
Abstract
Type 2 diabetes mellitus (T2DM) is a worldwide escalating health disorder resulting from insulin resistance and functional loss of insulin-producing beta cells that finally cause chronically elevated blood glucose concentrations. Here we review the role of ubiquitously expressed antioxidant protein DJ-1 in the pathogenesis of T2DM. In beta cells, DJ-1 protects against oxidative stress, endoplasmic reticulum stress, and streptozotocin- and cytokine-induced stress and preserves beta cell viability and insulin secretion. In skeletal muscle, DJ-1 controls energy metabolism and efficient fuel utilization, whereas in adipose tissue a role in adipogenesis and obesity-induced inflammation has been reported. This suggests that DJ-1 plays multiple roles in many cell types under metabolically challenging conditions as seen in obesity, insulin resistance, and T2DM.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
11.1 Introduction
Diabetes mellitus, a state of chronically increased blood glucose concentrations, is a worldwide escalating health concern affecting around 411 million people worldwide (Welters and Lammert 2014). Type 2 diabetes mellitus (T2DM), the most frequent type of diabetes, results from insulin resistance, i.e., when insulin signaling is blunted in important glucose-metabolizing tissues, and from dysfunction and loss of the insulin-producing beta cells located in the pancreatic islets or islets of Langerhans (Welters and Lammert 2014). Insulin resistance is closely related to obesity, a major risk factor for T2DM (Samuel et al. 2010; Shulman 2014; Sun et al. 2011). In obese individuals, maximally stretched adipocytes insufficiently store lipids, leading to ectopic lipid accumulation in hepatocytes and skeletal muscle (Samuel et al. 2010; Shulman 2014; Sun et al. 2011). It is thought that excess lipid species contribute to impairment of insulin signaling in peripheral tissues, thereby limiting glucose uptake and causing beta cells to produce more insulin to maintain normoglycemia (Shulman 2014; Remedi and Emfinger 2016; Welters and Lammert 2014). However, during this extended period of insulin overproduction, the beta cells are challenged by oxidative and endoplasmic reticulum stress or low-grade inflammation leading to dysfunction and loss of beta cells (Bensellam et al. 2012; Lenzen 2008; Weir and Bonner-Weir 2013). Therefore, agents reducing insulin resistance and agents protecting beta cells from dysfunction and cell death have been in the focus of development of diabetes therapies (Remedi and Emfinger 2016; Welters and Lammert 2014).
The DJ-1 protein is a multifunctional, ubiquitously expressed protein, which was first discovered as an oncogene in conjunction with ras, but later the DJ-1 gene emerged as a causative gene (called Park7) of inherited Parkinson’s disease (Ariga et al. 2013; Kahle et al. 2009). Among many functions reported, DJ-1 protects from oxidative stress insults, increases cell viability, acts as chaperone and protease, maintains mitochondrial integrity, and contributes to transcriptional regulation, although it does not bind to DNA itself (Ariga et al. 2013; Kahle et al. 2009). The number of proteins, which directly interact or are closely associated with DJ-1, is enormous and includes transcription factors and signal transduction factors.
The activity of DJ-1, which acts as a homodimer, is thought to be modulated by oxidation and posttranslational modifications (sumoylation and S-nitrosylation). DJ-1 contains three cysteine residues which can be oxidized sequentially to SOH, SO2H, and SO3H (Ariga et al. 2013). A lower degree of oxidation is thought to correspond to activated DJ-1, whereas complete oxidation renders the protein inactive, possibly even irreversibly (Duan et al. 2008; Choi et al. 2006; Ariga et al. 2013; Kahle et al. 2009). Accumulation of oxidized DJ-1 has been brought in connection with many diseases in which oxidative stress plays a pivotal role. For example, (over-)oxidized DJ-1 forms have been found in the brains of patients with Parkinson’s and Alzheimer’s disease (Choi et al. 2006).
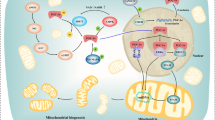
Oxidative stress, the presence of supraphysiological levels of reactive oxygen species (ROS), which can damage DNA and proteins, has also been linked to the pathogenesis of T2DM (Finkel 2011; Houstis et al. 2006). For example, ROS levels rise in response to high glucose exposure in rodent and human islets (Bensellam et al. 2012) and increase in response to a high-fat diet (HFD) or obesity in skeletal muscle (Anderson et al. 2009). However, as ROS are also important signaling molecules, ROS levels may not cause but just correlate with the disease state and may not be harmful under all conditions (Finkel 2011; Tiganis 2011). Here we review the literature about the role of DJ-1 in T2DM in different tissues, including skeletal muscle, adipose tissue, liver, and the islet of Langerhans/pancreatic islets (Fig. 11.1).

Suggested functions of DJ-1 in tissues relevant for glucose tolerance (Organ icons taken from Welters and Lammert 2014)
11.2 Role of DJ-1 in Glucose Homeostasis In Vivo
Impaired glucose tolerance results from reduced glucose uptake in peripheral tissues and/or insufficient amount of insulin due to islet dysfunction or beta cell loss. This involves many different tissues – e.g., skeletal muscle, liver, adipose tissue, and beta cells – which in concert define the degree of glucose intolerance. The role of DJ-1 in glucose tolerance in vivo has been studied using at least three different DJ-1 deficient mouse models, all harboring a constitutive DJ-1 gene deletion (Shi et al. 2015; Seyfarth et al. 2015; Jain et al. 2015, 2012; Kim et al. 2014) and leading to different results (Table 11.1). A first study suggested that DJ-1 deficiency decreases insulin secretion from isolated pancreatic islets (Jain et al. 2012). Consistently, 12-week-old male DJ-1-deficient mice were less glucose tolerant compared to controls, and this was accompanied with elevated ROS levels in DJ-1 KO (knockout) islets, an effect that was age dependent (Jain et al. 2012). The beta cell protective effect of DJ-1 in vivo was confirmed by a follow-up study, showing that injection of DJ-1 KO mice with the beta cell toxin streptozotocin (STZ) led to severe glucose intolerance, dramatically decreased insulin concentrations, as well as decreased beta cell viability and beta cell mass in comparison to STZ-injected control mice (Jain et al. 2015).
A potential role of DJ-1 in diet-induced obesity, i.e., high-fat diet (HFD) feeding, a model to study insulin resistance and glucose tolerance in mice, was analyzed using different experimental strategies (Table 11.1). For example, glucose intolerance was reported in DJ-1 KO mice in combination with a 12-week-long HFD challenge compared to controls (Kim et al. 2014). This phenotype was accompanied with reduced inflammation in adipose tissue (Kim et al. 2014), considered to be beneficial for insulin sensitivity due to reduced levels of circulating pro-inflammatory cytokines. However, as insulin sensitivity was unaltered in DJ-1-deficient mice compared to controls, dysfunctional pancreatic islets may again explain the observed glucose intolerance (Kim et al. 2014).
However, glucose tolerance was not affected in DJ-1 KO mice, fed either with a chow or HFD for 14 weeks (Seyfarth et al. 2015), in another study using mice derived from the same ES cell clone as a previous study with differences in glucose tolerance (Seyfarth et al. 2015; Kim et al. 2014; Manning-Bog et al. 2007; Pham et al. 2010). In contrast to all other studies, a markedly improved glucose tolerance was reported using a third DJ-1 KO mouse model under HFD conditions (Kim et al. 2005; Shi et al. 2015). The effect was explained by higher energy expenditure (“energy wasting”) by skeletal muscle of DJ-1 KO mice leading to lower adiposity, improved insulin sensitivity and glucose tolerance (Shi et al. 2015). The adiposity phenotype was in contrast to the phenotype of another DJ-1 model (Kim et al. 2014; Seyfarth et al. 2015), which displayed a transient increase in body fat mass (adjusted to body mass) in male and female DJ-1 KO mice, possibly caused by reduced physical activity of these DJ-1 KO mice (Seyfarth et al. 2015).
The reasons for the discrepancies in glucose homeostasis in the different DJ-1 KO mouse models may be explained by differences in the experimental setup (gender, fasting time, age, length of HFD challenge, or microbiomes). The use of organ- and cell-specific deletions of the DJ-1 gene, as well as mice overexpressing DJ-1, will allow more comprehensive insights in the tissue- and cell-specific role of DJ-1 mice in the future.
11.3 The Role of DJ-1 in the Islet of Langerhans
11.3.1 Protective Effect of DJ-1 in Beta Cells
Dysfunction and progressive loss of the insulin-producing beta cells lead to chronic hyperglycemia (Cnop et al. 2005; Weir and Bonner-Weir 2013; Remedi and Emfinger 2016; Welters and Lammert 2014), which is preceded by a stage of insulin resistance in T2DM. During mild insulin resistance, insulin overproduction by beta cells is still sufficient to maintain normoglycemia. However, even mild hyperglycemia is thought to reduce glucose-stimulated insulin secretion (Weir and Bonner-Weir 2013). With increasing insulin resistance and hyperglycemia, beta cells face additional stresses: Firstly, beta cells are exposed to high levels of ROS (Bensellam et al. 2012) that are predominately produced in mitochondria and quenched by an antioxidant defense system under normal conditions. However, as beta cells only express low levels of antioxidant proteins, the antioxidative capacity is limited in beta cells, thus more easily leading to oxidative stress (Lenzen 2008). Secondly, prolonged periods of insulin overproduction trigger ER stress leading to the recruitment of chaperones that support the folding or degradation of proteins. Excessive and prolonged ER stress, however, also triggers apoptotic pathways causing beta cell death (Weir and Bonner-Weir 2013).
In addition to oxidative stress and ER stress, beta cells in prediabetic subjects and individuals with established T2DM patients encounter low-grade inflammation, i.e., elevated levels of pro-inflammatory cytokines that affect islet function and survival (Abdulreda and Berggren 2013). The triggers for islet inflammation in T2DM still need to be clarified though (Abdulreda and Berggren 2013). Some of the inflammatory pathways may be shared with type 1 diabetes mellitus, in which immune cells infiltrate the islets and secrete cytotoxic pro-inflammatory cytokines promoting beta cell death (Abdulreda and Berggren 2013; Eizirik et al. 2009; Jorns et al. 2014).
A role of DJ-1 in oxidative stress/glucotoxic conditions in the islets of Langerhans was first suggested in a large quantitative proteomic analysis when isolated mouse islets were exposed to high glucose concentrations leading to a twofold upregulation of the DJ-1 protein (Waanders et al. 2009). Thereafter, the beta cell protective role of DJ-1 was shown for many different experimental stress conditions including oxidative stress (Inberg and Linial 2010; Jain et al. 2012; Jo et al. 2016a; Waanders et al. 2009), ER stress (Inberg and Linial 2010) or pro-inflammatory cytokine-induced stress (Jain et al. 2015; Jo et al. 2016b). Moreover, DJ-1 was shown to preserve mitochondrial integrity (Jain et al. 2012, 2015) and insulin secretion in beta cells and experimental beta cell lines (Inberg and Linial 2010; Jain et al. 2012, 2015). In line with increased beta cell viability and function, DJ-1 mRNA and protein are upregulated in beta cell lines or isolated mouse and human islets in response to experimental stress conditions, including treatment with H2O2, exposure to high glucose concentrations (Inberg and Linial 2010; Jain et al. 2012; Waanders et al. 2009), or treatment with thapsigargin, an ER stress inducer (Inberg and Linial 2010). Likewise, MIN6 cells with silenced DJ-1 levels are highly sensitive to H2O2-mediated oxidative insult compared to cells with normal DJ-1 expression (Inberg and Linial 2010).
Consistent with the DJ-1 loss-of-function experiments described, beta cell survival can be enhanced by DJ-1 overexpression in several beta cell lines. For example, adenoviral overexpression of DJ-1 in mouse insulinoma cells (MIN6) significantly preserved cell viability after H2O2 or thapsigargin treatment (Inberg and Linial 2010). Moreover, Jo et al. (2016a, b) demonstrated the protective effect of a cell permeable DJ-1 protein (Tat-DJ-1), which protected rat insulinoma cells (RINm5F) from H2O2 and also from pro-inflammatory cytokine-induced cell stress (Jo et al. 2016a, b).
The protective effects of DJ-1 were also demonstrated in vivo by challenging DJ-1-deficient mice with multiple low doses of STZ (Jain et al. 2015). STZ is a glucose analogue causing DNA alkylation and NAD+ depletion (due to hyperactivity of poly (ADP-ribose) polymerase, PARP) in beta cells resulting in insulitis and beta cell death. STZ treatment led to a doubled rate of beta cell death, decreased plasma insulin levels, increased fasting blood glucose concentrations, and a dramatically reduced glucose tolerance in DJ-1 KO mice compared to controls. Moreover, the mitochondrial network was reduced and less insulin granules were observed in STZ-treated DJ-1 KO beta cells compared to STZ-treated controls (Jain et al. 2015). Increased cell death rates were also observed ex vivo, when isolated islets of DJ-1 KO mice were treated with either a pro-inflammatory cytokine cocktail (interleukin-1β, tumor necrosis factor α, and interferon γ) or with STZ (Jain et al. 2015).
Most importantly, in human islets, an upregulation of DJ-1 protein was also observed after exposure to high glucose concentrations, indicating a protective role for DJ-1 in human beta cells (Jain et al. 2012). Interestingly, DJ-1 mRNA expression in human islets appears to be age dependent, i.e., DJ-1 expression was increased in islets of elderly humans (with an average age of 74 years) compared to islets of younger humans (with an average age of 44 years) (Jain et al. 2012). Thus, in human beta cells, DJ-1 expression may increase and adjust these cells to an age-related increase in ROS. The finding that DJ-1 expression is significantly reduced in the islets of elderly human subjects with T2DM (Jain et al. 2012) suggests that failure to upregulate DJ-1 weakens the cell stress defense in human islets making them more susceptible to oxidative stress followed by beta cell dysfunction and death.
11.3.2 Molecular Basis of the DJ-1 Protective Effect in Beta Cells
Different scenarios have been proposed to explain the beta cell protective effects of DJ-1 at a molecular level. Firstly, DJ-1 may quench ROS species by oxidation of its cysteine residues (Ariga et al. 2013). However, this capacity is limited (Junn et al. 2005), suggesting that ROS scavenging by DJ-1 alone is insufficient to normalize cellular redox homeostasis. More likely, DJ-1 may act as a stress sensor, which enables transcription factors to translocate to the nucleus, activates survival pathways, or reduces the activity of pro-apoptotic signaling pathways. The ability to serve as a redox sensor is attributed to at least one of its three cysteine residues (C106) thought to activate DJ-1 upon its oxidation (Ariga et al. 2013). In mouse islets and MIN6 cells, DJ-1 isoforms shift from basic to more acidic (oxidized) forms after exposure to H2O2 (Inberg and Linial 2010), suggesting that DJ-1 also serves as an oxidative sensor in beta cells as is seen in neurons (Ariga et al. 2013; Kahle et al. 2009). Consistent with this notion, the beta cell protective effect of DJ-1 is lost when a DJ-1 mutant lacking the oxidative sensitive cysteine residue C106 was used (Jo et al. 2016a).
Several downstream pathways of DJ-1 have been proposed to mediate protection of beta cells. For example, it has been reported that DJ-1 regulates nuclear factor erythroid 2-related factor (Nrf2), a master regulator of cellular oxidative stress defense, in cancer cell lines and mouse fibroblasts (Clements et al. 2006; Ma 2013). Under oxidative stress conditions, DJ-1 sequesters Keap-1 (Kelch-like ECH-associated protein 1), a cytosolic Nrf2-binding protein, which enables free Nrf2 to translocate to the nucleus and induce the expression of antioxidative genes restoring ROS levels to normal (Clements et al. 2006; Ma 2013). Although a potential link between DJ-1 and Nrf2 activation in the beta cell remains to be shown, the Keap1-Nrf2 system was shown to play an important role in beta cell maintenance in response to toxic levels of reactive species (Dinic et al. 2016; Yagishita et al. 2014).
A link between DJ-1 and other pathways affecting beta cell viability, the NF-κB and the mitogen-activated protein kinases (MAPK) pathways, has been suggested under oxidative stress conditions in rat insulinoma cells (Jo et al. 2016a). NF-κB is a transcription factor, which undergoes nuclear translocation, e.g., after oxidative stress or cytokine exposure in many different cell types. In beta cells, a potential activation of NF-κB by ROS or high glucose levels has been debated (Cnop et al. 2005). However, peroxide-challenged RINm5F cells displayed activated NF-κB, and this activation is reduced (less phosphorylation of p65 and IκBα) if the cells are treated with a cell permeable Tat-DJ-1 protein. This effect was abolished if cells were treated with a mutant DJ-1 protein lacking the oxidation-sensitive cysteine C106 (Jo et al. 2016a). Besides its effects on NF-κB, the functional Tat-DJ-1 protein also reduced the phosphorylation of MAP kinases p38, JNK (c-Jun N-terminal kinases), and ERK (extracellular signal-regulated kinases) (Jo et al. 2016a) and attenuated the apoptotic pathway (e.g., resulting in less cleaved caspase-3) after exposure to pro-inflammatory cytokines interleukin-1β, tumor necrosis factor α, and interferon γ (Jo et al. 2016b). As Tat-DJ-1 could reduce both, the peroxide and cytokine-induced high ROS levels, (Jo et al. 2016a, b), the DJ-1 protective effect in beta cells may primarily ground on its antioxidative effect.
Regarding ER stress, another DJ-1 protective mechanism was proposed by Inberg et al. who identified the transcription factor TFII-I as a cytosolic interaction partner of DJ-1 (Inberg and Linial 2010). TFII-I activates the expression of the chaperone BiP, which is part of the unfolded protein response (UPR) in ER stress (Inberg and Linial 2010). Interestingly, under conditions of high DJ-1 expression, BiP expression was less increased after thapsigargin treatment (Inberg and Linial 2010). It was suggested that a high amount of cytosolic DJ-1 reduces TFII-I nuclear translocation, thereby restraining the UPR response (including BiP). However, DJ-1 may also act via an independent mechanism to reduce ER stress.
Finally, DJ-1 protects mitochondrial integrity and function in beta cells via normalizing ROS levels (Jain et al. 2012, 2015). Mitochondria are an important source of ROS, and high ROS levels cause mitochondrial dysfunction and insulin secretion defects in beta cells (Supale et al. 2012). MIN6 cells silenced for DJ-1 display increased mitochondrial ROS levels, which can be restored to normal by transfection with a DJ-1 expression plasmid (Jain et al. 2012). Moreover, significantly more fragmented mitochondria are observed in DJ-1 silenced MIN6 cells and islets of DJ-1 KO mice aged 12–13 weeks (Jain et al. 2012), an effect that could be reversed by the antioxidant N-acetyl-L-cysteine (NAC) (Jain et al. 2012). Moreover, ATP production, an important mediator of insulin secretion in response to high glucose, was decreased in DJ-1-deficient mouse islets compared to control islets, consistent with the mitochondrial phenotype (Jain et al. 2012). The molecular basis of the protective effect of DJ-1 in mitochondria remains to be further explored. In this context, however, it is noteworthy that expression levels of dynamin-like protein (DLP1/DRP1), a regulator of mitochondrial fission, were shown to depend on DJ-1 expression in human neuroblastoma cells (Wang et al. 2012).
In conclusion, DJ-1 protects beta cells from various cell stresses and preserves mitochondrial homeostasis and insulin secretion. However, a common molecular mechanism explaining all cytoprotective effects of DJ-1 has not been proposed so far.
11.4 Adipose Tissue
Adipose tissue plays a central role in the development of insulin resistance (Shulman 2014; Welters and Lammert 2014; McArdle et al. 2013). Adipocytes store excess energy as triglycerides and also contribute to appetite and metabolic control by secreting hormones (McArdle et al. 2013). In obesity, adipose tissue expands due to adipocyte hypertrophy and differentiation of new adipocytes from precursor cells (adipogenesis) (Sun et al. 2011). In severe obesity, hypoxia and a low-grade inflammation introduced by resident and infiltrating immune cells lead to release of free fatty acids and pro-inflammatory cytokines, negatively affecting insulin signaling in other tissues (Sun et al. 2011; Samuel et al. 2010; Shulman 2014; Welters and Lammert 2014; McArdle et al. 2013).
The role of DJ-1 in adipogenesis and inflammation has been studied in vitro and in diet-induced obesity in rodents (Kim et al. 2014; Shi et al. 2015). DJ-1 is upregulated during adipogenic differentiation of fibroblast-like 3T3-L1 cells toward adipocytes in vitro. Interestingly, silencing of DJ-1 in 3T3-L1 reduced adipogenic differentiation and decreased markers of mature adipocytes (PPARγ, LPL, Glut4), showing that DJ-1 is required for adipogenesis in vitro (Kim et al. 2014). However, the role of DJ-1 in adipogenesis in vivo as investigated by Kim et al. (2014) may be different or at least multifaceted, as the expression of adipogenic genes, adipocyte number and size were not different between DJ-1 KO and control mice (Kim et al. 2014). This was partly confirmed for another DJ-1 mouse model that showed no change in adipogenic and lipogenic gene expression in the adipose tissue (Shi et al. 2015). However, the DJ-1 KO mice in this model were leaner and had smaller adipocytes compared to controls (Shi et al. 2015). Interestingly, mononuclear cell infiltration and interleukin-6 serum levels, a measure of adipose tissue inflammation in obesity, were decreased in DJ-1 KO mice suggesting that DJ-1 contributes to inflammation in adipose tissue, which however did not change insulin resistance or glucose tolerance in this mouse (Kim et al. 2014).
11.5 Skeletal Muscle
In humans and rodents, skeletal muscle accounts for most of the postprandial glucose uptake. In response to obesity or HFD, ROS levels rise in skeletal muscle (Anderson et al. 2009; Shi et al. 2015). In contrast to beta cells where extreme ROS levels are deleterious (see section on beta cells), in skeletal muscle, high ROS levels may rather be beneficial, especially as elevated ROS levels are found in conditions associated with increased life span, e.g., during exercise (Tiganis 2011).
In line with this, low DJ-1 expression in skeletal muscle causes a rise in ROS levels, which however did not lead to oxidative stress or mitochondrial disarrangement (Shi et al. 2015). Instead, DJ-1, which is upregulated under HFD conditions, seems to be involved in energy metabolism in the skeletal muscle, and DJ-1 deficiency was reported to be favorable for insulin sensitivity and glucose tolerance in vivo (Shi et al. 2015). It was suggested that elevated mitochondrial ROS levels in DJ-1 KO skeletal muscle induce the expression of uncoupling protein 3 (UCP3), thus facilitating proton leakage (mitochondrial uncoupling), increasing O2 consumption, but decreasing ATP production. As a consequence of emerging energy depletion, AMPK, a central mediator of cellular energy levels, is activated leading to increased glycolysis, generating a futile cycle thereby “wasting” energy (Shi et al. 2015). In line with this, DJ-1 KO mice consumed more oxygen and energy (even though no change in body temperature could be detected) and displayed a decreased body weight and adiposity. Consistently, the mice were more insulin sensitive and glucose tolerant compared to controls in response to a HFD (Shi et al. 2015). It remains to be resolved if other DJ-1 KO models share this phenotype, as no weight changes (Kim et al. 2014) or even transiently increased adiposity (Seyfarth et al. 2015) was reported for other DJ-1 KO mouse models in response to a HFD.
11.6 Liver
Hepatic insulin resistance leads to an increased glucose output contributing to chronic hyperglycemia (Samuel et al. 2010; Welters and Lammert 2014; Perry et al. 2014). Little is known about a potential function of DJ-1 in hepatic insulin resistance or nonalcoholic fatty liver disease (NAFLD), a condition closely related to hepatic insulin resistance (Samuel et al. 2010; Perry et al. 2014). Even though hepatic ROS levels rise in response to a HFD in mice (Lohr et al. 2016; Shi et al. 2015), DJ-1 mRNA expression is not found to increase (Shi et al. 2015). In addition, ROS levels in the liver of DJ-1 KO mice were not different compared to control mice, and the degree of lipid accumulation as well as expression of genes controlling hepatic lipid metabolism were unchanged, indicating no major role of DJ-1 in hepatic insulin resistance under the experimental conditions studied (Shi et al. 2015).
11.7 Conclusions and Implications for Future Research
DJ-1 is a ubiquitously expressed antioxidative protein with multiple functions. Low levels of DJ-1 are frequently associated with increasing ROS levels, especially under challenging conditions like hyperglycemia. Most likely, elevated DJ-1 levels will help to protect insulin- producing beta cells as well as other cells vulnerable to oxidative stress. Increasing DJ-1 by pharmacological treatment may be one way forward to treat diabetes and its complications, although adverse events of elevated DJ-1 activity have to be carefully considered, since ROS levels may correlate rather than contribute to the pathogenesis of T2DM in some tissues (Finkel 2011). One way forward to increase DJ-1 levels may be the clinically used chemical chaperone 4-phenyl butyric acid (PBA), which increases DJ-1 expression (Zhou et al. 2011) and improves insulin resistance in T2DM rodent mouse model (Ozcan et al. 2006). Alternatively, substances designed to maintain the activity of DJ-1 by preventing its hyperoxidation and inactivation (Inden et al. 2011; Kitamura et al. 2011) could be tested under conditions of T2DM.
References
Abdulreda MH, Berggren PO (2013) Islet inflammation in plain sight. Diabetes Obes Metab 15(Suppl 3):105–116. https://doi.org/10.1111/dom.12160
Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW 3rd, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD (2009) Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119(3):573–581. https://doi.org/10.1172/JCI37048
Ariga H, Takahashi-Niki K, Kato I, Maita H, Niki T, Iguchi-Ariga SM (2013) Neuroprotective function of DJ-1 in Parkinson’s disease. Oxidative Med Cell Longev 2013:683920. https://doi.org/10.1155/2013/683920
Bensellam M, Laybutt DR, Jonas JC (2012) The molecular mechanisms of pancreatic beta-cell glucotoxicity: recent findings and future research directions. Mol Cell Endocrinol 364(1–2):1–27. https://doi.org/10.1016/j.mce.2012.08.003
Choi J, Sullards MC, Olzmann JA, Rees HD, Weintraub ST, Bostwick DE, Gearing M, Levey AI, Chin LS, Li L (2006) Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J Biol Chem 281(16):10816–10824. https://doi.org/10.1074/jbc.M509079200
Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP (2006) DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc Natl Acad Sci U S A 103(41):15091–15096. https://doi.org/10.1073/pnas.0607260103
Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL (2005) Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54(Suppl 2):S97–107
Dinic S, Grdovic N, Uskokovic A, Dordevic M, Mihailovic M, Jovanovic JA, Poznanovic G, Vidakovic M (2016) CXCL12 protects pancreatic beta-cells from oxidative stress by a Nrf2-induced increase in catalase expression and activity. Proc Jpn Acad Ser B Phys Biol Sci 92(9):436–454. https://doi.org/10.2183/pjab.92.436
Duan X, Kelsen SG, Merali S (2008) Proteomic analysis of oxidative stress-responsive proteins in human pneumocytes: insight into the regulation of DJ-1 expression. J Proteome Res 7(11):4955–4961. https://doi.org/10.1021/pr800295j
Eizirik DL, Colli ML, Ortis F (2009) The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol 5(4):219–226. https://doi.org/10.1038/nrendo.2009.21
Finkel T (2011) Signal transduction by reactive oxygen species. J Cell Biol 194(1):7–15. https://doi.org/10.1083/jcb.201102095
Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J (2005) Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 45(4):489–496. https://doi.org/10.1016/j.neuron.2005.01.041
Houstis N, Rosen ED, Lander ES (2006) Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440(7086):944–948. https://doi.org/10.1038/nature04634
Inberg A, Linial M (2010) Protection of pancreatic beta-cells from various stress conditions is mediated by DJ-1. J Biol Chem 285(33):25686–25698. https://doi.org/10.1074/jbc.M110.109751
Inden M, Kitamura Y, Takahashi K, Takata K, Ito N, Niwa R, Funayama R, Nishimura K, Taniguchi T, Honda T, Taira T, Ariga H (2011) Protection against dopaminergic neurodegeneration in Parkinson’s disease-model animals by a modulator of the oxidized form of DJ-1, a wild-type of familial Parkinson’s disease-linked PARK7. J Pharmacol Sci 117(3):189–203
Jain D, Jain R, Eberhard D, Eglinger J, Bugliani M, Piemonti L, Marchetti P, Lammert E (2012) Age- and diet-dependent requirement of DJ-1 for glucose homeostasis in mice with implications for human type 2 diabetes. J Mol Cell Biol 4(4):221–230. https://doi.org/10.1093/jmcb/mjs025
Jain D, Weber G, Eberhard D, Mehana AE, Eglinger J, Welters A, Bartosinska B, Jeruschke K, Weiss J, Path G, Ariga H, Seufert J, Lammert E (2015) DJ-1 protects pancreatic beta cells from cytokine- and streptozotocin-mediated cell death. PLoS One 10(9):e0138535. https://doi.org/10.1371/journal.pone.0138535
Jo HS, Cha HJ, Kim SJ, Yeo HJ, Cho SB, Park JH, Lee CH, Yeo EJ, Choi YJ, Eum WS, Choi SY (2016a) Tat-DJ-1 inhibits oxidative stress-mediated RINm5F cell death through suppression of NF-kappaB and MAPK activation. Med Chem Res 25(11):2589–2598. https://doi.org/10.1007/s00044-016-1698-4
Jo HS, Yeo HJ, Cha HJ, Kim SJ, Cho SB, Park JH, Lee CH, Yeo EJ, Choi YJ, Eum WS, Choi SY (2016b) Transduced Tat-DJ-1 protein inhibits cytokines-induced pancreatic RINm5F cell death. BMB Rep 49(5):297–302
Jorns A, Arndt T, Meyer zu Vilsendorf A, Klempnauer J, Wedekind D, Hedrich HJ, Marselli L, Marchetti P, Harada N, Nakaya Y, Wang GS, Scott FW, Gysemans C, Mathieu C, Lenzen S (2014) Islet infiltration, cytokine expression and beta cell death in the NOD mouse, BB rat, Komeda rat, LEW.1AR1-iddm rat and humans with type 1 diabetes. Diabetologia 57(3):512–521. https://doi.org/10.1007/s00125-013-3125-4
Junn E, Taniguchi H, Jeong BS, Zhao X, Ichijo H, Mouradian MM (2005) Interaction of DJ-1 with Daxx inhibits apoptosis signal-regulating kinase 1 activity and cell death. Proc Natl Acad Sci U S A 102(27):9691–9696. https://doi.org/10.1073/pnas.0409635102
Kahle PJ, Waak J, Gasser T (2009) DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free Radic Biol Med 47(10):1354–1361. https://doi.org/10.1016/j.freeradbiomed.2009.08.003
Kim RH, Smith PD, Aleyasin H, Hayley S, Mount MP, Pownall S, Wakeham A, You-Ten AJ, Kalia SK, Horne P, Westaway D, Lozano AM, Anisman H, Park DS, Mak TW (2005) Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci U S A 102(14):5215–5220. https://doi.org/10.1073/pnas.0501282102
Kim JM, Jang HJ, Choi SY, Park SA, Kim IS, Yang YR, Lee YH, Ryu SH, Suh PG (2014) DJ-1 contributes to adipogenesis and obesity-induced inflammation. Sci Rep 4:4805. https://doi.org/10.1038/srep04805
Kitamura Y, Watanabe S, Taguchi M, Takagi K, Kawata T, Takahashi-Niki K, Yasui H, Maita H, Iguchi-Ariga SM, Ariga H (2011) Neuroprotective effect of a new DJ-1-binding compound against neurodegeneration in Parkinson’s disease and stroke model rats. Mol Neurodegener 6(1):48. https://doi.org/10.1186/1750-1326-6-48
Lenzen S (2008) Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans 36(Pt 3):343–347. https://doi.org/10.1042/BST0360343
Lohr K, Pachl F, Moghaddas Gholami A, Geillinger KE, Daniel H, Kuster B, Klingenspor M (2016) Reduced mitochondrial mass and function add to age-related susceptibility toward diet-induced fatty liver in C57BL/6J mice. Physiol Rep 4(19). 10.14814/phy2.12988
Ma Q (2013) Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53:401–426. https://doi.org/10.1146/annurev-pharmtox-011112-140320
Manning-Bog AB, Caudle WM, Perez XA, Reaney SH, Paletzki R, Isla MZ, Chou VP, McCormack AL, Miller GW, Langston JW, Gerfen CR, Dimonte DA (2007) Increased vulnerability of nigrostriatal terminals in DJ-1-deficient mice is mediated by the dopamine transporter. Neurobiol Dis 27(2):141–150. https://doi.org/10.1016/j.nbd.2007.03.014
McArdle MA, Finucane OM, Connaughton RM, McMorrow AM, Roche HM (2013) Mechanisms of obesity-induced inflammation and insulin resistance: insights into the emerging role of nutritional strategies. Front Endocrinol (Lausanne) 4:52. https://doi.org/10.3389/fendo.2013.00052
Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313(5790):1137–1140. https://doi.org/10.1126/science.1128294
Perry RJ, Samuel VT, Petersen KF, Shulman GI (2014) The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 510(7503):84–91. https://doi.org/10.1038/nature13478
Pham TT, Giesert F, Rothig A, Floss T, Kallnik M, Weindl K, Holter SM, Ahting U, Prokisch H, Becker L, Klopstock T, Hrabe de Angelis M, Beyer K, Gorner K, Kahle PJ, Vogt Weisenhorn DM, Wurst W (2010) DJ-1-deficient mice show less TH-positive neurons in the ventral tegmental area and exhibit non-motoric behavioural impairments. Genes Brain Behav 9(3):305–317. https://doi.org/10.1111/j.1601-183X.2009.00559.x
Remedi MS, Emfinger C (2016) Pancreatic beta-cell identity in diabetes. Diabetes Obes Metab 18(Suppl 1):110–116. https://doi.org/10.1111/dom.12727
Samuel VT, Petersen KF, Shulman GI (2010) Lipid-induced insulin resistance: unravelling the mechanism. Lancet 375(9733):2267–2277. https://doi.org/10.1016/S0140-6736(10)60408-4
Seyfarth K, Poschmann G, Rozman J, Fromme T, Rink N, Hofmann A, Wurst W, Stuhler K, Klingenspor M (2015) The development of diet-induced obesity and associated metabolic impairments in Dj-1 deficient mice. J Nutr Biochem 26(1):75–81. https://doi.org/10.1016/j.jnutbio.2014.09.002
Shi SY, Lu SY, Sivasubramaniyam T, Revelo XS, Cai EP, Luk CT, Schroer SA, Patel P, Kim RH, Bombardier E, Quadrilatero J, Tupling AR, Mak TW, Winer DA, Woo M (2015) DJ-1 links muscle ROS production with metabolic reprogramming and systemic energy homeostasis in mice. Nat Commun 6:7415. https://doi.org/10.1038/ncomms8415
Shulman GI (2014) Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med 371(23):2237–2238. https://doi.org/10.1056/NEJMc1412427
Sun K, Kusminski CM, Scherer PE (2011) Adipose tissue remodeling and obesity. J Clin Invest 121(6):2094–2101. https://doi.org/10.1172/JCI45887
Supale S, Li N, Brun T, Maechler P (2012) Mitochondrial dysfunction in pancreatic beta cells. Trends Endocrinol Metab 23(9):477–487. https://doi.org/10.1016/j.tem.2012.06.002
Tiganis T (2011) Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends Pharmacol Sci 32(2):82–89. https://doi.org/10.1016/j.tips.2010.11.006
Waanders LF, Chwalek K, Monetti M, Kumar C, Lammert E, Mann M (2009) Quantitative proteomic analysis of single pancreatic islets. Proc Natl Acad Sci U S A 106(45):18902–18907. https://doi.org/10.1073/pnas.0908351106
Wang X, Petrie TG, Liu Y, Liu J, Fujioka H, Zhu X (2012) Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J Neurochem 121(5):830–839. https://doi.org/10.1111/j.1471-4159.2012.07734.x
Weir GC, Bonner-Weir S (2013) Islet beta cell mass in diabetes and how it relates to function, birth, and death. Ann N Y Acad Sci 1281:92–105. https://doi.org/10.1111/nyas.12031
Welters A, Lammert E (2014) Diabetes Mellitus. In: Lammert E, Zeeb M (eds) Metabolism of human diseases: organ physiology and pathophysiology. Springer, Vienna, pp 163–173
Yagishita Y, Fukutomi T, Sugawara A, Kawamura H, Takahashi T, Pi J, Uruno A, Yamamoto M (2014) Nrf2 protects pancreatic beta-cells from oxidative and nitrosative stress in diabetic model mice. Diabetes 63(2):605–618. https://doi.org/10.2337/db13-0909
Zhou W, Bercury K, Cummiskey J, Luong N, Lebin J, Freed CR (2011) Phenylbutyrate up-regulates the DJ-1 protein and protects neurons in cell culture and in animal models of Parkinson disease. J Biol Chem 286(17):14941–14951. https://doi.org/10.1074/jbc.M110.211029
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Eberhard, D., Lammert, E. (2017). The Role of the Antioxidant Protein DJ-1 in Type 2 Diabetes Mellitus. In: Ariga, H., Iguchi-Ariga, S. (eds) DJ-1/PARK7 Protein. Advances in Experimental Medicine and Biology, vol 1037. Springer, Singapore. https://doi.org/10.1007/978-981-10-6583-5_11
Download citation
DOI: https://doi.org/10.1007/978-981-10-6583-5_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-6582-8
Online ISBN: 978-981-10-6583-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)