
Abstract
Influenza is a negative-sense single-stranded RNA virus with segmented genome. Each segment is encapsidated by a ribonucleoprotein (RNP) complex composed of RNA-dependent RNA polymerase (RdRP) and multiple copies of nucleoprotein (NP). The RNP complex plays a crucial role in viral life cycle, supporting and regulating transcription and replication of viral genome in infected cells. The structural characterization of RdRP and RNP in recent years has shed light on its functions and mechanism of action. In this review, we summarize current understanding on the structure of RNP complex, as well as the structure of each subunit. Crucial functions of RNP are also discussed.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Influenza
- RNA-dependent RNA polymerase
- Ribonucleoprotein
- PA
- PB1
- PB2
- Nucleoprotein
- Viral transcription and replication
Introduction
The influenza virus is a highly contagious pathogen that causes annual epidemics and sporadic pandemics, leading to substantial human morbidity and mortality and imposing heavy burden on our healthcare system. Although vaccines and antivirals have been developed to counteract influenza virus, its rapid mutation rate results in the efficient generation of resistant strains. Reassortment of virus strains within reservoir species produces novel influenza virus that posesses serious threat to humankind.
Influenza belongs to the Orthomyxoviridae family. It is a negative-sense single-stranded RNA virus ((−)ssRNA virus). Its genome is divided into eight segments. Each negative-sense RNA segment is encapsidated into a ribonucleoprotein (RNP) molecule, consisting of a trimeric RNA-dependent RNA polymerase complex (including PA, PB1 and PB2) and numerous NP proteins. RNP plays a central role in the life cycle of influenza virus. In the virion, genetic information is stored in the form of RNP. In infected cells, RNP is required for the transcription and replication of viral genome. In the generation of viral progenies, newly synthesized viral RNA has to be assembled into RNP before being packaged into a new viral particle.
In this chapter, we summarize current understanding on the structure of RNP complex, as well as the structure of each subunit. In addition, we also incorporate the latest findings on how the viral transcription and replication are carried out. Besides, other assistive and modulatory functions of each subunit are discussed.
Structure of RNP and Its Subunits
RNP is composed of an RNA-dependent RNA polymerase (RdRP) with three subunits – PA, PB1, and PB2, nucleoprotein (NP), and vRNA. RNP has a rod-shaped structure. Its head contains the RdRP complex. The vRNA segment extends from the head and is encapsidated by a number of NP molecules (Reviewed in Eisfeld et al. 2014).
Several low-resolution 3D structures of recombinant influenza RNP and RdRP were determined with electron microscopy and image processing (Martín-Benito et al. 2001; Area et al. 2004; Torreira et al. 2007; Coloma et al. 2009; Resa-Infante et al. 2010). Various crystal structures of RNP subunits started to emerge around in 2007 (Ye et al. 2006; Ng et al. 2008; He et al. 2008; Obayashi et al. 2008; Guilligay et al. 2008; Sugiyama et al. 2009; Yuan et al. 2009; Dias et al. 2009). The structure of native RNP was also determined by cryo-EM to around 20 Å in 2012 (Moeller et al. 2012; Arranz et al. 2012).
In 2014, crystal structure of vRNA promoter-bound influenza RdRP complex was determined using bat-derived H17N10 influenza virus and influenza B virus (Pflug et al. 2014; Reich et al. 2014). Soon after, the structures of 5’ cRNA-bound flu B RdRP and apo form of flu C RdRP were also solved (Hengrung et al. 2015; Thierry et al. 2016). Recently, the structure of an initiation state-resembling form of flu B RdRP was determined (Reich et al. 2017).
Structure of Influenza RNA-Dependent RNA Polymerase (RdRP) Complex
RdRP is a heterotrimeric complex with a U-shaped architecture (Fig. 5.1) (Pflug et al. 2014; Reich et al. 2014). PB1 is located at the center. Top region of the U contains two protruding arms formed by the PA-N endonuclease domain and the PB2 cap-binding domain. Both arms are in close proximity to the primer-entry/product-exit channel. The PA-C terminal domain and the promoter binding site are located at the bottom region of the U. The linker connecting PA-N and PA-C lies on the surface of PB1, forming extensive interaction with it. A large catalytic cavity is identified within PB1. There are four channels connecting the PB1 catalytic cavity from the exterior: the (1) template entry channel, (2) product exit channel, (3) NTP entry channel, and (4) template exit channel.

Structure of influenza RdRP heterotrimer (PDB: 4WSB). (a) Schematic diagram showing the architecture of RdRP heterotrimer. RdRP subunits assemble a U-shaped architecture. Major domains of each subunit are shown in oval. (b) Surface representation of RdRP showing a compact structure of RdRP heterotrimer. PA, PB1, and PB2 are colored in pink, pale blue, and pale green, respectively. Extensive inter-subunit interactions can be identified in the structure. The open cleft at the top of the U-shape is the location for cap-snatching. A capped RNA is captured by the PB2 cap-binding domain and is directed to the PA-N endonuclease for cleavage at 10–13 nt downstream of the cap. As a result, a capped primer is generated for viral protein transcription
PB1 forms the central scaffold of RdRP. It interacts with PA and PB2 through its N-terminal and C-terminal region, respectively, echoing previous mapping studies and co-crystal structure determination of RdRP subunits (González et al. 1996; Ohtsu et al. 2002; He et al. 2008; Obayashi et al. 2008; Sugiyama et al. 2009). However, the inter-subunit interactions are much more extensive than previously thought.
Various structures of RdRP reveal that the RdRP can undergo substantial conformational change (Resa-Infante et al. 2011; Pflug et al. 2014; Reich et al. 2014; Thierry et al. 2016). Two radically different configurations of PB2 could be observed (Thierry et al. 2016). The PA-N domain is also capable of in situ rotation (Reich et al. 2015; Hengrung et al. 2015; Thierry et al. 2016). This might be related to the activation of the cap-snatching mechanism or to the switch between replication and transcription mode of RdRP. In contrast, the structures of PB1, PA-C, and PA-linker remain relatively stable among various RdRP structures.
Structure of PA Subunit
PA is 716 aa long, and limited tryptic digestion reveals that it consists of two domains: (1) N-terminal (PA-N) domain and (2) C-terminal (PA-C) domain (Guu et al. 2008). PA-N contains the first 196 residues and is the smaller subunit (~25 kDa). The larger PA-C domain (~55 kDa) contains residues from 277 to 716. The two domains are connected by a long linker (residues 197–276).
PA-N Domain
PA-N forms a novel global fold. The PA-N structure has an α/β architecture, where five mixed β-strands form a twisted plane surrounded by seven α-helices. Structural alignment with type II restriction endonucleases reveals that PA-N contains a PD-(D/E)XK motif, a structural motif characteristic of many nucleases (Dias et al. 2009; Yuan et al. 2009).
A negatively charged cavity surrounded by helices α2–α5 houses one or two divalent cations (Fig. 5.2a). The structure by Yuan et al. includes one Mg2+, while the structure by Dias et al. includes two Mn2+. The divalent metal ions are coordinated by the same residues (H41, D108, E119, and K134) in both structures. The Mg2+ ion is coordinated by five ligands: E80, D108, and three water molecules that are stabilized by H41, E119, and the carbonyl oxygens of L106 and P107. For the Mn2 + -bound structure, one ion is coordinated by E80, D108, and two water molecules and the other ion by H41, D108, E119, and the carbonyl oxygen of I120. D108 acts as a bridge for both Mn2+ ions (Fig. 5.2b).

Structure of PA-N domain (PDB: 2W69). (a) Surface representation of PA-N showing its electrostatic potential. The catalytic site is indicated by a black arrow; it is strongly negatively charged. (b) Detailed structure showing the active site with a bound AMP. Crucial catalytic residues, H41, E80, D108, E119, and K134 are shown in white. H41, D108, E119, and K134 are involved in positioning the divalent ions. The positions of divalent ions Mg2+ or Mn2+ are marked in the structure. The AMP molecule reveals the nucleotide-binding site (N-site), ribose binding site (R-site), and the phosphate-binding site (P-site). Drawn using PDB: 3HW5
Co-crystal structures of PA-N with various nucleoside monophosphates (NMP) were determined by Zhao et al. (Zhao et al. 2009). NMPs were shown to bind at the catalytic center of PA-N, in close proximity with the divalent cations. These complexes represent the scenario nuclease cleavage reaction. E119, K134, K137, and Y130 form the phosphate-binding site (P site). H41 also contributes in stabilizing the phosphate moiety. Main chain atoms of A37, I38, C39, and the side chain of H41 form the ribose-binding site (R-site). A20, L38, L42, and E80 form the nucleoside-binding site (N site), which bind the nucleobase in a flexible manner (Fig. 5.2b).
The structure of residues 55–66 is highly variable. A truncated form of PA-N, where residues 52–64 were replaced by a single glycine, is able to fold correctly and can be crystallized. Besides, another study shows that the deletion of residues of 51–72 can retain normal endonuclease activity and proper folding (DuBois et al. 2012). The exact function of this loop is unclear.
PA-C Domain
PA-C forms a novel fold and can be subdivided into two parts, the “brain” domain and the “mouth” domain (Fig. 5.3). The “brain” domain contains a deep semicircular basic groove, hypothesized to be a putative RNA-binding groove (Obayashi et al. 2008; He et al. 2008). Highly conserved basic residues (K328, K539, R566, and K574) are found lining in the groove.

Structure of PA-C (PDB: 2ZNL). Cartoon representation of PA-C showing the “brain” domain (colored in pink) and “mouth” domain (colored in dark red). The N-terminus of PB1 (colored in purple) inserts between the jaws of the mouth domain. A putative RNA-binding groove resides at the brain domain
On the other hand, the “mouth” domain of PA-C interacts with PB1-N. The first 15 residues of PB1-N bind obliquely to a hydrophobic cavity of PA-C, located between the jaws (Obayashi et al. 2008; He et al. 2008). Mutagenic study shows that the PB1 LLFL motif (residues 7–10) is crucial for the binding of PB1-N to PA-C (Perez and Donis 2001). The apo form of PA-C without PB1-N also shows a highly similar structure (Moen et al. 2014).
Full-Length PA
In the context of RdRP heterotrimer, the overall folds of PA-N and PA-C are highly similar to the structures determined from individual domain.
The previously uncharacterized flexible linker region (residues 197–276) was revealed to be lying across the PB1 surface and contributes to multiple inter-subunit interactions (Fig. 5.4).

Full length PA subunit in the context of RdRP (PDB: 4WSB). PA is shown in cartoon representation, while PB1 and PB2 are shown in surface diagram. PA-N (colored in pink) locates at the top of the U structure of RdRP, while PA-C (colored in dark red) locates at the bottom. The linker region (colored in bright red) lies on the surface of PB1. The previously uncharacterized PA arch (residue 366–397) locates at the bottom and wraps around a β-hairpin of PB1. The RdRP promoter binding site is located in close proximity to the PA arch
New inter-subunit interactions were also identified. PA-N was shown to interact with both PB1 and PB2 through its helix α4. M92 and S93 form polar interactions with PB2, while E77 and T89 form polar interactions with PB1. On the other hand, the endonuclease flexible loop (residue 67–74) packs on PB1 helix α22, where K73 interacts with PB1 E731.
In PA-C domain, a formerly disordered loop (PA arch) was characterized in the full structure of PA (Fig. 5.5). The PA arch consists of residues 366–397 (residues of influenza A bat strain; corresponding to residues 371–402 of other human/avian strains). This loop forms an arch that allows a PB1 β-hairpin (residues 353–370) to insert. The PA arch is in close proximity to the 5’-vRNA hook. Residues 366–370 on the PA arch form a phosphate-binding loop, interacting with the backbone of 5’-vRNA promoter A10–A11.

Structure of the PA arch. PA is shown in cartoon representation, while PB1 and PB2 are shown in surface diagram. The orange ribbon represents 5’ vRNA. The hooked structure of 5’ vRNA is held by both PB1 and PA-C. The PA arch forms a loop that encloses a PB1 hairpin (residue 353–370). On its own, the PA arch is disordered
On the other hand, a previously disorganized region consisting of residues 544–553 (550-loop, corresponding to residues 549–558 of other human/avian strains) was shown to be a β-hairpin loop in the full-length PA structure. A recent co-crystal structure of bat influenza A full RdRP and serine-5-phosphorylated Pol II C-terminal domain (CTD) peptide shows that the 550-loop is in close proximity to the bound Pol II CTD peptide (Lukarska et al. 2017).
Furthermore, in various structures of full RdRP (flu A, flu B, and flu C), PA-N was shown to be capable of in situ rotation (Pflug et al. 2014; Reich et al. 2014; Hengrung et al. 2015; Thierry et al. 2016). However, the linker region remains at the same position. This suggested that the flexible loop region at the beginning of the linker region might play a role in the orientation of the PA-N domain. This might be related to the regulation between transcription and replication of the viral genome.
Structure of PB1 Subunit
Structure of PB1 has remained elusive for a long time due to its structural instability when expressed alone (Swale et al. 2016). Two co-crystal structures, PA-PB1 and PB1-PB2, have been determined and reveal the structure of the 15 N-terminal and 80 C-terminal residues of PB1 (Obayashi et al. 2008; He et al. 2008; Sugiyama et al. 2009).
The PB1 N-terminal (1–15) forms a 310 helix that inserts into the PA-C hydrophobic groove. The 310 helix contains a LLFL hydrophobic motif which is crucial for the interaction. Mutation of the first 12 residues could significantly disrupt PA-PB1 interaction (Perez and Donis 2001; Wunderlich et al. 2011).
The PB1 C-terminal (678–757) forms three α-helices that interacts with another three α-helices from the N-terminal region of PB2. Both polypeptides were unable to form tertiary structure on its own. PB1-PB2 interacts mainly through polar interaction, where PB1 K698 and D725 form salt bridges with PB2 (Sugiyama et al. 2009).
Full structure of PB1 was only determined in the context of the vRNA-bound full RdRP (Fig. 5.6) (Pflug et al. 2014; Reich et al. 2014). PB1 shows a typical right-handed RdRP fold, with the fingers, fingertips, palm, and thumb domains that are present in other RNA-dependent RNA polymerases (reviewed in te Velthuis 2014; Reguera et al. 2016).

Structure of PB1 subunit (PDB: 4WSB). The structure of PB1 shows a typical right-handed fold of RNA-dependent RNA polymerase, with both N-terminal and C-terminal extensions. The right-handed fold of PB1 (marked by dotted lines) consists of the fingers, palm, and thumb domain (colored in green, orange, and red, respectively). The N-terminus of PB1 (colored in gold) forms a short helix that interacts with PA-C, and the C terminus of PB1 (colored in light green) forms several helices that interact with PB2-N. A long, flexible β-ribbon carrying a bipartite NLS (colored in cyan) protrudes from the finger domain. It can interact with RanBP5 and is in close proximity with the viral promoter. A loop termed fingertips (colored in purple) is near the NTP entry channel. It is likely to be involved in NTP channeling and binding. Furthermore, a priming loop (colored in blue) is located in the PB1 cavity. It is crucial for de novo replication of viral genome. Lastly, the β-ribbon that inserts into PA arch (colored in dark yellow) is located on the exterior of the fingers domain
A large cavity can be found within PB1, in conjunction with the PB2 N-terminal domain. This forms the active site and it is accessible through several channels. The NTP entry channel involves highly conserved PB1 basic residues located mainly in the palm domain and the fingertip region, in close proximity to PA-C. The template entry channel involves all three subunits and is located near the promoter binding region. The product exit site is at the opposite site template entry channel, where the PA endonuclease, PB2 lid domain, and PB2 cap-binding domain are present. The template exit channel is located near the PB2 lid domain, which was proposed to be involved in separating the template-product duplex. The four-tunnel structure resembles other viral RdRP (Reguera et al. 2016).
The active site contains residues D305 on motif A and D445/D446 on motif C that play a crucial role in coordinating divalent metal ions and promote catalysis. Mutation of these residues abolish polymerase activity of PB1 (Pflug et al. 2014; Biswas and Nayak 1994). A parallel β-loop protruding into the active site from the thumb domain forms a priming loop (residues 641–657). This loop resembles the HCV priming loop and is important for de novo RNA synthesis (Pflug et al. 2014; Appleby et al. 2015).
The full RdRP structure confirms the roles of PB1 N-terminus and C-terminus in the interaction with PA and PB2, respectively. However, other regions were also revealed to be involved in inter-subunit interactions: (1) the surface of the finger domain is wrapped by the PA-linker, (2) the C-terminal thumb domains interact with both PB2-N and PA-C, and (3) the palm-domain interacts with the C-terminal domain of PB2 (Pflug et al. 2014). In the cRNA promoter-bound full RdRP structure, other inter-subunit interactions involving PB1 are detected due to conformational change (Thierry et al. 2016).
A long, flexible β-ribbon (residues 177–212; bat strain residues) protrudes out from the finger domain of PB1 (Pflug et al. 2014). It contains the bipartite NLS that was previously proven to be involved in RanBP5 binding and nuclear import of the PA-PB1 heterodimer.
Another β-hairpin at the finger domain protrudes into an extended loop in PA called the PA arch (Pflug et al. 2014). Both regions are involved in 5′ promoter anchoring. PB1 was also found to bind 3′ and 5′ viral promoter with the help of PB2/PA and PA, respectively.
Structure of PB2 Subunit
PB2 subunit is 759 residues in size. The first one-third (residues 1–247) constitute a N-terminal domain, and the remaining two-third make up a C-terminal domain (PB2-C). Individual domain structures generally correlate well when aligned onto the holo complex. The structure displays a high degree of flexibility and adopts different conformations in order to sustain steps in replication and transcription (Fig. 5.7).

Structure of PB2 subunit (PDB: 4WSB). The PB2 subunit consists of ten regions and adopts extended conformations when bound on PB1. PB2-N can be divided into an extreme N-domain and a “lid” domain, while PB2-C comprises a middle domain, the cap-binding domain, “627-domain” and a nuclear localization signal (NLS) domain. Cap-binding domain and 627-domain are linked up by a “cap-627 linke.” Note that the NLS (738–755) is not visible in this structure and is assumed to be flexible
PB2 N-Terminal Domain (1–247)
Structure of first 37 residues of PB2-N was obtained at first as a complex structure with PB1-C. In fact, apo-PB2 binds like a clamp on the PB1 core. The first and major point of anchor is via PB2-N binding onto the C-terminus of PB1. The PB1-C-PB2-N structure by Sugiyama et al. (Sugiyama et al. 2009) largely correlated with later published heterotrimeric flu A, flu B, and flu C polymerase structures (Pflug et al. 2014; Reich et al. 2014; Hengrung et al. 2015). Briefly, the first helix (residues 1–22) on PB2 distorts at around residue 15 and bends to about 90°. As a result, the first two-thirds of this helix leans against a “helix-duo” at the extreme C-terminus of PB1 (residues 712–753), and the remaining one-third lies to cover this three-helix ensemble. This motif is stabilized mainly via salt bridges. The rest of PB2 molecule then folds onto the PB1 core via various linkages spanning the whole molecule.
Residues between 40–110, composed of three helices, three strands, and loops, make up the N1 domain which mainly serves the structural role. This area makes extensive contact with PB1.
Residues beyond 110 to 247 make up a N2 linker domain, where a subdomain termed lid domain (residues 155–212) resides in. The lid domain exists as four intertwining helices and covers the PB1 core including the template and primer channels, which will otherwise become unprotected and more exposed to solvent. The lid domain is also close to the putative template exit channel.
PB2 Cap-Binding Domain (320–483)
The cap-binding domain comes after a small middle domain which has four intertwining helices spanning residues 251–316. This middle domain is neither in close contact with PB1 nor PA but is involved in a global domain reorientation. Hence it serves as a connection between the PB2-N and PB2-C.
The cap-binding domain consists of four helices folded against a beta sheet structure made up of five strands. Additional strands are found beyond the N- and C-termini of the beta sheet. The overall fold resembles a yacht made up of strands with the sailor (helices) on one side and m7GTP ligand at the base of the ensemble. m7GTP binding site is stabilized by a hydrophobic cluster of four phenylalanines. Structures of apo- and m7GTP-bound flu A cap-binding domains are generally similar (Guilligay et al. 2008; Liu et al. 2013). Binding of m7GTP is stabilized by partial aromatic stacking and salt bridges. Notably, H432 and K339/R355/N439 form salt bridges with α- and γ-phosphate groups, respectively. N439 is on a so-called 424-loop. The binding of m7-GTP is not rigid, and deviation in its orientation is allowed (Guilligay et al. 2008; Tsurumura et al. 2013).
Flu B cap-binding domain was found to bind not only methylated cap but also unmethylated ones or even GDP substrates. It appears that in flu B, certain degree of conformational variations can be tolerated, leading to differential side chain orientations which allow interaction with a wide range of substrates (Wakai et al. 2011; Liu et al. 2015a; Xie et al. 2016).
PB2 627-Domain (538–680) and NLS-Domain (690–759)
Although the holo-heterotrimeric polymerase complex structure is already available, various X-ray structures of these domains harboring mutations are still valuable sources for the investigation into host determinants. These structures include PDB: 2VY7, 2VY8, 3KC6, 3KHW, 2GMO (solution structure of NLS-domain). Structures composing of both domains are also available (PDB: 2VY6, 3CW4) (Tarendeau et al. 2007; Tarendeau et al. 2008; Kuzuhara et al. 2009; Yamada et al. 2010).
The first two-thirds of 627-domain is composed of eight helices (six α-helices and two 310 helices), while the remaining one-third mainly consists of turns and strands. The domain is compactly folded so that the helices reside on one side of the molecule, while the strands occupy the other side, resembling a blanket sheet covering on the bundle of helices. The 627-domain and NLS-domain are linked by a flexible linker. This linker was found unstructured and buried in the interface between 627-domain and the NLS-domain.
The NLS-domain itself adopts a globular fold with three antiparallel β-strands in the middle hydrophobic core, surrounded by unstructured loops and three helices. In pdb structure 2GMO, the bipartite NLS (738–755) is held close to this globular fold by a putative salt bridge D701-R753. The bipartite NLS shows different secondary structures. When 627-NLS domains were expressed alone, the bipartite NLS is unstructured. The bipartite NLS nevertheless adopts a well-formed helix as shown in apo flu C, or promoter bound flu A or flu B heterotrimeric complexes. This helical conformation may be implicated during replication and transcription, but its relevance in the context of nuclear import is not evidenced, since it has been established that PB2 is imported to the nucleus by importin-alpha, upon binding to which the NLS adopts an extended conformation (Pumroy et al. 2015).
Oligomerization of RdRP
RdRp has been shown to be capable of forming higher order oligomer. PB1 and PB2 were proposed to be the interacting site for oligomerization (Jorba et al. 2008). Consistent to this finding, a newer study showed that while the PA-PB1 heterodimer forms homogeneous monomeric and stable particle, the addition of truncated PB2 (residues 1–250) would result in the formation of dimerized RdRP particles (Swale et al. 2016).
The RdRP oligomer could be disrupted by the addition of vRNA. When vRNA is present, the RdRP oligomer will be dissociated into a monomeric RdRP form (Resa-Infante et al. 2010; Swale et al. 2016). This finding is contested by another study, where RdRP with a truncated PB2 (PB2 1–130) is capable of forming tetramer when vRNA or cRNA promoter is added to the dimerized RdRP. Furthermore, a further truncated PB2 (1–86) could not support either dimerization or tetramerization. This suggested that residues 86–130 might be crucial for the oligomerization of RdRP (Chang et al. 2015).
The EM structure of RdRP oligomer reveals two interacting interface (Fig. 5.8). The first interface is stronger, involving PA-C and the PB1-C/PB2-N binding interface, presumably covering PB2 residues 86–130. The second interface is more flexible. The PB1 finger domain is predicted to be involved in this interaction (Chang et al. 2015).

Dimer structure of RdRP revealed by electron microscopy (PDB: 3J9B). Cartoon representation of RdRP dimer structure determined by electron microscopy. PA, PB1, and PB2 are colored in pink, pale blue, and pale green, respectively. The interacting interface is located at PA-C and PB2 N-terminal region
Structure of RNP RdRP Complex and NP
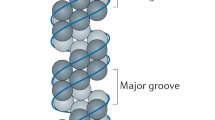
Early electron microscopy studies showed that RNP has a rod-shaped structure. The RNP strand exhibits a double-helical arrangement, with a terminal loop at one end, folding back on itself (Pons et al. 1969; Compans et al. 1972). The RdRP is located at the other end of the rod structure (Murti et al. 1988). In the absence of RdRP, vRNA can form complex with NP resembling the structure of native RNP (Yamanaka et al. 1990), while RNase-treated RNP retains its native conformation (Ruigrok and Baudin 1995). These suggest that the RNP conformation is primarily maintained by NP. A RNA chain wraps on the exterior of the NP helix. The 5′ and 3′ ends of the RNA forms a panhandle structure and are in contact with the polymerase. This architecture is consistent with the observation that influenza vRNA on the RNP is susceptible to RNase cleavage. Early studies estimated 15–20 nucleotides per NP molecules. Individual vRNPs are 10–15 nm in diameter and 30–120 nm long depending on the length of the gene segment (Noda et al. 2006).
Electron microscopy 3D reconstruction of mini-RNP (RNP with short vRNA-like genome) reveals a NP ring structure attached to the RdRP complex. RdRP interacts with two NP molecules at two sites (Martín-Benito et al. 2001; Coloma et al. 2009). This is consistent with previous interaction study, where NP was found to interact with PB1 and PB2 (Biswas et al. 1998, Poole et al. 2004). Since vRNA extends from the RdRP and folds back onto itself, where the extremities of both ends are bound by RdRP, it is natural that two molecules of NP, one near the 5′ end and the other one near the 3′ end, interact with RdRP at two locations.
3D Cryo-EM structures by Arranz et al. and Moeller et al. revealed structural characteristics of the RNP: (1) NP coiled to a double helix forming major and minor grooves as in a DNA molecule. Intra-strand NP-NP contact is via tail-loop-mediated oligomerization, whereas inter-strand NP molecules also contact at minor groove via a separate interface. (2) Orientation of intra-strand NP leads to a continuous path of positive residues allowing RNA molecules to attach onto the exterior of the RNP. (3) N-terminus of NP containing a nuclear localization signal (NLS1) is exposed on the surface and is accessible to host factors (Arranz et al. 2012; Moeller et al. 2012). Moreover, the structures constructed by Arranz et al. and Moeller et al. also show that two NP molecules attach to the RdRP complex (Fig. 5.9), in agreement with previous finding.

Low-resolution structure of native RNP revealed by cryo-EM. Cryo-EM structure reconstructed to a resolution of 20 Å showing the polymerase end of native RNP. The RdRP heterotrimer is colored in pink. A large domain and a smaller arm domain can be identified. Moeller et al. has regarded the small domain to be the PA-C domain, due to the matching between their sizes and shapes. Comparison with mini-RNP EM structure reveals that the arm domain can undergo conformational change. However, in various high-resolution RdRP structures recently determined, the conformational change of RdRP is caused by substantial rearrangement of PB2 or in situ rotation of PA-N and PB2 cap-binding domain. PA-C domain remains at the same position in all determined crystal structures. Therefore, it is unsure whether the small arm domain, capable of conformational change, represents the PA-C domain. Two NP molecules are shown to interact with the large domain of RdRP, inconsistent with previous studies. An additional mass is observed at the top of the large domain. Moeller et al. suggested that it represents an additional NP molecule
Further to the cryo-EM structures, a NP-NP dimer structure was published which implicates additional binding interface plausibly accounting for inter-strand interactions in RNP context. The dimeric NP interface lies on the “back” of the NP crescent and spans through residues 149–167 and 482–498. (PDB: 4IRY, Ye et al. 2012).
The periodicity of vRNA on NP strand is 20–32 nucleotides on average. The terminal loop comprises 3 to 8 NP molecules (Arranz et al. 2012; Moeller et al. 2012). In Moeller and coworkers’ reconstructed structures, an extra NP molecule can be found adjacent to the RdRP complex, in addition to the two NPs identified from previous studies (Fig. 5.9) (Moeller et al. 2012).
Structure of NP
Flu A, flu B, and flu C NP are 498, 560, and 565 amino acid residues in length, respectively. Flu B NP (BNP) contains an extra N-terminal domain of 72 residues which is not found in Flu A NP. The N-terminus of Flu C NP resembles its Flu A counterpart, but its C-terminus is about 51 residues longer. X-ray structures of ANP and BNP are available (PDB: 2IQH, 2Q06, 3TJ0, 4IRY, 3ZDP) (Ye et al. 2006; Ng et al. 2006; Ng et al. 2012; Ye et al. 2012; Chenavas et al. 2013a). The structure of NP is largely conserved among the Orthomyxoviridae family and is formed predominantly with helices, which fold to form a compact structure. The N-terminal region of BNP evolves to become more extended than the flu A counterpart and has been found to be implicated in a number of functions (Liu et al. 2015b).
The structure of NP has been extensively reviewed in Ng et al. 2009, Chenavas et al. 2013b and Yang et al. 2014. Briefly, the NP molecule comprises a head domain, a body domain, and a protruding tail loop (Fig. 5.10) (Ng et al. 2008; Ye et al. 2006; Ng et al. 2012; Ye et al. 2012; Chenavas et al. 2013a). Flu A H5N1 NP composes of a continuous fold of 19 helices and 7 beta-strands which are interconnected with flexible loop regions. Three regions are especially flexible, namely, the regions 1–20, 75–90, and 390–438, the third of which starts from the end of a β-strand (residue 390) followed by a long unstructured peptide to the tail loop (residues 402–428) and overlaps with a helix-loop-helix starting at residue 420. The overall arrangement of helices and strands renders the molecule a crescent shape, with head and body domains at two ends and a few linkers in the middle part serving like a hinge. As a result, unlike in PA and PB2 wherein distinct domains are linked up by linkers, domains in NP cannot be defined by a single continuous amino acid sequence (Fig. 5.10). In order to form homo-oligomers, tail-loop of a NP molecule is inserted into the binding groove of a neigherboring protomer.

Structure of NP (PDB: 2Q06). The NP molecule consists of a head domain, a body domain, and a tail-loop structure which are crucial for NP-NP homo-oligomerization. Homo-oligomerization is achieved by insertion of the tail loop to the binding groove at the body domain of a neighboring protomer. Conformational flexibility is conferred by the tail-loop and the unstructured regions which form a hinge at the middle of the molecule. This conformational flexibility is important for maintaining vRNP structure especially at the terminal polymerase end and loop end of the vRNP
The general features of BNP are similar to ANP. The two molecules shared backbone root-mean-square deviation of only 1.457 Å despite considerably different orientations of the tail loops.
The NP molecule demonstrates certain extent of flexibility. First the tail loop is flanked and followed by unstructured linker regions making it flexible. In fact, the tail loop of H1N1 NP deviates from the H5N1 tail loop for about 75°, while flu B tail loop deviates from H5N1 tail loop for about 60°. This renders tail-loop insertion at a range of angles possible and thus would also allow the formation of rigid (e.g., trimers or rings) and loose oligomers (e.g., higher-order helical oligomers). Second, the head domain and body domain should allow some degree of rocking. This could relate to possible conformational changes upon polymerase binding or during replication and transcription wherein the template RNA is slightly displaced from the NP.
Functions of RNP
RNP plays a major role in influenza life cycle. vRNP encapsidates the segmented genome of influenza inside virion. It is also involved in the transcription and replication of the viral genome in infected cells. RNP is also capable of nuclear import and export. The overview of RNP functions in influenza life cycle are shown in Fig. 5.11. Its various roles will be described in details below.

Roles of RNP in influenza life cycle. The segmented genomes of influenza virus are packaged into RNP molecules inside the virion. After virus attachment and endocytosis, the virion-derived RNPs are released into host cytoplasm. With the help of host transport machinery, these vRNPs are transported into nucleus for transcription and replication. Transcription of negative-sense vRNA produces capped mRNA with poly-A tails that is translated by host translational machinery. Newly synthesized RNP subunits are then transported back into nucleus for assembly. Current model strongly suggests that PA-PB1 enters nucleus as a heterodimer complex, while PB2 and NP enter nucleus on their own. On the other hand, replication of negative-sense vRNA produces full length positive-sense replica of the viral genome. cRNA is stabilized by the newly produced RNP subunits, forming an intermediate complex called cRNP. Full length negative-sense vRNA is synthesized using cRNA as template. The cRNA-to-vRNA process has to be mediated by a trans-acting or trans-activating RdRP. The vRNA products are then encapsidated by RdRP and NP, resulting in progeny vRNP. Lastly, these vRNPs are exported from the nucleus, assembled in the cytoplasm and packaged into viral progenies
Transcription
Transcription of viral genome results in the production of viral proteins. RNP can steal cap from Pol II transcripts through its cap-snatching mechanism. This is followed by transcription initiation, realignment, and elongation. Poly-A tail is added to the nascent transcript at the end before the mRNA is processed by host translational machinery. The overview of RNP transcription is shown in Fig. 5.12.

Transcription of viral genome. Influenza RNP requires host Pol II-derived capped RNAs as primer for transcription. Initiating Pol II with phosphorylated serine at position 5 can recruit capping enzymes and add cap structure to its nascent transcript. Influenza RNP can bind specifically to the initiating form of Pol II, causing it to arrest at this stage, and employ cap-snatching mechanism to steal the cap structure from the nascent transcript. The PB2 cap-binding domain can capture the cap structure, while the PA-N endonuclease domain can cleave the RNA at 10–13 nt downstream of the cap, usually after a G or A base. Rotation of PB2 cap-binding domain facilitates the insertion of the cleaved primer into the catalytic cavity of PB1. The 3′ end of cleaved primers can interact with vRNA template at the penultimate or last nucleotide, triggering transcription initiation. After the addition of nucleotides until position 4, the nascent transcript can either undergo realignment or proceed on with elongation. Elongation continues until it stutters over a poly-U track near the 5′ terminus of vRNA
Binding of Capped Cellular RNAs
The cap-binding activity resides on the PB2 cap-binding domain. This domain first captures a capped nascent RNA from Pol II and then directs it to the nearby PA-N endonuclease domain. It is unclear whether PA-N captures the capped RNA by chance or it is aided by cellular factors.
The “424-loop” on cap-binding domain was originally found to exert allosteric regulation on PB1 activity (Guilligay et al. 2008). Aligning 2VQZ (flu A m7GTP bound form) to the recent flu B mRNA primer bound structure (PDB: 5MSG, Reich et al. 2017) reveals that this “424-loop” must be displaced outward if a primer is bound instead of m7GTP; otherwise the loop will clash with primer RNA. This loop again is in close proximity to the incoming primer RNA within 6 Å. Hence its role in directing correct priming is expected.
On the other hand, as nascent Pol II transcripts are targeted for cap-snatching (Gu et al. 2015; Koppstein et al. 2015), the interaction between RdRP and Pol II is also crucial for cap-binding. In vitro direct binding of RdRP to phosphoserine 5 Pol II CTD has been proven (Engelhardt et al. 2005; Martínez-Alonso et al. 2016). Moreover, a co-crystal structure of RdRP with CTD peptide was determined recently (Lukarska et al. 2017). This structure reveals that the phosphoserine 5 Pol II CTD binds directly to PA-C, where residues K289, R454, K635, and R638 are involved.
Cap-Snatching by Endonuclease Cleavage
The influenza RNP requires a unique cap-snatching mechanism to enable successful transcription of viral proteins. 5′ capped oligonucleotides derived from host RNA are captured, cleaved, and used as primer for initiating transcript elongation. The cap-snatching endonuclease function was originally thought to be residing in PB1 or PB2 subunit (Li et al. 2001). The determination of PA-N structure unquestionably established that the endonuclease function resides in PA (Yuan et al. 2009; Dias et al. 2009).
PA-N contains a PD-(D/E)XK nuclease motif that is conserved in all influenza virus. The negatively charged catalytic site contains H41, E80, D108, and E119 for coordinating the metal ions. Another residue, K134, is proposed to be the catalytic lysine. PA-N endonuclease cleaves captured cellular RNAs at 10–13 nt downstream of the cap structure. Besides, it also displays preference for cleavage after a dinucleotide CA or a G (Rao et al. 2003; Datta et al. 2013; Sikora et al. 2014; Koppstein et al. 2015). Divalent metal ion is required for endonuclease activity, and the presence of different ions affects its endonuclease activity (Doan et al. 1999; Crepin et al. 2010; Xiao et al. 2014; Kotlarek and Worch 2016).
Initiation, Elongation, and Termination of Viral mRNA
PB1 was originally misidentified as having endonucleolytic actvitiy (Li et al. 2001); it was later revealed that the endonuclease active site resides in PA (Yuan et al. 2009; Dias et al. 2009). The present model proposed that after cap-binding and cleavage, PB2 rotates and inserts the capped primer into the active site of PB1 (Pflug et al. 2014; Reich et al. 2014), where it forms base pairing with the template. Primer with A or G at their 3′ end could form A:U or G:U base pairing with the 3′-ultimate U nucleotide of the viral template, initiating transcription at position 2 of the vRNA template. Besides, cleaved primers with G at the end can also pair with the 3′-penultimate C nucleotide (Koppstein et al. 2015), thus initiating transcription at position 3. Furthermore, a prime-and-realign phenomenon during mRNA synthesis was observed by several studies (Geerts-Dimitriadou et al. 2011a; Geerts-Dimitriadou et al. 2011b; Sikora et al. 2014; Koppstein et al. 2015). This happens when the nascent transcript slips back and reiterates transcription of the first template nucleotides. Recently, it was identified that the PB1 priming loop is involved in this prime-and-realign mechanism of capped primer, although its importance in mRNA synthesis remains unknown (Oymans and te Velthuis 2017).
PB2 may also play a role in assisting transcription initiation. A recent study by Hara et al. (Hara et al. 2017) showed that R142A abolished both replication and transcription. R142 is in direct contact with PB1 N276 on the palm, forming a hydrogen bond (bat flu A, PDB: 4WSB). In comparison, flu B capped-mRNA primer bound structure (PDB: 5MSG) has two arginines at the equivalent position, forming hydrogen bonds with PB1 (R142/R144, equivalent to K140/R142 in flu A). K145/R146 in flu B (R143/R144 in flu A) is within 6 Å to the incoming capped-mRNA primer. Thus it is highly plausible that the first two positive side chains (R142/R144 in flu B, K140/R142 in flu A) hold the strand by anchoring to PB1 and the following two interact with incoming primers to direct them to PB1 active site. Besides, another study proposed that 627-domain is required for accurate cleavage of capped primer and the transcription initiation at correct position (Nilsson et al. 2017).
After transcription initiation, current model proposes that elongation of mRNA proceeds in a template-dependent manner. Template vRNA and nascent mRNA separate and leave the active site by their respective exit channels. The 5′ promoter of vRNA is anchored to the RdRP. Stuttering occurs when the vRNA reaches its 5′-terminus, resulting in the poly-A tails of the mRNA products (Poon et al. 1999; Poon et al. 2000). The fate of the template vRNA after transcription remains unclear.
Replication
Replication of viral genome results in full copy of viral genome. The replicated viral genomes can be encapsidated into new vRNPs and packaged into viral progenies. Influenza virus replicates its genome in two steps: (1) copying a negative-sense vRNA into an intermediate positive-sense cRNA and (2) copying the positive-sense cRNA back into a negative-sense vRNA. In both steps, RNP performs de novo synthesis of nucleotides. It involves the de novo synthesis of a pppApG dinucleotide, followed by elongation and read through of the entire template. Furthermore, regarding to the second step of replication, a trans-activating model and a trans-acting model were proposed. Besides, a realignment mechanism has also been observed during the second step of replication. The overview of vRNA-to-cRNA replication is shown in Fig. 5.13, while the overview of cRNA-to-vRNA replication is shown in Fig. 5.14.

Replication of vRNA to cRNA. (a) PB1 priming loop facilitates de novo synthesis of pppApG dinucleotide using the vRNA as template at position 1 and 2 of the promoter. (b) Elongation of nascent transcript is accompanied by the breaking of interactions between 5′ and 3’ vRNA at the panhandle base pairing region. (c) As the nascent transcript leaves the catalytic cavity, it is stabilized by free RdRP and NP. Unlike transcription, the 5’-vRNA promoter has to be released in order to ensure a full read through of the viral genome. It is unclear when the release of 5’-vRNA promoter takes place. (d) The newly synthesized cRNA is assembled into a cRNP

Replication of cRNA to vRNA. The second step of replication (cRNA to vRNA) is different from the first step (vRNA to cRNA) as it requires the assistance of a trans-acting/trans-activating RdRP. Two models have been proposed. (a) Trans-activating model: the resident cRNP is responsible for polymerase activity. But this activity has to be activated by a trans-activating RdRP. (b) Trans-acting model: another RdRP is responsible for polymerase activity in trans. The trans-acting RdRP binds to the cRNP and replicates the cRNA template on the resident cRNP. (c) Replication is initiated by the synthesis of pppApG using positions 4 and 5 of the cRNA 3′ terminus. The pppApG dinucleotide is then realigned to positions 1 and 2, followed by elongation of nascent transcript. (d) In the trans-activating model, either the trans-activating RdRP encapsidates the newly synthesized vRNA and forms a progeny vRNP or another free RdRP is needed for the encapsidation. (e) In the trans-acting model, a free RdRP is required for the encapsidation of the nascent vRNA transcript
De Novo Synthesis and PB1 Priming Loop
The cRNA/vRNA replication process can be initiated de novo, meaning it can be achieved without primers. De novo replication is initiated by the synthesis of pppApG dinucleotide (Deng et al. 2006). Different mechanisms of pppApG production were observed between cRNA and vRNA synthesis. When vRNA is used as template, primer-independent initiation occurs at the 3′-ultimate position. After the synthesis of pppApG using position 1 and 2 of vRNA, elongation will follow immediately at position 3 (Deng et al. 2006). This process is termed terminal initiation. Another study suggested that terminal initiation with vRNA starts at position 2 (instead of positions 1 and 2), thus proposing that an additional purine nucleotide has to be added to the 3′-terminus of vRNA at position 1 by host factors in order to ensure the replication of full length viral genome (Zhang et al. 2010). On the other hand, when cRNA is used as a template, pppApG is synthesized using positions 4 and 5 of the template. This pppApG will then be realigned to positions 1 and 2, followed by elongation in a template-dependent manner (Deng et al. 2006; Zhang et al. 2010). This process is termed internal initiation. One study showed that terminal initiation is significantly faster than the internal initiation that involves a primer-and-realign mechanism (Reich et al. 2017).
A recent finding showed that the synthesis of the first three nucleotides from vRNA templates can compensate for the breaking of base pairing at the panhandle region. In contrast, the synthesis of nucleotides from cRNA 3′ extremity cannot compensate for the energy requirement, thus a prime-and-realign mechanism is needed (Reich et al. 2017).
Kinetic study showed that synthesis of pppApG is a rate-limiting step. Addition of ApG dinucleotide or uncapped primer ending with AG-3′ significantly accelerate the level of RNA synthesis. Capped primer accelerates the rate of reaction even further, indicating RNA with 5′ cap enhances efficiency (Reich et al. 2017).
Both the initiation of RNA synthesis and the prime-and-realign mechanism are closely related to the priming loop of PB1. RdRps capable of de novo synthesis usually contain a loop structure that can act as stacking platform for incoming NTP to catalyze the formation of the first phosphodiester bond (Butcher et al. 2001; Tao et al. 2002; Caillet-Saguy et al. 2014; Appleby et al. 2015). In the full structure of RdRP, this priming element can be identified in PB1. PB1 residues 641–657 form a conserved antiparallel β-loop that resembles HCV priming loop (Pflug et al. 2014).
Recent study confirmed that this priming loop is crucial for terminal initiation of de novo synthesis using vRNA as template. The priming loop does not affect internal initiation when cRNA is used as template. This loop might have to undergo conformational change and play a role in determining the rate of priming, initiation, and elongation. P651 on the loop was discovered to interact with the initiating NTP during de novo initiation (te Velthuis et al. 2016). By deletion study, the priming loop was observed to stimulate realignment during vRNA synthesis and suppresses internal extension. In contrast, during primer-dependent mRNA synthesis, priming loop was shown to suppress prime-and-realign mechanism (Oymans and te Velthuis 2017).
Trans-Acting or Trans-Activating Cis-Acting RNP
Using a genetic trans-complementation experiment, it was discovered that, with a template-bound replication-defective (i.e., PB2 R142A or R130A) RNP, even though it is not capable of synthesizing new RNA by itself, progeny vRNA could be produced in the presence of free RdRPs that have normal replication function. This indicates that the replication of viral genome occurs in trans. In contrast, the inability of mRNA synthesis by a template-bound transcription-defective (i.e., PB2 E361N) RNP cannot be rescued by a trans RdRP, indicating that mRNA synthesis occurs in cis (Jorba et al. 2009).
Previous study has demonstrated that the virion-derived vRNP is able to produce both mRNA and cRNA (Vreede et al. 2007); it is at least possible for the RNP to synthesize cRNA in cis. Thus the trans-acting model suggests that during the cRNP-to-vRNP phase, free RdRP first interacts with template-bound cRNP, presumably through the RdRP. The RdRP then translocates to the 3′-end of the cRNA and initiates replication in trans. Yet another free RdRP is required for the encapsidation of the newly synthesized vRNA. It is then assembled into a progeny vRNP (Jorba et al. 2009). This trans-acting model is consistent with two other findings: (1) oligomerization of RdRP can be observed (Jorba et al. 2008; Chang et al. 2015; Swale et al. 2016), and (2) electron micrographs of negatively stained RNPs reveal branched RNP structures (Moeller et al. 2012).
Recent study has shown that purified vRNP can perform mRNA and cRNA synthesis in vitro, while purified cRNP cannot synthesize vRNA either de novo or in the presence of an ApG primer. Only when purified RdRP is added to cRNP, synthesis of vRNA can be observed (York et al. 2013). This further supports the notion that free, trans-acting RdRP is required for successful cRNA-to-vRNA synthesis.
York et al. did not support the trans-acting model. Instead, a trans-activating model was proposed. The trans-activating model suggests that the trans RdRP only activates the resident RdRP, but the actual synthesis of vRNA is carried out in cis. This is supported by the finding that even an RdRP with nonfunctional polymerase activity could produce newly synthesized vRNA in conjunction with the resident cRNP (York et al. 2013). Therefore, the resident cRNP is responsible for RNA synthesis instead of the trans RdRP. In this model, it is possible that the trans-activating RdRP also fulfills the role of binding the nascent 5’ vRNA. This allows the trans-activating RdRP to assemble into a progeny vRNP (te Velthuis and Fodor 2016).
Stabilization of cRNAs
Stabilization of replicative intermediate cRNAs has been reported to be important for the replication of viral genome. A recent study reported that the PB2 627-domain is required for the stabilization of cRNA (Nilsson et al. 2017). On the other hand, the RNA binding capability of NP is also essential for stabilizing newly synthesized cRNA, preventing the replicative intermediate being degraded by cellular nuclease (Vreede et al. 2004; Vreede and Brownlee 2007). The stabilization of cRNA has been proposed as a mechanism to regulate the transition of transcription in early infection to replication in late infection (Vreede et al. 2004; Vreede and Brownlee 2007). The exact mechanism of cRNA stabilization by PB2 and NP remains unclear. However, the 627-domin is located close to the exit channel of nascent RNA during replication. It is plausible that this domain is involved in guiding incoming NP to newly synthesized vRNA for encapsidation. In fact, evidence supports that PB2 627-domain and NP interact with each other directly. Ng et al. demonstrated that this interaction is related to the region around R150 on NP, while Hsia et al. further showed that a peptide from NP spanning residues S145 to G185 is sufficient to cause chemical shift perturbation on PB2 627-domain, with PB2 D605 and V606 playing vital roles in the interaction (Ng et al. 2012; Hsia et al. 2018).
vRNA & cRNA Promoter Binding
Previous studies indicated that PB1 is highly involved in promoter binding (González and Ortín 1999a; González and Ortín 1999b; Jung and Brownlee 2006). Full RdRP structure confirmed the importance of PB1 in promoter binding. PB1 interacts with both 5′ and 3′ of vRNA and cRNA (Figs. 5.15a and 5.15b) (Pflug et al. 2014; Reich et al. 2014; Thierry et al. 2016). 5′ promoter region is held by both PA and PB1 (Fig. 5.15a), while 3′ promoter region interacts with all three subunits (Fig. 5.15b). A corkscrew conformation of the viral promoter could be observed, confirming previous studies and models (Flick et al. 1996; Hobom and Flick 1999; Brownlee and Sharps 2002; Tomescu et al. 2014).

Structure of vRNA promoter and its interaction with RdRP (PDB: 4WSB). (a) 5’-vRNA promoter (colored in red) is shown to interact with both PA and PB1 (colored pink and pale blue, respectively). 5′ Terminus of vRNA forms a hook structure, inconsistent with the previously proposed corkscrew model. PA-C RNA-binding groove is involved in binding 5’ vRNA, and the PA arch encircles both the 5’ vRNA and a PB1 β-ribbon. The 5’ vRNA is also situated near the NTP entry channel (located at the right side of the diagram). (b) 3’ vRNA promoter (colored in blue) is shown to interact with all three subunits (PA, PB1, and PB2 colored in pink, pale blue, and pale green, respectively). The 3′ extremity of vRNA is not shown in this structure. The long and flexible PB1 NLS β-ribbon is shown to be in close proximity with both 5′ and 3′ promoters
PB1 N-terminal H32, T34, and Y38 residues are involved in the interaction with the 5’ vRNA. Besides, a PB1 β-hairpin (residues 353–370; bat strain residues) inserts through the PA arch and interacts with the 5’ vRNA, where R365 forms multivalent interactions with the RNA backbone (Pflug et al. 2014). On the other hand, PB1 interacts with the 5′-3′ duplex region with the NLS-containing β-ribbon (residues 177–212) and C-terminal region of PB1 (residues 672–676). From the structure of Flu A RdRP, it is unclear how R571 and R572 mentioned by Li et al. could affect 5’ vRNA binding (Pflug et al. 2014; Li et al. 1998).
Although PB1 plays an important role in the binding of the 5′ and 3′ viral promoters, PA and PB2 are also essential.
PA-PB1 heterodimer has been shown to involve in 5′-promoter binding (Lee et al. 2002; Deng et al. 2005). Recently, the Kd between PA-PB1 dimer and 5’-vRNA promoter was determined to be as low as 0.2–0.4 nM, whereas their interaction to 3′ promoter is much weaker (Swale et al. 2016). The structure of PA-C reveals a putative RNA-binding groove. Highly conserved basic residues (K328, K539, R566, and K574) are found in the groove (Obayashi et al. 2008; He et al. 2008). The elucidation of full RdRp structure confirms that the putative RNA-binding groove indeed interacts with RNA. It binds specifically to the 5′ viral promoter (Fig. 5.15a). The first 10 nucleotides of the 5′ promoter form a hook that is sandwiched in a pocket formed on one side by strands β17–β18 and β20 of PA. The other side is formed by the PA arch, with a PB1 β-hairpin (bat influenza residues 353–370) inserting through the arch. The conserved basic residues located in the RNA groove form polar interactions with the 5′ hook (Pflug et al. 2014). Co-crystal of flu B RdRP with viral promoters shows that both 5’-vRNA promoter and 5’ cRNA promoter bind to PA in a highly similar mode (Thierry et al. 2016).
Also, it was recently discovered that RanBP5 regulates the binding of 5’-vRNA promoter to the PA-PB1 dimer. The formation of PA-PB1-RanBP5 heterotrimer prevents the binding of 5′ promoter to PA-PB1 dimer (Swale et al. 2016).
For PB2, its N-terminal region is in close contact with the promoter via residues 36–49 as revealed in the full RdRP structure (Pflug et al. 2014). Agreeing with this, during polymerase assembly, the binding of PA-PB1 dimer to 3’ vRNA necessitates the presence of PB2-N (Swale et al. 2016).
Hara et al. identified R124 in PB2-N and established by cross-linking experiment that this is important for both vRNA and cRNA promoter binding (Hara et al. 2017). Structurally the side-chain conformation of this residue is conserved in all available polymerase structures, either apo or promoter bound. In capped-mRNA primer bound flu B polymerase structure, it is apparent that R124 lies within the N2-linker, pointing toward incoming primer or template vRNA. However, its direct interaction with RNA is unlikely since several loops and helices on PB2 cause steric hindrances. It is thus conceivable that R124 assumes a structural role related to the lid domain instead of directly in contact with RNA.
Furthermore, two configurations of 3’ vRNA promoter have been observed, one represents the pre-initiation state, while the other one resembles the initiation state (Fig. 5.16). In the pre-initiation state, the 3’ vRNA promoter extremity interacts with PB1 at the exterior of RdRP; in the initiation-resembling state, it is inserted into the PB1 catalytic cavity (Reich et al. 2014; Reich et al. 2017).

Two configurations of 3’ vRNA promoter. The structure of flu B 3’ vRNA promoter in pre-initiation state (PDB, 4WRT; red, 5′ promoter; cyan, 3′ promoter) and initiation-resembling state (PDB, 5MSG; pale red, 5′ promoter; blue, 3′ promoter) is shown as cartoon representation. PB1 is shown as surface diagram (colored in pale blue). In pre-initiation state, the 3′ extremity of vRNA lies at the exterior of PB1 and interacts with its NLS-containing β-ribbon. In initiation-resembling state, the 3′ extremity of vRNA inserts into the catalytic cavity of PB1 through a narrow template entry channel. The conformation of 5’ vRNA remains the same in both states, whereas the PB1 NLS-containing β-ribbon shows substantial displacement. In the pre-initiation state, the ribbon (colored in pink) is “pulled” toward the 3’ vRNA extremities. In the initiation-resembling state, the same ribbon (colored in green) moves further away from the promoter
Conformational Change of RdRP Triggered by Promoter Binding
Promoter binding has been implicated in the activation of cap-binding and endonuclease activity (Lee et al. 2003; Rao et al. 2003). This observation is supported by recent discovery of two conformations of RdRP bound by 5’-vRNA promoter and 5’ cRNA promoter (Fig. 5.17) (Thierry et al. 2016).

Radical repacking of PB2 subunit. PB2 was shown to undergo radical conformational change in two crystal structures of flu B RdRP (PDB: 4FMZ & 5EPI). (a) The vRNA-bound form (PDB: 4FMZ) resembles the RdRP structure of bat influenza and flu B RdRP interacting with both 5′ and 3′ of vRNA (PDB: 4WSB & 4WSA). In this form, the PB2 cap-binding domain is situated at the top of the U-structure and faces the PA-N domain. Whereas the PB2 627-domain is located near PA-C and the palm domain of PB1, while the NLS domain is not visible in the structure. (b) In the cRNA-bound form (PDB: 5EPI), PB2 cap-binding domain and PB2 627 domain switch their positions. The cap-binding domain is situated near the palm domain of PB1, while the 627-domain moves nearer to the PA-N domain. Furthermore, the PB2 NLS helix becomes visible and is shown to interact directly with PA-N. The apo form of flu C RdRP (PDB: 5D98) resembles the cRNA-bound form. (c) and (d) Schematic diagrams of RdRP corresponding to (a) and (b)
In an apo (resting) state (flu C, PDB: 5D98), PB2 subunit exists in a compact state wherein the lid domain and the cap-binding domain exhibit extensive contact with PB1. Entry of capped mRNA is obscured. 627-domain is situated out of the protein core. NLS sequence forms a helix packed against PA-N domain, presumably resulting in blocking of the endonuclease activity. This conformation is similar to a cRNA promoter-bound form (flu B, PDB: 5EPI) with two obvious differences in the latter state: (1) although NLS-peptide remains bound to PA-N, the whole NLS-PA-N is rotated by around 90°; (2) the 627-domain is protruded out with a partially unfolded cap-627 linker, now further away from the protein core, leading to shifts of the lid domain and the cap-binding domain. The overall conformation becomes more extended. The orientation of 627-domain as such should register biological significance, since it is now in close proximity of the vRNA exit channel.
In situ rotation of cap-binding domain was observed in vRNA promoter bound flu A and flu B structures (PDB: 4WSB and 4WSA) (Fig. 5.18). This is particularly important for the cap-binding domain to direct the primer to PA-N domain for cleavage. It is currently unknown whether the binding of capped mRNA alone is sufficient to cause the drastic domain rearrangement. It is likely that more than one conformation may exist in equilibrium when in solution, as Thierry et al. suggested. Drastic rearrangement of the 627-domain is registered, albeit the polymerase shape becomes compact again. The overall relocation concerns lid domain, cap-binding domain, cap-627 linker, and the 627-domain so that (1) cap-627 linker is now closely packed against lid domain and (2) 627-domain is now situated next to the “back” of cap-binding domain, leaving the primer entry site open. Endonuclease domain is no longer bound by NLS-peptide. Indeed the electron density for the NLS-domain is not registered in any of available vRNA promoter-bound structures.

Rotation of cap-binding domain. In situ rotation of cap-binding domain was observed in several crystal structures (PDB: 4WSB, 4WSA, and 5MSG). The cap-binding cavity of PB2 can either point toward PA-N for cap-snatching, or it can point toward PB1 catalytic cavity for priming and transcription initiation
However, no significant structural change could be observed near the 5′ promoter binding site on both PA and PB1, thus the exact mechanism of conformational change and the activation of cap-snatching mechanism remains obscure.
Lastly, the difference between the tails of mRNA and cRNA products are affected by the binding of 5′ promoter to RdRP. Transcription of vRNA requires the 5′ promoter to associate tightly to RdRP, this steric constraint would hinder elongation and cause stuttering at the poly-U stretch, resulting in a poly-A tail in the mRNA product (Pritlove et al. 1998; Poon et al. 1998). On the other hand, replication of vRNA requires the release of 5′ promoter in order to allow read through of the entire genome. The exact mechanism of this switch is not clear, but PA arch and the PA RNA-binding groove might play a role due to their close proximity to 5′ promoter.
Conclusion
The RNP complex plays multiple crucial roles in influenza virus life cycle. In this article, the functions of RNP in transcription and replication are described in detail. It is worthwhile to note that much research has also been done on the role of RNP in host specificity and pathogenicity. Furthermore, several interactome studies have revealed the extensive interaction network of RNP with host factors. Lastly, RNP is also a promising target for drug discovery; numerous small molecules or peptides have been found to disrupt RNP assembly and activity.
From the perspective of structural research, there are several important problems remained unsolved. Firstly, although high-resolution structures of RdRP have been determined, the exact structure of RNP complex remains unknown. NP has long been identified to interact with RdRP (presumably through PB1 and PB2), yet its exact mode of binding has not been revealed. Secondly, it has been observed that RdRP undergoes substantial conformational change (particularly PB2). However, the cause and regulation of this phenomenon is not understood. Moreover, it would be fruitful to understand the effect of such conformational change on the activity of RNP. One would conjecture such structural change might play a role in the regulation between replication and transcription. Thirdly, several regions in the RdRP were unrecognized before the elucidation of full RdRP structure, e.g., PA arch, PB1 β-ribbon (residues 352–398 of bat flu A), and the 550-loop of PA. These regions are mostly uncharacterized at present, and their functions are not known. Fourthly, the actual initiation state of RdRP is not yet resolved. Base pairing cannot be observed in the PB1 catalytic cavity in all present structures. Structural determination of the initiation state could provide us with valuable insight on the mechanism of nucleotide synthesis by RdRP. Fifthly, regardless of the trans-acting model or trans-activating model, both involve dimerization or oligomerization of RdRP. In depth characterization of their modes of interaction would be much helpful and would give us understanding on the role and mechanism of the trans RdRP in RNA synthesis. Finally, numerous residues on the RdRP have been discovered to affect host specificity. However, their exact mechanism remains unclear and the existing structure of RdRP could not explain their phenotypes.
Although many host factors have been identified as interacting partners with RdRP or RNP, most of these interactions remain uncharacterized. Only the binding mode of Pol II CTD domain with RdRP and importin-α with NP and PB2 were structurally determined (Pumroy et al. 2015; Nakada et al. 2015; Lukarska et al. 2017). The characterization of interaction between host factors and RNP and structural determination of these complexes could give us a full picture on the function of RNP.
Altogether, previous research has provided us with profound understanding on the role of influenza RNP. However, some “black boxes” still remain at present, and efforts in clarifying the full mechanism of RNP, its role in host specificity, and its interactions with host factors are much needed. These basic scientific knowledge on RNP could assist us in further understanding the central process of viral growth and for developing measures to combat the virus infection.
References
Appleby TC, Perry JK, Murakami E et al (2015) Structural basis for RNA replication by the hepatitis C virus polymerase. Science 347:771–775
Area E, Martín-Benito J, Gastaminza P et al (2004) 3D structure of the influenza virus polymerase complex: localization of subunit domains. Proc Natl Acad Sci U S A 101:308–313
Arranz R, Coloma R, Chichón FJ et al (2012) The structure of native influenza virion ribonucleoproteins. Science 338:1634–1637
Biswas SK, Nayak DP (1994) Mutational analysis of the conserved motifs of influenza a virus polymerase basic protein 1. J Virol 68:1819–1826
Brownlee GG, Sharps JL (2002) The RNA polymerase of influenza a virus is stabilized by interaction with its viral RNA promoter. J Virol 76:7103–7113
Butcher SJ, Grimes JM, Makeyev EV et al (2001) A mechanism for initiating RNA-dependent RNA polymerization. Nature 410:235–240
Caillet-Saguy C, Lim SP, Shi P-Y et al (2014) Polymerases of hepatitis C viruses and flaviviruses: structural and mechanistic insights and drug development. Antivir Res 105:8–16
Chang S, Sun D, Liang H et al (2015) Cryo-EM structure of influenza virus RNA polymerase complex at 4.3 Å resolution. Mol Cell 57:925–935
Chenavas S, Crépin T, Delmas B et al (2013a) Influenza virus nucleoprotein: structure, RNA binding, oligomerization and antiviral drug target. Future Microbiol 8:1537–1545
Chenavas S, Estrozi LF, Slama-Schwok A et al (2013b) Monomeric nucleoprotein of influenza a virus. PLoS Pathog 9:e1003275
Coloma R, Valpuesta JM, Arranz R et al (2009) The structure of a biologically active influenza virus ribonucleoprotein complex. PLoS Pathog 5:e1000491
Compans RW, Content J, Duesberg PH (1972) Structure of the ribonucleoprotein of influenza virus. J Virol 10:795–800
Crepin T, Dias A, Palencia A et al (2010) Mutational and metal binding analysis of the endonuclease domain of the influenza virus polymerase PA subunit. J Virol 84:9096–9104
Datta K, Wolkerstorfer A, Szolar OHJ et al (2013) Characterization of PA-N terminal domain of influenza a polymerase reveals sequence specific RNA cleavage. Nucleic Acids Res 41:8289–8299
Deng T, Sharps J, Fodor E, Brownlee GG (2005) In vitro assembly of PB2 with a PB1-PA dimer supports a new model of assembly of influenza a virus polymerase subunits into a functional trimeric complex. J Virol 79:8669–8674
Deng T, Vreede FT, Brownlee GG (2006) Different de novo initiation strategies are used by influenza virus RNA polymerase on its cRNA and viral RNA promoters during viral RNA replication. J Virol 80:2337–2348
Dias A, Bouvier D, Crépin T et al (2009) The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458:914–918
Doan L, Handa B, Roberts NA, Klumpp K (1999) Metal ion catalysis of RNA cleavage by the influenza virus endonuclease. Biochemistry 38:5612–5619
DuBois RM, Slavish PJ, Baughman BM et al (2012) Structural and biochemical basis for development of influenza virus inhibitors targeting the PA endonuclease. PLoS Pathog 8:e1002830
Eisfeld AJ, Neumann G, Kawaoka Y (2014) At the centre: influenza a virus ribonucleoproteins. Nat Rev Microbiol 13:28–41
Engelhardt OG, Smith M, Fodor E (2005) Association of the influenza a virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J Virol 79:5812–5818
Flick R, Neumann G, Hoffmann E et al (1996) Promoter elements in the influenza vRNA terminal structure. RNA 2:1046–1057
Geerts-Dimitriadou C, Goldbach R, Kormelink R (2011a) Preferential use of RNA leader sequences during influenza a transcription initiation in vivo. Virology 409:27–32
Geerts-Dimitriadou C, Zwart MP, Goldbach R, Kormelink R (2011b) Base-pairing promotes leader selection to prime in vitro influenza genome transcription. Virology 409:17–26
González S, Ortín J (1999a) Characterization of influenza virus PB1 protein binding to viral RNA: two separate regions of the protein contribute to the interaction domain. J Virol 73:631–637
González S, Ortín J (1999b) Distinct regions of influenza virus PB1 polymerase subunit recognize vRNA and cRNA templates. EMBO J 18:3767–3775
González S, Zürcher T, Ortín J (1996) Identification of two separate domains in the influenza virus PB1 protein involved in the interaction with the PB2 and PA subunits: a model for the viral RNA polymerase structure. Nucleic Acids Res 24:4456–4463
Gu W, Gallagher GR, Dai W et al (2015) Influenza a virus preferentially snatches noncoding RNA caps. RNA 21:2067–2075
Guilligay D, Tarendeau F, Resa-Infante P et al (2008) The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat Struct Mol Biol 15:500–506
Guu TSY, Dong L, Wittung-Stafshede P, Tao YJ (2008) Mapping the domain structure of the influenza a virus polymerase acidic protein (PA) and its interaction with the basic protein 1 (PB1) subunit. Virology 379:135–142
Hara K, Kashiwagi T, Hamada N, Watanabe H (2017) Basic amino acids in the N-terminal half of the PB2 subunit of influenza virus RNA polymerase are involved in both transcription and replication. J Gen Virol 98:900–905
He X, Zhou J, Bartlam M et al (2008) Crystal structure of the polymerase PAC–PB1N complex from an avian influenza H5N1 virus. Nature 454:1123–1126
Hengrung N, El Omari K, Serna Martin I et al (2015) Crystal structure of the RNA-dependent RNA polymerase from influenza C virus. Nature 527:114–117
Hobom G, Flick R (1999) Interaction of influenza virus polymerase with viral RNA in the “corkscrew” conformation. J Gen Virol 80:2565–2572
Hsia H-P, Yang Y-H, Szeto W-C, Nilsson BE, Lo C-Y, Ng AK-L, Fodor E, Shaw P-C, Menéndez-Arias L (2018) Amino acid substitutions affecting aspartic acid 605 and valine 606 decrease the interaction strength between the influenza virus RNA polymerase PB2 ‘627’ domain and the viral nucleoprotein. PLOS ONE 13(1):e0191226
Huet S, Avilov SV, Ferbitz L et al (2010) Nuclear import and assembly of influenza a virus RNA polymerase studied in live cells by fluorescence cross-correlation spectroscopy. J Virol 84:1254–1264
Jorba N, Area E, Ortin J (2008) Oligomerization of the influenza virus polymerase complex in vivo. J Gen Virol 89:520–524
Jorba N, Coloma R, Ortín J (2009) Genetic trans-complementation establishes a new model for influenza virus RNA transcription and replication. PLoS Pathog 5:e1000462
Jung TE, Brownlee GG (2006) A new promoter-binding site in the PB1 subunit of the influenza a virus polymerase. J Gen Virol 87:679–688
Koppstein D, Ashour J, Bartel DP (2015) Sequencing the cap-snatching repertoire of H1N1 influenza provides insight into the mechanism of viral transcription initiation. Nucleic Acids Res 43:5052–5064
Kotlarek D, Worch R (2016) New insight into metal ion-driven catalysis of nucleic acids by influenza PA-Nter. PLoS One 11:e0156972
Kuzuhara T, Kise D, Yoshida H et al (2009) Structural basis of the influenza a virus RNA polymerase PB2 RNA-binding domain containing the pathogenicity-determinant lysine 627 residue. J Biol Chem 284:6855–6860
Lee MTM, Bishop K, Medcalf L et al (2002) Definition of the minimal viral components required for the initiation of unprimed RNA synthesis by influenza virus RNA polymerase. Nucleic Acids Res 30:429–438
Lee M-TM, Klumpp K, Digard P, Tiley L (2003) Activation of influenza virus RNA polymerase by the 5′ and 3′ terminal duplex of genomic RNA. Nucleic Acids Res 31:1624–1632
Li M-L, Ramirez BC, Krug RM (1998) RNA-dependent activation of primer RNA production by influenza virus polymerase: different regions of the same protein subunit constitute the two required RNA-binding sites. EMBO J 17:5844–5852
Li M-L, Rao P, Krug RM (2001) The active sites of the influenza cap-dependent endonuclease are on different polymerase subunits. EMBO J 20:2078–2086
Liu Y, Qin K, Meng G et al (2013) Structural and functional characterization of K339T substitution identified in the PB2 subunit cap-binding pocket of influenza a virus. J Biol Chem 288:11013–11023
Liu M, Lam MK-H, Zhang Q et al (2015a) The functional study of the N-terminal region of influenza B virus nucleoprotein. PLoS One 10:e0137802
Liu Y, Yang Y, Fan J et al (2015b) The crystal structure of the PB2 cap-binding domain of influenza B virus reveals a novel cap recognition mechanism. J Biol Chem 290:9141–9149
Lukarska M, Fournier G, Pflug A et al (2017) Structural basis of an essential interaction between influenza polymerase and pol II CTD. Nature 541:117–121
Martín-Benito J, Area E, Ortega J et al (2001) Three-dimensional reconstruction of a recombinant influenza virus ribonucleoprotein particle. EMBO Rep 2:313–317
Martínez-Alonso M, Hengrung N, Fodor E (2016) RNA-free and ribonucleoprotein-associated influenza virus polymerases directly bind the Serine-5-phosphorylated carboxyl-terminal domain of host RNA polymerase II. J Virol 90:6014–6021
Moeller A, Kirchdoerfer RN, Potter CS et al (2012) Organization of the influenza virus replication machinery. Science 338:1631–1634
Moen SO, Abendroth J, Fairman JW et al (2014) Structural analysis of H1N1 and H7N9 influenza a virus PA in the absence of PB1. Sci Rep 4:5944
Murti KG, Webster RG, Jones IM (1988) Localization of RNA polymerases on influenza viral ribonucleoproteins by immunogold labeling. Virology 164:562–566
Nakada R, Hirano H, Matsuura Y (2015) Structure of importin-alpha bound to a non-classical nuclear localization signal of the influenza a virus nucleoprotein. Sci Rep 5:15055
Ng AK-L, Zhang H, Tan K et al (2008) Structure of the influenza virus a H5N1 nucleoprotein: implications for RNA binding, oligomerization, and vaccine design. FASEB J 22:3638–3647
Ng AK-L, Wang J-H, Shaw P-C (2009) Structure and sequence analysis of influenza a virus nucleoprotein. Sci China Ser C Life Sci 52:439–449
Ng AK-L, Lam MK-H, Zhang H et al (2012) Structural basis for RNA binding and homo-oligomer formation by influenza B virus nucleoprotein. J Virol 86:6758–6767
Nilsson BE, te Velthuis AJW, Fodor E (2017) Role of the PB2 627 domain in influenza a virus polymerase function. J Virol 91:e02467-16–e02e02467
Noda T, Sagara H, Yen A et al (2006) Architecture of ribonucleoprotein complexes in influenza a virus particles. Nature 439:490–492
Obayashi E, Yoshida H, Kawai F et al (2008) The structural basis for an essential subunit interaction in influenza virus RNA polymerase. Nature 454:1127–1131
Ohtsu Y, Honda Y, Sakata Y et al (2002) Fine mapping of the subunit binding sites of influenza virus RNA polymerase. Microbiol Immunol 46:167–175
Oymans J, te Velthuis A (2017) Correct And efficient initiation of viral RNA synthesis by the influenza A virus RNA polymerase bioRvix 138487
Perez DR, Donis RO (2001) Functional analysis of PA binding by influenza a virus PB1: effects on polymerase activity and viral infectivity. J Virol 75:8127–8136
Pflug A, Guilligay D, Reich S, Cusack S (2014) Structure of influenza a polymerase bound to the viral RNA promoter. Nature 516:355–360
Pons MW, Schulze IT, Hirst GK, Hauser R (1969) Isolation and characterization of the ribonucleoprotein of influenza virus. Virology 39:250–259
Poon LL, Pritlove DC, Fodor E, Brownlee GG (1999) Direct evidence that the poly(A) tail of influenza A virus mRNA is synthesized by reiterative copying of a U track in the virion RNA template. J Virol 73:3473–3476
Poon LL, Fodor E, Brownlee GG (2000) Polyuridylated mRNA synthesized by a recombinant influenza virus is defective in nuclear export. J Virol 74:418–427
Pritlove DC, Poon LL, Fodor E et al (1998) Polyadenylation of influenza virus mRNA transcribed in vitro from model virion RNA templates: requirement for 5′ conserved sequences. J Virol 72:1280–1286
Pumroy RAA, Ke S, Hart DJJ et al (2015) Molecular determinants for nuclear import of influenza a PB2 by importin α isoforms 3 and 7. Structure 23:374–384
Rao P, Yuan W, Krug RM (2003) Crucial role of CA cleavage sites in the cap-snatching mechanism for initiating viral mRNA synthesis. EMBO J 22:1188–1198
Reguera J, Gerlach P, Cusack S (2016) Towards a structural understanding of RNA synthesis by negative strand RNA viral polymerases. Curr Opin Struct Biol 36:75–84
Reich S, Guilligay D, Pflug A et al (2014) Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature 516:361–366
Reich S, Guilligay D, Cusack S (2017) An in vitro fluorescence based study of initiation of RNA synthesis by influenza B polymerase. Nucleic Acids Res 45:3353–3368
Resa-Infante P, Recuero-Checa MA, Zamarreño N et al (2010) Structural and functional characterization of an influenza virus RNA polymerase-genomic RNA complex. J Virol 84:10477–10487
Ruigrok RW, Baudin F (1995) Structure of influenza virus ribonucleoprotein particles. II. Purified RNA-free influenza virus ribonucleoprotein forms structures that are indistinguishable from the intact influenza virus ribonucleoprotein particles. J Gen Virol 76(Pt 4):1009–1014
Sikora D, Rocheleau L, Brown EG, Pelchat M (2014) Deep sequencing reveals the eight facets of the influenza a/HongKong/1/1968 (H3N2) virus cap-snatching process. Sci Rep 4:6181
Sugiyama K, Obayashi E, Kawaguchi A et al (2009) Structural insight into the essential PB1-PB2 subunit contact of the influenza virus RNA polymerase. EMBO J 28:1803–1811
Swale C, Monod A, Tengo L et al (2016) Structural characterization of recombinant IAV polymerase reveals a stable complex between viral PA-PB1 heterodimer and host RanBP5. Sci Rep 6:24727
Tao Y, Farsetta DL, Nibert ML, Harrison SC (2002) RNA synthesis in a cage-structural studies of reovirus polymerase lambda3. Cell 111:733–745
Tarendeau F, Boudet J, Guilligay D et al (2007) Structure and nuclear import function of the C-terminal domain of influenza virus polymerase PB2 subunit. Nat Struct Mol Biol 14:229–233
Tarendeau F, Crepin T, Guilligay D et al (2008) Host determinant residue lysine 627 lies on the surface of a discrete, folded domain of influenza virus polymerase PB2 subunit. PLoS Pathog 4:e1000136
Thierry E, Guilligay D, Kosinski J et al (2016) Influenza polymerase can adopt an alternative configuration involving a radical repacking of PB2 domains. Mol Cell 61:125–137
Tomescu AI, Robb NC, Hengrung N et al (2014) Single-molecule FRET reveals a corkscrew RNA structure for the polymerase-bound influenza virus promoter. Proc Natl Acad Sci U S A 111:E3335–E3342
Torreira E, Schoehn G, Fernández Y et al (2007) Three-dimensional model for the isolated recombinant influenza virus polymerase heterotrimer. Nucleic Acids Res 35:3774–3783
Tsurumura T, Qiu H, Yoshida T et al (2013) Conformational polymorphism of m7GTP in crystal structure of the PB2 middle domain from human influenza a virus. PLoS One 8:e82020
te Velthuis AJW, Fodor E (2016) Influenza virus RNA polymerase: insights into the mechanisms of viral RNA synthesis. Nat Rev Microbiol 14:479–493
te Velthuis AJW, Robb NC, Kapanidis AN et al (2016) The role of the priming loop in influenza a virus RNA synthesis. Nat Microbiol 1:16029
Vreede FT, Brownlee GG (2007) Influenza virion-derived viral ribonucleoproteins synthesize both mRNA and cRNA in vitro. J Virol 81:2196–2204
Vreede FT, Jung TE, Brownlee GG (2004) Model suggesting that replication of influenza virus is regulated by stabilization of replicative intermediates. J Virol 78:9568–9572
Wakai C, Iwama M, Mizumoto K, Nagata K (2011) Recognition of cap structure by influenza B virus RNA polymerase is less dependent on the methyl residue than recognition by influenza a virus polymerase. J Virol 85:7504–7512
Wunderlich K, Juozapaitis M, Ranadheera C et al (2011) Identification of high-affinity PB1-derived peptides with enhanced affinity to the PA protein of influenza a virus polymerase. Antimicrob Agents Chemother 55:696–702
Xiao S, Klein ML, LeBard DN et al (2014) Magnesium-dependent RNA binding to the PA endonuclease domain of the avian influenza polymerase. J Phys Chem B 118:873–889
Xie L, Wartchow C, Shia S et al (2016) Molecular basis of mRNA cap recognition by influenza B polymerase PB2 subunit. J Biol Chem 291:363–370
Yamada S, Hatta M, Staker BL et al (2010) Biological and structural characterization of a host-adapting amino acid in influenza virus. PLoS Pathog 6:e1001034
Yamanaka K, Ishihama A, Nagata K (1990) Reconstitution of influenza virus RNA-nucleoprotein complexes structurally resembling native viral ribonucleoprotein cores. J Biol Chem 265:11151–11155
Yang Y, Tang Y-S, Shaw P-C (2014) Structure and function of nucleoprotein from Orthomyxoviruses. Biodesign 2:91–99
Ye Q, Krug RM, Tao YJ (2006) The mechanism by which influenza a virus nucleoprotein forms oligomers and binds RNA. Nature 444:1078–1082
Ye Q, Guu TSY, Mata DA et al (2012) Biochemical and structural evidence in support of a coherent model for the formation of the double-helical influenza a virus ribonucleoprotein. MBio 4:e00467–e00412
York A, Hengrung N, Vreede FT et al (2013) Isolation and characterization of the positive-sense replicative intermediate of a negative-strand RNA virus. Proc Natl Acad Sci 110(45):E4238
Yuan P, Bartlam M, Lou Z et al (2009) Crystal structure of an avian influenza polymerase PAN reveals an endonuclease active site. Nature 458:909–913
Zhang S, Wang J, Wang Q, Toyoda T (2010) Internal initiation of influenza virus replication of viral RNA and complementary RNA in Vitro. J Biol Chem 285:41194–41201
Zhao C, Lou Z, Guo Y et al (2009) Nucleoside monophosphate complex structures of the endonuclease domain from the influenza virus polymerase PA subunit reveal the substrate binding site inside the catalytic center. J Virol 83:9024–9030
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Lo, CY., Tang, YS., Shaw, PC. (2018). Structure and Function of Influenza Virus Ribonucleoprotein. In: Harris, J., Bhella, D. (eds) Virus Protein and Nucleoprotein Complexes. Subcellular Biochemistry, vol 88. Springer, Singapore. https://doi.org/10.1007/978-981-10-8456-0_5
Download citation
DOI: https://doi.org/10.1007/978-981-10-8456-0_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-8455-3
Online ISBN: 978-981-10-8456-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)