
Abstract
During hematopoiesis, a variety of cells are generated from stem cells through successive rounds of cell fate determination processes. Studies in the last two decades have demonstrated the involvement of Runx transcription factor family members in differentiation of multiple types of hematopoietic cells. Along with evolutionary conservation, the Runx
family is considered to be one of the ancestral regulators of hematopoiesis. It is conceivable that the Runx family is involved in shaping the immune system, which is then comprised of innate and acquired lymphoid cells in vertebrates. In this chapter, we will first summarize roles of Runx proteins during the development of T- and B-lymphocytes, which appeared later during evolution and express antigen specific receptors as a result of DNA recombination processes. We also discuss the recent findings that have unraveled the functions of Runx during differentiation of innate lymphoid cells (ILCs).
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
As expected from many studies showing multiple roles of Runx family in the control of development of many types of cells, genetic ablation of either Runx1 or Cbfβ resulted in lack of definitive hematopoiesis (Okuda et al. 1996; Wang et al. 1996a, b), placing Runx1/Cbfβ as one of the top regulators in the development of hematopoietic cells. Studies in invertebrates such as Drosophila also found that Runx family proteins play essential roles in hematopoiesis (Fossett and Schulz 2001; Lebestky et al. 2000), suggesting the ancestral function of Runx as an important regulator that controls the differentiation of hematopoietic cells (Braun and Woollard 2009). In this chapter, we summarize the roles of the Runx family in development of immune cells including lymphocytes and innate lymphoid cells (ILCs) in vertebrates.
2 Roles of Runx Complex in T Lymphocyte Development
2.1 Early Thymocyte Differentiation
After T-cell homing, precursors migrate from the fetal liver to the thymus rudiments around embryonic day 11.5–12.5 in mice, they colonize there and receive environmental cues to begin development into T-lymphoid cells. The expression of Notch ligands such as Delta-like 4 by thymic epithelial cells (TEC) is known to be a critical environmental signal for early thymocyte development (Hozumi et al. 2008; Koch et al. 2008). These early T cell progenitors (ETP) undergo sequential processes, which are controlled by transcription factor networks, in order to fully commit to the T cell lineage (Fig. 24.1) (Rothenberg et al. 2008). Since ETPs do not express CD4 and CD8 co-receptors, they are referred to as CD4−CD8− double negative (DN) thymocytes, which are further divided into four subsets by the expression of CD25 and CD44 or CD117 (c-Kit). CD25−CD44+ DN1 subset is relatively heterogeneous (Porritt et al. 2004) and contains ETPs that differentiate into CD25+CD44+ DN2 cells. Recent studies addressing the transcriptional regulation of early T cell development have proposed that the DN2 stage is further divided into DN2a and DN2b, and that complete commitment to the T cell lineage occurs at the DN2a and DN2b transition in a Bcl11b dependent manner, based on the observation that T cell development was arrested at the DN2a stage in Bcl11b-deficient mouse (Li et al. 2010; Ikawa et al. 2010). Importantly, in host mice where T cell development is reconstituted from Runx1-deficient hematopoietic stem cells (HSCs), a similar developmental block at the DN2 stage was observed (Fig. 24.1) (Ichikawa et al. 2004). Although the precise mechanism of how lack of Runx1 is involved in DN2a arrest is not clear, it is possible that Runx1 is necessary for induction of Bcl11b expression . Indeed association of Runx with a 3′ downstream enhancer that plays an essential role to drive Bcl11b gene expression in T-cells was recently reported (Li et al. 2013).

Runx and T cell development. Upon pre-thymic acquisition of thymus homing property at some point during differentiation of hematopoietic stem cells (HSC), early thymocyte progenitors (ETP) colonize at the thymus and begin to develop into T lymphocyte -lineage by cascading activation of T cell programs as well as erasing developmental potency to alternative lineages, referred to as commitment process. Full commitment to T-lineage occurs at transition from DN2 to DN3 stage, and Runx1 is essential for this transition. γδT cells are differentiated from DN2/DN3 cells and development of skin-specific γδT cells, known as dendritic epidermal T cells (DETC), requires Runx3. Runx1 is important to pass β-selection. CD4+CD8+ DP thymocyte precursors are selected according to affinity of TCR with self-peptide (positive/negative selection) and positively selected thymocytes differentiate into CD4 helper (Th) or CD8 cytotoxic (Tc) T cells (CD4/CD8 lineage choice). Cross-antagonism between Runx3 and ThPOK plays a central role to fine separation of two fates as well as to couple MHC specificity of TCRs with appropriate fate. Some DP precursors develop into regulatory T cells (Treg), natural killer T cells (NKT) and CD8aa intra epithelial cells (IEL) through process known as agonistic selection, during which Runx1 and Runx3 play important roles for their differentiation. In the periphery, upon encountering antigens and depending on environmental cues, Th cells differentiate into distinct types of effector cells such as Th1, Th2 and Th17, each of which secrete signature cytokine, IFNγ, IL-4 and IL-17, respectively. Roles of Runx proteins during effector Th subset differentiation are discussed in the Sect. 24.2.3.
There are two types of T-lymphocytes that express distinct T-cell antigen receptors, αβTCR and γδTCR. Although how and when the progenitors decide to become αβT cells or γδT cells remains controversial, there are ample studies addressing how the genes encoding α, β, δ and γ chains of TCR are activated and undergo DNA recombination, known as V(D)J recombination mediated by RAG-1/2 recombinase complexes at the antigen receptor loci. V(D)J recombination is a unique property endowed only to lymphocytes among somatic cells and is strictly controlled. Since there are other excellent reviews discussing the mechanisms of V(D)J recombination (Majumder et al. 2015), we simply focus on the effects of enhancer inactivation of each T-cell antigen gene. It has been shown that the Eα enhancer, Eβ enhancer, Eδ enhancer, and Eγ enhancer at the Tcra, Tcrb, Tcrd, and Tcrg genes, respectively, are essential for this recombination. Interestingly, each enhancer contains Runx recognition motifs (Hsiang et al. 1993; Redondo et al. 1991; Takeda et al. 1990; Hernandez-Munain and Krangel 1995) and is bound by Runx complexes (Tani-Ichi et al. 2011; Oestreich et al. 2006; Hollenhorst et al. 2007). Among these enhancers, Runx sites often locate closely to Ets binding sites (Wotton et al. 1994; Hernandez-Munain et al. 1998). For instance, in the Eβ enhancer, two ETS-Runx elements have been identified. Targeted mutations abrogating two Runx sites within the Eβ enhancers eliminated the enhancer function (Majumder et al. 2015), suggesting that Runx binding is essential for enhancer activation (Fig. 24.1). However, due to the arrest of DN2 stage through lack of Runx1 activity, the role of Runx1 function in the regulation of Tcrb gene activation remains unclear. It is noteworthy, however, that in addition to enhancers in the T- cell antigen receptor loci, a μ enhancer in the Igh locus that encodes the B-cell antigen receptor also contains ETS-Runx composite elements (Erman et al. 1998). Thus, there might be a common regulatory mechanism by which antigen receptor loci are activated through co-binding of ETS and Runx to essential enhancers in these loci. Consistent with this note, skin specific γδT cells, referred to as DETC (dendritic epidermal T cells) expressing the invariant Vγ3 chain, were absent in the epidermis of Runx3-deficient mice (Fig. 24.1) (Woolf et al. 2007). However, fewer precursors expressing the Vγ3 chain were observed in the fetal thymus of Runx3-deficient mice, suggesting that Runx3 is also involved in the processes of expansion driven by interleukin 2 (IL-2) and migration guided by integrin CD103 for DETC differentiation after γδTCR expression .
2.2 Differentiation of αβT Cell Subsets from CD4+CD8+ DP Precursors
2.2.1 Overview
During αβT cell differentiation, there is a “check point” stage to monitor whether V(D)J rearrangement at the Tcrb locus successfully generates a functional TCRβ chain, known as β-selection. Cells lacking TCRβ chain expression fail to form pre-TCR complexes, consisting of the common pre-Tα protein and the TCRβ protein, are not allowed to differentiate to the next stage and are arrested at the CD25+CD44− DN3 stage. On the other hand, signals through the pre-TCR complexes lead to cell proliferation and activate developmental programs including V(D)J rearrangement at the Tcra locus and the expression of CD4 and CD8 co-receptors. Thus CD4−CD8− DN thymocytes become CD4+CD8+ DP thymocytes expressing the mature αβTCR with diverse antigen-specificity and face another selection process, known as positive/negative selection, which evaluates the quality of αβTCR in terms of their affinity to self-peptides presented on the major histocompatibility complex (MHC). Lack of TCR-mediated signals due to insufficient affinity to self-peptide–MHC complexes causes apoptosis, designated as ‘death by neglect’. Cells with TCRs that react too strongly to self-peptide–MHC complexes , are thereby thought to be potentially self-reactive lymphocytes and are eliminated through a ‘negative selection’ process, to reduce the risk of auto-immunity . Only a few DP cells that express TCRs of appropriate affinity with self-peptide–MHC complexes are selected in a process known as ‘positive selection’, and proceed to develop into mature thymocytes. The CD4 and CD8 co-receptors help the TCRs to recognize self-peptides on MHC-class-II and MHC-class-I molecules via specific interaction with class-II and class-I, respectively. After positive selection, two major αβT-lineages, helper cells and cytotoxic cells are generated. It is well known that cells selected through MHC-class-II (thereby their TCRs are MHC-class-II specific) become helper cells and lose CD8 expression, while those selected by MHC-class-I differentiate into the cytotoxic lineage and lose CD4 expression. Thus, in addition to the specificity of TCRs to MHC classes, CD4/CD8 expression profiles also show a perfect match with helper/cytotoxic lineage choice. In addition to the helper/cytotoxic lineage dichotomy, at least three types of αβT cells, iNKT cells, regulatory T cells (Treg), and CD8aa intraepithelial lymphocytes (IEL) are generated from the CD4+CD8+ DP thymocytes through ‘agonist selection’ (Fig. 24.1) (Kronenberg and Rudensky 2005; Lambolez et al. 2007).
2.2.2 Runx and Cd4/Cd8 Gene Regulation
As mentioned above, lineage specific expression of CD4/CD8 co-receptors after positive selection stimulated studies addressing the regulation of this lineage specific expression (Ellmeier et al. 1999). The search for critical cis-regulatory elements in the Cd4 and Cd8 loci first resulted in the identification of a transcriptional silencer in the Cd4 locus as a critical regulatory region for helper-lineage specific CD4 expression (Sawada et al. 1994; Siu et al. 1994). A 434 bp transcriptional silencer, located at the first intron in the Cd4 gene, is sufficient to repress reporter transgene expression in DN thymocytes and cytotoxic-lineage cells. Furthermore, following studies that removed the Cd4 silencer from the mouse genome confirmed its relevance by revealing de-repression of CD4 in CD8+ T cells at a comparable level to that in CD4+ T cells (Leung et al. 2001; Zou et al. 2001). These results from mouse genetics clearly indicated that a single transcriptional silencer is responsible for helper-lineage specific CD4 expression by repressing Cd4 in alternative cytotoxic-lineage T cells.
On the other hand, functionally negative regulatory elements have not been identified in the Cd8 locus . Instead, at least six enhancer elements, termed as E8I to E8VI, have been isolated (Hostert et al. 1998; Ellmeier et al. 1998; Sakaguchi et al. 2015). The effects of each enhancer deletion on Cd8 expression revealed redundant function within the enhancers. Along with the CD8-lineage specific activity of E8I in a reporter transgene expression assay (Ellmeier et al. 1997), a combinatory regulation of enhancers rather than active repression by silencer(s) is supposed to control the lineage specificity of Cd8 expression .
Runx proteins are known to directly regulate both Cd4 and Cd8 expression. Runx1 protein was isolated from a search for a Cd4 silencer binding protein using the yeast one-hybrid screen (Taniuchi et al. 2002). Along with the essential requirement for the Runx recognition site for Cd4 silencer activity, conditional inactivation of Runx1 in DN2/3 thymocytes by the Lck-Cre transgene resulted in CD4 expression in DN3 cells as was observed by lack of Cd4 silencer (Taniuchi et al. 2002). On the other hand, loss of Runx3 but not Runx1, severely affected Cd4 silencer activity in CD8-lineage cells (Taniuchi et al. 2002) as was manifested by de-repression of CD4 in mature Runx3-deficient CD8+ T cells. Such a distinct role of two Runx proteins in Cd4 gene repression at two stages may reflect different expression patterns of Runx1 and Runx3. While Runx1 is most highly expressed in immature DN and DP thymocytes, expression of Runx3 was nearly specific to CD8 SP thymocytes (Egawa et al. 2007). In addition to the involvement of Runx proteins in Cd4 gene regulation, the binding of Runx3 to some enhancers in Cd8 gene was also reported (Sato et al. 2005). Recently, the functional contribution of Runx to Cd8 gene expression was demonstrated by the inefficient maintenance of CD8 expression in the absence of Runx3 (Hassan et al. 2011). Such dual roles of Runx3 in co-receptor gene expression, Cd4 silencing, and Cd8 reactivation, which comprise a key feature of cytotoxic-lineage cells predicted that induction of Runx3 expression might be a key event to activate programs that induce a cytotoxic fate. It should be noteworthy that transgene-mediated ectopic expression of Runx3 in immature thymocytes only partially redirected MHC-class-II-specific T cells to CD8+ T-cells (Kohu et al. 2005; Grueter et al. 2005), at least in part via low CD4 expression on precursor cells as a result of prolonged Cd4 silencer activity in the DN stage.
2.2.3 Antagonistic Interplay Between Runx and ThPOK
ThPOK is a member of the BTB/POZ transcription factor family; many members of this family have been shown to play essential roles in immune cell development (Ellmeier and Taniuchi 2014). Gain- and loss-of-function studies of ThPOK in mice have revealed that ThPOK is a master transcription factor for CD4+ helper T cell development (Kappes and He 2006). For instance, a natural mutation referred to as the hd mutation that results in substitution of glycine for arginine in the putative second zinc-finger domain of the ThPOK protein, led to a loss of CD4+ T cell development through fate conversion of MHC-class-II specific cells into CD4−CD8+ T cells (He et al. 2005). On the contrary, ectopic expression of ThPOK from DP precursors onwards resulted in lack of CD8+ T cells due to redirection of MHC-class-I-specific thymocytes to CD4+CD8− T cells (He et al. 2005).
This striking finding that the presence or absence of a single transcription factor, ThPOK, serves as a major determinant for CD4 helper versus CD8 cytotoxic lineage separation, raised the profound question of how helper-lineage-specific expression of ThPOK is controlled. The answer was revealed during characterization of Runx mutant mice. One issue to be considered while interpreting phenotypes caused by single Runx protein ablation is the redundancy between Runx family proteins. In particular, cross-regulation between Runx1 and Runx3 sometimes underestimates the effect caused by the lack of either protein. Given that the product of a single gene Cbfβ is the sole common subunit for all Runx proteins in mammals, inactivation of Cbfβ has the advantage in terms of avoiding redundancy between Runx proteins. It should be noted that the Cre/loxP-mediated recombination system occasionally suffers from the expansion of a leaky population that escapes Cre-mediated recombination. In addition, protein stability is another factor to be considered in Cre/loxP mediated conditional gene inactivation. Compared to Runx proteins that could be rapidly degraded by proteasomes, the Cbfβ protein seemed to be present longer after the inactivation of its gene. Thus, in thymocyte differentiation, inactivation of Cbfβ gene at the DN stage by Lck-Cre nearly recapitulated the compound inactivation of both Runx1 and Runx3 at the DP stage by Cd4-Cre. Analyses of T-cell development in these mice showed that loss of the Runx complex function in DP thymocytes led to severe reduction of mature thymocyte generation. Most importantly, in the remaining mature T-cell pool, CD8+ T cells were almost absent (Setoguchi et al. 2008). Further analyses using mice in which Runx3 inactivation was combined over Runx1 mutation, causing the deletion of the VWRPY motif at C-terminal end, revealed that redirection of MHC-class-I-specific T cells to CD4+ T-cells was the reason for the loss of CD8+ T cells. This was a phenocopy of the ThPOK transgenic mice, and prompted analyses of Cbfβ F/F : Cd4-Cre mice that retain a substantial number of CD8+ T cells that abnormally de-repress CD4. These CD4+CD8+ cells developed under the gradual loss of Cbfβ protein after positive selection, expressed Thpok gene, providing supportive evidence that redirection of MHC-class-I-specific thymocytes to the CD4+ T-cell lineage in mice lacking functional Runx proteins is due to inappropriate de-repression of Thpok. These observations clearly indicate that Runx proteins are involved in Thpok repression.
Naturally, the next question would be how Runx proteins are involved in Thpok regulation and whether Runx proteins directly regulate the regulatory regions of the Thpok gene. By using a ChIP-on-chip approach , two regions in the Thpok gene were identified as Runx binding sequences (RBS-1 and RBS-2). Further functional analysis of RBS-1 in a reporter transgene expression assay identified transcriptional silencer activity in this sequence. At the same time, a group led by Dr. Kappes also characterized the regulatory regions in Thpok and identified a silencer activity in a their distal regulatory element (DRE), that perfectly overlapped with RBS-1 (He et al. 2008). These observations indicate that silencer activity in DRE/RBS-1, hereafter referred to as Thpok silencer, is responsible for helper lineage specific expression via repression of Thpok expression in cytotoxic-lineage cells. The physiological relevance of Thpok silencer was confirmed by full Thpok de-repression as well as by the loss of CD8+ T cells upon its removal from mouse genomes (Setoguchi et al. 2008). Importantly, the analytical ChIP assay detected Runx binding to Thpok silencer in both ThPOK-expressing helper and non-expressing cytotoxic cells (Setoguchi et al. 2008). This observation indicates that Runx binding is essential but not sufficient for Thpok silencer activity and that uncharacterized mechanisms beyond Runx binding may serve as the switch that controls the specificity of Thpok silencer activity.
Thus two silencers at different loci, the Cd4 silencer and the Thpok silencer, require Runx protein binding to exert their silencer activity. Interestingly, albeit this common feature, both silencers show distinct dependency for VWRPY motifs at the C-terminal end of Runx proteins . While Cd4 silencer activity depends completely on the VWRPY motif, Thpok silencer still represses the Thpok gene to a significant extent without the VWRPY motif (Seo et al. 2012b).
Collectively, one of the most important functions of Runx proteins during cell fate decision by CD4+CD8+ DP precursors is the repression of Thpok as well as Cd4 genes. On the other hand, characterization of ThPOK function revealed that ThPOK acts to restrain Runx3 expression in CD4+ T cells. Thus, ThPOK and Runx3 repress the expression of each other, forming an antagonistic interplay. Since such antagonism between two transcription factors, which play a central role in the development of alternative lineages, is often observed at developmental branch point (Laiosa et al. 2006), the cross antagonism between ThPOK and Runx3 serves as a central mechanism in CD4 helper/CD8 cytotoxic dichotomy (Fig. 24.1). Given that CD8-lineage specific expression of Runx3 reflects a CD8-lineage specific expression from the distal P1 promoter-driven Runx3 transcript (Egawa et al. 2007), ThPOK should be involved in the regulatory mechanism that controls the lineage specific activity of the distal P1 promoter. IL-7 signals are known to be important for the generation of CD8-lineage cells in the thymus and can activate Runx3 (Park et al. 2010). In line with this finding, it was proposed that ThPOK is indirectly involved in Runx3 repression through induction of the SOCS protein family, a strong inhibitor of the IL7 cytokine signals, thereby preventing Runx3 induction (Luckey et al. 2014). Unfortunately, at this moment, little is known about the regulatory regions that control Runx3 expression in T cells. Further studies are necessary to precisely understand the regulation of CD8-lineage specific Runx3 expression.
2.2.4 Roles of Runx in Differentiation of Treg and iNKT Cells
Beyond their role in regulating the CD4/CD8 lineage separation, Runx proteins are known to have other important roles in development of T cell subsets. Results of Runx1 inactivation alone showed that the efficiency of β-selection and positive selection was impaired (Egawa et al. 2007). In addition, generation of invariant natural killer T (NKT) cells expressing the invariant Vα14 chain and the reactive lipid antigen on MHC class –I related CD1d was lost by lack of Runx1 (Egawa et al. 2005). Given that Runx3 is also expressed in iNKT cells, Runx3 is not likely to possess a compensatory function to support iNKT cell differentiation.
Another αβT cell subset generated from CD4+CD8+ DP precursors includes the regulatory T (Treg) cells (Ohkura et al. 2013; Josefowicz et al. 2012). Treg cells have a suppressive function and play an essential role in immune tolerance. FoxP3, a member of the forkhead box transcription factor family, is essential for the generation and function of Treg cells. Mutation of X chromosome-linked human FOXP3 gene result in the IPEX (immunodysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome, showing multi-organ autoimmune inflammatory disease, whereas mutations in the murine Foxp3 gene, for instance, a natural mutation in the scurfy strain or engineered loss of the functional mutation, are known to cause severe postnatal lethal autoimmune disorders (Josefowicz et al. 2012). Thus, how the expression and functions of Foxp3 are regulated, is an essential and profound question in immunology. A study by the Sakaguchi’s group has shown that RUNX1 can interact with FOXP3 protein by the immunoprecipitation assay (Ono et al. 2007). RUNX1 seemed to bind to the IL2 promoter and enhance IL-2 production upon TCR stimulation, which is repressed by FOXP3. In addition to regulation at the protein level, Runx proteins were shown to regulate Foxp3 gene expression through binding to promoters and CNS2 enhancers (Bruno et al. 2009; Kitoh et al. 2009; Rudra et al. 2009; Klunker et al. 2009). A CNS2 enhancer undergoes Treg specific DNA demethylation processes and contributes to stable expression of the Foxp3 gene (Zheng et al. 2010). Interestingly, CNS2 remained methylated in Treg cells lacking Cbfβ and Foxp3 expression level was reduced (Rudra et al. 2009; Kitoh et al. 2009). Results of Treg specific conditional inactivation of Runx1 or Runx3 genes showed that Runx1/Cbfβ complexes are responsible for maintaining Foxp3 expression levels and thereby preventing immunological disorders such as gastritis, serum IgE elevation, and lymphadenopathy (Kitoh et al. 2009).
The third αβT cells subset generated through agonistic selection of CD4+CD8+ DP thymocytes include the CD8αα IEL cells due to their unique expression profile of CD8αα homodimers but not CD8αβ heterodimers as well as the tissue localization in the space between gut epithelial cells. Distinct from the CD8αβ heterodimer, CD8αα homodimers interact with the MHC class I like TL molecule (Leishman et al. 2001). By using a soluble TL tetramer, the sole reagent that is useful to separately detect CD8αα from CD8αβ on cells expressing both forms, it was shown that a proportion of DP thymocytes already expressed the CD8aa homodimer, thereby referred to as CD4+CD8αβ+CD8αα+ triple positive (TP) thymocytes (Gangadharan et al. 2006). TP thymocytes are supposed to be precursors for CD8αα IEL, and they become CD4−CD8− DN thymocytes after agonistic selection. Runx3 is essential for the re-expression of CD8αα (Pobezinsky et al. 2012), presumably through direct activation of E8I enhancers .
2.3 Role of Runx in Differentiation of Effector T Cells
Advances in the last decade have identified and characterized novel effector CD4+ T cell subsets beside the classical Th1 and Th2 cells. In addition, these subsets retain a plasticity that allows them to occasionally modulate their identity (Nakayamada et al. 2012). A characteristic difference in the functions of these subsets is the pattern of cytokine secretion conferred by the induction of specific transcription factors, as is known in the case of classical Th1/Th2 subsets, these signature cytokines and transcription factors are IFNγ/Il-4 and T-bet/Gata3, respectively. Runx proteins have also been shown to regulate effector T cell subsets and cytokine expression. During Th1 cell differentiation, expression of Runx1 and Runx3 proteins exhibited unique reciprocal expression kinetics. Runx1 expression is downregulated while Runx3 expression is induced. Importantly, in differentiated Th1 cells, Runx3 represses Il4 gene transcription through binding to an Il4 silencer (Naoe et al. 2007; Djuretic et al. 2007).
Discovery of Th17 cells, whose characteristic cytokine is IL17, led to a renewed view of helper T cell differentiation as well as pathogenesis of autoimmune diseases (Harrington et al. 2005; Park et al. 2005). Detailed characterization of this IL17 producing cell subset including a comparison of gene expression profiles with other immune cells, combined with mouse genetics, identified RORγt (retinoic-acid-receptor-related orphan receptor-γt) as the master driver of Th17 differentiation (Ivanov et al. 2006). Enforced expression of Runx1 has been shown to induce IL17 expression, and vice versa, diminished Runx1expression by shRNA based knockdown showed an inhibitory effect against Th17 differentiation (Zhang et al. 2008). This Runx1 activity was reported to be mediated through direct regulation of the promoter/enhancer after protein complex formation with RORγt.
3 Roles of Runx Complexes in Early B Lymphocyte Development
B cell development occurs in the bone marrow in adult mice and initiates from the common lymphoid progenitors (CLPs) (Matthias and Rolink 2005). When CLPs adopt the B cell fate by starting to express the B cell lineage marker B220 and Rag recombinases , they are referred to as pre-pro-B cells. Once V(D)J fragments of immunoglobulin heavy chain locus (Igh) are successfully rearranged during the pro-B cell stage, the pre-B cell receptor is formed with the surrogate light chain and drives the transition from pro-B cells to pre-B cells. Like other lineages of blood cell types, it is no exception that early B cell development requires the expression of a series of transcription factors to activate B cell programming as well as to erase the potentials for alternative lineages. A widely-accepted model for transcription factor networks for B cells involves E2A, EBF, and Pax5, and a cascading cross-regulation among these three factors is central to the network. From a simplistic point of view, E2A activates EBF (Kee and Murre 1998), and EBF activates Pax5 (Decker et al. 2009).
Disruption of E2A or EBF (or both) results in developmental arrest at the pre-pro-B cell stage (Zhuang et al. 1994; Bain et al. 1994; Lin and Grosschedl 1995). Since E2A and EBF directly regulate genes specific to early B cell progenitors such as Rag1/2, Igαβ, VpreB, and λ5, these two factors have been considered as the main specification factors. On the other hand, inactivation of Pax5 results in blockade of B cell development at the pro-B-cell stage and such pro-B cells show the potential to follow other lineages after in vivo transplantation (Nutt et al. 1999; Rolink et al. 1999). Combined with the fact that Pax5 actively represses non-B-lineage genes (Mikkola et al. 2002), Pax5 has been considered to be the main commitment factor for B lymphoid-lineages. However, during recent years, other transcription factors important for B cell development in addition to E2A, EBF, and Pax5 have been identified, which include but are not limited to Bcl11a, Runx1, and Foxo1.
By using inducible-targeting strategies to overcome embryonic lethality, Runx1 was shown to have a pivotal role mainly in priming the lymphoid lineage (both B and T cells) during hematopoiesis (Ichikawa et al. 2004; Growney et al. 2005). More specifically, these studies clearly demonstrated that Runx1 is involved in the regulation of B cell generation since almost no B cells were developed in the absence of Runx1. A clue to the molecular mechanisms underlying these observations was obtained from a study showing that E2A and EBF cooperate with Runx1 to activate B cell programs before the involvement of Pax5 (Maier et al. 2004). Specifically, progressive demethylation of the mb-1 gene, which encodes a critical signaling component of the pre-B cell receptor CD79a, requires synergistic activities of both EBF and Runx1. This poised epigenetic change in the mb-1 gene is necessary for Pax5 to activate the mb-1 promoter.
A further attempt was made to decipher the precise relationship between Runx1 and EBF during B lymphopoiesis by conditionally inactivating Runx1, Runx3, and Cbfβ genes by mb1-cre transgene (Seo et al. 2012a). Consistent with previous reports, inactivation of Runx1 and Cbfβ (but not Runx3) resulted in defective B cell generation from early B cell progenitors. An interesting observation from this study was that Runx1 seems to directly activate Ebf transcription, since the amounts of Ebf mRNA was dramatically reduced from the progenitors in the absence of Runx1. Importantly, B cell development of Runx1-deficient cells was rescued by over-expression of EBF at least in vitro, suggesting that the major reason for developmental arrest in Runx1 F/F : mb-1Cre mice could be the reduced EBF expression. Therefore, it is possible that Runx1, together with E2A, is an upstream factor for EBF even though they seem to function together once expressed at the later stages (Lukin et al. 2010; Lin et al. 2010). Further studies will be required to dissect the extent to which E2A , Runx1, and EBF cooperate and the unique functions endowed to each transcription factor.
4 Roles of Runx Complexes in Development of Innate Lymphoid Cells (ILCs) and Conventional NK Cells
4.1 Overview of ILCs
Innate lymphoid cells (ILCs) are lymphocytes that reside in the mucosa and produce innate cytokines independently of antigen specificity upon infection or allergic stimulation to maintain the epithelial barrier (Sonnenberg and Artis 2015; Eberl et al. 2015; Cortez et al. 2015). ILCs do not have rearranged antigen-specific receptors but are dependent on the common γ chain of the cytokine receptor IL-2R (γc) for their differentiation . Based on their cytokine production and transcription factor requirements, ILCs are comprised of three groups, type I ILC (ILC1 and conventional NK: cNK), type II ILC (ILC2) and type III ILC (ILC3) populations. ILC populations generally express a dimeric IL-7 receptor α chain (CD127)/γc complex with some exceptions as described below. However, IL-7 is required only for ILC2 and ILC3 but not for the type I ILC population which expresses an IL-2/IL-15R β chain (CD122)/γc complex and is dependent on IL-15 instead of IL-7 for differentiation (Sonnenberg and Artis 2015; Fuchs et al. 2013; Klose et al. 2014).
The type I ILC population expresses the transcription factor T-bet and produces the type I cytokine IFNγ in response to IL-12, IL-15, and IL-18 (Sonnenberg and Artis 2015; Fuchs et al. 2013; Klose et al. 2013, 2014). In mice, CD3− NK1.1+ NKp46 (NCR)+ cells in tissues are recognized as type I ILC populations. Given that the type I ILC population is defined by IFNγ-producing cells , cNK cells in the spleen could also be categorized as type I ILC populations. However, recent studies suggest that cNK cells are a distinct population from ILC1 cells that are resident in mucosal tissues (Sojka et al. 2014; Klose et al. 2014; Gasteiger et al. 2015; Constantinides et al. 2014). The transcription factor Eomes is expressed only in cNK cells and is required for their differentiation while ILC1 cells in mucosal tissues are negative for Eomes (Gordon et al. 2012; Daussy et al. 2014). cNK cells do not express CD127 which is one of the markers for ILC1 cells (Cella et al. 2014). Thus, we hereafter term cNK cells as the Eomes+ tissue-nonresident type I ILC population and ILC1 cells as the Eomes− tissue-resident type I ILC population.
ILC2 cells respond to IL-25, IL-33 and TSLP derived from epithelial cells and produce type II cytokines, IL-5, IL-9, and IL-13 (Sonnenberg and Artis 2015; Moro et al. 2010; Neill et al. 2010). ILC2 cells are characterized by high GATA-3 expression, which is required for their differentiation and secretion of type II cytokines (Hoyler et al. 2012). In contrast, ILC1 and ILC3 cells express GATA-3 at intermediate levels (Serafini et al. 2014; Klose et al. 2014). Thus, GATA-3 deficiency affects all ILC populations. Other transcription factors such as RORα, Gfi1, and Bcl11b are also necessary for ILC2 differentiation (Wong et al. 2012; Spooner et al. 2013; Califano et al. 2015).
ILC3 cells react to IL-1β and IL-23 from dendritic cells and produce IL-17 and IL-22 (Sonnenberg and Artis 2015; Cella et al. 2009; Satoh-Takayama et al. 2008). RORγt and AHR regulate cytokine production by ILC3 cells and their differentiation (Sawa et al. 2010, 2011; Sanos et al. 2009; Lee et al. 2012; Kiss et al. 2011). According to CD4 and NCR expression, ILC3 cells in the adult intestine were originally sub-grouped into NCR+ ILC3 (NK-22, ILC22), CD4+ ILC3 and CD4− NCR− ILC3 cells (Sonnenberg and Artis 2015). More recently, it has been reported that ILC3 cells are comprised of two distinct populations, T-bet− CCR6+ ILC3 (Lymphoid tissue inducer-like cells: LTi-like cells) and T-bet+ CCR6− ILC3, because T-bet− CCR6+ ILC3 cells do not give rise to T-bet+ CCR6− ILC3 cells, and vice versa (Klose et al. 2013). While T-bet− CCR6+ ILC3s are CD4+ or CD4−, T-bet+ CCR6− ILC3 cells are mostly NCR+. LTi cells in the fetus highly express RORγt and contribute to lymphoid tissue formation including Peyer’s patches and lymph nodes. LTi cells are also recognized as members of ILC3 cells (Sonnenberg and Artis 2015).
4.2 Developmental Pathway of ILCs
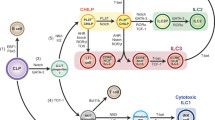
All ILC subsets develop from CLPs in the bone marrow and fetal liver (Sonnenberg and Artis 2015; Possot et al. 2011). CLPs also give rise to T- and B-lymphocytes as well as ILCs including cNK cells (Fig. 24.2). Some ILC progenitors downstream of CLPs were identified in the Lin− α4β7+ fraction of bone marrow cells. CXCR6+α4β7+ lymphoid progenitors (αLP) and early ILC progenitors (EILPs) lose the potential to become lymphocytes but maintain the capacity to differentiate into cNK, ILC1, ILC2, and ILC3 cells (Yang et al. 2015; Yu et al. 2014). EILPs are marked by the expression of the transcription factor TCF-1. Deficiency of TCF-1 leads to absence of all ILCs. Downstream of EILPs, two ILC-specific progenitor cells are designated as common helper ILC progenitors (CHILPs) expressing the transcription factor Id2, and the common precursor to ILCs (ILCPs) expressing the transcription factor PLZF (Klose et al. 2014; Constantinides et al. 2014). CHILPs give rise to ILC1, ILC2, and ILC3 cells but not cNK cells. Along with the expression of PLZF in a small proportion of CHILPs, CHILPs are supposed to be upstream of ILCPs. In line with this model, ILCPs have a limited capacity to differentiate into ILCs because LTi-like cells as well as cNK cells do not develop from ILCPs. ILC2 progenitors (ILC2Ps) are ILC2-specific progenitor cells that are downstream of ILCPs in the bone marrow. ILC1- and ILC3-specific ILC progenitor cells are ILC -lineage negative (ILCLN) cells that are characterized as Lin− CD127+ lacking ILC markers (ILC1: NK1.1, ILC2: KLRG1, ILC3: RORγt) and reside in the adult intestine. ILCLN cells were suggested to be the progenitor cells that migrate to the gut and lose their homing receptor α4β7. Apparently, ILCLN cells are likely to be a heterogeneous population because they differentiate into NCR+ ILC3 and LTi-like cells, each of which arises from a different developmental pathway (Fig. 24.2).

Runx and ILC development. All ILC populations are the progeny of common lymphoid progenitors (CLPs) in the fetal liver and bone marrow. The ILC-committed precursor cells are CXCR6+α4β7+ lymphoid progenitors (αLPs) and early ILC progenitors (EILPs) that give rise to all ILC populations, but not T or B cells. The common helper ILC progenitors (CHILPs) do not differentiate into conventional NK (cNK) cells in spleen, but maintain a capacity for other tissue-resident ILC populations. The common precursor to ILCs (ILCPs) is downstream of CHILPs and do not differentiate into LTi-like cells. ILC -lineage negative (ILCLN) cells in the intestine are the progenitor cells specific to ILC1 and type III ILC3. All ILCs are differentially characterized by the levels of P1-Runx3 transcripts. Roles of Runx3 during ILC specification are discussed in the Sect. 24.4.4
4.3 Runx Expression in ILC Progenitor Cells and ILC Subsets
Expression of Runx1, Runx2, and Runx3 is relatively low in CLPs, αLPs, and CHILPs. However, both Runx1 and Runx3 are expressed at extremely high levels in PLZF+ ILCPs (Ebihara et al. 2015). Runx3 transcript expression from the P1 promoter (P1-Runx3) is also very high in ILCPs and is correlated to PLZF expression during differentiation into CHILP, suggesting a possible cross-regulation between Runx3 and PLZF expression. However, the functions of Runx complexes in ILCPs remain to be elucidated. Given that ILC2Ps in the bone marrow and ILC2 cells in the intestine do not express Runx3 from the P1 transcripts, it is possible that downregulation of Runx3 expression is important for ILC2 differentiation. For ILC1 and ILC3 differentiation from ILCPs, P1-Runx3 transcripts are reduced to intermediate level in ILCLN cells in the adult intestine. Then, upregulation of P1-Runx3 occurs for ILC1 cells, while ILC3 cells maintain the intermediate level of it (Ebihara et al. 2015).
cNK cells develop from αLP and EILPs through NK progenitor cells (Lin− CD122+ NK1.1− DX5−). P1-Runx3 transcripts are generally low throughout cNK cell differentiation compared to those in ILC1 cells in the intestine and liver (Ebihara et al. 2015; Ohno et al. 2008; Levanon et al. 2014).
ILC subsets in the intestine and cNK cells in the spleen have been well studied regarding Runx expression and function. In the intestine, cNK cells are relatively rare whereas other ILC subsets are dominant. The intestinal intraepithelial layer is enriched with ILC1 cells. ILC1, ILC2 and ILC3 cells are evenly distributed in the intestinal lamina propria and Peyer’s patches. All ILC subsets including ILC1, ILC2 and ILC3 cells in the intestine predominantly express Runx3 transcripts (Ebihara et al. 2015). However, ILC2 cells in the intestine use the P2, but not the P1 promoter although ILC1 and ILC3 cells in the intestine express only P1-Runx3 transcripts. cNK cells express Runx3 from both promoters, but the major transcript is the P2-Runx3 (Ebihara et al. 2015; Levanon et al. 2014). When P1-Runx3 transcripts are deleted in CD8+ T cells, Runx3 protein is barely detected in αβT cells (Egawa et al. 2007; Egawa and Littman 2008) due to the inefficient activity of Kozak sequences to initiate translation for P2-Runx3 protein (Kim et al. 2015). When P1-Runx3 transcript level was examined by a reporter allele that specifically reflects Runx3 P1-promoter activity, reporter expression was found to be very high in ILC1 cells, intermediate in ILC3 cells , low in cNK cells, and undetected in ILC2 cells. Given that P1-Runx3 transcripts are correlated to Runx3 protein expression at least in T cells , differential expression pattern of P1-Runx3 mRNA should be associated with Runx3 protein expression in ILCs. However, P2-Runx3 transcripts in ILC2 cells might be translated into protein at least to some extent because high level of Runx3 protein can be detected in cNK cells when P2-Runx3 transcripts are abundant (Levanon et al. 2014).
Runx3 is also expressed in ILC1 or cNK cells in other tissues. Liver-resident NK cells turned out to be ILC1 cells in which the P1-Runx3 mRNA amount was as high as those in the intestinal ILC1 cells (Ebihara et al. 2015). Runx3 expression in skin-resident NK cells is as low as that in skin-nonresident NK cells. In the uterus, DBA+ NK cells also express Runx3 mainly from the P1 promoter (Levanon et al. 2014). Thus, among all Runx family members, Runx3 is the dominant Runx protein in all ILC subsets and is expressed highly in most type I ILC population and intermediately in ILC3 cells.
4.4 Roles of Runx Complexes in ILCs Development
4.4.1 Type I ILC
Several genetic approaches have clarified that Runx complexes are involved in cNK cell function and differentiation. Mice harboring the hypomorphic allele of Cbfβ exhibited absence of NKPs and cNK cells. When Cbfβ is deleted in hematopoietic cells with Vav1-Cre mice , LMPPs and CLPs do not emerge (Satpathy et al. 2014), suggesting the requirement of Cbfβ for early differentiation of lymphocytes before commitment to cNK cells . Recently, cNK cell-specific Cbfβ function has been examined by conditional Cbfβ gene inactivation using NCR-iCre mice. Cbfβ deletion in cNK cells leads to great reduction of NK cells in the spleen and an immature NK cell phenotype including low expression of Ly49, low DX5, and Eomes, and inefficient IFNγ production in response to IL-12 and IL-18 stimulation (Ebihara et al. 2015). Conditional deletion of Runx3 in cNK cells recapitulates the phenotypes of Cbfβ-deficiency in cNK cells. However, probably due to compensation by Runx1, the phenotypes of Runx3-deficient cNK cells are generally milder than those of Cbfβ-deficient cNK cells (Ebihara et al. 2015; Levanon et al. 2014). ChIP-seq and transcriptome analysis showed that products of Runx3-bound genes seemed to be associated with survival, proliferation, maturation, and migration of cNK cells (Levanon et al. 2014). Runx3 appears to function downstream of IL-15 signaling and contributes to cNK cell survival. However, the precise mechanism of Runx3 induction through IL-15 signaling is still unclear. Runx3 also positively regulates CD96 and Crtam, both of which are involved in cNK cell activity (Levanon et al. 2014). Thus, the Runx3/Cbfβ complex regulates cNK cell survival and functions.
ILC1 cells in the intestine and liver also require Runx3/Cbfβ complexes for their survival and IFNγ response to IL-12 (Ebihara et al. 2015). Normal levels of T-bet expression in Cbfβ-deficient ILC1 cells in the intestine suggested that T-bet might be upstream regulator for Runx3 expression in ILC1 cells as was observed in CD8+ T cells . Type I ILC populations in the skin and salivary gland were reduced in the absence of Runx3 or Cbfβ. Taken together, the Runx3/Cbfβ complex is indispensable to all type I ILC populations.
4.4.2 Type II ILC
ILC2 cells in the intestine express only the P2-Runx3 transcript. Runx3 is dispensable for ILC2 differentiation in the intestine (Ebihara et al. 2015). During the differentiation of effector CD4+ T cell subsets, GATA-3 antagonizes Runx3 through protein-protein interactions to promote TH2 skewing (Yagi et al. 2010), whereas Runx3 blocks GATA-3 activity for TH1 differentiation (Kohu et al. 2009). This balance between Runx3 and GATA-3 seems to be one of the determinant factors for ILC1 and ILC2 function and differentiation as well. Runx3 expression might overwhelm GATA3 expression in ILC1 cells and GATA-3 could suppress Runx3 in ILC2 cells. Further studies will be necessary to clarify the physiological roles of Runx complexes in ILC2s .
4.4.3 Type III ILC
An early study showed that LTi cells in the fetal gut are reduced in mice lacking P1-Runx1 transcripts or Cbfβ2 variants, resulting in a severe deficit in secondary lymphoid organ formation (Tachibana et al. 2011). LTi cells express less RORγt in the fetal gut of Cbfβ2-deficient mice. However, the counterparts of LTi cells in the adult intestine normally express RORγt in those mice, suggesting the possible association of RORγt with Runx complexes in ILC3s. Recently, LTi cells turned out to be the progeny of CLPs which require the Runx1/Cbfβ complex for differentiation (Possot et al. 2011; Sonnenberg and Artis 2015; Constantinides et al. 2014; Klose et al. 2014; Cherrier et al. 2012; Satpathy et al. 2014). Therefore, it should be considered that reduction of LTi cells in Cbfβ2-deficient mice might reflect impaired CLP differentiation. Recently, as the ILC differentiation process has become more characterized, the roles of Runx complexes in ILC3s have been revealed (Ebihara et al. 2015). Among the Runx family members, Runx3 is predominantly expressed by all ILC3 subsets at intermediate levels, which is less than in ILC1 cells and more than in cNK cells and ILC2 cells. Runx3 ablation in all hematopoietic cells showed normal differentiation from CLPs to ILCPs stages, but resulted in accumulation of ILCLN cells in the intestine, reduction of ILC1 cells, and absence of ILC3 cells in the intestine. Runx3-deficient ILCLN cells are not apoptotic and cannot give rise to ILC3 cells in vivo when transferred into alymphoid mice. Fewer ILC1 cells were also developed from Runx3-deficient ILCLN cells than Runx3-competent ILCLN cells. Thus, Runx3 is necessary for ILCLN cells to differentiate into ILC1 and ILC3 cells.
Mechanistically, Runx3 directly contributes to RORγt expression in ILC3 cells (Ebihara et al. 2015). A reporter assay using the human NK cell line exhibited that Runx3 enhances RORγt promoter activity through the Runx binding site in the RORγt promoter. In addition, Runx3 binding to the RORγt promoter in ILC3 cells was confirmed by the ChIP assay, indicating that Runx3 regulates RORγt in ILC3 cells (Ebihara et al. 2015). The aryl hydrocarbon receptor (AHR) is another ILC3-related transcription factor that was shown to regulate ILC3 differentiation and IL-22 production together with RORγt. In the absence of Runx3, expression of AHR was also undetected in ILC3 cells (Ebihara et al. 2015). AHR expression in ILC3 cells was reduced to half in mice harboring half dosage of the RORγt gene, suggesting that RORγt is also involved in the mechanisms that regulate AHR expression in ILC3 cells. Although direct binding of RORγt to the AHR promoter in ILC3 cells has not been examined yet, RORγt binds to enhancer regions in the AHR promoter of TH17 cells, counterpart αβT cells that share many features with ILC3 cells (Ebihara et al. 2015; Ciofani et al. 2012). These data support that Runx3 regulates RORγt and its downstream AHR. Collectively, Runx3 is indispensable for ILCLN cells to acquire two ILC3 transcription factors, RORγt and AHR, for final differentiation into ILC3 cells.
References
Bain, G., Maandag, E. C., Izon, D. J., Amsen, D., Kruisbeek, A. M., Weintraub, B. C., et al. (1994). E2A proteins are required for proper B cell development and initiation of immunoglobulin gene rearrangements. Cell, 79(5), 885–892.
Braun, T., & Woollard, A. (2009). RUNX factors in development: Lessons from invertebrate model systems. Blood Cells, Molecules & Diseases, 43(1), 43–48. doi:10.1016/j.bcmd.2009.05.001.
Bruno, L., Mazzarella, L., Hoogenkamp, M., Hertweck, A., Cobb, B. S., Sauer, S., et al. (2009). Runx proteins regulate Foxp3 expression. The Journal of Experimental Medicine, 206(11), 2329–2337. doi:10.1084/jem.20090226.
Califano, D., Cho, J. J., Uddin, M. N., Lorentsen, K. J., Yang, Q., Bhandoola, A., et al. (2015). Transcription factor Bcl11b controls identity and function of mature Type 2 innate lymphoid cells. Immunity, 43(2), 354–368. doi:10.1016/j.immuni.2015.07.005.
Cella, M., Fuchs, A., Vermi, W., Facchetti, F., Otero, K., Lennerz, J. K., et al. (2009). A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature, 457(7230), 722–725. doi:10.1038/nature07537.
Cella, M., Miller, H., & Song, C. (2014). Beyond NK cells: The expanding universe of innate lymphoid cells. Frontiers in Immunology, 5, 282. doi:10.3389/fimmu.2014.00282.
Cherrier, M., Sawa, S., & Eberl, G. (2012). Notch, Id2, and RORgammat sequentially orchestrate the fetal development of lymphoid tissue inducer cells. The Journal of Experimental Medicine, 209(4), 729–740. doi:10.1084/jem.20111594.
Ciofani, M., Madar, A., Galan, C., Sellars, M., Mace, K., Pauli, F., et al. (2012). A validated regulatory network for Th17 cell specification. Cell, 151(2), 289–303. doi:10.1016/j.cell.2012.09.016.
Constantinides, M. G., McDonald, B. D., Verhoef, P. A., & Bendelac, A. (2014). A committed precursor to innate lymphoid cells. Nature, 508(7496), 397–401. doi:10.1038/nature13047.
Cortez, V. S., Robinette, M. L., & Colonna, M. (2015). Innate lymphoid cells: New insights into function and development. Current Opinion in Immunology, 32, 71–77. doi:10.1016/j.coi.2015.01.004.
Daussy, C., Faure, F., Mayol, K., Viel, S., Gasteiger, G., Charrier, E., et al. (2014). T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. The Journal of Experimental Medicine, 211(3), 563–577. doi:10.1084/jem.20131560.
Decker, T., Pasca di Magliano, M., McManus, S., Sun, Q., Bonifer, C., Tagoh, H., & Busslinger, M. (2009). Stepwise activation of enhancer and promoter regions of the B cell commitment gene Pax5 in early lymphopoiesis. Immunity, 30(4), 508–520. doi:10.1016/j.immuni.2009.01.012.
Djuretic, I. M., Levanon, D., Negreanu, V., Groner, Y., Rao, A., & Ansel, K. M. (2007). Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nature Immunology, 8(2), 145–153.
Eberl, G., Colonna, M., Di Santo, J. P., & McKenzie, A. N. (2015). Innate lymphoid cells. Innate lymphoid cells: A new paradigm in immunology. Science, 348(6237), aaa6566. doi:10.1126/science.aaa6566.
Ebihara, T., Song, C., Ryu, S. H., Plougastel-Douglas, B., Yang, L., Levanon, D., et al. (2015). Runx3 specifies lineage commitment of innate lymphoid cells. Nature Immunology, 16(11), 1124–1133. doi:10.1038/ni.3272.
Egawa, T., & Littman, D. R. (2008). ThPOK acts late in specification of the helper T cell lineage and suppresses Runx-mediated commitment to the cytotoxic T cell lineage. Nature Immunology, 9(10), 1131–1139. doi:10.1038/ni.1652.
Egawa, T., Eberl, G., Taniuchi, I., Benlagha, K., Geissmann, F., Hennighausen, L., et al. (2005). Genetic evidence supporting selection of the Valpha14i NKT cell lineage from double-positive thymocyte precursors. Immunity, 22(6), 705–716. doi:10.1016/j.immuni.2005.03.011.
Egawa, T., Tillman, R. E., Naoe, Y., Taniuchi, I., & Littman, D. R. (2007). The role of the Runx transcription factors in thymocyte differentiation and in homeostasis of naive T cells. The Journal of Experimental Medicine, 204(8), 1945–1957. doi:10.1084/jem.20070133.
Ellmeier, W., & Taniuchi, I. (2014). The role of BTB-zinc finger transcription factors during T cell development and in the regulation of T cell-mediated immunity. Current Topics in Microbiology and Immunology, 381, 21–49. doi:10.1007/82_2014_374.
Ellmeier, W., Sunshine, M. J., Losos, K., Hatam, F., & Littman, D. R. (1997). An enhancer that directs lineage-specific expression of CD8 in positively selected thymocytes and mature T cells. Immunity, 7(4), 537–547.
Ellmeier, W., Sunshine, M. J., Losos, K., & Littman, D. R. (1998). Multiple developmental stage-specific enhancers regulate CD8 expression in developing thymocytes and in thymus-independent T cells. Immunity, 9(4), 485–496.
Ellmeier, W., Sawada, S., & Littman, D. R. (1999). The regulation of CD4 and CD8 coreceptor gene expression during T cell development. Annual Review of Immunology, 17, 523–554.
Erman, B., Cortes, M., Nikolajczyk, B. S., Speck, N. A., & Sen, R. (1998). ETS-core binding factor: A common composite motif in antigen receptor gene enhancers. Molecular and Cellular Biology, 18(3), 1322–1330.
Fossett, N., & Schulz, R. A. (2001). Functional conservation of hematopoietic factors in Drosophila and vertebrates. Differentiation; Research in Biological Diversity, 69(2–3), 83–90. doi:10.1046/j.1432-0436.2001.690202.x.
Fuchs, A., Vermi, W., Lee, J. S., Lonardi, S., Gilfillan, S., Newberry, R. D., et al. (2013). Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity, 38(4), 769–781. doi:10.1016/j.immuni.2013.02.010.
Gangadharan, D., Lambolez, F., Attinger, A., Wang-Zhu, Y., Sullivan, B. A., & Cheroutre, H. (2006). Identification of pre- and postselection TCRalphabeta+ intraepithelial lymphocyte precursors in the thymus. Immunity, 25(4), 631–641. doi:10.1016/j.immuni.2006.08.018.
Gasteiger, G., Fan, X., Dikiy, S., Lee, S. Y., & Rudensky, A. Y. (2015). Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science, 350(6263), 981–985. doi:10.1126/science.aac9593.
Gordon, S. M., Chaix, J., Rupp, L. J., Wu, J., Madera, S., Sun, J. C., et al. (2012). The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity, 36(1), 55–67. doi:10.1016/j.immuni.2011.11.016.
Growney, J. D., Shigematsu, H., Li, Z., Lee, B. H., Adelsperger, J., Rowan, R., et al. (2005). Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood, 106(2), 494–504. doi:10.1182/blood-2004-08-3280.
Grueter, B., Petter, M., Egawa, T., Laule-Kilian, K., Aldrian, C. J., Wuerch, A., Ludwig, Y., Fukuyama, H., Wardemann, H., Waldschuetz, R., Moroy, T., Taniuchi, I., Steimle, V., Littman, D. R., & Ehlers, M. (2005). Runx3 regulates integrin alpha E/CD103 and CD4 expression during development of CD4−/CD8+ T cells. Journal of Immunology (Baltimore, Md: 1950), 175(3), 1694–1705. doi:175/3/1694 [pii].
Harrington, L. E., Hatton, R. D., Mangan, P. R., Turner, H., Murphy, T. L., Murphy, K. M., & Weaver, C. T. (2005). Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nature Immunology, 6(11), 1123–1132. doi:10.1038/ni1254.
Hassan, H., Sakaguchi, S., Tenno, M., Kopf, A., Boucheron, N., Carpenter, A. C., Egawa, T., Taniuchi I., & Ellmeier, W. (2011). Cd8 enhancer E8I and Runx factors regulate CD8alpha expression in activated CD8+ T cells. Proceedings of the National Academy of Sciences of the United States of America, 108(45), 18330–18335. doi:1105835108 [pii] 10.1073/pnas.1105835108.
He, X., Dave, V. P., Zhang, Y., Hua, X., Nicolas, E., Xu, W., Roe, B. A., & Kappes, D. J. (2005). The zinc finger transcription factor Th-POK regulates CD4 versus CD8 T-cell lineage commitment. Nature, 433(7028), 826–833. doi:nature03338 [pii] 10.1038/nature03338.
He, X., Park, K., Wang, H., He, X., Zhang, Y., Hua, X., et al. (2008). CD4-CD8 lineage commitment is regulated by a silencer element at the ThPOK transcription-factor locus. Immunity, 28(3), 346–358.
Hernandez-Munain, C., & Krangel, M. S. (1995). c-Myb and core-binding factor/PEBP2 display functional synergy but bind independently to adjacent sites in the T-cell receptor delta enhancer. Molecular and Cellular Biology, 15(6), 3090–3099.
Hernandez-Munain, C., Roberts, J. L., & Krangel, M. S. (1998). Cooperation among multiple transcription factors is required for access to minimal T-cell receptor alpha-enhancer chromatin in vivo. Molecular and Cellular Biology, 18(6), 3223–3233.
Hollenhorst, P. C., Shah, A. A., Hopkins, C., & Graves, B. J. (2007). Genome-wide analyses reveal properties of redundant and specific promoter occupancy within the ETS gene family. Genes & Development, 21(15), 1882–1894. doi:10.1101/gad.1561707.
Hostert, A., Garefalaki, A., Mavria, G., Tolaini, M., Roderick, K., Norton, T., et al. (1998). Hierarchical interactions of control elements determine CD8alpha gene expression in subsets of thymocytes and peripheral T cells. Immunity, 9(4), 497–508.
Hoyler, T., Klose, C. S., Souabni, A., Turqueti-Neves, A., Pfeifer, D., Rawlins, E. L., et al. (2012). The transcription factor GATA-3 controls cell fate and maintenance of Type 2 innate lymphoid cells. Immunity, 37(4), 634–648. doi:10.1016/j.immuni.2012.06.020.
Hozumi, K., Mailhos, C., Negishi, N., Hirano, K., Yahata, T., Ando, K., et al. (2008). Delta-like 4 is indispensable in thymic environment specific for T cell development. Journal of Experimental Medicine, 205(11), 2507–2513. doi:10.1084/jem.20080134.
Hsiang, Y. H., Spencer, D., Wang, S., Speck, N. A., & Raulet, D. H. (1993). The role of viral enhancer “core” motif-related sequences in regulating T cell receptor-gamma and -delta gene expression. Journal of Immunology (Baltimore, Md: 1950), 150(9), 3905–3916.
Ichikawa, M., Asai, T., Saito, T., Seo, S., Yamazaki, I., Yamagata, T., et al. (2004). AML-1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nature Medicine, 10(3), 299–304. doi:10.1038/nm997.
Ikawa, T., Hirose, S., Masuda, K., Kakugawa, K., Satoh, R., Shibano-Satoh, A., et al. (2010). An essential developmental checkpoint for production of the T cell lineage. Science, 329(5987), 93–96. doi:10.1126/science.1188995.
Ivanov, I. I., McKenzie, B. S., Zhou, L., Tadokoro, C. E., Lepelley, A., Lafaille, J. J., et al. (2006). The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell, 126(6), 1121–1133. doi:10.1016/j.cell.2006.07.035.
Josefowicz, S. Z., Lu, L. F., & Rudensky, A. Y. (2012). Regulatory T cells: Mechanisms of differentiation and function. Annual Review of Immunology, 30, 531–564. doi:10.1146/annurev.immunol.25.022106.141623.
Kappes, D. J., & He, X. (2006). Role of the transcription factor Th-POK in CD4:CD8 lineage commitment. Immunological Reviews, 209, 237–252. doi:IMR344 [pii] 10.1111/j.0105-2896.2006.00344.x.
Kee, B. L., & Murre, C. (1998). Induction of early B cell factor (EBF) and multiple B lineage genes by the basic helix-loop-helix transcription factor E12. The Journal of Experimental Medicine, 188(4), 699–713.
Kim, B., Sasaki, Y., & Egawa, T. (2015). Restriction of nonpermissive RUNX3 protein expression in T lymphocytes by the Kozak sequence. Journal of Immunology, 195(4), 1517–1523. doi:10.4049/jimmunol.1501039.
Kiss, E. A., Vonarbourg, C., Kopfmann, S., Hobeika, E., Finke, D., Esser, C., & Diefenbach, A. (2011). Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science, 334(6062), 1561–1565. doi:10.1126/science.1214914.
Kitoh, A., Ono, M., Naoe, Y., Ohkura, N., Yamaguchi, T., Yaguchi, H., et al. (2009). Indispensable role of the Runx1-Cbfbeta transcription complex for in vivo-suppressive function of FoxP3+ regulatory T cells. Immunity, 31(4), 609–620.
Klose, C. S., Kiss, E. A., Schwierzeck, V., Ebert, K., Hoyler, T., d’Hargues, Y., et al. (2013). A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature, 494(7436), 261–265. doi:10.1038/nature11813.
Klose, C. S., Flach, M., Mohle, L., Rogell, L., Hoyler, T., Ebert, K., et al. (2014). Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell, 157(2), 340–356. doi:10.1016/j.cell.2014.03.030.
Klunker, S., Chong, M. M., Mantel, P. Y., Palomares, O., Bassin, C., Ziegler, M., et al. (2009). Transcription factors RUNX1 and RUNX3 in the induction and suppressive function of Foxp3+ inducible regulatory T cells. The Journal of Experimental Medicine, 206(12), 2701–2715. doi:10.1084/jem.20090596.
Koch, U., Fiorini, E., Benedito, R., Besseyrias, V., Schuster-Gossler, K., Pierres, M., et al. (2008). Delta-like 4 is the essential, nonredundant ligand for Notch1 during thymic T cell lineage commitment. The Journal of Experimental Medicine, 205(11), 2515–2523. doi:10.1084/jem.20080829.
Kohu, K., Sato, T., Ohno, S., Hayashi, K., Uchino, R., Abe, N., Nakazato, M., Yoshida, N., Kikuchi, T., Iwakura, Y., Inoue, Y., Watanabe, T., Habu, S., & Satake, M. (2005). Overexpression of the Runx3 transcription factor increases the proportion of mature thymocytes of the CD8 single-positive lineage. Journal of Immunology (Baltimore, Md: 1950), 174(5), 2627–2636. doi:174/5/2627 [pii].
Kohu, K., Ohmori, H., Wong, W. F., Onda, D., Wakoh, T., Kon, S., et al. (2009). The Runx3 transcription factor augments Th1 and down-modulates Th2 phenotypes by interacting with and attenuating GATA3. Journal of Immunology, 183(12), 7817–7824. doi:10.4049/jimmunol.0802527.
Kronenberg, M., & Rudensky, A. (2005). Regulation of immunity by self-reactive T cells. Nature, 435(7042), 598–604. doi:10.1038/nature03725.
Laiosa, C. V., Stadtfeld, M., & Graf, T. (2006). Determinants of lymphoid-myeloid lineage diversification. Annual Review of Immunology, 24, 705–738. doi:10.1146/annurev.immunol.24.021605.090742.
Lambolez, F., Kronenberg, M., & Cheroutre, H. (2007). Thymic differentiation of TCR alpha beta(+) CD8 alpha alpha(+) IELs. Immunological Reviews, 215, 178–188. doi:10.1111/j.1600-065X.2006.00488.x.
Lebestky, T., Chang, T., Hartenstein, V., & Banerjee, U. (2000). Specification of Drosophila hematopoietic lineage by conserved transcription factors. Science, 288(5463), 146–149.
Lee, J. S., Cella, M., McDonald, K. G., Garlanda, C., Kennedy, G. D., Nukaya, M., et al. (2012). AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nature Immunology, 13(2), 144–151. doi:10.1038/ni.2187.
Leishman, A. J., Naidenko, O. V., Attinger, A., Koning, F., Lena, C. J., Xiong, Y., et al. (2001). T cell responses modulated through interaction between CD8alphaalpha and the nonclassical MHC class I molecule, TL. Science, 294(5548), 1936–1939. doi:10.1126/science.1063564.
Leung, R. K., Thomson, K., Gallimore, A., Jones, E., Van den Broek, M., Sierro, S., et al. (2001). Deletion of the CD4 silencer element supports a stochastic mechanism of thymocyte lineage commitment. Nature Immunology, 2(12), 1167–1173.
Levanon, D., Negreanu, V., Lotem, J., Bone, K. R., Brenner, O., Leshkowitz, D., & Groner, Y. (2014). Transcription factor Runx3 regulates interleukin-15-dependent natural killer cell activation. Molecular and Cellular Biology, 34(6), 1158–1169. doi:10.1128/MCB.01202-13.
Li, L., Leid, M., & Rothenberg, E. V. (2010). An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science, 329(5987), 89–93. doi:10.1126/science.1188989.
Li, L., Zhang, J. A., Dose, M., Kueh, H. Y., Mosadeghi, R., Gounari, F., & Rothenberg, E. V. (2013). A far downstream enhancer for murine Bcl11b controls its T-cell specific expression. Blood, 122(6), 902–911. doi:10.1182/blood-2012-08-447839.
Lin, H., & Grosschedl, R. (1995). Failure of B-cell differentiation in mice lacking the transcription factor EBF. Nature, 376(6537), 263–267. doi:10.1038/376263a0.
Lin, Y. C., Jhunjhunwala, S., Benner, C., Heinz, S., Welinder, E., Mansson, R., et al. (2010). A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nature Immunology, 11(7), 635–643. doi:10.1038/ni.1891.
Luckey, M. A., Kimura, M. Y., Waickman, A. T., Feigenbaum, L., Singer, A., & Park, J. H. (2014). The transcription factor ThPOK suppresses Runx3 and imposes CD4(+) lineage fate by inducing the SOCS suppressors of cytokine signaling. Nature Immunology, 15(7), 638–645. doi:10.1038/ni.2917.
Lukin, K., Fields, S., Lopez, D., Cherrier, M., Ternyak, K., Ramirez, J., et al. (2010). Compound haploinsufficiencies of Ebf1 and Runx1 genes impede B cell lineage progression. Proceedings of the National Academy of Sciences of the United States of America, 107(17), 7869–7874. doi:10.1073/pnas.1003525107.
Maier, H., Ostraat, R., Gao, H., Fields, S., Shinton, S. A., Medina, K. L., et al. (2004). Early B cell factor cooperates with Runx1 and mediates epigenetic changes associated with mb-1 transcription. Nature Immunology, 5(10), 1069–1077. doi:10.1038/ni1119.
Majumder, K., Bassing, C. H., & Oltz, E. M. (2015). Regulation of Tcrb gene assembly by genetic, epigenetic, and topological mechanisms. Advances in Immunology, 128, 273–306. doi:10.1016/bs.ai.2015.07.001.
Matthias, P., & Rolink, A. G. (2005). Transcriptional networks in developing and mature B cells. Nature Reviews Immunology, 5(6), 497–508. doi:10.1038/nri1633.
Mikkola, I., Heavey, B., Horcher, M., & Busslinger, M. (2002). Reversion of B cell commitment upon loss of Pax5 expression. Science, 297(5578), 110–113. doi:10.1126/science.1067518.
Moro, K., Yamada, T., Tanabe, M., Takeuchi, T., Ikawa, T., Kawamoto, H., et al. (2010). Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature, 463(7280), 540–544. doi:10.1038/nature08636.
Nakayamada, S., Takahashi, H., Kanno, Y., & O’Shea, J. J. (2012). Helper T cell diversity and plasticity. Current Opinion in Immunology, 24(3), 297–302. doi:10.1016/j.coi.2012.01.014.
Naoe, Y., Setoguchi, R., Akiyama, K., Muroi, S., Kuroda, M., Hatam, F., et al. (2007). Repression of interleukin-4 in T helper type 1 cells by Runx/Cbf beta binding to the Il4 silencer. The Journal of Experimental Medicine, 204(8), 1749–1755.
Neill, D. R., Wong, S. H., Bellosi, A., Flynn, R. J., Daly, M., Langford, T. K., et al. (2010). Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature, 464(7293), 1367–1370. doi:10.1038/nature08900.
Nutt, S. L., Heavey, B., Rolink, A. G., & Busslinger, M. (1999). Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature, 401(6753), 556–562. doi:10.1038/44076.
Oestreich, K. J., Cobb, R. M., Pierce, S., Chen, J., Ferrier, P., & Oltz, E. M. (2006). Regulation of TCRbeta gene assembly by a promoter/enhancer holocomplex. Immunity, 24(4), 381–391. doi:10.1016/j.immuni.2006.02.009.
Ohkura, N., Kitagawa, Y., & Sakaguchi, S. (2013). Development and maintenance of regulatory T cells. Immunity, 38(3), 414–423. doi:10.1016/j.immuni.2013.03.002.
Ohno, S., Sato, T., Kohu, K., Takeda, K., Okumura, K., Satake, M., & Habu, S. (2008). Runx proteins are involved in regulation of CD122, Ly49 family and IFN-gamma expression during NK cell differentiation. International Immunology, 20(1), 71–79. doi:10.1093/intimm/dxm120.
Okuda, T., van Deursen, J., Hiebert, S. W., Grosveld, G., & Downing, J. R. (1996). AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell, 84(2), 321–330.
Ono, M., Yaguchi, H., Ohkura, N., Kitabayashi, I., Nagamura, Y., Nomura, T., et al. (2007). Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature, 446(7136), 685–689. doi:10.1038/nature05673.
Park, H., Li, Z., Yang, X. O., Chang, S. H., Nurieva, R., Wang, Y. H., et al. (2005). A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nature Immunology, 6(11), 1133–1141. doi:10.1038/ni1261.
Park, J. H., Adoro, S., Guinter, T., Erman, B., Alag, A. S., Catalfamo, M., Kimura, M. Y., Cui, Y., Lucas, P. J., Gress, R. E., Kubo, M., Hennighausen, L., Feigenbaum, L., & Singer, A. (2010). Signaling by intrathymic cytokines, not T cell antigen receptors, specifies CD8 lineage choice and promotes the differentiation of cytotoxic-lineage T cells. Nature Immunology, 11(3), 257–264. doi:ni.1840 [pii] 10.1038/ni.1840.
Pobezinsky, L. A., Angelov, G. S., Tai, X., Jeurling, S., Van Laethem, F., Feigenbaum, L., et al. (2012). Clonal deletion and the fate of autoreactive thymocytes that survive negative selection. Nature Immunology, 13(6), 569–578. doi:10.1038/ni.2292.
Porritt, H. E., Rumfelt, L. L., Tabrizifard, S., Schmitt, T. M., Zuniga-Pflucker, J. C., & Petrie, H. T. (2004). Heterogeneity among DN1 prothymocytes reveals multiple progenitors with different capacities to generate T cell and non-T cell lineages. Immunity, 20(6), 735–745. doi:10.1016/j.immuni.2004.05.004.
Possot, C., Schmutz, S., Chea, S., Boucontet, L., Louise, A., Cumano, A., & Golub, R. (2011). Notch signaling is necessary for adult, but not fetal, development of RORgammat(+) innate lymphoid cells. Nature Immunology, 12(10), 949–958. doi:10.1038/ni.2105.
Redondo, J. M., Pfohl, J. L., & Krangel, M. S. (1991). Identification of an essential site for transcriptional activation within the human T-cell receptor delta enhancer. Molecular and Cellular Biology, 11(11), 5671–5680.
Rolink, A. G., Nutt, S. L., Melchers, F., & Busslinger, M. (1999). Long-term in vivo reconstitution of T-cell development by Pax5-deficient B-cell progenitors. Nature, 401(6753), 603–606. doi:10.1038/44164.
Rothenberg, E. V., Moore, J. E., & Yui, M. A. (2008). Launching the T-cell-lineage developmental programme. Nature Reviews Immunology, 8(1), 9–21. doi:10.1038/nri2232.
Rudra, D., Egawa, T., Chong, M. M., Treuting, P., Littman, D. R., & Rudensky, A. Y. (2009). Runx-CBFbeta complexes control expression of the transcription factor Foxp3 in regulatory T cells. Nature Immunology, 10(11), 1170–1177. doi:10.1038/ni.1795.
Sakaguchi, S., Hombauer, M., Hassan, H., Tanaka, H., Yasmin, N., Naoe, Y., et al. (2015). A novel Cd8-cis-regulatory element preferentially directs expression in CD44hiCD62L+ CD8+ T cells and in CD8alphaalpha+ dendritic cells. Journal of Leukocyte Biology, 97(4), 635–644. doi:10.1189/jlb.1HI1113-597RR.
Sanos, S. L., Bui, V. L., Mortha, A., Oberle, K., Heners, C., Johner, C., & Diefenbach, A. (2009). RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nature Immunology, 10(1), 83–91. doi:10.1038/ni.1684.
Sato, T., Ohno, S., Hayashi, T., Sato, C., Kohu, K., Satake, M., & Habu, S. (2005). Dual functions of Runx proteins for reactivating CD8 and silencing CD4 at the commitment process into CD8 thymocytes. Immunity, 22(3), 317–328.
Satoh-Takayama, N., Vosshenrich, C. A., Lesjean-Pottier, S., Sawa, S., Lochner, M., Rattis, F., et al. (2008). Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity, 29(6), 958–970. doi:10.1016/j.immuni.2008.11.001.
Satpathy, A. T., Briseno, C. G., Cai, X., Michael, D. G., Chou, C., Hsiung, S., et al. (2014). Runx1 and Cbfbeta regulate the development of Flt3+ dendritic cell progenitors and restrict myeloproliferative disorder. Blood, 123(19), 2968–2977. doi:10.1182/blood-2013-11-539643.
Sawa, S., Cherrier, M., Lochner, M., Satoh-Takayama, N., Fehling, H. J., Langa, F., et al. (2010). Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science, 330(6004), 665–669. doi:10.1126/science.1194597.
Sawa, S., Lochner, M., Satoh-Takayama, N., Dulauroy, S., Berard, M., Kleinschek, M., et al. (2011). RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nature Immunology, 12(4), 320–326. doi:10.1038/ni.2002.
Sawada, S., Scarborough, J. D., Killeen, N., & Littman, D. R. (1994). A lineage-specific transcriptional silencer regulates CD4 gene expression during T lymphocyte development. Cell, 77(6), 917–929.
Seo, W., Ikawa, T., Kawamoto, H., & Taniuchi, I. (2012a). Runx1-Cbfbeta facilitates early B lymphocyte development by regulating expression of Ebf1. The Journal of Experimental Medicine, 209(7), 1255–1262. doi:10.1084/jem.20112745.
Seo, W., Tanaka, H., Miyamoto, C., Levanon, D., Groner, Y., & Taniuchi, I. (2012b). Roles of VWRPY motif-mediated gene repression by Runx proteins during T-cell development. Immunology and Cell Biology, 90(8), 827–830. doi:10.1038/icb.2012.6 icb20126 [pii].
Serafini, N., Klein Wolterink, R. G., Satoh-Takayama, N., Xu, W., Vosshenrich, C. A., Hendriks, R. W., & Di Santo, J. P. (2014). Gata3 drives development of RORgammat+ group 3 innate lymphoid cells. The Journal of Experimental Medicine, 211(2), 199–208. doi:10.1084/jem.20131038.
Setoguchi, R., Tachibana, M., Naoe, Y., Muroi, S., Akiyama, K., Tezuka, C., Okuda, T., & Taniuchi, I. (2008). Repression of the transcription factor Th-POK by Runx complexes in cytotoxic T cell development. Science, 319(5864), 822–825. doi:319/5864/822 [pii] 10.1126/science.1151844.
Siu, G., Wurster, A. L., Duncan, D. D., Soliman, T. M., & Hedrick, S. M. (1994). A transcriptional silencer controls the developmental expression of the CD4 gene. The EMBO Journal, 13(15), 3570–3579.
Sojka, D. K., Plougastel-Douglas, B., Yang, L., Pak-Wittel, M. A., Artyomov, M. N., Ivanova, Y., et al. (2014). Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. eLife, 3, e01659. doi:10.7554/eLife.01659.
Sonnenberg, G. F., & Artis, D. (2015). Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nature Medicine, 21(7), 698–708. doi:10.1038/nm.3892.
Spooner, C. J., Lesch, J., Yan, D., Khan, A. A., Abbas, A., Ramirez-Carrozzi, V., et al. (2013). Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nature Immunology, 14(12), 1229–1236. doi:10.1038/ni.2743.
Tachibana, M., Tenno, M., Tezuka, C., Sugiyama, M., Yoshida, H., & Taniuchi, I. (2011). Runx1/Cbfbeta2 complexes are required for lymphoid tissue inducer cell differentiation at two developmental stages. Journal of Immunology, 186(3), 1450–1457. doi:10.4049/jimmunol.1000162.
Takeda, J., Cheng, A., Mauxion, F., Nelson, C. A., Newberry, R. D., Sha, W. C., et al. (1990). Functional analysis of the murine T-cell receptor beta enhancer and characteristics of its DNA-binding proteins. Molecular and Cellular Biology, 10(10), 5027–5035.
Tani-Ichi, S., Satake, M., & Ikuta, K. (2011). The pre-TCR signal induces transcriptional silencing of the TCRgamma locus by reducing the recruitment of STAT5 and Runx to transcriptional enhancers. International Immunology, 23(9), 553–563. doi:10.1093/intimm/dxr055.
Taniuchi, I., Osato, M., Egawa, T., Sunshine, M. J., Bae, S. C., Komori, T., et al. (2002). Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell, 111(5), 621–633.
Wang, Q., Stacy, T., Binder, M., Marin-Padilla, M., Sharpe, A. H., & Speck, N. A. (1996a). Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proceedings of the National Academy of Sciences of the United States of America, 93(8), 3444–3449.
Wang, Q., Stacy, T., Miller, J. D., Lewis, A. F., Gu, T. L., Huang, X., et al. (1996b). The CBFbeta subunit is essential for CBFalpha2 (AML1) function in vivo. Cell, 87(4), 697–708.
Wong, S. H., Walker, J. A., Jolin, H. E., Drynan, L. F., Hams, E., Camelo, A., et al. (2012). Transcription factor RORalpha is critical for nuocyte development. Nature Immunology, 13(3), 229–236. doi:10.1038/ni.2208.
Woolf, E., Brenner, O., Goldenberg, D., Levanon, D., & Groner, Y. (2007). Runx3 regulates dendritic epidermal T cell development. Developmental Biology, 303(2), 703–714. doi:10.1016/j.ydbio.2006.12.005.
Wotton, D., Ghysdael, J., Wang, S., Speck, N. A., & Owen, M. J. (1994). Cooperative binding of Ets-1 and core binding factor to DNA. Molecular and Cellular Biology, 14(1), 840–850.
Yagi, R., Junttila, I. S., Wei, G., Urban Jr., J. F., Zhao, K., Paul, W. E., & Zhu, J. (2010). The transcription factor GATA3 actively represses RUNX3 protein-regulated production of interferon-gamma. Immunity, 32(4), 507–517. doi:10.1016/j.immuni.2010.04.004.
Yang, Q., Li, F., Harly, C., Xing, S., Ye, L., Xia, X., et al. (2015). TCF-1 upregulation identifies early innate lymphoid progenitors in the bone marrow. Nature Immunology, 16(10), 1044–1050. doi:10.1038/ni.3248.
Yu, X., Wang, Y., Deng, M., Li, Y., Ruhn, K. A., Zhang, C. C., & Hooper, L. V. (2014). The basic leucine zipper transcription factor NFIL3 directs the development of a common innate lymphoid cell precursor. eLife, 3. doi:10.7554/eLife.04406.
Zhang, F., Meng, G., & Strober, W. (2008). Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nature Immunology, 9(11), 1297–1306. doi:10.1038/ni.1663.
Zheng, Y., Josefowicz, S., Chaudhry, A., Peng, X. P., Forbush, K., & Rudensky, A. Y. (2010). Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature, 463(7282), 808–812. doi:10.1038/nature08750.
Zhuang, Y., Soriano, P., & Weintraub, H. (1994). The helix-loop-helix gene E2A is required for B cell formation. Cell, 79(5), 875–884.
Zou, Y. R., Sunshine, M. J., Taniuchi, I., Hatam, F., Killeen, N., & Littman, D. R. (2001). Epigenetic silencing of CD4 in T cells committed to the cytotoxic lineage. Nature Genetics, 29(3), 332–336. doi:10.1038/ng750.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Ebihara, T., Seo, W., Taniuchi, I. (2017). Roles of RUNX Complexes in Immune Cell Development. In: Groner, Y., Ito, Y., Liu, P., Neil, J., Speck, N., van Wijnen, A. (eds) RUNX Proteins in Development and Cancer. Advances in Experimental Medicine and Biology, vol 962. Springer, Singapore. https://doi.org/10.1007/978-981-10-3233-2_24
Download citation
DOI: https://doi.org/10.1007/978-981-10-3233-2_24
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-3231-8
Online ISBN: 978-981-10-3233-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)