
Abstract
This review focuses on the impact of nickel on human health. In particular, the dual nature of nickel as an essential as well as toxic element in nature is described, and the main forms of nickel that can come in contact with living systems from natural sources and anthropogenic activities are discussed. Concomitantly, the main routes of nickel uptake and transport in humans are covered, and the potential dangers that nickel exposure can represent for health are described. In particular, the insurgence of nickel-derived allergies, nickel-induced carcinogenesis as well as infectious diseases caused by human pathogens that rely on nickel-based enzymes to colonize the host are reviewed at different levels, from their macroscopic aspects on human health to the molecular mechanisms underlying these points. Finally, the importance of nickel as a beneficial element for human health, especially being essential for microorganisms that colonize the human guts, is examined.
Please cite as: Met. Ions Life Sci. 13 (2013) 321–357
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction: The Double Face of Nickel in Biological Systems
Nickel is the 24th most abundant element regarding the natural abundance in the Earth’s crust [1]. This metal exists in nature either in insoluble particles, which are components of fumes and dusts, like nickel sulfides (NiS, Ni3S2), oxides (NiO), and silicates, or in water-soluble nickel compounds, such as nickel acetate, nickel chloride, and nickel sulfate. Natural sources of nickel include dusts from volcanic emissions and weathering of rocks and soils [2]. Soluble and insoluble nickel compounds are also found in soils and in waters [3]. In water, nickel ions are generally divalent, present as the greenish hexa-hydrated [Ni(H2O)6]2+ ion.
The unique physical and chemical properties of nickel – low thermal and electrical conductivities, high resistance to corrosion and oxidation, excellent strength and toughness at elevated temperatures, and capability of being magnetized – make this metal and its compounds suitable materials for many applications widely found in modern industry [4]. Human activities, such as emission of nickel-containing fuel, industrial nickel production and utilization of nickel compounds, concur to the environmental release of nickel and to pollution by nickel and its products.
Human exposure to nickel occurs primarily via inhalation, ingestion, and dermal absorption [5]. Insoluble particulate nickel enters the vertebrate cells by phagocytosis, whereas nickel carbonyl is soluble in lipids and permeates the plasma membrane. Soluble nickel is transported into cells of vertebrate organisms by diffusion or through calcium channels and/or divalent cation transporters (DMT-1), involved in iron uptake [6]. Transport of nickel in blood plasma is mediated by binding to albumin and some small ligands, such as amino acids (e.g., histidine) and small peptides [7,8]. The Ni2+-L-histidine complex is the major form of nickel transport across the cell membrane, and the Ni2+-albumin complex is the form for systemic transport [9].
Exposure to nickel compounds yields a variety of adverse effects on humans. Nickel immune reaction, as a form of dermatitis, is one of the most common allergies in the modern world [10]. In addition, chronic nickel exposure can produce serious respiratory, cardiovascular, and kidney diseases. Some alterations in immune response in animal models have been observed as a result of nickel contact [11]. Nickel induces the production of reactive oxygen species (ROS), like the superoxide radical (O −·2 ), hydrogen peroxide (H2O2), and hypochlorous acid (HOCl) by several cells, such as in neutrophils and monocytes. This ultimately causes apoptosis in a number of cellular types, including human neutrophils and T-cells [12,13]. High exposure to nickel impairs the normal homeostasis of essential metal ions, decreasing the levels of calcium, magnesium, manganese, and zinc in different tissues [14] and possibly interfering with normal iron cofactor binding to specific proteins [15–17]. Finally, the most serious concerns of nickel for human health is the nickel-induced teratogenicity and carcinogenesis, documented by the International Agency for Research on Cancer (IARC) in 1990 [18].
Despite its poisoning potential, nickel plays a fundamental role in living organisms, revealing its double faced nature of both as an essential and toxic element [19]. The importance of nickel for plants, bacteria, archaea, and unicellular eukaryotes is well documented. In these organisms, the choice of nickel as catalyst for important biological reactions is related to its flexible coordination geometry, which makes this metal a very versatile element for many biological applications [1]. Nickel is a necessary component in the active site of several essential metallo-enzymes in bacteria and lower eukaryotes. So far, eight microbial nickel-containing enzymes have been identified, which include urease, hydrogenase, CO dehydrogenase, acetyl-CoA synthase, methyl-CoM reductase, Ni-superoxide dismutase, acireductone dioxygenase, and glyoxalase I, while a few other possible nickel-dependent enzymes are emerging [20]. The majority of known nickel-dependent enzymes have been structurally determined and nickel ions have been demonstrated to play an essential role in their enzymatic catalysis. In higher eukaryotes, the only known nickel-depending enzyme is plant urease. Some plant species that live in serpentine soils have evolved to hyperaccumulate nickel ions, creating complex systems of metal detoxification and homeostasis that constitute appealing systems for phytoremediation of contaminated environments [21].
So far, no nickel-containing enzyme or cofactor is known in higher animals. However, this metal has been included in the group of “possibly essential elements” for animals and humans as early as the 1970s [19]. Many experiments on animal models showed that nickel could be beneficial, if not essential, for optimal reproductive function, bone composition and strength, energy metabolism, and sensory function. The reasons of this essentiality remain obscure, even if some hypotheses have been suggested [5, 22].
The double face of nickel, being both a poison and an essential micronutrient, implies that nickel-dependent organisms have developed tightly regulated systems for nickel handling, delivery it into the correct cellular location, and avoiding its dangerous potential. Bacteria that rely on nickel ions for environmental colonization and growth represent a good model to study nickel metabolism and homeostasis in nickel-dependent organisms. Due to the relative paucity of known nickel-dependent enzymes, the study of intracellular nickel homeostasis may represent a paradigm to study the general handling of dangerous and essential metal ions in vivo.
In this chapter we review the present knowledge on the impact that nickel ions have on human health, focusing on the dangerous potential of nickel, leading to allergy, carcinogenesis, and infectious diseases, and the proofs of its essentiality, and on what is currently known on the molecular mechanisms underlying these points.
2 Nickel Hazard for Human Health
The most diffuse hazardous health effects caused by nickel exposure are nickel-induced carcinogenesis and allergy. They are both mediated by active changes in metabolic pathways that underlie inflammation, stress response, oxidative stress, cell proliferation, and cell death [23]. As no protein specific for nickel homeostasis is known in mammals, one would not expect a specific nickel-mediated change of gene expression and metabolism. Indeed, many of the nickel effects on cells are triggered by non-specific interactions of nickel ions with macromolecules and general formation of reactive compounds that mediate cellular damage. Furthermore, the cellular response to nickel is related to signal transduction cascades such as second messengers, protein kinases, phosphatases or transcription factors, which are involved in general metal ion response. Notably, nickel exposure produces a rather specific pattern of gene expression. Nickel-driven alteration of transcription of genes involved in oxygen deficiency has been extensively studied in vitro for its relevance to nickel carcinogenesis [23].
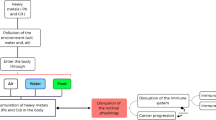
On the other hand, the activation of the inflammatory response, with the induction of genes for chemokines and cytokines correlated with nickel-induced allergy and asthma, has been studied mostly in vivo [23]. Additionally, few proteins with nickel-binding motifs have been identified and the effect of nickel binding to these proteins can be related to the specific cellular response to nickel exposure. A schematic representation of nickel uptake routes, intracellular distribution, and major effects on human cells metabolism, which will be discussed in detail in the following sections, is presented in Figure 1.

Schematic representation of known nickel uptake routes, intracellular distribution, and its major effects on cellular metabolism in humans. ROS = reactive oxide species; HIF = hypoxia-inducible factor; HRE = hypoxia-responsive enhancer; GSH = reduced glutathione; NF-κB = nuclear factor κ-B; AP-1 = activating protein 1; TGF-β = transforming growth factor β; NF-AT = nuclear factor of activated T cells; DMT-1 = divalent cation transporter 1; NDRG-1 = N-myc downstream regulated gene 1, DAN = differential screening-selected gene aberrative in neuroblastoma.
2.1 Nickel-Induced Carcinogenesis
2.1.1 The Carcinogenic Potential of Nickel
The propensity of nickel workers to develop cancers in the respiratory tract was firstly reported in 1933 [24]. Subsequent epidemiological studies and carcinogenic assays in animals corroborated the carcinogenicity of nickel compounds, which is now generally accepted [18]. Epidemiological studies have reported an increased risk of lung and nasal cancers among nickel mining, smelting, and refinery workers [25]. For many years, it was believed that only water-insoluble nickel components of fumes and dusts, like nickel sulfides and oxides, were carcinogenic. Subsequent epidemiological studies indicated instead that aerosols of water-soluble nickel compounds, such as nickel acetate, nickel chloride and sulfate, are also carcinogenic in vivo, although with a lower potential [24]. Nickel present in endoprostheses, bone-fixing plates and screws, and other implanted medical devices made of metal alloys, has been suspected, but not proven, to be the cause of sporadic local tumors. There is no epidemiological evidence on possible cancer risk from general environmental and dietary nickel exposures.
In animal models, nickel compounds induce tumors at virtually all sites of applications. Rats are more susceptible to nickel poisoning effects than mice, hamsters, or rabbits. The reasons for these differences may reflect different abilities of the phagocytes to ingest and metabolize nickel-containing particles, the different mechanisms of nickel uptake, transport, distribution, and retention in the animal body, and the differences in the capacity of antioxidant systems among animals of different species and strains [23]. This suggests that genetic predispositions, including metabolic variations of different species and strains of animals, may also play an important role in nickel carcinogenesis. Similar genetic predispositions possibly also occur in human populations.
The routes of administration that were shown to produce tumors include inhalation, intramuscular, intrarenal, intraperitoneal, intraocular, and subcutaneous applications [23]. The injection of crystalline Ni2S3 or NiS into experimental animals resulted in a high incidence of tumors in the application sites, while no tumor was induced in animals treated with soluble NiSO4. In rats, intraocular and intramuscular injections generated the highest tumor incidences, yielding tumors in all animals, often readily metastasized to the lung and other organs [23]. Similarly, only water-insoluble nickel compounds were found to be carcinogenic in rats via the inhalation route, while soluble NiSO4 was not found to be carcinogenic [26]. Notably, intrarenal nickel injection, beside causing kidney tumors in rats, also caused erythrocytosis due to induction of erythropoietin, a part of the hypoxia-mimicking effect of nickel [23].
Insoluble nickel compounds are generally considered stronger carcinogens than soluble compounds. The difference of their carcinogenic activity is related to the faster clearance of soluble nickel ions from the tissues, which in turn retain insoluble nickel particles longer [5]. These data indicate that prolonged exposure to a nickel carcinogen is critical for tumor development. Nickel compounds also transform human cells in vitro [27]. Crystalline nickel particles, phagocytized by cultured cells, exist in intracellular vacuoles with internal acid pH that facilitates nickel solubilization, creating local high concentrations of soluble nickel inside the cell. Nickel-filled vacuoles easily migrate and reach the cellular nucleus, potentially exerting their effect on DNA and nuclear proteins. On the contrary, exposure of cells to water-soluble salts results in low nuclear and high cytosolic nickel contents [28].
Nickel compounds have been shown to produce a significant synergistic effect, when co-administrated with other carcinogens, in enhancing cell transformation both in vitro and in vivo. On the other hand, essential metals such as manganese, magnesium, and zinc, co-administered with Ni3S2 to animal models, reduced local tumor incidence in a dose-dependent manner. Magnesium was the strongest and zinc was the weakest inhibitor. Separate administration of the essential metals did not produce this effect [23].
2.1.2 Molecular Mechanisms for Nickel-Induced Neoplastic Transformation
The molecular basis of nickel carcinogenesis remains elusive, as data obtained by different groups are difficult to reconcile, and a general consensus on the mechanisms has still to be reached. To be defined carcinogenic, a compound should provoke two effects fundamental for tumor genesis, that is, heritable changes in gene expression and cell proliferation. In most instances, carcinogenic compounds provide the first condition by interacting with DNA and DNA-binding proteins, changing DNA structure and inducing mutations in its sequence, usually during replication. Often, these sequence changes alter the expression level or function of tumor suppressor genes or oncogenes. However, nickel does not behave like a typical mutagen, because it does not show high affinity for DNA nor displays mutagenic potential in most assays on bacteria, fruit fly, mammalian cells, and whole animals [29–33]. Therefore, alternative routes for nickel to change the gene expression levels and cellular phenotypes have been described. Nickel has been found to act at the DNA level, mostly through epigenetic mechanisms. Nickel promotion of tumors, on the other hand, occurs mostly at the protein level, and involves DNA-binding proteins such as transcription factors, metal-binding proteins, and proteins participating in important cellular pathways. The result is a change of the general cellular metabolism and a deregulation of cellular homeostasis inducing carcinogenic transformations in the cells.
2.1.2.1 Effects of Nickel on DNA Structure and Gene Expression
(i) Genotoxic effects. Nickel compounds have been reported to be mildly clastogenic, causing extensive DNA damage and chromosome aberrations, particularly in the heterochromatic region of the genome [5]. Nickel generates oxides and reactive species that produce DNA-protein cross-links and oxidative DNA damages [34]. This mechanism is typical of several transition metals that can generate reactive oxygen species in biological fluids at physiological pH. However, this effect cannot fully explain the high carcinogenic potential of nickel: this metal is a weaker generator of redox-active species, but it is as good a carcinogen as chromium, which is very active in ROS production [35]. In addition, highly redox-active metals, such as copper, which also binds DNA more avidly than nickel, are only weakly or not carcinogenic [36]. The ability of carcinogenic metals to facilitate DNA damage through inhibition of DNA-repair enzymes or binding to histones can also explain its genotoxic activity [37,38].
(ii) Epigenetic effects. Further evidence from epidemiological, animal, and cellular studies shows a role of epigenetics in nickel carcinogenesis, in addition to genetic-based mechanisms. One major requirement for nickel carcinogenicity is the prolonged action on the target tissue, performed by compounds with limited solubility and long retention in biologic fluids [24], which is typical of tumor promoters acting through epigenetic mechanisms, rather than tumor initiators, which are usually mutagenic. Indeed, nickel was found to synergistically increase the tumorigenic potential of several carcinogenic agents.
The nickel-induced epigenetic changes include silencing of genes for DNA repair and tumor suppressors, mostly occurring through nickel-driven DNA methylation, which can modify the chromatin structure and eventually the genetic expression. In DNA of higher eukaryotes the methylation of CpG dinucleotides is an important modification that leads to modulation of gene expression. In general, increased cytosine methylation represses transcription, and it is thus more abundant in the heterochromatin regions of chromosomes [39]. Exposure to nickel compounds enhances DNA methylation, leading to inactivation of gene expressions [40]. The nickel-induced silencing of a tumor suppressor gene, occurring through promoter hypermethylation, has been associated with cellular transformations in vivo [41]. Although the mechanisms by which nickel induces DNA hypermethylation are presently unknown, a possible model includes the ability of Ni2+ to substitute Mg2+, normally bound to DNA, seeding chromatin condensation and triggering de novo DNA methylation by methylases that recognize newly generated heterochromatic and unmethylated DNA [40]. This property is related to the same charge and very similar ionic radius of Ni2+ (69 pm) and Mg2+ (71 pm).
In addition to DNA methylation, nickel epigenetic effects are related to the ability of nickel to affect the global levels of histone modifications, implicating global deregulation of gene expression. Indeed, acetylation in the N-terminal tail of histone proteins is a well-known mechanism to regulate the chromatin transcriptional state, important to control the access of regulatory proteins to DNA. Conversely, histone methylation results in more pronounced chromatin and gene silencing. Nickel decreases the acetylation levels of histones H2A, H2B, H3 and H4, and increases H3K9 dimethylation, and the ubiquitination of H2A and H2B [42]. The higher methylation of the histones occurs through the nickel-induced inhibition of specific Fe2+ and α-ketoglutarate-dependent demethylases belonging to the family of Jmjc-domain containing histone demethylases (JHDM), such as JHMD1 and JMJD1A [43,44]. The inhibited acetylation of histone H4 is thought to occur through the direct interaction of the metal ion with His18 of the histone itself, which would prevent its interaction with the specific acetyl-transferase enzyme [45]. Low levels of histone acetylation in nickel-exposed cells can also result from low levels of acetyl-CoA, a universal donor of acetyl groups, due to the nickel-dependent inhibition of pyruvate dehydrogenase kinase that converts pyruvate into acetyl-CoA [42]. The increase in H2A/H2B ubiquitination has been correlated to nickel up-regulation of the ubiquitin-conjugating enzyme H6 (UbcH6) E2 ligase or the inhibition of an unidentified histone de-ubiquitinating activity [45]. Moreover, nickel induces the truncation, at the N-terminus of the histone H2A, and, both at N- and C-termini, of the histone H2B, possibly through the activation of specific nuclear proteolytic enzymes belonging to the calpain family [46]. The importance of deregulation of histone modifications by nickel has been demonstrated by observing that nickel-transformed cells, treated with the histone deacetylase inhibitor, namely trichostatin A, revert the phenotype to that of untransformed cells along with the gene expression profile, suggesting that histone acetylation might be inhibited during nickel-induced transformation [47].
In addition to changing gene expression levels, epigenetic effects also promote cell proliferation: recent studies have demonstrated that the unlimited proliferative character of a nickel-transformed cell line could be reversed by induction of senescence. The senescence gene is located in the donor X chromosome, and its expression is regulated by DNA methylation. These data indicate that transformation by nickel may involve DNA methylation and silencing of a senescence gene in the X chromosome [48].
2.1.2.2 Effects of Nickel on Proteins, Transcriptional Regulators, and Metabolic Pathways
(i) Disruption of calcium homeostasis. Nickel blocks calcium channels and disturbs intracellular calcium homeostasis. This results in the experimentally observed rapid proliferation of nickel-transformed cells in low-calcium media [49]. Since cytoplasmic calcium levels regulate expression of genes associated with cell growth, differentiation, and apoptosis, derangement of calcium regulation would impact the entire cellular metabolism [49]. In particular, nickel was shown to increase intracellular calcium levels: nickel likely uses calcium channels to enter the cells, and induces calcium release from intracellular stores, possibly through a cell surface receptor. Therefore, changes of calcium homeostasis invoked by nickel exposure may change the cellular expression induced by other signalling pathways, eventually leading to malignant transformation [23].
(ii) Oxidative damage. Soluble and insoluble nickel compounds can be redox-active at physiological pH, although to a lesser extent than iron and copper complexes, and they can generate reactive oxygen species. This is possible when the redox couple Ni3+/Ni2+ is formed, which usually only occurs when nickel is bound by certain natural ligands like peptides and proteins, especially those forming square planar nickel complexes. Reactions of such complexes with O2 or H2O2 yield the hydroxyl radical ·OH and other radicals. The oxidation of water-insoluble nickel sulfides may involve both nickel and sulfur and lead to generation of not only nickel-, but also sulfur-derived ROS and other reactive intermediates (e.g. the sulfite anion). This enables the sulfides to produce a wider spectrum of oxidative damage than other nickel compounds and may be responsible for their high carcinogenic activity. In addition, nickel can deplete some important antioxidant ligands, such as ascorbate and reduced glutathione (GSH). Nickel is capable of depleting intracellular ascorbate through catalytic oxidation and hydrolysis of both ascorbic and dehydroascorbic acid, and inhibition of ascorbic acid transporters [23]. GSH depletion is likely the result of a cellular response to the ROS species generated by nickel. Coherently, resistance to nickel toxicity is usually associated to high levels of GSH [50]. Finally, the enzymatic components of cellular antioxidant defence, such as superoxide dismutase, catalase, glutathione peroxidase, and glutathione reductase, are also affected by nickel exposition [23].
The nickel-induced oxidative stress can activate some transcriptional pathways through some oxidation-sensitive transcription factors. ROS created by nickel exposition result in lipid peroxidation, whose products can create adducts with DNA, thus altering gene expression. Protein oxidation, leading to protein fragmentations and cross-linking with other molecules (e.g., with DNA) and oxidative DNA and chromatin damage are also consequences of ROS generated by nickel. The presence of cross-links in chromatin may lead to morphologic aberrations of chromosomes. In vitro, nickel was found to promote DNA cleavage by H2O2 predominantly at the cytosine, thymine, and guanine bases [51]. ROS attack on DNA’s sugar moiety produces apurinic sites in DNA and mediates in vitro hydrolysis of 2′-deoxyguanosine. The depurination occurs concurrently with DNA strand scission and fragmentation. Some compounds generated by oxidative stress, like 8-oxoguanine, may also misdirect DNA methylation and perturb orderly binding of transcription factors to DNA.
(iii) Activation of hypoxia signalling. Nickel exposure produces a rather specific pattern of gene expression, which involves the same activation pathways of the response to hypoxia [52], and in particular the activation of the HIF-1 transcription factor. This protein exists as a HIF-1α/HIF-1β (ARNT) hetero-dimer, with the α subunit being the regulatory unit, formed in response to low oxygen tension in the cells. Under normal oxygen concentrations, HIF-1α is virtually undetectable in most cells [53]. In these conditions, the protein interacts with the tumor suppressor protein VHL, a part of the ubiquitin-ligase complex that induces the ubiquitination of HIF-1α and its rapid degradation. The structural basis for specific interaction of HIF-1α and VHL is provided by the introduction of the hydroxyl group at the C4 position of Pro402 and Pro564 [54], which facilitates hydrogen bonding with Ser111 and His115 in VHL. On the other hand, hydroxylation of Asp803 prevents complex formation between HIF-1α and the transcriptional co-activators CBP and P300, providing a second mechanism by which HIF-mediated transcription is regulated [55]. The hydroxylation reactions are carried out by specific hydroxylases that employ both Fe2+ and ascorbate as cofactors to split dioxygen into two oxygen atoms, one of them converted into hydroxide. Ascorbate is a reducing agent needed to avoid iron oxidation, and to maintain the metal ion bound to the enzyme as Fe2+. Hypoxia signalling reduces the HIF-1α hydroxylation, therefore stabilizing HIF-1α and allowing it to join HIF-1α. The heterodimer translocates into the nucleus, where it binds the hypoxia-responsive enhancers (HREs) and recruits the co-activator acetyltransferase P300 [56].
Similarly to hypoxia, nickel was found to be a strong stabilizer of the HIF-1α protein and an activator of HIF-dependent transcription, inhibiting its enzymatic hydroxylation [56]. This likely occurs through a depletion of intracellular ascorbate that follows nickel-driven oxidation and/or uptake inhibition [57]. This results in the inactivation of the hydroxylases, followed by the induction of HIF-1 and activation expression of hypoxia-inducible genes [56]. The activation of the hypoxic signalling pathway and the switch of cellular metabolism to a state that mimics permanent hypoxia may be a part of nickel-induced carcinogenesis [42]. Indeed, hypoxia is a common state in tumors because transformed cells grow faster than the blood vessels providing them with oxygen. This state can activate genes that enable cells to overcome nutritive deprivation, to escape from the hostile metabolic microenvironment, and to stimulate angiogenesis. Additionally, cellular responses to hypoxic stress include inhibition of cell proliferation and, when cell damage is irreversible, apoptosis. Therefore, imitation of the state of hypoxia by nickel may provide the conditions for the selection of cells that have altered energy metabolism, changed growth control and/or have become resistant to apoptosis. A result of nickel-induced hypoxia response is the induction of numerous genes involved in glucose transport and glycolysis, coding for carbonic anhydrase IX, ceruloplasmin, erythropoietin, inducible nitric oxide synthase, vascular endothelial growth factor (VEGF), and many others [23].
(iv) Additional alterations of other signalling pathways. Exposure of cells to nickel also induces a change of gene expression that yields cells with spectra of expressed genes similar to cancer cells. The proteins responsible for this effect are some transcription factors, involved in different metabolic pathways, such as oxidative stress defence (NF-κB, AP-1), inflammatory response (NF-κB, TGF-β), apoptosis (p53), angiogenesis (ATF-1), calcium homeostasis (NF-AT), all activated by nickel [23]. Nickel also induces activation of the K-ras oncogene, and inhibits tumor suppressor genes, such as Rb, p16, FHIT, Zac-1, and Gas-1 [23].
(v) Interaction with proteins. The gene coding for the Cap43 protein (also called N-myc downstream-regulated gene 1 (NDRG1)) is induced by nickel through the hypoxia pathway [58]. This protein is usually expressed at low levels in normal tissues, but it is over-expressed in a variety of cancers, including lung, brain, melanoma, liver, prostate, breast and renal cancers, and has been often used as a marker of tumor progression [59]. The physiological function of this protein is not clear so far, but likely it acts as a tumor suppressor protein. The fact that this protein is ubiquitously expressed and highly conserved in all multicellular organisms, and that its expression is regulated by different central pathways that respond to several stress and growth signals, such as cellular proliferation, differentiation, growth arrest, neoplasia, tumor progression and metastasis, hypoxia, heavy metal response, favors a central role in cellular metabolism [60]. Impairing of calcium homeostasis, also triggered by nickel, induces Cap43/NDRG1 production. Interestingly, three repeats of the GTRSRSHTSE sequence are found in the C-terminal tail of Cap43/NDRG1, which are not present in the other family members, and have been postulated to serve as nickel binding motifs, and can bind one nickel ion each [61,62]. It is noteworthy that these repeats fall into a region that in all NDRG family members is predicted to be unfolded [63], therefore possibly involved in regulation through interaction with a partner or cofactor such as nickel. The fact that the 30-amino acid fragment of the C-terminal tail of the nickel-induced Cap43/NDRG1 is able to bind up to three nickel ions could shed new light onto the complex mechanism of nickel toxicity, in particular in relation to the physiological role exerted by the stress protein genes up-regulated by metal exposure, suggesting a possible role of Cap43/NDRG1 as a detoxification agent and a feedback mode of nickel sensing. The crystal structure of a homologue, NDRG2, only expressed in adult skeletal muscle and brain, has been recently reported, and the protein was thought to serve as molecular interactor for regulating cell proliferation and cell differentiation [64].
Beside Cap43/NDRG1 protein and histones, nickel has been reported to form complexes with other proteins, possibly significantly altering their conformations, interactions, functionality, and eventually cellular homeostasis, triggering the observed adverse effects. At physiological pH, the strength of Ni2+ interactions with proteins depends on the type of amino acid residues, their positions relative to each other, and their accessibility in the protein molecule. The greatest affinity for Ni2+ is shown by histidine and cysteine residues in proteins. One of these proteins, named DAN, possessing a Ni2+ binding motif at the C-terminal region, shows a tumor suppressor activity. Interaction of DAN with Ni2+ can impair its activity triggering the tumorigenic phenotype [65]. Similarly, cullin-2, a component of the complex that serves HIF-1α ubiquitination, features three Ni2+ and Co2+ binding sites [66]. Metal binding prevents ubiquitination, therefore contributing to the typical hypoxia response. Nickel can also bind the iron regulatory protein 1 (IRP-1), a central regulator of iron homeostasis [67]. Replacement of one Fe2+ with Ni2+ in the 4Fe-4S cluster inactivates the protein enzymatic activity and may contribute to the nickel-induced hypoxic signalling. The ability of Ni2+ and Cu2+ to bind neuromedin C, a neuropeptide, may represent the overlap between the metabolisms of these two metal ions, and may imply that Ni2+ is able to impair copper homeostasis in the brain [68].
2.2 The Different Faces of Nickel Allergy
2.2.1 Nickel Effects on Immune Response
The immune response triggered by nickel exposure is generally important, and responsible of allergic contact dermatitis (ACD), one of the most significant consequence of occupational exposure to nickel. The first event in nickel immunological response is a silent sensitization phase, which is initiated upon the first contact with the antigen, leading to the generation of allergen-specific T-cells, and lymphocyte activation. Upon re-exposure to the allergen, an elicitation phase occurs, resulting in clinically apparent inflammation. In this second phase, the T-cells exert cytotoxic functions and secrete inflammatory mediators, such as chemokines and cytokines, to amplify the inflammatory response and produce eczematous skin reaction [69].
As many ACDs, nickel allergy requires two events for being established both for sensitization and elicitation, that is (i) the activation of antigen-specific T-cells, and (ii) a non-specific proinflammatory microenvironment necessary for the development of a hypersensitivity response, also called innate immune signal [70]. In humans, nickel ions can trigger both these events, directly activating proinflammatory pathways [70].
2.2.1.1 How Nickel Activates Antigen-Specific T-Cells
T-cells are the major effectors in Ni2+ hypersensitivity. These cells are usually activated when a peptide, recognized as non-self, is bound to a major histocompatibility complex (MHC) protein of the membrane of an antigen-presenting cell (APC), and interacts with a T-cell receptor (TCR) [71]. This is the signal that activates the lymphocytes and subsequently the immune cascade. Ni2+ ions, as many other immunologically active low molecular chemicals, are defined as haptens, that is antigens generally invisible to the immune system by themselves, becoming visible only when bound to proteins or peptides [72]. While, generally, hapten recognition by T-cells requires covalent hapten attachment to MHC-associated peptides, transition metal ions such as Ni2+ do not form stable covalent protein modifications, they rather produce geometrically highly defined coordination complexes with reversible binding [73].
During the immune response, Ni2+ has been found to be reversibly bound to a particular MHC-II molecule, called HLA-DR52c, and an unknown MHC-bound peptide via a pH-sensitive interaction, consistent with metal ion coordination to amino acid side chains of histidine or acidic residues [74]. The structure of HLA-DR52c in complex with a self-peptide deriving from the Tu elongation factor (pTu), has been reported [75]. Although the HLA-DR52c/pTu complex is not able to bind Ni2+, this structure is important to understand how Ni2+ can be presented in this complex. In particular, four residues (His81, Asp55, Gln70, and Gln74) have been indicated as being potentially involved in metal coordination. However, these are not sufficient to allow Ni2+ binding, coherently with the observation that a specific peptide is needed to form the Ni/HLA-DR52c/peptide complex [75]. How the metal ion influences TCR binding to the Ni/HLA-DR52c/peptide complex is still unknown. It has been proposed that the metal ion binds to the interface between MHC and TCR proteins, sterically influencing the interaction [76]. Based on current biochemical data, hypothetically the metal ion can bind the MHC and become the ligand of T-cells at least in four different ways [77]: (a) Ni2+ could bind the MHC only; (b) Ni2+ can bind both the presented peptide and the MHC molecule; (c) Ni2+ can interact with the presented peptide only; (d) Ni2+ can induce a conformational change of the peptide and/or of the MHC, which are therefore recognized as neoantigens. A further possibility is that Ni2+ ions interfere with the processing of self-proteins in the APC and the exposure of cryptic self-peptides, which can be recognized as non-self by TCR [78].
Besides activating T-cells in a Ni/MHC/peptide ternary complex, Ni2+ appears to serve as a direct and peptide-independent linker between TCR and MHC, bearing certain similarities to superantigen-mediated activation [79]. Potential Ni2+ contacts both in the TCR and in the specific MHC molecule are responsible for Ni2+ reactivity independently from protein processing by antigen-presenting cells and peptide binding to MHC. This new type of Ni2+-induced TCR-MHC crosslinking might explain the high frequency of Ni-reactive T cells in the human population [79].
2.2.1.2 How Nickel Activates the Innate Immune Signal
Ni2+ is the only allergen for which a direct mechanism of innate immune activation has conclusively been demonstrated and unravelled at the molecular level. In vitro and in vivo experiments showed that treatment of human endothelial cells with Ni2+ triggers rapid expression of the surface molecules VCAM1, ICAM1, and E-selectin and monocyte-attracting chemokine MCP-1 [80–82], required for leukocyte adhesion and inflammation. On the other hand, expression of lymphocyte-attracting cytokines such as IP10, Mig, MDC, PARC, and TARC, occurs at later stages and correlates well with the infiltration of T-cells into the dermis and epidermis [82].
Molecular analysis revealed that, in humans, Ni2+ activates the IKK2/NF-κB and the MAPK/p38 signalling pathways, both regulating gene expression leading to inflammation [83,84]. These activations occur through Ni2+ interaction with the membrane toll-like receptor 4 (TLR4), in combination with its co-receptor MD2, which induces receptor dimerization probably bridging two TLR4 monomers. Coherently, human cells expressing TLR4 and MD2 including macrophages, fibroblasts, and dendritic cells were able to induce a proinflammatory response upon Ni2+ treatment [85]. The non-conserved His456 and His458 residues in human TLR4 are critically required for Ni2+-induced proinflammatory gene expression, possibly acting as specific ligands for the metal ion [85]. Notably, mouse TLR4, which lacks these histidine residues, is not able to produce the proinflammatory pathway in response to Ni2+ exposition. Activation of innate immunity in mice rather needs co-stimulating adjuvants, such as the bacterial cell wall component lipopolysaccharide (LPS).
Another pathway responsible for Ni2+-induced ACD is the death receptor-mediated or extrinsic apoptosis pathway. It was reported that Ni2+ transcriptionally represses expression of cFLIP, a cellular antagonist of the pro-apoptotic caspase-8, in both primary human keratinocytes and endothelial cells [86], which, as a result, are strongly sensitized to apoptosis. Coherently, there is evidence for an increased occurrence of death ligand-mediated keratinocyte apoptosis in the course of Ni2+-induced ACD in sensitized individuals [87]. Enhanced cell death of keratinocytes is predicted to impair the barrier function of the skin. Hence, Ni2+-mediated cFLIP down-regulation might tip the balance towards increased apoptosis of certain skin cell populations, which successively may augment the severity of the ACD response during the elicitation phase by increasing the efficient concentration of Ni2+ arriving in the epidermis [70]. In addition, nickel is able to induce apoptosis in a number of immune cells, including human neutrophils and T-cells, through the mitochondrial pathway that activates caspase-3, likely as a response to Ni2+-induced oxidative stress [12,13]. The production of ROS by Ni2+ exposition also acts in concert with the mechanisms described above to produce and amplify the inflammatory response. In particular, ROS act as signalling molecules and are recognized as important inducers of the proinflammatory response [69].
2.2.2 The Impact of Nickel-Induced Dermatitis
The immunological effects of nickel described above are responsible for allergic contact dermatitis (ACD), which is the most spread dermatitis over the world and is constantly increasing, reaching 20% of the human population. It was discovered for the first time in 1930 [88]. It is caused by Ni2+ ions solubilized from nickel-containing alloys by sweat and other body fluids that serve as sensitizing allergen and come in contact with skin. Although the risk of occupational disability is an issue for a relatively large group of professionals, such as metal workers, cashiers, or hairdressers, the major problem associated with Ni2+-induced contact hypersensitivity is the wide presence of this metal ion in modern industrial products, so that it is very common to come in contact with the allergen. Ni2+ is released from coins, earrings, watches, belt buckles, bras, mobile phones, dental and orthopedic implants, and cardiovascular stents [70]. In Europe, currently around 65 million individuals are sensitized to Ni2+ [89]. Legislative interventions limiting the amount of Ni2+ release from products intended for prolonged skin contact, such as the Danish nickel regulation in 1990 [90] and the EU directive 94/27/EG in 2001 (also known as “Nickel Directive”), result in decreasing sensitization rates, but prevalence of contact eczema to Ni2+ are still considerably high [91]. In its classic description, nickel ACD involves the hands and forearms mainly after occupational exposure. Sensitized individuals generally experience a predictable localized response following cutaneous exposure to nickel, including erythema, vesicle formation, scaling, and pruritus [92]. A nickel-free diet remains controversial in the treatment of ACD. In addition, inhaled nickel in ambient air might be a risk factor for nickel sensitization [93]. The most important way of preventing nickel allergy is the avoidance of exposure. Rubber gloves are inefficient for prevention of nickel contact, as metal ions penetrate through them. Polyurethane coating of metal items can be beneficial in protection from nickel contact, as are barrier creams containing sodium EDTA [94].
Nickel is present in most dietary items, and food is considered to be a major source of exposure to nickel for the general population. Certain foods, such as green beans, broccoli, peas, canned vegetables and spaghetti, canned fruit, dried fruit, nuts, cocoa, and chocolate are routinely found to be high in nickel content. Nickel present in the diet of a nickel-sensitive person can provoke systemic reactions, called systemic contact dermatitis (SCD). SCD is manifested as generalized eczematous reactions, maculopapular rash and vasculitis-like skin, along with systemic symptoms such as headache, malaise, diarrhea, fever, and arthralgia [95]. The exact mechanisms of systemic contact dermatitis are still not known. The eczema flare at sites of previous exposure suggests T cells resting at the area or homing the site upon systemic allergen exposure [96]. Circulating immune complexes together with non-specific cytokine release by the hapten stimulation can be responsible for the generalized reactions [95,96].
In patients with SCD, adherence to a low-nickel diet and avoidance of local exposure to metal objects result in the disappearance of skin symptoms [92]. Another therapeutic approach, studied for both ACD and SCD, is nickel hypo-sensitization, that is the procedure allowing sensitized patients affected by ACD to safely make contact with nickel containing objects, and those with SCD to freely eat nickel-containing foods. This aim is pursued through oral administration of capsules containing nickel to ACD and SCD patients. For ACD, the clinical studies, performed with oral supplementation of high nickel doses, are still preliminary, as they have been conducted with a limited number of subjects and for a limited period of treatment, but have given promising results [97]. In the case of SCD, the studies of oral hypo-sensitization with very low doses of nickel for a prolonged time led to an improvement of the symptoms and a complete remission in 50% of the patients. No side effects are observed during and after the treatment. The hypo-sensitization treatment exerts its clinical effects through a modulation of the allergic immune reaction specific for nickel [98].
3 Nickel-Dependent Infectious Diseases
Nickel in the active site of several enzymes is responsible for the catalysis of important biological reactions, such as urea hydrolysis, hydrogen metabolism, methane formation, CO/CO2 inter-conversion, superoxide metabolism, and detoxification of methylglyoxal [99]. For many bacteria, archaea and unicellular eukaryotes these nickel-dependent processes render possible the colonization of environments that are inhospitable and hostile. These include parts of human and animal bodies, where these nickel-obligate pathogens grow and survive in virtue of nickel-catalyzed reactions. Consistently, some nickel-dependent enzymes are virulence factors for pathogenic organisms. As no nickel-dependent enzyme is known in vertebrates, catalyzed nickel-dependent processes and mechanisms of nickel delivery into specific enzymes can be regarded and employed as possible selective targets to control the pathogenesis of nickel-obligate organisms.
3.1 Nickel-Dependent Enzymes in Pathogenic Microorganisms
The biological role of nickel is essentially defined by its use as a cofactor of enzymes found in all phyla of life (plants, fungi, eubacteria, and archaea). All known nickel enzymes involve the transformation and/or production of gases (ammonia, carbon monoxide, carbon dioxide, methane, dihydrogen, and dioxygen) involved in the geo-biological cycles of carbon, nitrogen, and oxygen. The nickel ion in the active sites of these enzymes exhibits a very large flexibility in metal coordination and redox chemistry, spanning coordination numbers from 4 to 6 and oxidation states from +1 to +3, with potentials ranging from +900 to −600 mV [100]. In these enzymes, nickel often is inserted in a multinuclear metal cluster that also includes modified amino acids and/or exogenous ligands [20].
The importance of nickel enzymes for human health is related to the fact that, among eight known nickel-dependent enzymes, four (glyoxalase I, acireductone dioxygenase, hydrogenase, and urease) are present in pathogenic microorganisms and are often essential for their growth and pathogenesis. For example, infections by urease-dependent organisms represent a widespread source of diseases, ranging from tuberculosis to urinary tract infections, hepatic coma, and kidney stones [101]. The glyoxalase enzyme of the protozoan parasites has been regarded as a potential chemotherapeutic target against eukaryotic pathogens, such as Trypanosoma or Leishmania [102].
3.1.1 Glyoxalase I
Glyoxalase (Glx) I and GlxII catalyze the conversion of methylglyoxal, a toxic species that forms covalent adducts with DNA, to lactate (Figure 2) [103]. For a long time, all GlxI enzymes were believed to be Zn2+ enzymes, like the human GlxI. However, it was later shown that some GlxI enzymes are more active with Ni2+ in vitro. These include the GlxI from some bacteria, such as the pathogen Pseudomonas aeruginosa, and trypanosomatids, responsible for several diseases, such as Chagas’ disease and skin sores called leishmaniasis. It is not clear whether this metal ion is functional for GlxI in vivo [104]. A single Ni2+ ion in an octahedral coordination environment composed by 2 His, 2 Glu, and two water molecules, acts as a Lewis acid catalyst and remains in the 2+ oxidation state throughout the isomerization reaction (Figure 2). On the other hand, the inactive Zn2+ enzyme is five-coordinated [103].

Reaction catalyzed by GlxI and GlxII, and schematic structure of the nickel-containing active site.
3.1.2 Acireductone Dioxygenase
Acireductone dioxygenase (ARD) catalyzes the incorporation of two oxygen atoms into the substrate acireductone, the penultimate intermediate of the methionine salvage pathway (Figure 3) [105,106]. The latter is a ubiquitous process that regulates methionine levels, thus playing an essential role in several biosynthetic processes.

Reaction catalyzed by acireductone dioxygenase, and schematic structure of the nickel-containing active site.
In Klebsiella ATCC 8724, this enzyme is a monomeric protein, showing a classic jellyroll structural motif. The active site, relatively well exposed to the solvent, contains Ni2+ in an octahedral high-spin coordination center, involving three His, one Asp, and two water molecules in equatorial positions (Figure 3). The Ni2+-bound enzyme catalyzes the so-called “off-pathway” reaction, leading to the formation of relatively large amounts of carbon monoxide. Peculiarly, this enzyme can also bind Fe2+ in the same coordination site. Fe2+ binding stabilizes a different protein form that catalyzes the “on-pathway” reaction, leading to production of methionine. The Ni2+- and Fe2+-bound forms differ constitutionally only in the identity of the metal ion bound. Interestingly, this difference is sufficient to induce significant structural changes resulting from differential packing of a compact hydrophobic core of the protein, accompanied by extensive changes in secondary structure and ordering of nearby structural features [107].
3.1.3 Urease
Urease is key to the global nitrogen cycle as it catalyzes the hydrolysis of urea, produced by vertebrates, to ammonia and carbamate, which then spontaneously decomposes to give a second molecule of ammonia and bicarbonate (Figure 4) [108].

Reaction catalyzed by urease, and schematic structure of the nickel-containing active site.
The subsequent hydrolysis of the reaction products induces an overall pH increase that has negative implications both in human and animal health, as it is used by several pathogens, such as Helicobacter pylori, to colonize hostile acid habitats. In addition, this reaction represents a nitrogen source for several organisms that infect humans. Therefore, urease is a virulence factor for pathogens in the animal gut, urinary tract, and stomach.
Microbial ureases are generally heteropolymeric proteins with a quaternary structure (αβγ)3. In some bacteria, such as those of the genus Helicobacter, the trimer is of the type (αβ)3, with the β subunit corresponding to the fused β and γ subunits normally found in other bacteria, and presents a higher level of oligomerization that leads to an enzyme with a quaternary structure ((αβ)3)4. On the other hand, plant ureases are hexameric proteins α6, each α subunit being highly homologous to each (αβγ) assembly of microbial ureases [109].
Several structures of ureases are available. In all cases, the active site contains two Ni2+ ions bridged by the carboxylate group of a carbamylated lysine and by a hydroxide ion (Figure 4). Each Ni is also coordinated by two histidines and one water molecule, whereas Ni(2) is further bound to an aspartate. This results in a penta-coordinate Ni(1) and hexa-coordinate Ni(2). In the resting state of the enzyme from Bacillus pasteurii, the active site accommodates a fourth water molecule, completing a tetrahedral cluster of solvent molecules [110]. The access to the active site is regulated by a flexible helix-loop-helix segment, with the position of amino acids involved in the catalysis being critically affected by the flap movement [111].
The structures of B. pasteurii urease (BPU), in the native hydrated form and complexed with several inhibitors of different chemical classes, suggest a structure-based reaction mechanism for urease, and highlight the importance of both metal ions in the reactivity of the enzyme [109]. The structures of BPU bound with borate [112], a substrate analogue, and with diamidophosphate [110], a transition state analogue, are particularly significant to understand the reaction mechanism [109,113].
3.1.4 [NiFe]-Hydrogenase
Hydrogenase enzymes catalyze the formation of hydrogen gas from protons and electrons and/or the reverse reaction, oxidizing H2 as a source of reducing power, and coupling it to the reduction of various terminal electron acceptors (e.g., O2, NO −3 , SO 2 −4 , CO2, and fumarate), in energy-conserving pathways (Figure 5). They can act as sensors for the availability of hydrogen gas [114].

Reaction catalyzed by [NiFe]-hydrogenases, and schematic structure of the nickel-containing active site.
The [NiFe]-hydrogenases are a class of hydrogenase enzymes that contain nickel and iron in the active site and are widely distributed in bacteria and archaea, including some pathogens [115]. A [NiFe]-hydrogenase is required for efficient colonization by H. pylori and Salmonella enterica serovar Typhimurium, a food poison, Campylobacter jejuni, a bacterium closely related to Helicobacter, as well as many enteric bacteria (e.g., E. coli, Shigella, and Yersinia species) [20,116]. These infectious organisms use the hydrogen produced by other microorganisms in the body, a resource not used by the human host, as a supply of energy for their respiratory pathway. This permits the growth and maintains efficient virulence during animal infections [116].
[NiFe]-hydrogenases are usually composed of two subunits. The smaller β subunit contains three closely spaced FeS clusters (two [4Fe-4S] and one [3Fe-4S]), linearly arranged, that transfer electrons to and from the active site. The active site is constituted by a hetero-nuclear bimetallic [NiFe] center, buried at the bottom of a hydrophobic channel in the larger α subunit. The nickel-containing hydrogenases can be classified in two different categories, differing by ligation of the [NiFe] cluster found in the active site to either cysteine or seleno-cysteine.
In the oxidized form of the enzyme, the Ni and Fe atoms are bridged by two cysteines and a third, non-protein ligand, possibly a sulfide or an oxide (Figure 5). The Ni atom is bound to two additional cysteines, forming the equatorial plane of a distorted square pyramidal coordination geometry with the O/S bridging atom being the fifth axial ligand. In [NiFeSe]-hydrogenase, selenium, in the form of seleno-cysteine, is a ligand to Ni. The Fe atom resides in a pseudo-octahedral coordination geometry, additionally bound to CO, and two CN–.
In the structure of the reduced enzyme, the non-protein bridging ligand in the bimetallic center is absent (Figure 5), leading to the conclusion that activation of [NiFe]-hydrogenases by H2 involves the removal of such a bridge, thereby causing the opening of binding sites on both Ni and Fe atoms. The subsequent steps of the catalytic mechanism have not been fully elucidated to date, but they likely include nickel, iron, or cysteine thiolates as potential substrate-binding sites and redox centers [115].
3.2 Molecular Regulation of Nickel Homeostasis in Pathogenic Microorganisms
The essentiality of nickel for nickel-dependent organisms, together with the environmental scarcity of this metal ion, led nickel-obligate pathogens to evolve a tight system for nickel homeostasis with mechanisms that correlate Ni2+ availability with the expression of proteins involved in nickel transport and utilization. In particular, attaining sufficiently high intracellular nickel concentrations to meet the demand of the nickel enzymes and, at the same time, protecting the cell by the poisoning potential of nickel excess, requires (i) an efficient nickel uptake/efflux system, ii) molecular chaperones and metallo-chaperones, and (iii) nickel sensors (Figure 6).

Schematic representation of bacterial Ni2+ homeostasis, intracellular transport, and utilization. ABC = ATP-binding cassette; NiCoT = nickel cobalt transporter; RcnA = resistance to cobalt and nickel A; CznA = cadmium-zinc-nickel resistance A; MCR = methyl coenzyme-M reductase; ADR = acireductone dioxygenase; GlxI = glyoxalase I.
3.2.1 Nickel Membrane Transporters
Ni2+ ions should enter into the cytoplasm in order to be inserted into the active site of nickel-dependent enzymes. On the other hand, nickel excess must be extruded from the intracellular location, in order to prevent nickel-related danger at toxic metal concentrations. Therefore, the metal ions must be able to pass through the cytoplasmic membrane entering or exiting the cell according to the intracellular nickel abundance. Low levels of nickel can travel through the plasma membrane by way of multiple, possibly nonspecific, systems. However, in many of the microorganisms that require this metal ion, dedicated nickel uptake transporters have been identified that belong mainly to two different classes, the NikABCDE import pumps and the nickel/cobalt permeases (NiCoT). The former, firstly characterized in E. coli, belongs to the family of ATP-binding cassette (ABC) transporters, and couples the translocation of a substrate to the hydrolysis of ATP. NikB and NikC are trans-membrane proteins that form a nickel pore. NikD and NikE are proteins that bind to and hydrolyse ATP [117]. NikA is a periplasmic protein that binds one nickel per protein in the context of a nickelophore. The nature of this complex is controversial, but recent structural data have identified two free histidines coordinating Ni2+ in the metal binding site of NikA, suggesting that Ni2+-(L-His)2 is the nickelophore recognized by the periplasmic transporter [118]. This transport system has been found in several pathogenic organisms, such as Brucella suis, Vibrio parahemolyticus, Helicobacter hepaticus, Yersinia sp., and Staphylococcus aureus [119]. The NiCoT family is composed by monomeric single component permeases with eight trans-membrane helices, found in many bacteria, such as the pathogen H. pylori, as well as several archaea and fungi. While some family members are specific for Ni2+, other permeases transport both Ni2+ and Co2+, with the preference of one metal over the other [20].
In gram-negative bacteria, some nickel-specific importers have been found in the outer membrane. In particular, in H. pylori FecA3 and FrpB4 have been suggested to be involved in TonB-dependent transport of nickel [20].
The most common mechanism for metal resistance is metal efflux, made by exporters that, in several organisms, have been predicted and/or demonstrated to pump nickel out of the cytoplasm and of the periplasm. Only in a few cases the transporters are specific for nickel [120]. In particular, nickel exporters have been identified for H. pylori and E. coli. In H. pylori, CznABC is proposed to be a novel system that pumps Cd2+, Zn2+, and Ni2+ across both the inner and outer membranes [121]. In E. coli, RcnA is a membrane transporter with six trans-membrane segments, with only limited homology with other transporters families, involved in specific export of Ni2+ and Co2+ [122].
3.2.2 Nickel Molecular Chaperones and Metallo-Chaperones
Nickel activation pathways imply nickel delivery into the buried cavity of specific enzymes, usually synthesized as apo-proteins undergoing activation through metal ion incorporation in a post-translational regulation mechanism of the enzymatic activity. Some of these pathways, usually involving a multistep, tightly regulated mechanism with several accessory proteins, have been extensively studied in bacteria and in some cases they even overlap (e.g., hydrogenase and urease pathways) [99].
The functional, biochemical and structural properties of these chaperones have been investigated in the case of urease (UreDEFG proteins), hydrogenase (HypABCDEF and SlyD), carbon monoxide dehydrogenase (CooCTJ), acetyl-CoA decarbonylase synthase (AcsF), and superoxide dismutase (SodX and CbiXhp) [99]. No accessory protein is known for acireductone dioxygenase and for glyoxalase so far.
Even though many aspects of these processes remain largely obscure, some general aspects of the roles performed by the accessory proteins are maintained throughout the investigated systems. In particular, (i) nickel storage, (ii) nickel delivery, and (iii) nucleotide triphosphate hydrolysis have been found in several nickel-driven enzyme activations.
These three functions are carried out by specific protein domains, organized in a modular fashion, either in a single protein or in separated chaperones. The nickel storage role is normally carried out by a His-rich pattern: this motif may constitute a whole protein, such as in the case of Hpn or HspA, two nickel accumulators found in Helicobacter pylori [123–125], or may be located at the C- or at the N-terminus of a specific chaperone, as in the case of several UreE [126] and of UreG from Mycobacterium tuberculosis [127], Streptomyces coelicolor and Glycine max [128] for urease, HypB from Bradyrhizobium japonicum [129] and SlyD from Escherichia coli [130] for hydrogenase, and CooJ from Rhodospirillum rubrum [131] for CO dehydrogenase.
On the other hand, the nickel delivery function is performed using nickel binding sites on specific metallo-chaperones, such as UreE for urease [109,132–134], HypA, and possibly HypB, for hydrogenase [20], and CooJ for carbon monoxide dehydrogenase [131]. Finally, hydrolysis of nucleotide triphosphates is required to complete the biosynthesis of the enzyme with a reaction catalyzed by homologous P-loop GTPases: HypB intervenes in the activation of both hydrogenase and urease [135], AcsF plays its function in the activation of acetyl-CoA decarbonylase synthase [136], CooC is involved in the assembly of carbon monoxide dehydrogenase [137], while UreG is the GTPase essential for urease assembly [109].
UreG from many organisms, including the pathogenic bacteria M. tuberculosis and H. pylori, has been reported to be an intrinsically disordered protein, existing in a flexible behavior in vitro in the absence of other protein partners [127,138–141]. This observation reveals that, at least for urease, protein disorder plays a role in the protein-protein interaction network leading to enzyme activation, and represents a further post-translational mechanism to regulate the Ni2+-dependent enzymatic activity. Furthermore, intrinsic disorder has been found also in some portions of other urease proteins, such as in the C-terminal sequence of UreE involved in the protein interaction with metal ions [134,142], which in turn regulates UreE interaction with UreG [142], and in the C-terminal sequence of UreF, which is disordered in the free form of the protein and becomes correctly folded upon UreF-UreD interaction [143].
3.2.3 Nickel Sensors
The crucial players of nickel homeostasis networks are specific nickel-responsive transcriptional regulators, which specifically bind promoters of genes involved in Ni2+ homeostasis, such as nickel importers or efflux pumps, nickel storage proteins, nickel chaperones and nickel-dependent enzymes. These proteins couple specific metal ion binding with a change in their DNA binding affinity and/or specificity, thus translating the concentration of a certain metal ion into a change in transcriptional response. Several nickel sensors, belonging to different families of regulators, have been discovered and characterized, including NikR (NikR family), RcnR (RcnR/CsoR family), NmtR (ArsR/SmtB family), KmtR (ArsR/SmtB family), SrnRQ (ArsR/SmtB family), and Nur (Fur family) [144].
These proteins exist as homo-dimers or homo-tetramers and usually act as transcriptional repressors and negatively control mRNA synthesis, preventing RNA polymerase from initiating the transcription at the promoter. Ni2+ ions bind in regulatory sites, acting (i) as co-repressors, increasing the affinity of the protein repressor for the DNA operator sequences: this is the case of proteins that regulate genes for metal ion efflux, storage, trafficking, and tolerance (RcnR, NmtR, KmtR); (ii) as inducers, decreasing the affinity of the repressor for the DNA operator sequences, eventually leading to transcriptional activation of genes for membrane uptake systems (NikR, SrnRQ, Nur). Only in the case of NikR from H. pylori the metal-responsive transcriptional regulator act as pleiotropic regulator, exerting a dual control, both as activator and repressor, on different promoters [144].
Nickel-protein interactions, selectively driven by the coordination chemistry and geometry of metal binding sites, usually octahedral or square planar, are propagated away from the specific metal binding site through changes in protein structure and/or dynamics, along the protein backbone, resulting in a modification of the DNA binding affinity of the protein. For example, binding of metal ions to their specific coordination sites within the ArsR/SmtB family drives an allosteric change, with the stabilization of a protein conformer with low affinity for DNA, and decreases the internal dynamics of the protein backbone, leading to the unavailability, in energetic terms, of the conformer that features high affinity for DNA [145]. On the other hand, in case of HpNikR, the presence of bound Ni2+ [146,147] does not induce, by itself, the stabilization of a specific conformer, and increases the protein dynamics unlocking inter-domain motions, supporting the view that the likely mechanism of interaction of the protein with its operator DNA sequence involves a selection of the correct conformation coupled with an induced fit mechanism facilitated by the presence of bound Ni2+ [148,149]. These events produce a finely tuned metabolic response driven by Ni2+ ions, including the coordinated control of the entire machinery of metallo-enzyme synthesis and activation, as well as the systems of homeostasis that involve competitive Ni2+ uptake, intracellular accumulation, and extrusion.
3.3 Nickel-Obligate Microorganisms with Severe Impact on Human Health
3.3.1 Helicobacter pylori as a Nickel-Dependent Pathogen: A Possible Correlation between Nickel Intake and Cancer Development
Helicobacter pylori is a Gram-negative bacterium and the principal causative agent of many acute and chronic gastric pathologies, including peptic ulcer. It is a major factor of risk for the insurgence of gastric carcinomas and lymphomas [150]; accordingly, the WHO classified the bacterium as a class 1 carcinogen in 1994. After the colonization, untreated H. pylori infections persist for the entire life of the host because of the inability of the human immune response to efficiently counter the bacterium [151]. H. pylori is widespread worldwide, and infects up to 50% and 80% of adults in industrialized and developing countries, respectively.
Consistent with the hostility of the human stomach as a habitat for bacterial growth, H. pylori has set up a number of adaptive mechanisms, allowing the bacterium to promptly respond to environmental stresses, such as mild to strong acidity, fluctuating nutrient availability and osmolarity, oxygen tension and host immune responses. These adaptive responses rely on transcriptional regulatory networks that control coordinated expression of tolerance and virulence genes in space and time [152]. Key virulence determinants for the processes of bacterial colonization include two essential nickel enzymes, that is, Ni2+-dependent urease, which allows buffering of the acidic gastric environment, and a [NiFe]-hydrogenase, which allows consumption of energy-yielding hydrogen, freely available in the gastric niche. These enzymes were shown to be required for full colonization of mouse stomach.
The importance of Ni2+ ions in H. pylori physiology is emphasized by the number and diversity of proteins involved in nickel homeostasis, which control the activity of nickel enzymes at the cellular level with three different processes: (i) regulation of transcription and expression of genes encoding metallo-enzymes and metal trafficking proteins, performed by the pleiotropic Ni2+ sensor NikR; (ii) specific delivery of the metal ion cofactors to the protein active sites and metal-binding pockets, performed by the urease and hydrogenase chaperones; and (iii) intracellular metal ion accumulation and storage, performed by the outer membrane transporters FrpB4 and FecA3, the Ni2+ permease NixA, the Ni2+ efflux system CznABC, the cytoplasmic Ni2+ accumulators Hpn, Hpn-like, and HspA. Due to their importance for bacterial pathogenesis, these processes all represent possible targets for the development of alternative antibacterial strategies. Coherently, ureG-negative mutants (which can synthesize apo-urease but are unable to incorporate Ni2+ into the active site) are deficient in colonizing the gastric mucosa of nude mice, thereby highlighting a link between Ni activation of urease and host colonization [153].
As urease is one of the most abundant enzymes in H. pylori, representing up to 10% of the bacterial protein content, it is likely that the demand for nickel is high in this microorganism. Similarly to other pathogens, metal ion starvation triggers the expression of toxins and metal ion scavengers in H. pylori, allowing the pathogen to compete with the host for these essential nutrients. Accordingly, infections by H. pylori have been epidemiologically linked to certain forms of anemia and impaired iron metabolism in hosts, proving that bacterial infection can significantly impact on the balance of human metal ion homeostasis [154]. Nickel is naturally abundant in many types of food [5], therefore, it is plausible that H. pylori enters in contact with the necessary amount of Ni2+ to satisfy its metabolic demand in the stomach. The absence of known enzymes that require nickel as an essential cofactor in higher eukaryotes suggests that there is no competition for nickel between the host and the bacterium. However, the flux of nickel is probably not continuous and occurs in successive batch-like conditions rather than a constant stream of metals. In addition, nickel availability is also a function of pH, which fluctuates widely within the gastric compartments. As a consequence, H. pylori must be able to accumulate Ni2+ when relatively high exogenous concentrations are available. This intracellular nickel reservoir, most likely bound to Ni2+ storage proteins, would in turn be available for urease or/and hydrogenase maturation when Ni2+ concentrations are limited. This was demonstrated in a recent study, aimed to correlate Ni2+ levels introduced with diet, and colonization of H. pylori [155]. Even in Ni2+-depleted diet, wild-type H. pylori is able to colonize the host stomach because it can store Ni2+ ions in the intracellular environment, relying on Hpn and Hpn-like Ni2+ accumulators. If these proteins are mutated, the colonization levels of Ni-depleted animals are statistically lower than colonization levels of Ni-fed animals.
3.3.2 Nickel Homeostasis and Intracellular Parasitism: Eukaryotic and Prokaryotic Pathogens
Some nickel-dependent prokaryotic and eukaryotic microorganisms infect the human body as intracellular parasites. The most important eukaryotic pathogens are Trypanosomatids, such as Trypanosoma cruzi and Leishmania spp., which are protozoa causing Chagas’ disease and leishmaniasis, respectively. Their life cycle includes an intracellular stage in the mammalian host. These organisms produce a Ni2+-dependent glyoxalase I, which is essential for pathogen survival as it serves to detoxify methylglyoxal, a toxic product of glycolysis and other metabolic pathways (see Section 3.1) [156]. The use of Ni2+ ions for glyoxalase activity in trypanosomatids is exceptional, as in other eukaryotes the enzyme cofactor is zinc. This reflects distinctive substrate selectivity between the enzyme of the pathogen and human host, indicating that the enzyme could be a potential chemotherapeutic target against the protozoan parasite [157].
One of the most important intracellular infective bacteria worldwide is Mycobacterium tuberculosis, the etiological agent of tuberculosis. This bacterium establishes infection in adult humans primarily in the lungs by infecting macrophages, where it can remain asymptomatic or can become active manifesting the clinical symptoms. Currently, the WHO estimates that one-third of the world’s population is latently infected with tuberculosis, with 9.27 million new cases and 1.76 million deaths annually [158]. Upon infection, M. tuberculosis transfers into a bactericidal phagosome that is subsequently fused to a lysosome, creating a vacuole characterized by an acidic (pH 5.5), highly nitro-oxidative, nutrient-limiting, and possibly hypoxic microenvironment. In this hostile environment, M. tuberculosis relies on its exceptional metabolic flexibility and ability to adapt, replicate and/or persist within changing and adverse microenvironments. Initially, it has been proposed that M. tuberculosis Ni2+-dependent urease contributes to alkalize the mycobacterium-containing vacuole in macrophages, promoting a more favorable environment for the intracellular persistence of the bacilli. However, a recent study indicated that urease activity does not influence the general bacterial fitness in vitro. In addition, the alkalizing effect of the M. tuberculosis urease activity has only been found in resting macrophages, while it is not present in the more acidic microenvironment of the phagolysosomal compartment of activated macrophages, suggesting that the alkalizing effect provided by the mycobacterial urease activity is somehow modest [159]. On the other hand, the observation that M. tuberculosis urease expression and activity increase upon nitrogen deprivation [160], suggests that urea may be a potential source of nitrogen for M. tuberculosis. Consistently, this bacterium assimilates urea, as major nitrogen source, in an urease-dependent manner, this process generating ammonia, which is one of the key precursors for the biosynthesis of essential amino acids such as glutamate [159].
The apparent dispensability of urease activity in a mouse model suggests that other readily available nitrogen sources within the host cell could bypass the need for M. tuberculosis to metabolize urea, with a general functional redundancy, metabolic versatility, and compensatory mechanisms that characterize M. tuberculosis ability to adapt to virtually any microenvironment encountered in its host, in which carbon and nitrogen sources may vary qualitatively and quantitatively [159]. In addition, the ability of M. tuberculosis to infect extra-pulmonary sites may require the urease activity for bacillus persistence or replication at specific sites of infection within the host. In this case, the ability to utilize urea as a source of nitrogen could be critical at specific sites of infection where other sources of nitrogen are limited [159].
The mycobacterial urease activity may also have a role in evading the immune response of the host in response to the bacterial infection. Indeed, it is known that exogenous NH4Cl blocks phagosome-lysosome fusion and promotes phagosome-endosome fusion in mouse mononuclear phagocytes [161,162]. This suggests that ammonia production by M. tuberculosis may partly slow down the phagolysosome formation, providing the bacterium with less hostile environmental conditions. In addition, M. bovis was demonstrated to attenuate the MHC-II molecule expression in infected macrophages, with consequent inhibition of CD4+ T-cell activation and general suppression of the host immune system [163,164].
The ability of the mycobacterial urease to alkalize the microenvironment has been exploited to develop a novel vaccine candidate, based on M. bovis urease, consisting of a urease-inactive M. bovis bacterium that expresses the Listeria monocytogenes listeriolysin [165]. In the absence of the mycobacterial urease activity, the mild acidic pH of the bacillus-containing vacuole provides an ideal pH environment for listeriolysin perforation of the vacuole membrane. The phagosome lysis promotes mycobacterial translocation into the macrophage cytoplasm, leading to cellular apoptosis and stronger immune responses that provide better protection upon M. tuberculosis reinfection.
The importance of metal ion homeostasis for M. tuberculosis is underlined by the observation that its genome encodes an atypically large number (at least twelve) metal sensors of the ArsR-SmtB family [144], allowing to speculate that these proteins enable the pathogen to respond rapidly to metal fluxes in the phagosome [166]. Notably, two of these metal-dependent transcription factors, named NmtR and KmtR, specifically respond to Ni2+ and Co2+ concentrations and regulate the expression of membrane proteins – an ATPase efflux pump (NmtA) and a cation diffusion facilitator (CDF), respectively – that control metal ion efflux from the cell [166]. The reason of having two Ni2+/Co2+ sensors for metal ion efflux systems has been explained with the different affinity observed for these two proteins and their cognate metal ions, which allows a fine modulation of transcriptional response [166]. In particular, KmtR appears to have higher affinity for Ni2+ and Co2+, and it is therefore able to detect the basal level of the two metal ions, thus de-repressing the gene of CDF transport at low metal concentrations. Only a higher cytoplasmic metal ion threshold is able to bind NmtR, which presents a lower affinity for Ni2+ and Co2+ as compared to KmtR, allowing the expression of the NmtA efflux pump [166]. These data support the significance of these metal ions for this pathogen as well as the bacterial ability to respond to metal fluxes, depending on two levels of Ni2+ or Co2+ sensing.
4 Nickel Essentiality in Animals and Humans
Nickel is classified as a “possibly essential element” for animals and humans since the 1970s [19]. Nickel deficiency in humans has never been reported, as, in general, human nickel intake greatly exceeds the requirements, which have been estimated between 5 and 50 μg per day [22].
4.1 Effects of Nickel Depletion in Higher Organisms
The essentiality of Ni2+ for higher eukaryotes, firstly proposed in 1920s, is still debated [5]. The importance of this metal for animals and humans has been tested evaluating the effect of nickel deficiency in animals. Rats grown in the absence or in low-abundance of nickel exhibit very severe consequences, as depressed growth, low hemoglobin, red blood cell counts and activity of several liver and kidney enzymes, as well as the presence of urea, ATP, and glucose in serum, and also glucose, glycogen, and triglycerides in the liver [167]. The growth dependence on nickel was more significant in the second depleted generation, which also showed anemia, manifested in decreased hemoglobin and hematocrit values [5]. Nickel deprivation during reproduction in rats increased perinatal mortality [168], while in breeding goats it significantly decreased the success of first insemination and conception rate and increased the number of breeding attempts to achieve pregnancy and the abortion rate [169]. This effect seems to be related to the ability of nickel to be involved in cyclic nucleotide-gated (CNG) channel functions [170]. CNG channels are located in a number of organs, including the central nervous, urogenital, and reproductive systems. The CNG channels also have an important role in kidney function and, thus, in sodium metabolism [170].
Additionally, nickel deficiency impairs the absorption of iron from the intestine and the concentrations of several metals, such as iron, copper, and zinc, were also decreased in the liver of nickel-depleted animals [171,172]. Nickel deficiency also results in lowered specific activities of many enzymes involved in carbohydrate and amino acid metabolism. It was suggested that the essentiality of nickel for higher organisms is linked to its modulation of gene expression in eukaryotic cells [58]. Nickel has been proposed to be also involved in membrane stability or in lipid metabolism [173].
4.2 Influence of Nickel Availability on Gut’s Microflora
The wide presence of Ni2+-dependent enzymes in prokaryotes and unicellular eukaryotes raises the possibility that nickel, not required by the human and animal body itself, is rather needed for the normal development of the gut’s microflora, which impacts host metabolism in a variety of ways and is responsible for several metabolic and cardiovascular diseases [174]. The gut microbiota comprises the largest microbial community in the human body and represents one of the highest cellular densities in natural ecosystems, reaching 1011 to 1012 cells/mL of luminal content, and includes species of all kingdoms of life, Eukaryotes, Bacteria, and Archaea [175]. The bacterial composition is made mostly by the Firmicutes and by the Bacteroidetes phyla and includes Proteobacteria and Actinobacteria [176]. This population comes in contact with all the nutrients that are ingested through diet, creating a symbiotic equilibrium with the host for the utilization of micronutrients, including metal ion cofactors. In particular, in the human intestinal tract, there are large populations of bacterial cells with the potential to bind and sequester metals that enter the body, such as the ones from the Lactobacillus group [177]. Therefore, it has been postulated that gut microorganisms may play a role in protecting the host from the toxic potential of metal ions ingested with diet, preventing their entry to the body from the intestine, and this could also be the case with Ni2+ ions [177]. Accordingly, while Ni2+ ions administered by other routes, such as inhalation or dermal exposure, show a carcinogenic potential, there is no epidemiological evidence on possible cancer risk from dietary nickel exposures, and this may be due to the protective effect of the gut microflora.
Besides this function, it is plausible that Ni2+-dependent enzymes play important roles also for the intestine community, and that Ni2+ homeostasis in some of these microorganisms is important for developing a health-promoting gut microflora. Indeed, early work reported that several bacterial species found in the human and animal intestine show consistent urease activity [178]. Among them, some species of the genus Bifidobacterium, such as B. bifidum and B. longum, are noteworthy [179], because of the known probiotic effects of some types of Bifidobacteria, that for decades have been added to food to promote healthy effects [180]. Other urease-producing species, coming from the genus Lactobacillus, such as L. fermentum, are interesting as they codify an acid urease which possesses an optimum of pH at 3–4 [178].

Reaction catalysed by methyl coenzyme-M reductase, and schematic structure of the nickel-containing active site.
Methanogenic archaea compose an important component of the microorganisms living in human and animal guts, where they use dihydrogen as reductant to produce CH4 [181]. In humans, Methanobrevibacter smithii is the main actor of the methanogen population. Methanogens have been proposed to play a key role in the pathogenesis of irritable bowel syndrome and chronic constipation [181]. The last step of methane formation is catalyzed by methyl coenzyme-M reductase [182]. The reaction involves methane formation concomitantly with the formation of a disulfide bond between coenzyme M and coenzyme B (Figure 7). The active site of this enzyme contains coenzyme F430, a Ni tetrapyrrole hydrocorphin with the metal in the Ni(I) oxidation state in the active form. Structural data on the active site has been obtained for the inactive oxidized Ni(II) state (Figure 7). This correlates the presence of nickel in the diet with the healthy methanogenic activity in the guts.
5 Conclusions and Outlook
The wide use of nickel in many modern technologies and in objects of common use increases the amount of nickel release and accumulation into the environment and the possibility for humans to come in contact with this metal. Indeed, nickel-containing objects, such as coins, are the cause of severe occupational health problems, whose social costs have to be considered worldwide.
The double nature of nickel, that is, acting both as a toxic and a beneficial element for human health, was proposed already in the early 1900s. The requirement of nickel as a cofactor in the active sites of enzymes has been recognized in 1975 and since then several studies have been conducted, which were aimed to describe the molecular mechanisms of the effects of nickel on all forms of life, including higher organisms.
However, the rationale of nickel carcinogenesis and allergy in humans, as well as the cascade of events involving metal ion-dependent gene regulation and protein-protein interactions leading to nickel homeostasis in eukaryotes, is yet largely unknown. A full understanding of the molecular aspects of the effects of nickel on human health represents therefore an important challenge and a future task for bioinorganic chemistry, with the potential to have a significant impact for the human population.
- ABC:
-
ATP-binding cassette
- ACD:
-
allergic contact dermatitis
- acetyl-CoA:
-
acetyl-coenzyme A
- APC:
-
antigen-presenting cell
- Ard:
-
acireductone dioxygenase
- ARNT:
-
aryl hydrocarbon receptor nuclear translocator
- BPU:
-
Bacillus pasteurii urease
- CDF:
-
cation diffusion facilitator
- CNG:
-
cyclic nucleotide-gated
- DMT-1:
-
divalent cation transporter
- EDTA:
-
ethylenediamine-N,N,N′,N′-tetraacetate
- Glx:
-
glyoxalase
- GSH:
-
glutathione (reduced)
- GTP:
-
guanosine 5′-triphosphate
- HIF:
-
hypoxia-inducible factor
- HRE:
-
hypoxia-responsive enhancer
- IKK:
-
Iκ-B kinase
- IRP:
-
iron-regulatory protein
- LPS:
-
lipopolysaccharide
- MAPK:
-
mitogen-activated protein kinase
- MHC:
-
major histocompatibility complex
- NDRG:
-
N-myc downstream regulated gene 1
- NF-κB:
-
nuclear factor κ-B
- NiCoT:
-
nickel/cobalt permease
- ROS:
-
reactive oxygen species
- SCD:
-
systemic contact dermatitis
- TCR:
-
T-cell receptor
- TLR:
-
toll-like receptor
- VEGF:
-
vascular endothelial growth factor
- VHL:
-
von Hippel-Lindau tumor suppressor
References
S. Ciurli, S. Mangani, in Handbook on Metalloproteins, Eds. I. Bertini, A. Sigel, H. Sigel, Marcel Dekker, New York, USA, 2001, pp. 669–708.
E. Merian, Toxicol. Envir. Chem. 1984, 8, 9–38.
R. G. Garrett, in Metal Ions in Biology and Medicine, Eds J. A. Centeno, P. Collery, G. Fernet, R. B. Finkelman, H. Gibb, J.-C. Etienne; John Libbey Eurotext, Paris, 2000, Vol. 6, pp. 508–510.
C. L. Coyle, E. I. Stiefel, in The Bioinorganic Chemistry of Nickel, Ed J. R. Lancaster, Jr., VCH, New York, 1988, pp. 1–28.
E. Denkhaus, K. Salnikow, Crit. Rev. Oncol. Hematol. 2002, 42, 35–56.
D. G. Barceloux, J. Toxicol. Clin. Toxicol. 1999, 37, 239–258.
M. Van Soestbergen, F. W. Sunderman, Jr., Clin. Chem. 1972, 18, 1478–1484.
N. Asato, M. Soestbergen, F. W. Sunderman, Jr., Clin. Chem. 1975, 21, 521–527.
J. D. Glennon, B. Sarkar, Biochem. J. 1982, 203, 15–23.
F. Torres, M. das Gracas, M. Melo, A. Tosti, Clin. Cosmet. Investig. Dermatol. 2009, 2, 39–48.
R. B. Schiffer, F. W. Sunderman, Jr., R. B. Baggs, J. A. Moynihan, J. Neuroimmunol. 1991, 34, 229–239.
M. Freitas, P. Barcellos-de-Souza, C. Barja-Fidalgo, E. Fernandes, BioMetals 2013, 26, 13–21.
A. Au, J. Ha, M. Hernandez, A. Polotsky, D. S. Hungerford, C. G. Frondoza, J. Biomed. Mater. Res. A 2006, 79, 512–521.
M. Anke, A. Trüpschuch, G. Gunstheimer, in Trace Elements in Man and Animals 10, Eds A. M. Roussel, R. A. Anderson, A. E. Favrier, Kluwer Academic, 2002, pp. 685–686.
S. G. Schafer, W. Forth, Ecotoxicol. Environ. Saf. 1983, 7, 87–95.
J. Tallkvist, A. M. Wing, H. Tjalve, Pharmacol. Toxicol. 1994, 75, 244–249.
M. Muller-Fassbender, B. Elsenhans, A. T. McKie, K. Schumann, Toxicology 2003, 185, 141–153.
International Agency for Research on Cancer, "Chromium, Nickel and Welding", Proceedings of the Monographs on the Evaluation of Carcenigenic Risks to Humans, Lyon, France, 1990.
F. H. Nielsen, D. A. Ollerich, Fed. Proc. 1974, 33, 1767–1772.
Y. Li, D. B. Zamble, Chem. Rev. 2009, 109, 4617–4643.
M. Yusuf, Q. Fariduddin, S. Hayat, A. Ahmad, Bull. Environ. Contam. Toxicol. 2011, 86, 1–17.
EMEA, European Medicines Agency, 2008; http://www.ema.europa.eu/ema.
K. S. Kasprzak, K. Salnikow, in Nickel and Its Surprising Impact in Nature, Vol. 2 of Metal Ions in Life Sciences, Eds A. Sigel, H. Sigel, R. K. O. Sigel, Wiley, Chichester, 2007, Vol. 2, pp. 581–618.
K. S. Kasprzak, F. W. Sunderman, Jr., K. Salnikow, Mutat. Res. 2003, 533, 67–97.
H. M. Shen, Q. F. Zhang, Environ. Health Perspect. 1994, 102 (Suppl. 1), 275–282.
A. R. Oller, M. Costa, G. Oberdorster, Toxicol. Appl. Pharmacol. 1997, 143, 152–166.
K. A. Biedermann, J. R. Landolph, Cancer Res. 1987, 47, 3815–3823.
M. Costa, T. L. Davidson, H. Chen, Q. Ke, P. Zhang, Y. Yan, C. Huang, T. Kluz, Mutat. Res. 2005, 592, 79–88.
N. W. Biggart, M. Costa, Mutat. Res. 1986, 175, 209–215.
J. B. Little, J. M. Frenial, J. Coppey, Teratog. Carcinog. Mutagen. 1988, 8, 287–292.
T. Miura, S. R. Patierno, T. Sakuramoto, J. R. Landolph, Environ. Mol. Mutagen. 1989, 14, 65–78.
F. Z. Arrouijal, H. F. Hildebrand, H. Vophi, D. Marzin, Mutagenesis 1990, 5, 583–589.
B. Kargacin, C. B. Klein, M. Costa, Mutat. Res. 1993, 300, 63–72.
M. Freitas, A. Gomes, G. Porto, E. Fernandes, J. Biol. Inorg. Chem. 2010, 15, 1275–1283.
T. J. O’Brien, S. Ceryak, S. R. Patierno, Mutat. Res. 2003, 533, 3–36.
B. Desoize, In Vivo 2003, 17, 529–539.
A. Hartwig, T. Schwerdtle, Toxicol. Lett. 2002, 127, 47–54.
W. Bal, R. Liang, J. Lukszo, S. H. Lee, M. Dizdaroglu, K. S. Kasprzak, Chem. Res. Toxicol. 2000, 13, 616–624.
L. Lande-Diner, J. Zhang, T. Hashimshony, A. Goren, I. Keshet, H. Cedar, Cold Spring Harb. Symp. Quant. Biol. 2004, 69, 131–138.
Y. W. Lee, C. B. Klein, B. Kargacin, K. Salnikow, J. Kitahara, K. Dowjat, A. Zhitkovich, N. T. Christie, M. Costa, Mol. Cell. Biol. 1995, 15, 2547–2557.
B. Govindarajan, R. Klafter, M. S. Miller, C. Mansur, M. Mizesko, X. Bai, K. LaMontagne, Jr., J. L. Arbiser, Mol. Med. 2002, 8, 1–8.
K. Salnikow, A. Zhitkovich, Chem. Res. Toxicol. 2008, 21, 28–44.
H. Chen, Q. Ke, T. Kluz, Y. Yan, M. Costa, Mol. Cell. Biol. 2006, 26, 3728–3737.
H. Chen, N. C. Giri, R. Zhang, K. Yamane, Y. Zhang, M. Maroney, M. Costa, J. Biol. Chem. 2010, 285, 7374–7383.
R. Martinez-Zamudio, H. C. Ha, Epigenetics 2011, 6, 820–882.
A. A. Karaczyn, F. Golebiowski, K. S. Kasprzak, Chem. Res. Toxicol. 2005, 18, 1934–1942.
Q. Zhang, K. Salnikow, T. Kluz, L. C. Chen, W. C. Su, M. Costa, Toxicol. Appl. Pharmacol. 2003, 192, 201–211.
C. B. Klein, K. Conway, X. W. Wang, R. K. Bhamra, X. H. Lin, M. D. Cohen, L. Annab, J. C. Barrett, M. Costa, Science 1991, 251, 796–799.
S. Orrenius, B. Zhivotovsky, P. Nicotera, Nat. Rev. Mol. Cell. Biol. 2003, 4, 552–565.
K. Salnikow, M. Gao, V. Voitkun, X. Huang, M. Costa, Cancer Res. 1994, 54, 6407–6412.
W. Bal, A. M. Protas, K. S. Kasprzak, in Metal Ions in Toxicology: Effects, Interactions, Interdependencies, Vol. 8 of Metal Ions in Life Sciences, Eds A. Sigel, H. Sigel, R. K. O. Sigel, Royal Society of Chemistry, Cambridge, 2011, pp. 319–373.
K. Salnikow, M. V. Blagosklonny, H. Ryan, R. Johnson, M. Costa, Cancer Res. 2000, 60, 38–41.
S. Salceda, J. Caro, J. Biol. Chem. 1997, 272, 22642–22647.
W. C. Hon, M. I. Wilson, K. Harlos, T. D. Claridge, C. J. Schofield, C. W. Pugh, P. H. Maxwell, P. J. Ratcliffe, D. I. Stuart, E. Y. Jones, Nature 2002, 417, 975–978.
S. J. Freedman, Z. Y. Sun, F. Poy, A. L. Kung, D. M. Livingston, G. Wagner, M. J. Eck, Proc. Natl. Acad. Sci. USA 2002, 99, 5367–5372.
P. Maxwell, K. Salnikow, Cancer Biol. Ther. 2004, 3, 29–35.
K. Salnikow, S. P. Donald, R. K. Bruick, A. Zhitkovich, J. M. Phang, K. S. Kasprzak, J. Biol. Chem. 2004, 279, 40337–40344.
D. Zhou, K. Salnikow, M. Costa, Cancer Res. 1998, 58, 2182–2189.
H. Cangul, K. Salnikow, H. Yee, D. Zagzag, T. Commes, M. Costa, Cell. Biol. Toxicol. 2002, 18, 87–96.
T. P. Ellen, Q. Ke, P. Zhang, M. Costa, Carcinogenesis 2008, 29, 2–8.
M. A. Zoroddu, M. Peana, T. Kowalik-Jankowska, H. Kozlowski, M. Costa, J. Inorg. Biochem. 2004, 98, 931–939.
M. A. Zoroddu, M. Peana, S. Medici, R. Anedda, Dalton Trans. 2009, 5523–5534.
J. G. Burchfield, A. J. Lennard, S. Narasimhan, W. E. Hughes, V. C. Wasinger, G. L. Corthals, T. Okuda, H. Kondoh, T. J. Biden, C. Schmitz-Peiffer, J. Biol. Chem. 2004, 279, 18623–18632.
J. Hwang, Y. Kim, H. B. Kang, L. Jaroszewski, A. M. Deacon, H. Lee, W. C. Choi, K. J. Kim, C. H. Kim, B. S. Kang, J. O. Lee, T. K. Oh, J. W. Kim, I. A. Wilson, M. H. Kim, J. Biol. Chem. 2011, 286, 12450–12460.
K. Kondo, T. Ozaki, Y. Nakamura, S. Sakiyama, Biochem. Biophys. Res. Commun. 1995, 216, 209–215.
K. Kanaya, A. L. Tsai, T. Kamitani, Biochem. Biophys. Res. Commun. 2002, 290, 294–299.
S. Oshiro, K. Nozawa, M. Hori, C. Zhang, Y. Hashimoto, S. Kitajima, K. Kawamura, Biochem. Biophys. Res. Commun. 2002, 290, 213–218.
C. Harford, B. Sarkar, Biochem. Biophys. Res. Commun. 1995, 209, 877–882.
S. F. Martin, P. R. Esser, F. C. Weber, T. Jakob, M. A. Freudenberg, M. Schmidt, M. Goebeler, Allergy 2011, 66, 1152–1163.
M. Schmidt, M. Goebeler, J. Mol. Med. (Berl.) 2011, 89, 961–970.
P. S. Friedmann, Toxicol. Lett. 2006, 162, 49–54.
K. Landsteiner, J. Jacobs, J. Exp. Med. 1935, 61, 643–656.
H. J. Thierse, K. Gamerdinger, C. Junkes, N. Guerreiro, H. U. Weltzien, Toxicology 2005, 209, 101–107.
L. Lu, J. Vollmer, C. Moulon, H. U. Weltzien, P. Marrack, J. Kappler, J. Exp. Med. 2003, 197, 567–574.
S. Dai, F. Crawford, P. Marrack, J. W. Kappler, Proc. Natl. Acad. Sci. USA 2008, 105, 11893–11897.
F. Sinigaglia, J. Invest. Dermatol. 1994, 102, 398–401.
Y. Wang, S. Dai, Immunol. Res. 2013, 55, 83–90.
P. Griem, C. von Vultee, K. Panthel, S. L. Best, P. J. Sadler, C. F. Shaw, 3rd, Eur. J. Immunol. 1998, 28, 1941–1947.
K. Gamerdinger, C. Moulon, D. R. Karp, J. Van Bergen, F. Koning, D. Wild, U. Pflugfelder, H. U. Weltzien, J. Exp. Med. 2003, 197, 1345–1353.
O. Wildner, T. Lipkow, J. Knop, Exp. Dermatol. 1992, 1, 191–198.
M. Goebeler, G. Meinardus-Hager, J. Roth, S. Goerdt, C. Sorg, J. Invest. Dermatol. 1993, 100, 759–765.
M. Goebeler, A. Trautmann, A. Voss, E. V. Brocker, A. Toksoy, R. Gillitzer, Am. J. Pathol. 2001, 158, 431–440.
M. Goebeler, J. Roth, E. B. Brocker, C. Sorg, K. Schulze-Osthoff, J. Immunol. 1995, 155, 2459–2467.
M. Goebeler, R. Gillitzer, K. Kilian, K. Utzel, E. B. Brocker, U. R. Rapp, S. Ludwig, Blood 2001, 97, 46–55.
M. Schmidt, B. Raghavan, V. Muller, T. Vogl, G. Fejer, S. Tchaptchet, S. Keck, C. Kalis, P. J. Nielsen, C. Galanos, J. Roth, A. Skerra, S. F. Martin, M. A. Freudenberg, M. Goebeler, Nat. Immunol. 2010, 11, 814–819.
M. Schmidt, M. Hupe, N. Endres, B. Raghavan, S. Kavuri, P. Geserick, M. Goebeler, M. Leverkus, J. Cell. Mol. Med. 2010, 14, 1760–1776.
A. Trautmann, M. Akdis, D. Kleemann, F. Altznauer, H. U. Simon, T. Graeve, M. Noll, E. B. Brocker, K. Blaser, C. A. Akdis, J. Clin. Invest. 2000, 106, 25–35.
S. Rothman, Dermatol. Wochenschr. 1930, 2, 98–99.
R. Spiewak, J. Pietowska, K. Curzytek, Expert Rev. Clin. Immunol. 2007, 3, 851–859.
T. Menne, K. Rasmussen, Contact Dermatitis 1990, 23, 57–58.
J. P. Thyssen, K. Ross-Hansen, T. Menne, J. D. Johansen, Contact Dermatitis 2010, 63, 102–106.
Y. Yoshihisa, T. Shimizu, Dermatol. Res. Pract. 2012, 2012, 749561.
E. Mann, U. Ranft, G. Eberwein, D. Gladtke, D. Sugiri, H. Behrendt, J. Ring, T. Schafer, J. Begerow, J. Wittsiepe, U. Kramer, M. Wilhelm, Contact Dermatitis 2010, 62, 355–362.
R. Darlenski, J. Kazandjieva, K. Pramatarov, Int. J. Dermatol. 2012, 51, 523–530.
J. P. Thyssen, T. Menne, Chem. Res. Toxicol. 2010, 23, 309–318.
R. I. Nijhawan, M. Molenda, M. J. Zirwas, S. E. Jacob, Dermatol. Clin. 2009, 27, 355–364,
D. Bonamonte, A. Cristaudo, F. Nasorri, T. Carbone, O. De Pita, G. Angelini, A. Cavani, Contact Dermatitis 2011, 65, 293–301.
M. Minelli, D. Schiavino, F. Musca, M. E. Bruno, P. Falagiani, G. Mistrello, G. Riva, M. Braga, M. C. Turi, V. Di Rienzo, C. Petrarca, C. Schiavone, M. Di Gioacchino, Int. J. Immunopathol. Pharmacol. 2010, 23, 193–201.
Nickel and Its Surprising Impact in Nature, Eds A. Sigel, H. Sigel, R. K. O. Sigel, John Wiley & Sons, Chichester, Met. Ions Life Sci. 2007, 2, pp. 1–702.
S. W. Ragsdale, J. Biol. Chem. 2009, 284, 18571–18575.
H. L. Mobley, M. D. Island, R. P. Hausinger, Microbiol. Rev. 1995, 59, 451–480.
S. Wyllie, A. H. Fairlamb, Semin. Cell. Dev. Biol. 2011, 22, 271–277.
N. Sukdeo, E. Daub, J. F. Honek, in Nickel and Its Surprising Impact in Nature, Vol. 2 of Metal Ions in Life Sciences, Eds A. Sigel, H. Sigel, R. K. O Sigel, John Wiley & Sons, Chichester, 2007, pp. 445–472.
N. Sukdeo, J. F. Honek, Biochim. Biophys. Acta 2007, 1774, 756–763.
T. C. Pochapsky, T. Ju, M. Dang, R. Beaulieu, G. M. Pagani, B. OuYang, in Nickel and Its Surprising Impact in Nature, Vol. 2 of Metal Ions in Life Sciences, Eds A. Sigel, H. Sigel, R. K. O Sigel, John Wiley & Sons, Chichester, 2007, Vol. 2, pp. 473–500.
Q. Xu, R. Schwarzenbacher, S. S. Krishna, D. McMullan, S. Agarwalla, K. Quijano, P. Abdubek, E. Ambing, H. Axelrod, T. Biorac, J. M. Canaves, H.-J. Chiu, M.-A. Elsliger, C. Grittini, S. K. Grzechnik, M. DiDonato, J. Hale, E. Hampton, G. W. Han, J. Haugen, M. Hornsby, L. Jaroszewski, H. E. Klock, M. W. Knuth, E. Koesema, A. Kreusch, P. Kuhn, M. D. Miller, K. Moy, E. Nigoghossian, J. Paulsen, R. Reyes, C. Rife, G. Spraggon, R. C. Stevens, H. van den Bedem, J. Velasquez, A. White, G. Wolf, K. O. Hodgson, J. Wooley, A. M. Deacon, A. Godzik, S. A. Lesley, I. A. Wilson, Proteins: Structure, Function, and Bioinformatics 2006, 64, 808–813.
T. Ju, R. B. Goldsmith, S. C. Chai, M. J. Maroney, S. S. Pochapsky, T. C. Pochapsky, J. Mol. Biol. 2006, 363, 823–834.
S. Ciurli, in Nickel and Its Surprising Impact in Nature, Vol. 2 of Metal Ions in Life Sciences, Eds A. Sigel, H. Sigel, R. K. O Sigel, Wiley, Chichester, 2007, pp. 241–278.
B. Zambelli, F. Musiani, S. Benini, S. Ciurli, Acc. Chem. Res. 2011, 44, 520–530.
S. Benini, W. R. Rypniewski, K. S. Wilson, S. Miletti, S. Ciurli, S. Mangani, Structure Fold. Des. 1999, 7, 205–216.
F. Musiani, E. Arnofi, R. Casadio, S. Ciurli, J. Biol. Inorg. Chem. 2001, 3, 300–314.
S. Benini, W. R. Rypniewski, K. S. Wilson, S. Mangani, S. Ciurli, J. Am. Chem. Soc. 2004, 126, 3714–3715.
S. Ciurli, S. Benini, W. R. Rypniewski, K. S. Wilson, S. Miletti, S. Mangani, Coord. Chem. Rev. 1999, 190–192, 331–355.
W. Lubitz, M. van Gastel, W. Gartner, Nickel and Its Surprising Impact in Nature, Vol. 2 of Metal Ions in Life Sciences, Eds A. Sigel, H. Sigel, R. K. O Sigel, John Wiley & Sons, Chichester, 2007, pp. 279–322.
P. M. Vignais, B. Billoud, Chem. Rev. 2007, 107, 4206–4272.
R. J. Maier, Biochem. Soc. Trans. 2005, 33, 83–85.
C. Navarro, L.-F. Wu, M.-A. Mandrand-Berthelot, Mol. Microbiol. 1993, 9, 1181–1191.
H. Lebrette, M. Iannello, J. C. Fontecilla-Camps, C. Cavazza, J. Inorg. Biochem. 2013, 121, 16–18.
L. Macomber, R. P. Hausinger, Metallomics 2011, 3, 1153–1162.
D. H. Nies, FEMS Microbiol. Rev. 2003, 27, 313–339.
F. N. Stahler, S. Odenbreit, R. Haas, J. Wilrich, A. H. Van Vliet, J. G. Kusters, M. Kist, S. Bereswill, Infect. Immun. 2006, 74, 3845–3852.
A. Rodrigue, G. Effantin, M. A. Mandrand-Berthelot, J. Bacteriol. 2005, 187, 2912–2916.
R. Ge, Y. Zhang, X. Sun, R. M. Watt, Q. Y. He, J. D. Huang, D. E. Wilcox, H. Sun, J. Am. Chem. Soc. 2006, 128, 11330–11331.
R. J. Maier, S. L. Benoit, S. Seshadri, BioMetals 2007, 20, 655–664.
Y. B. Zeng, D. M. Zhang, H. Li, H. Sun, J. Biol. Inorg. Chem. 2008, 13, 1121–1131.
F. Musiani, B. Zambelli, M. Stola, S. Ciurli, J. Inorg. Biochem. 2004, 98, 803–813.
B. Zambelli, F. Musiani, M. Savini, P. Tucker, S. Ciurli, Biochemistry 2007, 46, 3171–3182.
R. Real-Guerra, F. Staniscuaski, B. Zambelli, F. Musiani, S. Ciurli, C. R. Carlini, Plant Mol. Biol. 2012, 78, 461–475.
J. W. Olson, R. J. Maier, J. Bacteriol. 2000, 182, 1702–1705.
C. Wulfing, J. Lombardero, A. Pluckthun, J. Biol. Chem. 1994, 269, 2895–2901.
R. K. Watt, P. W. Ludden, J. Biol. Chem. 1998, 273, 10019–10025.
H.-K. Song, S. B. Mulrooney, R. Huber, R. P. Hausinger, J. Biol. Chem. 2001, 276, 49359–49364.
H. Remaut, N. Safarov, S. Ciurli, J. Van Beeumen, J. Biol. Chem. 2001, 276, 49365–49370.
K. Banaszak, V. Martin-Diaconescu, M. Bellucci, B. Zambelli, W. Rypniewski, M. J. Maroney, S. Ciurli, Biochem. J. 2012, 441, 1017–1026.
N. Mehta, J. W. Olson, R. J. Maier, J. Bacteriol. 2003, 185, 726–734.
H. K. Loke, P. A. Lindahl, J. Inorg. Biochem. 2003, 93, 33–40.
R. L. Kerby, P. W. Ludden, G. P. Roberts, J. Bacteriol. 1997, 179, 2259–2266.
P. Neyroz, B. Zambelli, S. Ciurli, Biochemistry 2006, 45, 8918–8930.
B. Zambelli, M. Stola, F. Musiani, K. De Vriendt, B. Samyn, B. Devreese, J. Van Beeumen, P. Turano, A. Dikiy, D. A. Bryant, S. Ciurli, J. Biol. Chem. 2005, 280, 4684–4695.
B. Zambelli, P. Turano, F. Musiani, P. Neyroz, S. Ciurli, Proteins 2009, 74, 222–239.
B. Zambelli, N. Cremades, P. Neyroz, P. Turano, V. N. Uversky, S. Ciurli, Mol. Biosyst. 2012, 8, 220–228.
M. Bellucci, B. Zambelli, F. Musiani, P. Turano, S. Ciurli, Biochem. J. 2009, 422, 91–100.
Y. H. Fong, H. C. Wong, C. P. Chuck, Y. W. Chen, H. Sun, K. B. Wong, J. Biol. Chem. 2011, 286, 43241–43249.
B. Zambelli, F. Musiani, S. Ciurli, Met. Ions Life Sci. 2012, 10, 135–170.
Z. Ma, F. E. Jacobsen, D. P. Giedroc, Chem. Rev. 2009, 109, 4644–4681.
B. Zambelli, M. Bellucci, A. Danielli, V. Scarlato, S. Ciurli, Chem. Commun. 2007, 3649–3651.
B. Zambelli, A. Danielli, S. Romagnoli, P. Neyroz, S. Ciurli, V. Scarlato, J. Mol. Biol. 2008, 383, 1129–1143.
F. Musiani, B. Bertosa, A. Magistrato, B. Zambelli, P. Turano, V. Losasso, C. Micheletti, S. Ciurli, P. Carloni, J. Chem. Theor. Comp. 2010, 6, 3503–3515.
S. Benini, M. Cianci, S. Ciurli, Dalton Trans. 2011, 40, 7831–7833.
J. G. Kusters, A. H. van Vliet, E. J. Kuipers, Clin. Microbiol. Rev. 2006, 19, 449–490.
H. de Reuse, S. Bereswill, FEMS Immunol. Med. Microbiol. 2007, 50, 165–176.
A. Danielli, G. Amore, V. Scarlato, PLoS Pathog. 2010, 6, e1000938. DOI: 10.371/journal.ppat.1000938.
K. A. Eaton, S. Krakowka, Infect. Immun. 1994, 62, 3604–3607.
K. Muhsen, D. Cohen, Helicobacter 2008, 13, 323–340.
S. L. Benoit, E. F. Miller, R. J. Maier, Infect. Immun. 2013, 81, 580–584.
M. Sousa Silva, A. E. Ferreira, R. Gomes, A. M. Tomas, A. Ponces Freire, C. Cordeiro, Int. J. Med. Microbiol. 2012, 302, 225–229.
S. C. Chauhan, P. K. Padmanabhan, R. Madhubala, Curr. Drug Targets 2008, 9, 957–965.
WHO, "Global tuberculosis control report 2011", World Health Organization, Geneva, Switzerland, 2011.
W. Lin, V. Mathys, E. L. Ang, V. H. Koh, J. M. Martinez Gomez, M. L. Ang, S. Z. Zainul Rahim, M. P. Tan, K. Pethe, S. Alonso, Infect. Immun. 2012, 80, 2771–2779.
D. L. Clemens, B. Y. Lee, M. A. Horwitz, J. Bacteriol. 1995, 177, 5644–5652.
A. H. Gordon, P. D. Hart, M. R. Young, Nature 1980, 286, 79–80.
P. D. Hart, M. R. Young, J. Exp. Med. 1991, 174, 881–889.
K. Sendide, A. E. Deghmane, J. M. Reyrat, A. Talal, Z. Hmama, Infect. Immun. 2004, 72, 4200–4209.
T. Mukai, Y. Maeda, T. Tamura, Y. Miyamoto, M. Makino, FEMS Immunol. Med. Microbiol. 2008, 53, 96–106.
L. Grode, P. Seiler, S. Baumann, J. Hess, V. Brinkmann, A. Nasser Eddine, P. Mann, C. Goosmann, S. Bandermann, D. Smith, G. J. Bancroft, J. M. Reyrat, D. van Soolingen, B. Raupach, S. H. Kaufmann, J. Clin. Invest. 2005, 115, 2472–2479.
D. R. Campbell, K. E. Chapman, K. J. Waldron, S. Tottey, S. Kendall, G. Cavallaro, C. Andreini, J. Hinds, N. G. Stoker, N. J. Robinson, J. S. Cavet, J. Biol. Chem. 2007, 282, 32298–32310.
A. Schnegg, M. Kirchgessner, in In Trace Element Metabolism in Man and Animals-3, Ed M. Kirchgessner, Proceedings of the 3rd Intern. Symposium, Technische Universität, Munchen, Germany, 1978, pp. 236–243.
F. H. Nielsen, D. R. Myron, S. H. Givand, T. J. Zimmerman, D. A. Ollerich, J. Nutr. 1975, 105, 1620–1630.
M. Szilagyi, M. Anke, I. Balogh, Acta Vet. Hung. 1991, 39, 231–238.
K. Yokoi, E. O. Uthus, F. H. Nielsen, Biol. Trace Elem. Res. 2003, 93, 141–154.
F. H. Nielsen, T. R. Shuler, T. G. McLeod, T. J. Zimmerman, J. Nutr. 1984, 114, 1280–1288.
J. W. Spears, R. W. Harvey, L. J. Samsell, J. Nutr. 1986, 116, 1873–1882.
G. I. Stangl, M. Kirchgessner, J. Nutr. 1996, 126, 2466–2473.
V. Tremaroli, F. Backhed, Nature 2012, 489, 242–249.
C. Palmer, E. M. Bik, D. B. DiGiulio, D. A. Relman, P. O. Brown, PLoS Biol. 2007, 5, e177.
P. B. Eckburg, E. M. Bik, C. N. Bernstein, E. Purdom, L. Dethlefsen, M. Sargent, S. R. Gill, K. E. Nelson, D. A. Relman, Science 2005, 308, 1635–1638.
M. Monachese, J. P. Burton, G. Reid, Appl. Environ. Microbiol. 2012, 78, 6397–6404.
K. Suzuki, Y. Benno, T. Mitsuoka, S. Takebe, K. Kobashi, J. Hase, Appl. Environ. Microbiol. 1979, 37, 379–382.
F. Turroni, E. Foroni, P. Pizzetti, V. Giubellini, A. Ribbera, P. Merusi, P. Cagnasso, B. Bizzarri, G. L. de’Angelis, F. Shanahan, D. van Sinderen, M. Ventura, Appl. Environ. Microbiol. 2009, 75, 1534–1545.
I. S. Arvanitoyannis, M. Van Houwelingen-Koukaliaroglou, Crit. Rev. Food Sci. Nutr. 2005, 45, 385–404.
M. Pimentel, R. P. Gunsalus, S. S. Rao, H. Zhang, Am. J. Gastroenter. Suppl. 2012, 1, 28–33.
B. Jaun, R. K. Thauer, in Nickel and Its Surprising Impact in Nature, Vol. 2 of Metal Ions in Life Sciences, Eds A. Sigel, H. Sigel, R. K. O Sigel, John Wiley & Sons, Ltd., Chichester, UK, 2007, pp. 323–356.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Zambelli, B., Ciurli, S. (2013). Nickel and Human Health. In: Sigel, A., Sigel, H., Sigel, R. (eds) Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences, vol 13. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-7500-8_10
Download citation
DOI: https://doi.org/10.1007/978-94-007-7500-8_10
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-7499-5
Online ISBN: 978-94-007-7500-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)