide gel electrophoresis. Nucleic Acids Res 16(14):6597–6605
Wang PL, Bao Y, Yee MC et al (2014) Circular RNA is expressed across the eukaryotic tree of life. PloS one 9(3):e90859
You X, Vlatkovic I, Babic A (2015) Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci 18(4):603
Barrett SP, Wang PL, Salzman J (2015) Circular RNA biogenesis can proceed through an exon-containing lariat precursor. Elife 4
Jakobi T, Dieterich C (2018) Deep computational circular RNA analytics from RNA-seq data. Methods Mol Biol 1724:9–25
Reznichenko A (2012) Translational renal genetics. University Library Groningen Host. Humana Press, New York
Shapiro BA, Yingling YG, Kasprzak W et al (2007) Bridging the gap in RNA structure prediction. Curr Opin Struct Biol 17(2):157–165
Ebbesen KK, Kjems J, Hansen TB (2016) Circular RNAs: identification, biogenesis and function. Biochim Biophys Acta 1859:163–168
Hansen TB, Jensen TI, Clausen BH et al (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495:384–388
Memczak S, Jens M, Elefsinioti A et al (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495:333–338
Zheng Q, Bao C, Guo W et al (2016) Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun 7:11215
Ashwal-Fluss R, Meyer M, Pamudurti NR et al (2014) circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 56:55–66
Du WW, Yang W, Liu E et al (2016) Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res 44:2846–2858
Gao Y, Wang J, Zhao F (2015) CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol 16(1):4
Memczak S, Jens M, Elefsinioti A et al (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495(7441):333
Salzman J, Chen RE, Olsen MN et al (2013) Cell-type specific features of circular RNA expression. PLoS Genet 9(9):e1003777
Lasda E, Parker R (2014) CircularRNAs: diversity of form and function. RNA 20:1829–1842
Li Z, Huang C, Bao C et al (2015) Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 22(3):256
Conn SJ, Pillman KA, Toubia J (2015) The RNA binding protein quaking regulates formation of circRNAs. Cell 1(6):1125–1134
Hansen TB, Jensen TI, Clausen BH et al (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495(7441):384
Gao Y, Zhao F (2018) Computational strategies for exploring circular RNAs. Trends Genet 34(5):389–400
Zhang XO, Wang HB, Zhang Y et al (2014) Complementary sequence-mediated exon circularization. Cell 159(1):134–147
Szabo L, Morey R, Palpant NJ et al (2015) Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol 16(1):126
Glažar P, Papavasileiou P, Rajewsky N (2014) circBase: a database for circular RNAs. RNA 20(11):1666–1670
Meng X, Chen Q, Zhang P et al (2017) CircPro: anintegrated tool for the identification of circRNAs with protein-coding potential. Bioinformatics 33:3314–3316
Memczak S, Jens M, Elefsinioti A (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495(7441):333
Zhang XO, Wang HB, Zhang Y et al (2014) Complementary sequence-mediated exon circularization. Cell 159(1):134–147
Gao Y, Zhang J, Zhao F (2017) Circular RNA identification based on multiple seed matching. Brief Bioinform. https://doi.org/10.1093/bib/bbx014
Song X, Zhang N, Han P et al (2016) Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic Acids Res 44(9):e87–e87
Chuang TJ, Wu CS, Chen CY et al (2015) NCLscan: accurate identification of non-co-linear transcripts (fusion, trans-splicing and circular RNA) with a good balance between sensitivity and precision. Nucleic Acids Res 44(3):e29–e29
Szabo L, Morey R, Palpant NJ et al (2015) Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol 16(1):126
Baruzzo G, Hayer KE, Kim EJ et al (2017) Simulation-based comprehensive benchmarking of RNA-seq aligners. Nat Methods 14(2):135
Dobin A, Davis CA, Schlesinger F et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1):15–21
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25(9):1105–1111
Cheng J, Metge F, Dieterich C (2015) Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics 32(7):1094–1096
Jackson RJ (2013) The current status of vertebrate cellular mRNA IRESs. Cold Spring Harb Perspect Biol 5:a011569
Yang Y, Fan X, Mao M (2017) Extensive translation of circular RNAs driven by N-6-methyladenosine. Cell Res 27:626–641
Wang Y, Wang Z (2015) Efficient backsplicing produces translatable circular mRNAs, RNA-Publ. RNA Soc 21:172–179
Chen CY, Sarnow P (1995) Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science 268:415–417
Abe N, Matsumoto K, Nishihara M (2015) Rolling circle translation of circular RNA in living human cells. Sci Rep 5:16435
Guo JU, Agarwal V, Guo H et al (2014) Expanded identification and characterization of mammalian circular RNAs. Genome Biol 15:409
Aitken CE, Lorsch JR (2012) A mechanistic overview of translation initiation in eukaryotes. Nat Struct Mol Biol 19:568–576
Pamudurti NR, Bartok O, Jens M et al (2017) Translation of CircRNAs. Mol Cell 66:9–21
Legnini I, Di Timoteo G, Rossi F et al (2017) CircZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol Cell 66:22–37
Dominissini D, Moshitch-Moshkovitz S, Schwartz S (2012) Topology of the human and mouse m(6)A RNA methylomes revealed by m(6)A-seq. Nature 485:201–284
Meyer KD, SaletoreY ZP et al (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell 149:1635–1646
Meyer KD, PatilDP ZJ et al (2015) 5′ UTR m(6)A promotes cap-independent translation. Cell 163:999–1010
Fu Y, Dominissini D, Rechavi G (2014) Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet 15:293–306
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Awasthi, R. et al. (2018). An Overview of Circular RNAs. In: Xiao, J. (eds) Circular RNAs. Advances in Experimental Medicine and Biology, vol 1087. Springer, Singapore. https://doi.org/10.1007/978-981-13-1426-1_1
Download citation
DOI: https://doi.org/10.1007/978-981-13-1426-1_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-1425-4
Online ISBN: 978-981-13-1426-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)