
Abstract
CD4+ T helper cells regulate appropriate cellular and humoral immune responses to a wide range of pathogens and get involved in many diseases progress. The balance of the earliest determined CD4+ T helper cell subsets, Th1 and Th2, play an important role in allergy and autoimmune diseases. During the research, Animal models in immunology research are necessary and always the powerful tools for the basic scientific research. With the new sequence technologies, the finding of key gene mutation in Th1/Th2 cells has been proved to be related to human diseases. Here, we review four animal models about four key genes in Th1/Th2 cells to introduce the balance between Th1/Th2 cells. Furthermore, the related genetic mutations in human diseases and the new therapies are reviewed in this chapter, which show the importance of Th1/Th2 cells in human diseases further.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
3.1 Introduction
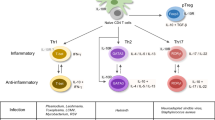
Th1/Th2 cells are stringently regulated as aberrant cell activity is involved in immunopathologies, such as allergic responses, immunodeficiencies, and lymphomas. The elucidation of the mechanisms that regulate Th1/Th2 cell differentiation, function, and fate should highlight targets for novel therapeutics. With gene-deficient mouse models, it is convenient to define the specific functions of the key genes in Th1/Th2 cell. Indeed, Tbx21-knockout mice exhibit more severe disease after virus infection, and get asthma-like phenotype independent of allergen exposure. In addition, conditional deletion of Gata3 in T cell results in impairment of both Il4-dependent and -independent Th2 differentiation, permitting Th1 differentiation in the absence of Ifng and Il12.
One of the most convincing lines of evidence establishing the importance of Th1/Th2 differentiation process is the existence of monogenic abnormalities in particular differentiation processes or effector functions resulting in disease. A growing set of human disorders in the Th differentiation/function pathways has been identified, which establish the importance of these differentiation processes in humans. In this chapter, we will summarize several key gene mutations in Th1/Th2 differentiation process, including master regulators, STAT family members, and other important transcription factors.
3.2 The Balance Between Th1/Th2 Cells in Allergy and Autoimmune Disease Models
The precise reaction of adaptive immune system protects hosts from many pathogenic and abnormal antigen infections, under the regulation of a very specific way. Animal models in immunology research are necessary, and always determine the level of the work.
3.2.1 Animal Model of T-bet
Finotto et al. [23] generated mice lacking the T-bet gene by targeted disruption. Heterozygous or homozygous knockout mice exhibited airway hyperresponsiveness in response to methacholine. T-bet homozygous knockout mice showed peribronchial and perivenular infiltration with eosinophils and lymphocytes compared with wild-type littermates. T-bet heterozygotes, which display only 50 % reduction of T-bet protein expression, displayed a phenotype very similar to that of mice with a complete absence of T-bet. T-bet heterozygotes and homozygote knockout mice also demonstrated thickening of the airway subbasement membrane collagen layer as well as increased expression of some cytokines. However, the asthma-like phenotype in these mice was independent of allergen exposure, and was not altered by allergen exposure. Finotto et al. concluded that mice with a targeted deletion of the T-bet gene and severe combined immunodeficient mice receiving CD4(+) cells from T-bet knockout mice spontaneously demonstrated multiple physiologic and inflammatory features characteristic of asthma.
By replacing the C terminus of T-bet, which contains the trans-activation domain, with the repression domain of the Drosophila “engrailed” protein, Mullen et al. [48] generated a dominant-negative (DN) T-bet cDNA. Introduction of DN T-bet into cells developing under Th1-inducing conditions, but not into mature Th1 cells, and substantially inhibited their capacity to express Ifng, and resulted in defective persistence of hypersensitivity site I chromatin within Ifng. Levels of Il12rb2, however, were reduced in both developing and mature Th1 cells by DN T-bet. Screening for homeobox factors expressed in Th1 cells by oligonucleotide array and RT-PCR analysis determined that only Hlx (142995) is expressed at higher levels in Th1 lymphocytes than in Th2 lymphocytes. Hlx appeared at a slower rate than T-bet, and could be induced by T-bet. Ectopic expression in Stat4 −/− Th2-like cells of both T-bet and Hlx allowed maximal and synergistic expression of Ifng in these cells. Introduction of DN T-bet into mature Th1 cells inhibited the expression of Hlx.
Svensson et al. [70] noted that both Il15 −/− mice, which lack NK and NKT cells, and Ifng −/− mice are highly susceptible to genital herpes simplex virus (HSV)-2 infection. CD4-positive T cells and IFN-γ are the most important components of acquired immunity to genital HSV-2 infection, and impaired HSV-2-specific IFN-γ responses in humans correlate with recurrent clinical disease. Svensson et al. vaginally infected T-bet −/− mice with HSV-2. T-bet −/− mice had increased vaginal and spinal cord viral titers, more severe disease, shorter time to death, reduced NK-cell activity, impaired Ifng production, lower specific antibody production, and fewer splenic B cells compared with wild-type mice. These differences were even more marked in mice, first vaccinated with an attenuated HSV-2 strain, and then challenged with virulent HSV-2. However, CD8-positive T cell-mediated cytotoxicity was actually stronger in T-bet −/− mice compared with wild-type mice. Svensson et al. concluded that T-bet is important in both innate defense and for generation of protective Th1 immunity against genital HSV-2 infection, primarily through its control of NK and CD4-positive T-cell function.
Ravindran et al. noted that T-bet-deficient mice resolve infection with the intracellular pathogen Listeria monocytogenes despite having only small numbers of Cd4-positive Ifng-producing T cells. In contrast, they found that challenge of T-bet −/− mice with an attenuated Salmonella strain resulted in death of most mice in less than a month. T-bet −/− mice failed to produce IFN-γ and to switch immunoglobulin isotypes. Spleen cells of infected T-bet −/− mice did not produce IFN-γ, but they did secrete increased levels of IL-10, but not IL-4. Ravindran et al. [61] concluded that CD4-positive T cells expressing T-bet are required for development of Salmonella-specific Th1 cells, regulation of IL-10 production, and resistance to Salmonella infection.
By using immunohistochemical analysis, Wang et al. found expression of T-bet in inflammatory infiltrates of human synovial tissue from patients with rheumatoid arthritis (RA). T-bet −/− mice with the collagen antibody-induced arthritis model of RA had markedly reduced joint inflammation at both early and late time points. Mice lacking both Rag2 and T-bet were resistant to disease. However, adoptive transfer of dendritic cells expressing T-bet reconstituted inflammation in both T-bet −/− and T-bet −/− Rag2 −/− mice. Wang et al. [77] concluded that T-bet has a vital role in DCs that links innate and adaptive immunity to regulate inflammatory responses.
3.2.2 Animal Model of GATA3
GATA3 was initially found with development functions. Lim et al. found that null mutations of Gata3 in mice led to a reduced accumulation of tyrosine hydroxylase (TH) and dopamine beta-hydroxylase (DPH) mRNA, whereas several other sympathetic nervous system (SNS) genes were unaffected. They showed that Th and Dbh deficiencies led to reduced noradrenalin in the SNS, and that noradrenaline deficiency was the proximal cause of death in mutants by feeding catechol intermediates to pregnant dams, thereby partially averting Gata3 mutation-induced lethality. The older, pharmacologically rescued mutants showed abnormalities that could not be detected in untreated mutants. These late embryonic defects included renal hypoplasia and developmental defects in structures derived from cephalic neural crest cells. Thus, Lim et al. [43] showed that Gata3 has a role in the differentiation of multiple cell lineages during embryogenesis.
To elucidate GATA3 function, Pandolfi et al. disrupted the mouse gene by homologous recombination in embryonic stem cells. Mice heterozygous for the Gata3 mutation were found to be fertile and appeared in all respects to be normal, whereas homozygous mutant embryos died between days 11 and 12 postcoitum and displayed massive internal bleeding, marked growth retardation, severe deformities of the brain and spinal cord, and gross aberrations in fetal liver hematopoiesis. The functions of GATA1 and GATA2 had previously been studied by comparable methods. The results in aggregate demonstrated that each GATA-binding protein has a unique and essential function during the development of the mouse embryo. In each case, targeted mutagenesis also revealed surprising roles for each factor, underscoring the power of this experimental approach: GATA1 is essential for erythroid cell development, while disruption of GATA2 indicates a function during very early events in the development of all blood cell lineages [55].
Using microarray analysis, Kaufman et al. identified Gata3 as an induced transcription factor in embryonic day-13 to -18.5 mouse skin. Whole-mount in situ hybridization analysis revealed Gata3 expression in early vibrissae follicles, and later in developing epidermis and in the cone of presumptive inner root sheath (IRS) precursor cells within hair follicles. Examination of pharmacologically rescued Gata3 −/− embryos and grafted Gata3 −/− skin showed aberrations in hair follicle morphogenesis that included not only structural defects in the IRS and hair shaft, but also molecular defects in cell lineage determination. Kaufman et al. [32] concluded that, along with LEF1 and WNTs, GATA3 is at the crossroads of both lymphocyte differentiation and of the IRS versus hair shaft cell fate decision in hair follicle morphogenesis.
Pai et al. [54] generated mice conditionally lacking Gata3 at early (double-negative) and late (double-positive) stages of thymic differentiation. They found that Gata3 was indispensable for thymocytes to pass through beta selection, the process by which T-cell receptor-beta is paired with pre-T-cell receptor-alpha, a requirement for double-negative stage-3 cell survival. Furthermore, Gata3 was required for single-positive Cd4 thymocyte development. Pai et al. concluded that continued expression of GATA3 is required at multiple stages of thymocyte differentiation.
Zhu et al. generated mice with a conditional deletion of Gata3 and Gata3-deficient mouse T-cell lines and found that both Il4-dependent and -independent Th2 differentiation was diminished, permitting Th1 differentiation in the absence of Ifng and Il12. Deletion of Gata3 from established Th2 cells abolished production of Il5 and Il13, but not of Il4. Mice lacking Gata3 produced Ifng rather than Th2 cytokines in response to infection with Nippostrongylus brasiliensis. Zhu et al. [85] concluded that Gata3 serves as a principal switch in determining Th1-Th2 responses.
Van der Wees et al. analyzed auditory brainstem response thresholds in heterozygous Gata3-knockout mice from 1 to 19 months of age and demonstrated a hearing loss of 30 dB compared to wild-type littermates. No physiologic or morphologic abnormalities were found in the brainstem, cerebral cortex, or the outer or middle ear. However, the cochleae of Gata3 +/− mice showed significant progressive morphologic degeneration starting with the outer hair cells at the apex and ultimately involving all hair cells and supporting cells in the entire cochlea. Van der Wees et al. concluded that hearing loss following GATA3 haploinsufficiency is peripheral in nature and that this defect is detectable from early postnatal development and continues through adulthood [2].
Kouros-Mehr et al. found that Gata3 was the most highly enriched transcription factor in mammary epithelium of pubertal mice. Conditional deletion of Gata3 led to severe defects in mammary development due to failure in terminal end bud formation during puberty. After acute Gata3 loss, adult mice exhibited undifferentiated luminal cell expansion with basement-membrane detachment, which led to caspase-mediated cell death [38].
3.2.3 Animal Model of STAT6
Kuperman et al. developed mice conditionally expressing STAT6 only in the lung epithelium and demonstrated that these mice were protected from all pulmonary effects of IL13, a critical mediator of allergic asthma [15]. Reconstitution of STAT6 only in epithelial cells was sufficient for IL13-induced airway hyperreactivity and mucus production in the absence of inflammation, fibrosis, or other lung pathology [40].
Bour-Jordan et al. [6] showed that T cells from double-knockout mice deficient in Ctla4 and Stat6 were skewed toward a Th2 phenotype in vitro and in vivo by bypassing the need for Stat6. Instead, induction of Gata3 occurred in vitro and Cd4-positive cells migrated to peripheral tissues in vivo. In addition, T-cell receptor cross-linking induced a relative increase of Nfatc1 versus Nfatc2 nuclear translocation and enhanced NF-KB activation compared with Stat6 −/− T cells. Bour-Jordan et al. proposed that CTLA4 regulates T-cell differentiation by controlling the overall strength of the T-cell activation signal, bypassing the cytokine dependency of Th2 differentiation.
Wang et al. noted that BALB/c mice are prone to develop Th2 rather than Th1 responses to antigen and are resistant to experimental myasthenia gravis. However, they found that after immunization with muscle acetylcholine receptor (AChR), BALB/c mice lacking Stat6 were susceptible to EMG and developed more anti-AChR antibodies and complement-fixing anti-AChR antibodies than wild-type or Stat4 −/− mice. Stat6 −/− mouse Cd4-positive T cells proliferated to AChR in a manner comparable to wild-type and Stat4 −/− mice, but Stat6 −/− mice had abundant AChR-specific Ifng-producing Th1 cells that were nearly absent in wild-type and Stat4 −/− mice. Wang et al. [78] concluded that anti-AChR Th1 cells are important in MG pathogenesis.
Chen et al. reported that mice lacking Stat6 were susceptible to virus infection. They found viruses or cytoplasmic nucleic acids trigger STING (also named MITA/ERIS) to recruit STAT6 to the endoplasmic reticulum, leading to STAT6 phosphorylation on Ser(407) by TBK1 and Tyr(641), independent of JAKs. Phosphorylated STAT6 then dimerizes and translocates to the nucleus to induce specific target genes responsible for immune cell homing. Virus-induced STAT6 activation is detected in all cell-types tested, in contrast to the cell-type specific role of STAT6 in cytokine signaling, and Stat6(−/−) mice are susceptible to virus infection. Thus, STAT6 mediates immune signaling in response to both cytokines at the plasma membrane, and virus infection at the endoplasmic reticulum [9].
Rosen et al. investigated the role of Stat6 in oxazolone colitis, a murine model of ulcerative colitis. Colitic wildtype mice had increased Stat6 phosphorylation in epithelial cells, T cells, macrophages, and NKT cells. Mice lacking Stat6 had reduced colitis and decreased induction of the pore-forming tight junction protein Cldn2. Likewise, STAT6 knockdown in human colon epithelial cells reduced CLDN2 induction. Wild-type mice, but not Stat6 −/− mice, had increased mRNA expression of the Th2-inducing cytokines Il33 and thymic stromal lymphopoietin (TSLP). Mesenteric lymph node (MLN) cells from Stat6 −/− mice with colitis exhibited reduced secretion of Il4, Il5, Il13, and Ifng. Il33 augmented secretion of Il5, Il6, Il13, and IFNg from both wild-type and Stat6 −/− MLN cells. Rosen et al. [65] concluded that STAT6 is involved in the pathogenesis of ulcerative colitis and has important roles in altering epithelial barrier function and regulating Th2-inducing cytokine production.
3.2.4 Animal Model of c-Maf
Kim et al. [33, 34] demonstrated that the homozygous null mutant Maf mouse embryo exhibits defective lens formation and microphthalmia.
Ring et al. [63] found that Maf −/− mouse embryos exhibited a slightly foreshortened head and abnormal lens development, and that nearly all died within a few hours of birth. The one surviving animal exhibited microphthalmia, followed by cutaneous closure of the ocular chamber. Fiber cell differentiation and elongation ceased by embryonic day 12.5 in Maf −/− lens, with persistence of a hollow lens vesicle and absence of alpha-crystallin expression. Cells at the equatorial zone of the lens withdrew from the cell cycle and began to express fiber cell-specific proteins, but they failed to elongate and differentiate normally. After embryonic day 16.5, the mutant vesicle became progressively deformed. Ring et al. identified functional MAF-binding sites in the promoter regions of mouse alpha-A-crystallin (CRYAA), mouse beta-B2 crystallin (CRYBB2), and human beta-A4-crystallin (CRYBA4). They concluded that Maf deficiency causes a profound defect in early maturation of primary and secondary lens fiber cells.
Lyon et al. [45] reported a mouse mutant that, in the heterozygous state, exhibits mild pulverulent cataract named “opaque flecks in lens” (Ofl). The mutant was shown to be allelic with a knockout of Maf. Homozygotes for Ofl and for Maf null mutations were similar except for the addition of renal tubular nephritis in surviving Ofl homozygotes. Sequencing identified the mutation as a 1803G-A transition, leading to an arg291-to-gln (R291Q) substitution in the basic region of the DNA-binding domain. Since mice heterozygous for Maf knockouts showed no cataracts, the authors suggested that the Ofl R291Q mutant protein may have a dominant effect. The mutation also resulted in a selective alteration in DNA binding affinities to target oligonucleotides containing variations in core CRE and TRE elements. The authors hypothesized that arginine-291 may be important for core element binding and suggested that the mutant protein may exert a differential downstream effect among its binding targets.
By ethylnitrosourea (ENU) mutagenesis, Perveen et al. [57] identified a semidominant mouse c-Maf mutation, resulting in an asp90-to-val (D90V) substitution at a highly conserved residue within the N-terminal minimal transactivation domain (MTD). The phenotype of D90V homozygotes was isolated cataract. Functional analysis revealed that the D90V mutation results in increased promoter activation and enhances p300 recruitment in a cell type-dependent manner. Perveen et al. observed similar enhancement of p300 interaction with the S50T mutation in the MTD of the NRL gene, which suggests a common mechanism of action.
Wende et al. [80] found that conditional knockout of Maf in mouse DRG cells disrupted the architecture and function of several rapidly adapting mechanoreceptor subtypes. Pacinian corpuscles, specialized to detect high-frequency vibrations, were severely atrophied, with loss of innervating axons. In vitro skin-saphenous nerve preparations of Maf-knockout mice revealed abnormal fire response to mechanical stimuli.
3.3 Human Disease Reports and New Therapies Regarding Th1/Th2 Cells
Because of the significant role in Th1 and Th2 cells in adaptive immune system, patients with genetic mutation with the key genes in Th1 and Th2 development or mutations of molecules in APC which will case impair activation signal transduction leads to serious hereditary disease [53]. Early in 1952, tyrosine kinase mutation was found in agammaglobulinemia patients, which is the first reported human gene mutation [7]. And with more and more discoveries in both basic immunology research and clinical disease research, the knowledge of these two fields reconfirmed each other’s conclusion and implies each other’s inspiration [8, 17].
The finding of key gene mutation and the relation with human disease develop with both knowledge growth in medical and immunology field, and most importantly, with the new sequence technologies. We are at the eve of affordable full sequence assay of individuals, so the main finding in sequence still need the hints from classic immunology research, to emphases on the target genes. The first step of target genes narrow down is identify the abnormal gene and the frequency of this mutation, with the help of the database of human genome. Lower than 1 % of frequency is considered significant, and among these genes, the mutation related to immunology functions is the most worthy focused on (Table 3.1). In other conditions, if the full sequence is not available or affordable, we need to stick on the suspected genes, in this case, the research of human gene sequence is a tool to demonstrate what happened in mouse genes regulation.
3.3.1 The Discovery of GATA3 Gene Mutation
GATA3 was found located on chromosome 10p15 by in situ hybridization in 1991 [30], and on mouse chromosome 2 in 1993 [14], 4 years before it was found necessary and sufficient for Th2 cytokine gene expression [84] and related to asthma disease [82].
Terminal deletions of chromosome 10p result in a DiGeorge-like phenotype that includes hypoparathyroidism, heart defects, immune deficiency, deafness, and renal malformations. One region that contributes to this complex phenotype is that for the syndrome of hypoparathyroidism, sensorineural deafness, and renal insufficiency. Van Esch et al. performed deletion-mapping studies in two HDRS patients and defined a critical 200-kb region that contains the GATA3 gene. Search for GATA3 mutations in 3 other HDR probands identified 1 nonsense mutation and two intragenic deletions that predicted a loss of function, as confirmed by absence of DNA binding by the mutant GATA3 protein. These results demonstrated that GATA3 is essential in the embryonic development of the parathyroids, auditory system, and kidneys, and showed that GATA3 haploinsufficiency causes human HDR syndrome [76].
Muroya et al. studied nine Japanese families with HDR syndrome. FISH and microsatellite analysis showed heterozygous deletions including GATA3 in four families. Sequence analysis showed heterozygous novel mutations in three families, including a missense mutation in exon 4, an insertion mutation, and a nonsense mutation in exon 6 [49].
In 10 patients with HDR syndrome from 7 unrelated families, Nesbit et al. identified and characterized 7 mutations in exons 3 through 6 of the GATA3 gene. Using electrophoretic mobility shift, dissociation, yeast 2-hybrid, and glutathione S-transferase pull-down assays, Nesbit et al. [51] demonstrated that mutations involving the C-terminal zinc finger (ZnF2) or adjacent basic amino acids result in a loss of DNA binding, but those of the N-terminal zinc finger (ZnF1) either lead to a loss of interaction with specific zinc finger proteins of FOG2 (ZFPM2) or alter DNA-binding affinity.
Hernandez et al. [27] reported a mother and daughter with HDR and female genital tract malformations in whom they identified a deletion in the GATA3 gene.
Chiu et al. sequenced the CASR and GATA3 genes in five unrelated Chinese families with familial hypoparathyroidism. They identified three novel mutations in the GATA3 gene responsible for familial hypoparathyroidism and deafness. Except for a previously described polymorphism, they found no genetic variants in the CASR gene [11].
Ali et al. analyzed the GATA3 gene in 21 HDR probands and 14 patients with isolated hypoparathyroidism (FIH); no mutations were found in the FIH patients, but 13 different heterozygous germline mutations were identified in the HDR probands, including 1 missense, 1 splice site, 3 nonsense, and 8 frameshift mutations. EMSA analysis revealed three classes of GATA3 mutations: those involving of loss of DNA binding due to loss of the C-terminal zinc finger, which represent over 90 % of mutations reported in GATA3; those resulting in reduced DNA-binding affinity; and those that do not alter DNA binding or affinity but likely alter the conformation change that occurs during binding in the DNA major groove, as predicted by three-dimensional modeling [4].
In a 14-year-old boy with neurologic symptoms in addition to the HDR triad of hypoparathyroidism, sensorineural deafness, and renal dysplasia, who did not have any microdeletion in the 22q11.2 or 10p14 regions by FISH analysis, Ferraris et al. identified a heterozygous de novo 2-bp deletion in exon2 of the GATA3 gene. The authors concluded that haploinsufficiency of GATA3 may be responsible for a complex neurologic picture in addition to the known triad of HDR syndrome [21].
In a 29-year-old Portuguese with severe hypoparathyroidism, bilateral mild neurosensory deafness, and agenesis of the vagina and uterus but no kidney abnormalities, Moldovan et al. analyzed the GATA3 gene and identified a heterozygous missense mutation. The authors noted that this patient, along with the mother and daughter with HDR and female genital tract malformations studied by Hernandez et al., seemed to confirm the role of GATA3 in regulating developmental mechanisms of the uterus and vagina [46].
3.3.2 The Discovery of Tbox21 Gene Mutation
TBX21 is a Th1-specific T-box transcription factor that controls the expression of the hallmark Th1 cytokine, interferon-gamma [71]. And cloned in the same year [83]. Tbx21 has six exons and is on chromosome 11D in an area showing homology of synteny with human chromosome 17.
Akahoshi et al. identified a -1993T-C SNP in the promoter region of the TBX21 gene and found that the substitution increases the affinity of an unknown nuclear protein for the binding site in that region, resulting in increased transcriptional activity of the TBX21 gene. There was a significant association between the promoter SNP and aspirin-induced asthma in a Japanese cohort (p = 0.004), with increased risk associated with a C allele (OR = 1.93; 95 % CI, 1.22–3.06). The association was confirmed in additional independent samples from patients with asthma and nasal polyposis regardless of aspirin hypersensitivity (p = 0.008) [3].
Sasaki et al. in 2004 screened for polymorphisms in the T-bet gene and detected two microsatellite repeat polymorphisms located in intron 1 and the 3′-flanking region, and two single nucleotide polymorphisms, including a His33Gln substitution within the coding region. In the Japanese population, polymorphisms in Tbx21 and Th1-related genes have been linked in humans to a greater risk of developing type 1 diabetes, which is Gln-positive phenotype and (CA)14 allele in 3′-flanking region of T-bet. Furthermore, Gln33 T-bet showed a significantly higher transcriptional activity of the IFN-g gene via a dual luciferase reporter assay, suggests the T-bet Gln33 polymorphism, which is present at a greater frequency in Japanese patients with type 1 diabetes, is responsible for more transcription from the IFN-g promoter, and suggests that T-bet-mediated control of IFN-g production is a contributing factor to the pathogenesis of this disease. Sasaki and colleagues study suggests the first evidence of an association between type 1 diabetes and polymorphisms in the T-bet gene, and that variation in T-bet transcriptional activity may play a role in the development of type 1 diabetes, possibly through the effect on IFN-gamma production in Th1 cells [67].
3.3.3 The Discovery of STAT1 Gene Mutation
Of STAT1, by using whole-exome sequencing, a JEM paper in 2011 identified heterozygous germline mutations in STAT1 in 47 patients from 20 kindreds with AD (autosomal dominant) CMCD (Chronic mucocutaneous candidiasis disease) [44], while this disease is used to be taken caused by IL-17F deficiency [36, 59] or AR (autosomal recessive) IL-17RA deficiency, because high titers of neutralizing auto antibodies against IL-17A, IL-17F, and IL-22 are found in CMCD patients [58]. But these genotype phenomena are not common enough to reach the etiology conclusion. Previously described heterozygous STAT1 mutant alleles are loss-of-function [19] and thus AD are easier to get bacterial disease caused by impaired STAT1-dependent immune system responses to IFN-γ. Other loss-of-function STAT1 alleles cause AR predisposition to intracellular bacterial and viral diseases, caused by impaired STAT1-dependent responses to IFN-α/β, IFN-γ, IFN-λ, and IL-27, as the expected results of STAT1 deficiency. One example of this alleles is MSMD-causing loss-of-function STAT1 allele L706S [19] K201N [37] and K211R [39] with the mutation on coiled-coil (CC) domain of STAT1, which plays a key role in unphosphorylated STAT1 dimerization and STAT1 nuclear dephosphorylation. Loss-of-function STAT1 mutations lead to severe viral and mycobacterial infections [5]. Not only impaired IL-12 and IL-23 signaling, STAT4 deficiency is also reported in STAT1 loss-of-function patients [68].
On the other side, the 12 AD CMCD-inducing STAT1 mutant alleles R274Q in 4 different families are gain-of-function and increase STAT1-dependent cellular responses to these cytokines, and to cytokines that predominantly activate STAT3, such as IL-6 and IL-21. All of these mutations affect the coiled-coil domain and impair the nuclear dephosphorylation of activated STAT1, accounting for their gain-of-function and dominance. Stronger cellular responses to the STAT1-dependent IL-17 inhibitors IFN-α/β, IFN-γ, and IL-27, and stronger STAT1 activation in response to the STAT3-dependent IL-17 inducers IL-6 and IL-21, hinder the development of T cells producing IL-17A, IL-17F, and IL-22. Gain-of-function STAT1 alleles therefore cause AD CMCD by impairing IL-17 immunity [64]. Another study researched 14 patients from five families with autosomal dominant CMC (Chronic mucocutaneous candidiasis), a disease characterized by susceptibility to candida infection of skin, nails, and mucous membranes [35], and poor production of interferon-γ, interleukin-17, and interleukin-22 [59], thus have mucosal antifungal immunity [12]. Th1–interferon-γ responses were defective in patients with autosomal dominant CMC [75], and Th17 responses in these patients were also found with this disorder [20]. With the significant finding in CD4+ T.cells subset in recent years, these patients cellular abnormality suggests that the defect lay within the interleukin-12 receptor and interleukin-23 receptor signaling pathways, and by array-based sequence capture followed by next-generation sequencing, heterozygous missense mutations in the DNA sequence encoding the coiled-coil domain of STAT1 in the patients [74]. Recent reports show that some STAT1 gain-of-function patients even have disseminated fungal infections or an IPEX (immunodysregulation polyendocrinopathy enteropathy X-linked syndrome)-like syndrome [73]. Some patients with disseminated Coccidioides immitis or Histoplasma capsulatum with heterozygous missense mutations in the STAT1 coiled-coil or DNA-binding domains, enhanced STAT1 phosphorylation, delayed dephosphorylation, enhanced DNA binding and transactivation, and enhanced interaction with protein inhibitor of activated STAT1. The mutations caused enhanced IFN-γ-induced gene expression, but we found impaired responses to IFN-γ restimulation, thus with severe, disseminated dimorphic yeast infections [66].
3.3.4 The Discovery of MST1 Gene Mutation
Mammalian sterile 20-like protein kinase 1 (MST1), also known as serine/threonine protein kinase 4 (STK4). MST1 is cloned and named by Creasy and Chernoff in [16]. One year after, Taylor found MST1 have kinase function, and rename it as STK4 [72]. DNA in eukaryotic cells is associated with histone proteins; hence, hallmark properties of apoptosis, such as chromatin condensation, may be regulated by posttranslational histone modifications. Cheung and colleagues reported that phosphorylation of histone H2B at ser14 correlates with cells undergoing programmed cell death in vertebrates. They identified a 34-kD apoptosis-induced H2B kinase as caspase-cleaved MST1. MST1 could phosphorylate H2B at ser14 in vitro and in vivo, and the onset of H2B ser14 phosphorylation was dependent upon cleavage of MST1 by caspase-3. These data revealed a histone modification uniquely associated with apoptotic chromatin in species ranging from frogs to humans and provided insights into a physiologic substrate for MST1. These data also provided evidence for a potential apoptotic histone code [10]. Using human and other mammalian cells found that MST1 phosphorylated FOXO transcription factors at a site conserved within the forkhead domain of FOXO proteins from mammals to Caenorhabditis elegans. Oxidative stress induced MST1-mediated phosphorylation of FOXO3 at ser207, which disrupted interaction of FOXO3 with other proteins, promoted FOXO3 nuclear translocation, and induced cell death. Knockdown of the C. elegans MST1 ortholog Cst1 shortened life span and accelerated tissue aging, whereas Cst1 overexpression promoted life span and delayed tissue aging. The Cst1-induced life span extension was dependent on the FOXO ortholog Daf16 [42].
In two consanguineous unrelated Turkish families with four patients affected by combined immunodeficiencies with multiple bacterial and viral infections, autoimmunity, and progressive CD4 and naive CD8 T-cell lymphopenia, Nehme et al. identified putative truncation mutations in the STK4 gene. The patient in the first family was homozygous for an arg117-to-ter substitution. The three affected sibs in the second family were homozygous for a 1-bp deletion that resulted in a frame-shift at residue 368 and a contiguous nonsense codon at residue 369. Parents and healthy sibs in both families were heterozygous for the mutations, indicating autosomal recessive inheritance. RT-PCR analysis showed loss of function and expression of STK4 in patients. Lymphoproliferative responses and lymphocyte survival were also impaired. FOXO1, IL7R, and BCL2 were poorly expressed in patient T cells, whereas FAS expression was upregulated. Nehme et al. [50] concluded that the STK4/FOXO1 pathway has a role in controlling the death of naive T cells.
In three members of a consanguineous Iranian kindred with T- and B-lymphopenia, neutropenia, cardiac malformations, and recurrent bacterial, viral, and fungal infections, warts, and abscesses, Abdollahpour et al. identified a homozygous premature termination mutation in the STK4 gene. Parents and healthy sibs were heterozygous, indicating autosomal recessive inheritance. Western blot analysis showed that patients with homozygous mutations expressed no STK4, whereas heterozygous carriers expressed intermediate levels compared to wildtype homozygous subjects. STK4-deficient lymphocytes and neutrophils exhibited enhanced loss of mitochondrial membrane potential and increased susceptibility to apoptosis [1].
Patients with MST1 deficiency have B cell and T cell lymphopenia, decreased numbers of CD62L+ (also known as L-selectin) CCR7+ central memory T cells, TCRs that show impaired variable-α (Vα) gene usage (which suggests that they have a restricted TCR repertoire), elevated EBV loads and EBV-positive lymphomas In addition, MST1 is crucial for thymic egress and lymphocyte migration [47].
3.3.5 The Discovery of STAT6 Gene Mutation
Quelle et al. [60] identified a number of expressed genes in the signal transducers and activators of a transcription (STAT) family. Human and murine full-length cDNA clones were obtained and sequenced. The sequence of the human cDNA was identical to the sequence published by Hou et al. [28] for the interleukin-4-induced transcription factor (called by them IL4 Stat), while the murine STAT6 amino acid and nucleotide sequences reported by Quelle et al. were 83 and 84 % identical to the human sequences, respectively.
By screening an embryonic lung fibroblast cDNA library with a wildtype STAT6 probe, Patel et al. identified two variant cDNAs, which they termed STAT6b and STAT6c, encoding an N-terminal 110-amino acid truncation and a 27-amino acid deletion in the SH2 domain, respectively. RNase protection analysis detected ubiquitous expression of all three variants with STAT6b expression greatest in spleen and STAT6c expression greatest in lung [56].
In the Chromosomes location research, Copeland et al. [13] found that seven mouse Stat genes map in three clusters, with each cluster located on a different autosome. They suggested that the Stat family arose by a tandem duplication of the ancestral Stat gene, followed by dispersion of the linked loci to different chromosomes. They mapped Stat6 and Stat2 to the distal region of chromosome 10. During an analysis of NAB2, Svaren et al. obtained the sequence adjacent to this gene by PCR of genomic DNA. They found that the STAT6 gene is located unusually close to the NAB2 gene, such that the 3-prime ends of their mRNAs overlap [69]. Since the human NAB2 gene was previously mapped to 12q13.3-q14.1, it is likely that STAT6 maps to the same position. By fluorescence in situ hybridization, Leek et al. [41] mapped STAT6 to 12q13.
Duetsch et al. [18] identified 13 single-nucleotide polymorphisms (SNPs) in STAT6 and tested them for linkage/association with asthma and related traits (total serum IgE level, eosinophil cell count, and SLOPE of the dose-response curve after bronchial challenge) in 108 Caucasian sib-pairs. Neither the SNPs nor a GT repeat in exon 1 showed linkage/association to asthma. A significant association was found between a SNP in intron 18 and an increase in total IgE levels (P = 0.0070), as well as an association between allele A4 of the GT repeat polymorphism and an increase in eosinophil cell count (P = 0.0010). The authors concluded that rather than contributing to the pathogenesis of asthma, the human STAT6 gene is more likely involved in the development of eosinophilia and changes in total IgE levels [18].
In a case-control association study of 214 white British subjects, Gao et al. demonstrated a significant association with asthma of an allele with a 13-GT repeat sequence in exon 1 of the STAT6 gene (OR, 1.52; 95 % CI, 1.02–2.28; p = 0.027), whereas the 16-GT allele showed an inverse association with asthma (p = 0.018). Furthermore, individuals with the 13-GT allele had higher IgE levels compared with individuals with the 16-GT allele (p = 0.004). Transient transfection assays of different alleles revealed significantly higher transcriptional activity with the 13-GT allele compared to the 16-GT allele in Jurkat, HMC-1, and BEAS-2B cell lines. Gao et al. [24] suggested that the GT repeat polymorphism of the STAT6 gene contributes to susceptibility to atopic asthma and total serum IgE levels, and that variation in the length of the GT repeat sequence influences the regulation of promoter activity.
Several studies have shown linkage of 12q13-q24 with atopy-related phenotypes. STAT6 is 1 of the candidate genes in this region, because of its involvement in Th2 cell differentiation, recruitment, and effector function. Studying a population-based cross-sectional cohort of 1,407 German adults, Weidinger et al. evaluated 6 polymorphisms of STAT6 for evidence of association with serum IgE levels and atopic disease. One polymorphism in intron 2 showed a significant association with total serum IgE (p = 0.015). A STAT6 risk haplotype for elevated IgE showed odds ratios of 1.54 (p = 0.032), 1.6 (p = 0.025), and 2.54 (p = 0.007) for IgE percentiles of 50, 60, and 90 %, respectively [79].
3.3.6 The Discovery of STAT4 Gene Mutation
STAT4 was cloned and located to chromosome 2q32 in 1997 by Yamamoto, and found not ubiquitously, but expressed in specific tissues, including spleen, heart, brain, peripheral blood cells, and testis [81].
Since linkage peaks containing the STAT4 gene had been reported in genome scans of patients with systemic lupus erythematosus, Remmers et al. included the genotyping of three series of patients with SLE and control subjects of European ancestry as part of a large case-control disease-association analysis of a linkage region on chromosome 2q associated with rheumatoid arthritis. They found that a haplotype marked by the STAT4 SNP rs7574865 was strongly associated with SLE [62], being present on 31 % of chromosomes of case patients and 22 % of those of controls (P = 1.87 × 10(−9); odds ratio for having the risk allele in chromosomes of patients versus those of controls, 1.55). Homozygosity for the risk allele, as compared with absence of the allele, was associated with a more than doubled risk for SLE and a 60 % increased risk for rheumatoid arthritis. Independently, Gateva et al. [25] and Han et al. [26] replicated the association of SLE susceptibility with one STAT4 SNP at Chromosome 2:191964633 (rs7574865).
To identify risk loci for SLE susceptibility, Gateva et al. [25] selected SNPs from 2,466 regions that showed nominal evidence of association with SLE (P less than 0.05) in a genomewide study and genotyped them in an independent sample of 1,963 cases and 4,329 controls. This new cohort replicated the association with STAT4 at rs7574865 (combined P value = 1.4 × 10(−41), OR = 1.57, 95 % confidence interval of 1.49–1.69).
Han et al. performed a genomewide association study of SLE in a Chinese Han population by genotyping 1,047 cases and 1,205 controls using Illumina-Human610-Quad BeadChips and replicating 78 SNPs in 2 additional cohorts (3,152 cases and 7,050 controls). Han et al. found association with the STAT4 gene at rs7574865 (combined P value = 5.17 × 10(−42), odds ratio = 1.51, 95 % confidence interval 1.43–1.61).
By measuring serum IFNa activity and IFNa-induced gene expression in PBMCs from a cohort of 270 SLE patients of different ethnic backgrounds, Kariuki et al. [31] showed that the T allele of the STAT4 SNP rs7574865 was simultaneously associated with both lower serum IFNa activity and greater IFNa-induced gene expression. Although the IRF5 SLE risk genotype was associated with higher serum IFNa activity, the influence of STAT4 was dominant on the sensitivity of PBMCs to serum IFNa. Kariuki et al. concluded that the risk variant of STAT4 is critical in the dysregulation of the IFNa pathway and SLE susceptibility.
3.3.7 The Discovery of c-Maf Gene Mutation
Maf was cloned by Nishizawa et al. [52] in human, and by Ring in mouse [63].
In five affected members of a three-generation family with autosomal dominant juvenile-onset cataract (CTRCT21), Jamieson et al. [29] identified a 1670G-C transversion in the MAF gene, resulting in an arg288-to-pro (R288P) substitution in the basic region of the DNA-binding domain of MAF, predicted to cause an abnormal helical conformation. The cataracts were cortical pulverulent opacities in a lamellar distribution. Nuclear pulverulent opacities were present in two cases. There was later progression with posterior subcapsular opacification that necessitated surgery in adult life. Two of the five affected individuals had microcornea, and one also had bilateral iris colobomas. The mutation was not found in 217 other subjects with a range of eye anomalies or in 496 normal control chromosomes.
Wende et al. [80] found that skin of carriers of the R288P substitution displayed reduced acuity to high-frequency vibration compared with normal controls. In 12 affected members of a three-generation family with congenital cerulean cataract, 6 of whom also had microcornea, Vanita et al. identified heterozygosity for an 890A-G transition in the MAF gene, resulting in the replacement of a highly conserved lys297 with arg (K297R) in a basic region of the DNA-binding domain of the protein. The mutation was not found in 106 unrelated controls.
3.3.8 Other Human Gene Mutation Reports
Very few report of gene mutation specifically block human Th1 cell development, but there are patients with mendelian susceptibility to mycobacterial diseases, and are easy to get clinical disease caused by weakly virulent mycobacterial species in otherwise healthy individuals. Since 1996, disease-causing mutations have been found in five autosomal genes (IFNGR1, IFNGR2, STAT1, IL12B, IL12BR1) and one X-linked gene (NEMO). These genes display a high degree of allelic heterogeneity, defining at least 13 disorders. Although genetically different, these conditions are immunologically related, as all result in impaired IL-12/23-IFN-gamma-mediated immunity. These atopic mutation shows important role of Th1 cells in anti-batetials immunity [22].
References
Abdollahpour H, Appaswamy G, Kotlarz D, Diestelhorst J, Beier R, Schaffer AA et al (2012) The phenotype of human STK4 deficiency. Blood 119:3450–3457
Aguayo S, Munoz MJ, de la Torre A, Roset J, de la Pena E, Carballo M (2004) Identification of organic compounds and ecotoxicological assessment of sewage treatment plants (STP) effluents. Sci Total Environ 328:69–81
Akahoshi M, Obara K, Hirota T, Matsuda A, Hasegawa K, Takahashi N et al (2005) Functional promoter polymorphism in the TBX21 gene associated with aspirin-induced asthma. Hum Genet 117:16–26
Ali A, Christie PT, Grigorieva IV, Harding B, Van Esch H, Ahmed SF et al (2007) Functional characterization of GATA3 mutations causing the hypoparathyroidism-deafness-renal (HDR) dysplasia syndrome: insight into mechanisms of DNA binding by the GATA3 transcription factor. Hum Mol Genet 16:265–275
Boisson-Dupuis S, Kong XF, Okada S, Cypowyj S, Puel A, Abel L et al (2012) Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol 24:364–378
Bour-Jordan H, Grogan JL, Tang Q, Auger JA, Locksley RM, Bluestone JA (2003) CTLA-4 regulates the requirement for cytokine-induced signals in T(H)2 lineage commitment. Nat Immunol 4:182–188
Bruton OC (1952) Agammaglobulinemia. Pediatrics 9:722–728
Casanova JL, Abel L (2007) Primary immunodeficiencies: a field in its infancy. Science 317:617–619
Chen H, Sun H, You F, Sun W, Zhou X, Chen L et al (2011) Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 147:436–446
Cheung WL, Ajiro K, Samejima K, Kloc M, Cheung P, Mizzen CA et al (2003) Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell 113:507–517
Chiu WY, Chen HW, Chao HW, Yann LT, Tsai KS (2006) Identification of three novel mutations in the GATA3 gene responsible for familial hypoparathyroidism and deafness in the Chinese population. J Clin Endocrinol Metab 91:4587–4592
Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ et al (2009) Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 206:299–311
Copeland NG, Gilbert DJ, Schindler C, Zhong Z, Wen Z, Darnell JE Jr et al (1995) Distribution of the mammalian Stat gene family in mouse chromosomes. Genomics 29:225–228
Copeland NG, Jenkins NA, Gilbert DJ, Eppig JT, Maltais LJ, Miller JC et al (1993) A genetic linkage map of the mouse: current applications and future prospects. Science 262:57–66
Cormier SA, Kolls JK (2011) Innate IL-13 in virus-induced asthma? Nat Immunol 12:587–588
Creasy CL, Chernoff J (1995) Cloning and characterization of a human protein kinase with homology to Ste20. J Biol Chem 270:21695–21700
Davis MM (2012) Immunology taught by humans. Sci Transl Med 4:117fs2
Duetsch G, Illig T, Loesgen S, Rohde K, Klopp N, Herbon N et al (2002) STAT6 as an asthma candidate gene: polymorphism-screening, association and haplotype analysis in a Caucasian sib-pair study. Hum Mol Genet 11:613–621
Dupuis S, Dargemont C, Fieschi C, Thomassin N, Rosenzweig S, Harris J et al (2001) Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science 293:300–303
Eyerich K, Foerster S, Rombold S, Seidl HP, Behrendt H, Hofmann H et al (2008) Patients with chronic mucocutaneous candidiasis exhibit reduced production of Th17-associated cytokines IL-17 and IL-22. J Invest Dermatol 128:2640–2645
Ferraris S, Del Monaco AG, Garelli E, Carando A, De Vito B, Pappi P et al (2009) HDR syndrome: a novel “de novo” mutation in GATA3 gene. Am J Med Genet A 149A:770–775
Filipe-Santos O, Bustamante J, Chapgier A, Vogt G, de Beaucoudrey L, Feinberg J et al (2006) Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol 18:347–361
Finotto S, Neurath MF, Glickman JN, Qin S, Lehr HA, Green FH et al (2002) Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science 295:336–338
Gao PS, Heller NM, Walker W, Chen CH, Moller M, Plunkett B et al (2004) Variation in dinucleotide (GT) repeat sequence in the first exon of the STAT6 gene is associated with atopic asthma and differentially regulates the promoter activity in vitro. J Med Genet 41:535–539
Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X et al (2009) A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet 41:1228–1233
Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z et al (2009) Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet 41:1234–1237
Hernandez AM, Villamar M, Rosello L, Moreno-Pelayo MA, Moreno F, Del Castillo I (2007) Novel mutation in the gene encoding the GATA3 transcription factor in a Spanish familial case of hypoparathyroidism, deafness, and renal dysplasia (HDR) syndrome with female genital tract malformations. Am J Med Genet A 143:757–762
Hou J, Schindler U, Henzel WJ, Ho TC, Brasseur M, McKnight SL (1994) An interleukin-4-induced transcription factor: IL-4 Stat. Science 265:1701–1706
Jamieson RV, Perveen R, Kerr B, Carette M, Yardley J, Heon E et al (2002) Domain disruption and mutation of the bZIP transcription factor, MAF, associated with cataract, ocular anterior segment dysgenesis and coloboma. Hum Mol Genet 11:33–42
Joulin V, Bories D, Eleouet JF, Labastie MC, Chretien S, Mattei MG et al (1991) A T-cell specific TCR delta DNA binding protein is a member of the human GATA family. EMBO J 10:1809–1816
Kariuki SN, Kirou KA, MacDermott EJ, Barillas-Arias L, Crow MK, Niewold TB (2009) Cutting edge: autoimmune disease risk variant of STAT4 confers increased sensitivity to IFN-alpha in lupus patients in vivo. J Immunol 182:34–38
Kaufman CK, Zhou P, Pasolli HA, Rendl M, Bolotin D, Lim KC et al (2003) GATA-3: an unexpected regulator of cell lineage determination in skin. Genes Dev 17:2108–2122
Kim JI, Ho IC, Grusby MJ, Glimcher LH (1999) The transcription factor c-Maf controls the production of interleukin-4 but not other Th2 cytokines. Immunity 10:745–751
Kim JI, Li T, Ho IC, Grusby MJ, Glimcher LH (1999) Requirement for the c-Maf transcription factor in crystallin gene regulation and lens development. Proc Natl Acad Sci USA 96:3781–3785
Kirkpatrick CH (1994) Chronic mucocutaneous candidiasis. J Am Acad Dermatol 31:S14–S17
Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV et al (2010) Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med 207:299–308
Kong XF, Ciancanelli M, Al-Hajjar S, Alsina L, Zumwalt T, Bustamante J et al (2010) A novel form of human STAT1 deficiency impairing early but not late responses to interferons. Blood 116:5895–5906
Kouros-Mehr H, Slorach EM, Sternlicht MD, Werb Z (2006) GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell 127:1041–1055
Kristensen IA, Veirum JE, Moller BK, Christiansen M (2011) Novel STAT1 alleles in a patient with impaired resistance to mycobacteria. J Clin Immunol 31:265–271
Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z et al (2002) Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med 8:885–889
Leek JP, Hamlin PJ, Bell SM, Lench NJ (1997) Assignment of the STAT6 gene (STAT6) to human chromosome band 12q13 by in situ hybridization. Cytogenet Cell Genet 79:208–209
Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB et al (2006) A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 125:987–1001
Lim KC, Lakshmanan G, Crawford SE, Gu Y, Grosveld F, Engel JD (2000) Gata3 loss leads to embryonic lethality due to noradrenaline deficiency of the sympathetic nervous system. Nat Genet 25:209–212
Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A et al (2011) Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 208:1635–1648
Lyon MF, Jamieson RV, Perveen R, Glenister PH, Griffiths R, Boyd Y et al (2003) A dominant mutation within the DNA-binding domain of the bZIP transcription factor Maf causes murine cataract and results in selective alteration in DNA binding. Hum Mol Genet 12:585–594
Moldovan O, Carvalho R, Jorge Z, and Medeira A (2011) A new case of HDR syndrome with severe female genital tract malformation: comment on “Novel mutation in the gene encoding the GATA3 transcription factor in a Spanish familial case of hypoparathyroidism, deafness, and renal dysplasia (HDR) syndrome with female genital tract malformations” by Hernandez et al. Am J Med Genet A 155A:2329–2330
Mou F, Praskova M, Xia F, Van Buren D, Hock H, Avruch J et al (2012) The Mst1 and Mst2 kinases control activation of rho family GTPases and thymic egress of mature thymocytes. J Exp Med 209:741–759
Mullen AC, Hutchins AS, High FA, Lee HW, Sykes KJ, Chodosh LA et al (2002) Hlx is induced by and genetically interacts with T-bet to promote heritable T(H)1 gene induction. Nat Immunol 3:652–658
Muroya K, Hasegawa T, Ito Y, Nagai T, Isotani H, Iwata Y et al (2001) GATA3 abnormalities and the phenotypic spectrum of HDR syndrome. J Med Genet 38:374–380
Nehme NT, Pachlopnik Schmid J, Debeurme F, Andre-Schmutz I, Lim A, Nitschke P et al (2012) MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood 119:3458–3468
Nesbit MA, Bowl MR, Harding B, Ali A, Ayala A, Crowe C et al (2004) Characterization of GATA3 mutations in the hypoparathyroidism, deafness, and renal dysplasia (HDR) syndrome. J Biol Chem 279:22624–22634
Nishizawa M, Kataoka K, Goto N, Fujiwara KT, Kawai S (1989) v-maf, a viral oncogene that encodes a “leucine zipper” motif. Proc Natl Acad Sci USA 86:7711–7715
Ochs HD, Hitzig WH (2012) History of primary immunodeficiency diseases. Curr Opin Allergy Clin Immunol 12:577–587
Pai SY, Truitt ML, Ting CN, Leiden JM, Glimcher LH, Ho IC (2003) Critical roles for transcription factor GATA-3 in thymocyte development. Immunity 19:863–875
Pandolfi PP, Roth ME, Karis A, Leonard MW, Dzierzak E, Grosveld FG et al (1995) Targeted disruption of the GATA3 gene causes severe abnormalities in the nervous system and in fetal liver haematopoiesis. Nat Genet 11:40–44
Patel BK, Pierce JH, LaRochelle WJ (1998) Regulation of interleukin 4-mediated signaling by naturally occurring dominant negative and attenuated forms of human Stat6. Proc Natl Acad Sci USA 95:172–177
Perveen R, Favor J, Jamieson RV, Ray DW, Black GC (2007) A heterozygous c-Maf transactivation domain mutation causes congenital cataract and enhances target gene activation. Hum Mol Genet 16:1030–1038
Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK et al (2011) Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332:65–68
Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C et al (2010) Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med 207:291–297
Quelle FW, Shimoda K, Thierfelder W, Fischer C, Kim A, Ruben SM et al (1995) Cloning of murine Stat6 and human Stat6, Stat proteins that are tyrosine phosphorylated in responses to IL-4 and IL-3 but are not required for mitogenesis. Mol Cell Biol 15:3336–3343
Ravindran R, Foley J, Stoklasek T, Glimcher LH, McSorley SJ (2005) Expression of T-bet by CD4 T cells is essential for resistance to Salmonella infection. J Immunol 175:4603–4610
Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW et al (2007) STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med 357:977–986
Ring BZ, Cordes SP, Overbeek PA, Barsh GS (2000) Regulation of mouse lens fiber cell development and differentiation by the Maf gene. Development 127:307–317
Romberg N, Morbach H, Lawrence MG, Kim S, Kang I, Holland SM et al (2013) Gain-of-function STAT1 mutations are associated with PD-L1 overexpression and a defect in B-cell survival. J Allergy Clin Immunol 131:1691–1693
Rosen MJ, Chaturvedi R, Washington MK, Kuhnhein LA, Moore PD, Coggeshall SS et al (2013) STAT6 deficiency ameliorates severity of oxazolone colitis by decreasing expression of claudin-2 and Th2-inducing cytokines. J Immunol 190:1849–1858
Sampaio EP, Hsu AP, Pechacek J, Bax HI, Dias DL, Paulson ML et al (2013) Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J Allergy Clin Immunol 131:1624–1634 248
Sasaki Y, Ihara K et al (2004) Identification of a novel type 1 diabetes susceptibility gene, T-bet. Hum Genet 115(3):177–184
Smeekens SP, Plantinga TS, van de Veerdonk FL, Heinhuis B, Hoischen A, Joosten LA et al (2011) STAT1 hyperphosphorylation and defective IL12R/IL23R signaling underlie defective immunity in autosomal dominant chronic mucocutaneous candidiasis. PLoS ONE 6:e29248
Svaren J, Apel ED, Simburger KS, Jenkins NA, Gilbert DJ, Copeland NA et al (1997) The Nab2 and Stat6 genes share a common transcription termination region. Genomics 41:33–39
Svensson A, Nordstrom I, Sun JB, Eriksson K (2005) Protective immunity to genital herpes simplex [correction of simpex] virus type 2 infection is mediated by T-bet. J Immunol 174:6266–6273
Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH (2000) A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100:655–669
Taylor LK, Wang HC, Erikson RL (1996) Newly identified stress-responsive protein kinases, Krs-1 and Krs-2. Proc Natl Acad Sci USA 93:10099–10104
Uzel G, Sampaio EP, Lawrence MG, Hsu AP, Hackett M, Dorsey MJ et al (2013) Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J Allergy Clin Immunol 131:1611–1623
van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C et al (2011) STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med 365:54–61
van der Graaf CA, Netea MG, Drenth IP, te Morsche RH, van der Meer JW, Kullberg BJ (2003) Candida-specific interferon-gamma deficiency and toll-like receptor polymorphisms in patients with chronic mucocutaneous candidiasis. Neth J Med 61:365–369
Van Esch H, Groenen P, Nesbit MA, Schuffenhauer S, Lichtner P, Vanderlinden G et al (2000) GATA3 haplo-insufficiency causes human HDR syndrome. Nature 406:419–422
Wang J, Fathman JW, Lugo-Villarino G, Scimone L, von Andrian U, Dorfman DM et al (2006) Transcription factor T-bet regulates inflammatory arthritis through its function in dendritic cells. J Clin Invest 116:414–421
Wang W, Ostlie NS, Conti-Fine BM, Milani M (2004) The susceptibility to experimental myasthenia gravis of STAT6−/− and STAT4−/− BALB/c mice suggests a pathogenic role of Th1 cells. J Immunol 172:97–103
Weidinger S, Klopp N, Wagenpfeil S, Rummler L, Schedel M, Kabesch M et al (2004) Association of a STAT 6 haplotype with elevated serum IgE levels in a population based cohort of white adults. J Med Genet 41:658–663
Wende H, Lechner SG, Cheret C, Bourane S, Kolanczyk ME, Pattyn A et al (2012) The transcription factor c-Maf controls touch receptor development and function. Science 335:1373–1376
Yamamoto K, Kobayashi H, Arai A, Miura O, Hirosawa S, Miyasaka N (1997) cDNA cloning, expression and chromosome mapping of the human STAT4 gene: both STAT4 and STAT1 genes are mapped to 2q32.2-->q32.3. Cytogenet Cell Genet 77:207–210
Zhang DH, Cohn L, Ray P, Bottomly K, Ray A (1997) Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J Biol Chem 272:21597–21603
Zhang WX, Yang SY (2000) Cloning and characterization of a new member of the T-box gene family. Genomics 70:41–48
Zheng W, Flavell RA (1997) The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89:587–596
Zhu J, Min B, Hu-Li J, Watson CJ, Grinberg A, Wang Q et al (2004) Conditional deletion of Gata3 shows its essential function in T(H)1-T(H)2 responses. Nat Immunol 5:1157–1165
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Zhang, Y., Zhang, Y., Gu, W., He, L., Sun, B. (2014). Th1/Th2 Cell’s Function in Immune System. In: Sun, B. (eds) T Helper Cell Differentiation and Their Function. Advances in Experimental Medicine and Biology, vol 841. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-9487-9_3
Download citation
DOI: https://doi.org/10.1007/978-94-017-9487-9_3
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-9486-2
Online ISBN: 978-94-017-9487-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)