
Abstract
This review focuses on recent studies showing that cardiolipin (CL), a unique mitochondrial phospholipid, regulates many cellular functions and signaling pathways, both inside and outside the mitochondria. Inside the mitochondria, CL is a critical target of mitochondrial generated reactive oxygen species (ROS) and regulates signaling events related to apoptosis and aging. CL deficiency causes perturbation of signaling pathways outside the mitochondria, including the PKC-Slt2 cell integrity pathway and the high osmolarity glycerol (HOG) pathway, and is a key player in the cross-talk between the mitochondria and the vacuole. Understanding these connections may shed light on the pathology of Barth syndrome, a disorder of CL remodeling.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Cardiolipin
- Phosphatidylglycerol
- Cellular signaling
- Apoptosis
- Cell wall biogenesis
- Mitophagy
- Mitochondria
- Vacuolar function
- Reactive oxygen species
- Anionic phospholipids
- Barth syndrome
11.1 Introduction
It is not unusual to find even current depictions of membranes as homogenous lipid matrices that function primarily to support the allegedly important protein molecules embedded within. This belies the fascinating discoveries in the past two decades of cellular and organelle-specific functions attributed to individual membrane lipids, and of the plethora of regulatory and signaling molecules derived from glycerolipids and sphingolipids. In this light, it is essential to elucidate the functions of specific membrane lipids and the cellular consequences of their depletion.
A phospholipid that has been the focus of considerable attention relatively recently – although it was first isolated and purified from beef heart in 1942 [1, 2], is cardiolipin (CL). CL is structurally unique. In contrast to the other membrane phospholipids, in which a single glycerol backbone is acylated to two fatty acid chains, CL contains two phosphatidyl groups (linked to a glycerol backbone) and four fatty acyl chains. It is enriched in energy harvesting membranes of mitochondria, chloroplasts, bacterial plasma membranes, and hydrogenosomes, underscoring the importance of this lipid in energy production [3–5]. CL is tightly associated with mitochondrial proteins and respiratory chain complexes and is essential for their optimal activity [6–10]. In the inner membrane, CL provides structural stability to membrane proteins through hydrophobic and electrostatic interactions.
In light of its association with the respiratory apparatus, the role of CL in mitochondrial bioenergetics was not entirely unexpected. Interestingly, however, recent studies carried out primarily in yeast indicate that CL is also required for cellular functions that are not directly associated with oxidative phosphorylation. In accordance with a broad definition of a ‘bioactive lipid’ as one in which changes in levels lead to functional consequences [11], perturbation of CL composition (including CL levels, acyl species, and degree of peroxidation) leads to dramatic cellular consequences: (1) Alterations in CL levels and acyl chain composition increases the recruitment to the mitochondria of cytosolic proteins that trigger apoptosis [12–14]. (2) Perturbation of CL synthesis or remodeling leads to increased production of reactive oxygen species (ROS), which induces aging [15–20]. (3) Blocking CL synthesis in yeast at the first step of the pathway deleteriously affects cell wall biogenesis and alters the response of two signaling pathways, the protein kinase C (PKC)-Slt2 mitogen activated protein kinase (MAPK) and the high osmolarity glycerol (HOG) pathways [21–23]. (4) The inability of yeast cells to synthesize CL leads to decreased vacuolar function and reduced V-ATPase activity, suggesting that CL mediates cross talk between mitochondria and the vacuole [24]. The current review focuses on the role of CL in regulating these cellular functions. We conclude with unanswered questions that remain exciting avenues for future studies, which may have implications for understanding the pathophysiology of Barth syndrome (BTHS), a genetic disorder of CL remodeling.
11.2 CL Biosynthesis and Remodeling
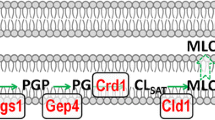
One of the most intriguing aspects of CL biosynthesis is that the lipid that is initially synthesized contains primarily saturated fatty acids, while the mature CL that is essential for normal cellular function contains unsaturated fatty acids. The distinct composition of acyl chains is achieved through a highly conserved pathway of synthesis and remodeling, as shown in Fig. 11.1. The first step is catalyzed by phosphatidylglycerolphosphate (PGP) synthase (Pgs1), which converts CDP-diacylglycerol (DAG) and glycerol-3-phosphate (G-3-P) to PGP [25, 33]. PGP is dephosphorylated to phosphatidylglycerol (PG) by PGP phosphatase (Gep4) [26, 34]. The mammalian homologue of the yeast GEP4 gene was recently identified as protein tyrosine phosphatase localized in the mitochondrion (PTPMT1) [34]. CL synthase (Crd1) catalyzes an irreversible condensation reaction in which the phosphatidyl group of CDP-DAG is linked to PG via cleavage of a high-energy anhydride bond to form CL [27–30, 35–38]. CL synthase does not show strong preference for specific fatty acyl chains [38–40]. How, then, is acyl specificity achieved? The newly synthesized CL undergoes deacylation by a CL-specific deacylase (Cld1), which is homologous to the mammalian phospholipase A2 [31, 41, 42]. Cld1 removes one saturated fatty acyl chain from CL to form monolysocardiolipin (MLCL) [31]. The transacylase tafazzin (Taz1) reacylates MLCL with an unsaturated fatty acid to form mature CL [32, 43, 44]. Taz1 carries out exchange of acyl chains between CL and phospholipids that primarily include phosphatidylcholine (PC), to sequentially replace the fatty acyl chains from all four acyl positions of CL [45, 46]. The end result of this exchange is molecular symmetry of CL molecules across the eukaryotic kingdom, from yeast to humans, which is characteristic of the organism and of specific tissues and organs [47]. For example, in yeast, the mature form of CL contains oleic acid, while CL in the normal human heart is primarily tetralinoleoyl-CL (L4-CL) [47]. A deficiency of tafazzin in humans leads to a complete absence of L4-CL, resulting in the severe cardiomyopathy observed in BTHS.

Synthesis and remodeling of CL in yeast. CL synthesis begins with the conversion of CDP-diacylglycerol (CDP-DG) to phosphatidylglycerolphosphate (PGP) by PGP synthase (encoded by PGS1) [25]. PGP is dephosphorylated to phosphatidylgylcerol (PG) by GEP4-encoded PGP phosphatase [26]. CL synthase (encoded by CRD1) converts PG to premature CL containing primarily saturated fatty acids (FA) [27–30]. CL is deacylated by CL deacylase (encoded by CLD1) to monolyso-CL (MLCL) [31], which is reacylated by the TAZ1-encoded enzyme tafazzin to mature CL containing unsaturated fatty acids [32]. The yeast gene names are depicted in green, while phospholipids and their intermediates are shown in red
While tafazzin is the only known yeast enzyme that adds fatty acyl chains to MLCL, two other enzymes in addition to tafazzin remodel CL in mammalian cells. MLCL acyltransferase-1 (MLC-LAT1), isolated and purified from pig liver mitochondria, shows specificity for linoleate [48]. Thus, over-expression of MLCLAT1 in tafazzin-deficient BTHS lymphoblasts increased incorporation of linoleic acid into CL, and RNAi knockdown of MLCLAT1 in HeLa cells showed reduced linoleic acid inclusion in CL [49]. The biological function of this enzyme is not clear. A second enzyme, acyl-CoA:lysoCL acyltransferase 1 (ALCAT1), identified in mouse, was initially thought to be located in the endoplasmic reticulum, but was subsequently determined to be present in the mitochondrial-associated membranes, where phospholipid traffic between the endoplasmic reticulum and the mitochondria takes place [17, 50]. In contrast to MLCLAT1, ALCAT1 shows no specificity for linoleic acid. ALCAT1 was shown to catalyze CL remodeling to incorporate long chain polyunsaturated fatty acyl chains such as docosahexaenoic acid (DHA) [17]. Enhanced incorporation of polyunsaturated fatty acyl chains in CL makes it more susceptible to oxidative damage by ROS, causing early peroxidation [51–53]. ALCAT1 null mutant mice exhibit elevated CL levels along with increased L4-CL [16, 17], whereas overexpression of ALCAT1 has been shown to decrease total CL levels and increase incorporation of long chain polyunsaturated fatty acyl chains [54]. These findings suggest that ALCAT1 may negatively regulate CL biosynthesis.
In light of the importance of CL in cellular function, it is not surprising that perturbation of CL synthesis leads to serious illness. The most direct example of this link is seen in BTHS, a life-threatening illness characterized by dilated cardiomyopathy and sudden death from arrhythmia [55, 56]. BTHS results from mutation in the CL remodeling enzyme tafazzin [44, 57]. This leads to an abnormal CL profile characterized by decreased total CL, increased MLCL, and aberrant CL acylation, most notably the loss of the predominant CL species in normal myocardium, L4-CL [58]. How these abnormalities cause the associated pathology in BTHS is not known [59].
CL abnormalities have also been observed in heart failure [60, 61]. Heart failure due to dilated cardiomyopathy is the primary cause of death in diabetic patients [62, 63]. Metabolic perturbations observed in diabetic cardiomyopathy include increased utilization of fatty acid substrates, decreased utilization of glucose, and mitochondrial dysfunction [64–66]. However, the molecular mechanism that leads to heart failure in diabetic patients is not known. Interestingly, a decrease in CL levels and alterations in CL acyl species were found in early stages of diabetes induced by streptozotocin in mice, suggesting that mitochondrial dysfunction and cardiomyopathy may be due to alterations in CL metabolism [67, 68]. The decrease in CL levels may result from remodeling of CL fatty acyl species with DHA, which is known to cause CL peroxidation by ROS [51–53]. In summary, depletion of CL content and alterations in CL fatty acyl species lead to BTHS, and may also contribute to pathological conditions and metabolic perturbations in other human disorders.
11.3 CL and Apoptosis
Perturbations in CL levels and acyl composition play a crucial role in regulating apoptosis, the complex process leading to programmed cell death. The role of CL in apoptosis derives from its interactions with cytochrome c (Cyt c) and with apoptotic proteins (Fig. 11.2).

Perturbation of CL metabolism triggers apoptosis. The binding of cytochrome c (Cyt c) to CL is essential to anchor it to the inner mitochondrial membrane, facing the intermembrane space [69, 70]. Peroxidation of CL (CL-OOH) by reactive oxygen species (ROS) leads to release of Cyt c to the cytosol, which serves as a signal to initiate apoptosis [71–73]. Caspase-8 cleaves Bid protein to its active form, truncated Bid (t-Bid) [74–76]. Binding of t-Bid to CL enhances translocation of CL to the outer mitochondrial membrane, which facilitates targeting of apoptotic proteins (Bak and Bax) to the outer membrane [77, 78]
11.3.1 CL and Cyt c
Interactions between CL and Cyt c are an important determinant of apoptosis. Cyt c, which transfers electrons from complex III to complex IV, is bound to the outer leaflet of the mitochondrion inner membrane through interactions with CL [69, 70]. The binding of Cyt c to CL is essential to anchor it to the inner membrane, and release of Cyt c to the cytosol serves as a signal to recruit apoptotic proteins to the mitochondria to initiate apoptosis [74, 75, 79, 80]. CL binds Cyt c in two different conformations – a loosely bound state that is facilitated by means of electrostatic interactions, and a tightly bound state that is mediated by hydrophobic interactions in which Cyt c is partially embedded in the inner membrane [69, 70]. Release of Cyt c from CL requires dissociation of both electrostatic and hydrophobic interactions [81]. The production of ROS may alter the CL-Cyt c association [80–82]. Alternatively, the peroxidation of CL by hydrogen peroxide generated in the mitochondria leads to the release of Cyt c from the tightly bound state into the intermembrane space [12, 76].
11.3.2 Recruitment of Apoptotic Proteins
An early trigger of apoptosis is the change in CL composition in the mitochondrial inner and outer membranes, followed by dissipation of the membrane potential and flipping of phosphatidyserine (PS) to the external surface of the plasma membrane [83, 84]. A diverse set of apoptotic proteins such as t-Bid, Bax, Bak, and caspase-8 are recruited to the mitochondrial surface of cells undergoing apoptosis in a CL-dependent manner [71–73]. Upon activation, caspase-8 migrates to the mitochondrial outer membrane in regions where CL is present. Caspase-8 is said to cleave Bid to its active form, tBid (truncated Bid). A significant amount of CL is translocated from the inner to the outer mitochondrial membrane, which likely serves as a signal for binding of the apoptotic proteins [14, 85, 86]. The binding of t-Bid to CL is thought to further increase CL transfer to the outer membranes. Alternatively, apoptotic proteins may be guided to the mitochondria by means of altering the outer membrane charge [87]. By increasing the CL content, the mitochondrial outer membrane may accrue a more negative charge, which serves as a targeting signal for recruiting polycationic apoptotic proteins to the mitochondria [77, 85]. Consistent with this, ectopic overexpression of a CL-binding protein masked the negative charge on the membrane and inhibited apoptosis [87]. The recruitment to and oligomerization of Bak-Bax in the outer mitochondrial membrane is a CL dependent process, which permeabilizes the outer mitochondria to trigger Cyt c release and progression of apoptosis [88, 89]. This suggests that CL-rich regions in the outer membrane serve as a key signal for targeting pro-apoptotic proteins of the Bcl2 family to bring about apoptosis [78, 85].
11.3.3 Translocation of CL
Early in apoptosis, CL translocation from the inner to the outer mitochondrial membrane may be carried out through several transport modes. First, the inner and outer membrane contact sites, which are enriched in CL through interactions with mitochondrial creatine kinase (MtCK), could facilitate the transfer of CL from the inner to outer membrane [90–95]. Second, phospholipid scramblase-3 (PLS-3) has been shown to translocate CL from the inner membrane to the outer membrane during the onset of apoptosis [96–98]. Consistent with this, cells overexpressing PLS-3 exhibit increased apoptosis, while inactivation of PLS-3 leads to increased resistance to UV-induced apoptosis [97]. CL and Bid interactions have been shown at the contact sites, which likely contribute to mitochondrial permeabilization to induce apoptosis [99]. Changes in CL content in the membrane may be mediated by Bid, as evidence suggests that Bid exhibits lipid transfer activity [100, 101]. Lymphoblastoid cells derived from BTHS and TAZ knockdown HeLa cells were more resistant to Fas-induced apoptosis [72]. Specifically, reduction of mature CL caused defective activation of caspase-8, suggesting that processing of caspase-8 on the mitochondrial membranes is CL-dependent. To summarize, CL in the mitochondria is an important mediator of apoptosis, and apoptotic proteins are directed to the mitochondria in a CL-dependent manner.
11.4 CL in Bioenergetics and Mitochondrial Dysfunction
The relationship between CL and ROS is complex. CL physically interacts with proteins of the mitochondrial respiratory chain complexes and other components of the membrane and forms lipid scaffolds for tethering and stabilizing mitochondrial membrane proteins to enhance their enzymatic activities [7, 8, 102–105]. Consistent with the role of CL in bioenergetics, mitochondria deficient in CL exhibit decreased activity of respiratory complexes and carrier proteins [106]. The generation of ROS in mitochondria, which is a byproduct of oxidative phosphorylation [107–109], is enhanced upon CL deficiency [20]. ROS, in turn, damages CL by peroxidation of the unsaturated fatty acids.
11.4.1 CL and Supercomplexes
For efficient substrate channeling between the individual complexes, the mitochondrial respiratory chain components are organized in supramolecular structures called supercomplexes [110]. In mammalian cells, complex I is associated with two units of complex III and multiple units of complex IV. In S. cerevisiae, which lacks complex I, two copies of complex III are bound to either one or two units of complex IV. CL deficiency in yeast leads to destabilization of the respiratory supercomplexes, indicating that CL functions to stabilize these complexes [10, 111, 112]. Similarly, tafazzin deficient human fibroblasts exhibit destabilization of the supercomplexes [113]. For efficient ADP/ATP exchange, CL is also required for the association of the ADP/ATP carrier protein with the supercomplexes [114].
11.4.2 CL Deficiency and ROS Generation
The role of CL in the supercomplexes may be that of a proton trap, to avoid leakage of protons and enhance the membrane potential for efficient oxidative phosphorylation [115–117]. Not surprisingly, defective supercomplex formation and CL deficiency lead to increased ROS production [20, 118].
Among the respiratory chain complexes, complexes I and III are prime sites for ROS generation [119–122]. Because of the proximity of CL to these ROS generating centers, the unsaturated fatty acyl chains of CL are susceptible to damage by peroxidation. Superoxide generated by respiratory complex III causes peroxidation of CL and alters the activity of Cyt c oxidase [123–125]. Optimal function of Cyt c oxidase, the terminal enzyme complex of the respiratory chain, is dependent on CL [6, 126–128]. Reduced activity of Cyt c oxidase from reperfused heart was restored specifically by exogenous supplementation of CL, but not by peroxidized CL or other phospholipids [129]. In addition, reduced activity and increased ROS generation by complexes I and III were also rescued by CL supplementation [125, 130]. These studies indicate that peroxidized CL cannot effectively carry out mitochondrial functions that are dependent on normal CL.
Peroxidation of CL by ROS is seen as the primary cause of CL mobilization to the outer leaflet of the inner membrane. Human leukemia cells treated with the apoptosis-inducing drug staurosporine rapidly underwent apoptosis along with an increase in CL content in the outer mitochondrial membrane [83]. However, the change in CL content was preceded by increased ROS production and CL peroxidation, suggesting that perturbation of CL metabolism could be an early step in mitochondria-induced apoptosis. Due to the high content of unsaturated fatty acyl chains, CL is particularly susceptible to peroxidation [131, 132]. Peroxidation of CL alters the molecular conformation leading to formation of non-bilayer hexagonal structures, which could serve as a marker for targeting the cytosolic apoptotic machinery to the mitochondria [77].
11.4.3 CL in Mitochondrial Dysfunction and Aging
Under normal physiological conditions, the damaged fatty acyl chains of CL may be replaced through the remodeling process [45]. Pathological remodeling of CL has been linked to mitochondrial dysfunction in human diseases [17, 18, 61, 67]. Recent studies have shown that ALCAT1 may be involved in the pathological remodeling of CL in cells undergoing oxidative stress. As mentioned earlier, ALCAT1 overexpression leads to a decrease in CL levels and aberrant remodeling of CL with long chain polyunsaturated acyl chains such as DHA, which are highly susceptible to oxidation by ROS [17, 52, 53]. The close proximity of CL to respiratory complexes in the inner membrane where ROS is generated increases exposure of these long chain unsaturated fatty acyl chains to ROS. Aberrant CL remodeling resulting from increased ALCAT1 expression leads to the mitochondrial dysfunction seen in pathological conditions such as hyperthyroid cardiomyopathy, diabetes, and diet-induced obesity in mice [15–17, 133]. ALCAT1 null mice exhibit increased expression of MLCAT1 along with elevated levels of CL containing linoleic acid. These findings underscore the significance of CL remodeling and the impact of this process on mitochondrial function and ROS generation.
A decline in CL levels appears to be a primary feature of aging [134–139]. In aging cells, CL is pathologically remodeled with polyunsaturated fatty acyl chains such as arachidonic and docosahexaenoic acids, which are more susceptible to peroxidation than linoleic acid in normal CL [18, 54]. Mitochondrial CL levels, along with oxidative capacity and ATP synthesis, decrease significantly with age [134, 140–143].
CL is required for the optimal function of several mitochondrial carrier proteins involved in the transport of essential metabolites into mitochondria [106]. In the heart, oxidation of pyruvate and β-oxidation of fatty acids are two major sources of ATP generation [144–146]. The transport of pyruvate into mitochondria by the pyruvate carrier and the exchange of carnitine esters by the carnitine:acylcarnitine translocase are, therefore, critical for energy metabolism. Studies have demonstrated that enzymatic activities of both the mitochondrial pyruvate carrier and carnitine:acylcarnitine translocase, which are dependent on CL [147, 148], are decreased in aging heart muscle [149, 150]. Interestingly, administration of acetyl-L-carnitine in aged rats restored decreased CL levels and the activities of the mitochondrial pyruvate carrier and carnitine:acylcarnitine translocase to levels found in young rats [149, 150]. Dietary supplementation of acetyl-L-carnitine also showed similar beneficial effects, increasing mitochondria membrane potential and, in turn, improving physical mobility in aged rats [141, 151]. These findings suggest that the supply of carnitine to the mitochondria may become limited during aging, hindering energy production through β-oxidation [150, 152]. Although acetyl-L-carnitine supplementation restored CL levels and improved mitochondrial metabolic functions in aged animals, the underlying molecular mechanism remains unresolved.
11.5 CL and the PKC-Slt2 Cell Integrity Pathway
Null mutants in yeast have been characterized for each step of the CL biosynthetic pathway, and mutants blocked earlier in the pathway have more severe phenotypes. Thus, the pgs1Δ mutant, which cannot synthesize CL or the precursor PG (Fig. 11.1), exhibits severe growth defects not only in non-fermentable carbon sources, which are metabolized by respiration, but also in fermentable carbon sources, in which respiration is not required [25, 153]. This observation indicated that PG and/or CL are required for cellular functions apart from mitochondrial bioenergetics [154]. Genetic studies to isolate spontaneous suppressors of the pgs1Δ mutant growth defect identified a loss of function mutation of KRE5, a gene involved in cell wall biogenesis [21]. Consistent with defective cell wall biogenesis, the pgs1Δ mutant exhibited enlarged cell size characteristic of cell wall mutants, reduced levels of β-1,3-glucan as a result of decreased activity of glucan synthase, and sensitivity to cell wall perturbing agents [155–157]. These defects were restored by disruption of KRE5 in pgs1Δ, which increased expression of the genes FKS1 and FKS2 encoding glucan synthase [22]. These findings were in agreement with the identification of PGS1 in a screen to identify genes involved in cell wall biogenesis [158].
Studies to gain insight into the mechanism linking CL to the cell wall focused on the PKC-Slt2 cell integrity pathway. Activation of the cell integrity pathway is triggered by signals generated from cell wall sensor proteins to Rom2, which, in turn, activates formation of the GTP-bound form of Rho1p. The activated Rho1 protein transmits a signal to Pkc1 to trigger the Mpk1/Slt2 MAPK signaling cascade, which results in dual phosphorylation of Slt2 [159, 160]. The dual phosphorylation of Slt2 is essential to activate transcription factors that up-regulate genes involved in cell wall remodeling, particularly in response to heat stress [156, 161, 162]. The pgs1Δ mutant exhibited defective activation of the PKC-Slt2 cell-integrity signaling cascade, indicated by decreased Slt2 phosphorylation levels [22]. Consistent with this, overexpression of individual genes in the PKC-Slt2 pathway rescued the growth defect of pgs1Δ at elevated temperature and improved resistance to the cell wall perturbing chemicals calcofluor white and caffeine. Interestingly, deletion of KRE5 in pgs1Δ also led to increased activation of the PKC-Slt2 cell-integrity pathway.
A mitochondrial connection to the cell wall is not new. Genome-wide screens have identified several yeast genes required for mitochondrial function that, when mutated, affect chemical components of the cell wall [158, 163, 164]. Furthermore, mitochondrial respiratory defects negatively impact the synthesis of cell wall components [158, 164]. The underlying mechanism whereby CL regulates cell wall remodeling is not known. One possibility is that CL is required for activity of one or more proteins that exhibit dual localization in the cell wall/plasma membrane and mitochondria [165]. Interesting possibilities include three proteins of the PKC-Slt2 cell integrity pathway, Fks1, Zeo1 and Rho1, which are found both in the mitochondria and the plasma membrane [166–168]. Mitochondrial targeting of these proteins may be CL-dependent. Alternatively, their stability in the mitochondrial membrane may be decreased in the absence of CL.
The yeast cell wall also plays an important role in regulating replicative life span [169]. Consistent with this, the pgs1Δ mutant, which exhibits cell wall defects, also has a decreased replicative life span [21, 23]. Intriguingly, experiments to elucidate the mechanism linking PG/CL to defects in the cell wall, PKC/Slt2 signaling and aging led to another signaling pathway – the HOG stress response pathway.
11.6 CL and the HOG Stress Response Pathway
In response to stress, cells are regulated by the opposing actions of the PKC-Slt2 and HOG signaling pathways [170–172]. Heat or low osmolarity stress leads to activation of the PKC-Slt2 pathway, resulting in increased expression of the cell wall remodeling genes leading to a decrease in turgor pressure [173–175]. In contrast, activation of the HOG signaling pathway causes an increase in turgor pressure [175, 176]. Because the pgs1Δ mutant exhibited defective activation of the PKC-Slt2 signaling cascade, it was hypothesized that growth defects of the mutant resulted from increased turgor pressure, which may be rescued by down-regulation of the HOG pathway (Fig. 11.3) [23]. This hypothesis was supported by the finding that deletion of SHO1, an upstream activator of HOG signaling, rescued growth defects, increased the replicative life span, and alleviated sensitivity to cell wall perturbing agents in pgs1Δ [23]. Interestingly, the mutant did not exhibit increased activation of the HOG pathway. It is possible that, in the absence of PKC-Slt2 activation, even wild type levels of HOG activation lead to turgor pressure levels that affect growth. These findings suggest that homeostasis achieved by these two signaling pathways is perturbed upon CL deficiency (Fig. 11.3).

CL deficiency leads to perturbation of PKC-Slt2 and HOG signaling pathways. The PKC-Slt2 and HOG signaling pathways coordinately regulate cell wall biogenesis and intracellular turgor pressure, respectively [173, 175]. Under hypertonic or cold stress conditions, extracellular osmolarity is increased, causing an efflux of intracellular water to reduce the turgor pressure on the cell wall. To counteract augmented extracellular osmolarity, the HOG pathway is activated, which leads to an increase in intracellular turgor pressure [170, 177]. In contrast, under heat or hypotonic stress, extracellular osmolarity is decreased, which causes an influx of water inside the cell to increase intracellular turgor pressure. To counteract the increased turgor pressure, the activated PKC-Slt2 pathway induces cell wall synthesis [172, 173]. We hypothesize that disruption of the CL pathway by mutation of PGS1 generates a signal that is detected by regulators or components of the PKC-Slt2 pathway, which, in turn, down-regulates the pathway [21, 22]. Under these conditions, an increase in intracellular turgor pressure by activation of the HOG pathway is deleterious in pgs1Δ cells [23]
11.7 CL Mediates Cross-Talk Between Mitochondria and Vacuole
The yeast crd1Δ mutant, which lacks CL, was shown to have defective vacuolar function [24]. CL deficiency caused decreased V-ATPase activity and proton pumping, reduced vacuolar acidification, and enlargement of the vacuole. The yeast vacuole plays a crucial role in adjusting to high external osmolarity and decreased turgor pressure, and in maintaining cytosolic ion concentrations [178, 179]. Consistent with perturbation of intracellular osmotic balance in the crd1Δ mutant, growth and vacuolar defects were rescued by supplementation of sorbitol [24].
In some genetic backgrounds, the crd1Δ mutant exhibits increased expression of RTG2, a critical sensor of mitochondrial dysfunction that relays metabolic defects to the nucleus via the retrograde signaling pathway [180, 181]. Consistent with overactivation of Rtg2, deletion of the RTG2 gene restored vacuolar acidification and V-ATPase activity and rescued the growth defect of the crd1Δ mutant at elevated temperature. However, deletion of the retrograde pathway activator RTG3 did not rescue the mutant, suggesting that the defects observed in crd1Δ resulted from Rtg2 functions unrelated to retrograde activation.
One possible explanation for the vacuolar defects in crd1Δ is that the loss of CL leads to intracellular osmotic imbalance, as suggested by the enlarged cell size of the mutant (Fig. 11.4). Consistent with this, deletion of the NHX1 gene (but not any of the other vacuole ion transporters) in crd1Δ restored vacuolar morphology to wild type levels [24]. Nhx1 is the Na+/H+ exchanger located in late endosomal/prevacuolar membranes, and is involved in the export of protons in exchange for cytosolic Na+ or K+ [182, 183].

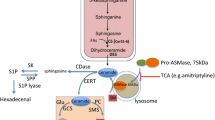
Proposed models to explain the role of CL in vacuolar function. It is likely that CL is transported to the vacuole through mitophagy, the selective degradation of mitochondria via the autophagosome, which delivers its cargo to the vacuole. (a) Under normal physiological conditions, CL may provide stability to the V-ATPase, which is essential to maintain its activity [24]. (b) CL deficiency may lead to perturbation of mitophagy, which results in decreased delivery of CL to the vacuole and, subsequently, to destabilization of the V-ATPase, decreased ATPase activity, and enlargement of the vacuole
Another possibility is that CL may regulate vacuolar function by directly activating the V-ATPase (Fig. 11.4). While CL is predominantly found in the mitochondrial membranes, significant amounts are also detected in the vacuolar membrane, and the levels vary depending on the carbon source of the growth media [184]. How does CL, which is synthesized in the mitochondria, get to the vacuole? The most likely mechanism is via selective degradation of the mitochondria by the autophagic process known as mitophagy, which is strongly induced in yeast by nutrient starvation and during the stationary growth phase [185–190]. CL that has integrated into the vacuolar membrane as a result of mitophagy may directly activate the V-ATPase and/or stabilize the protein. This possibility is highly speculative at this stage, as such interactions have not yet been reported.
Cross-talk between mitochondria and vacuole is further supported by a recent finding, which showed that the vacuolar pH is a determinant of mitochondrial function and aging in yeast cells [191]. Aging yeast cells exhibit a decline in vacuolar acidity, which causes mitochondrial dysfunction and a decrease in replicative life-span [191]. Consistent with this, enhancing vacuolar acidity by overexpressing VMA1 or VPH2, which encode proteins that regulate V-ATPase activity, suppressed mitochondrial dysfunction. The mechanisms underlying the interplay between vacuole and mitochondria, and the role of CL in this process, remain to be elucidated.
11.8 Unanswered Questions and Future Directions
The studies discussed here show that changes in the levels and species of CL affect not only mitochondrial function but also signaling pathways and other organelles. Elucidating the mechanisms whereby CL mediates these activities remains an exciting area for future investigation. In this regard, we pose the following questions.
During apoptosis, the CL content of the outer mitochondrial membrane increases [83, 87]. How is CL transferred from its site of synthesis in the inner mitochondrial membrane to the outer membrane?
Peroxidation of CL has been shown to be a major mechanism of free radical toxicity resulting from ischemia-reperfusion injury to cardiac myocytes [129, 192–194]. The degree of peroxidation is dependent upon the acyl composition of the lipid. What regulates the fatty acyl chain composition of CL? Is this regulation age dependent?
What is the mechanism whereby CL regulates vacuolar function and V-ATPase activity? Intere-stingly, enlargement of the lysosome (mammalian equivalent of the vacuole) was also observed in the mouse model of BTHS, suggesting that the role of CL in vacuole/lysosome function is highly conserved [195]. Are the vacuole/lysosome defects due to CL deficiency? Alternatively, are the defects an indirect consequence of perturbation of mitophagy?
How does CL regulate the PKC-Slt2 and HOG signaling pathways? The mammalian homolog of HOG1, p38, is also a signal relay protein that responds to osmotic stress. p38 is involved in the cardiac expression of proinflammatory cytokines and in the development of cardiac dysfunction relative to the inflammatory response [196]. A role for p38 in cardiomyopathy is suggested by the finding that depletion of p38α alleviates cardiomyopathy induced by overexpression of the α-adrenergic receptor [197]. However, the link between CL and p38 is speculative, as the effects of CL deficiency on mammalian p38 have not been studied. In addition to p38, members of the PKC family are also involved in maintaining cardiac structure and function [198, 199]. A recent study showed that PKCθ is expressed at significant levels in neonatal mouse ventricular myocytes and is specifically activated during stress [200]. Furthermore, PKCθ deficiency leads to dilation of heart muscle cells and decreased viability. Similarly, PKCε migrates to the mitochondria and plays a cardio-protective role in injuries arising from ischemia and reperfusion [201–203]. However, it should be noted that expression of only a few PKC isoforms exhibit beneficial effects in cardiac injury [204–206]. It would be interesting to determine if CL is involved in the modulation of PKC function in heart muscle.
Answering these questions will have important implications for understanding the pathophysiology of BTHS and other disorders in which CL deficiency plays a role. Although BTHS is a monogenic disorder, the clinical presentation is highly variable, even among patients with the same mutation, ranging from death in the newborn period to asymptomatic. This suggests that physiological modifiers may contribute to the clinical symptoms observed in BTHS patients. It is likely that additional deficiencies in cellular functions that require CL may exacerbate the symptoms of tafazzin deficiency in BTHS.
In conclusion, our understanding of the role of CL in essential cell functions and signaling networks has increased dramatically in recent years. However, it is probably safe to assume that we have only scratched the surface of this expanding field.
References
Pangborn MC (1942) Isolation and purification of a serologically active phospholipid from beed heart. J Biol Chem 143:247–256
Pangborn MC (1947) The composition of cardiolipin. J Biol Chem 168:351–361
Dowhan W (1997) Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu Rev Biochem 66:199–232
Depalo N, Catucci L, Mallardi A, Corcelli A, Agostiano A (2004) Enrichment of cardiolipin content throughout the purification procedure of photosystem II. Bioelectrochemistry 63:103–106
Corcelli A (2009) The cardiolipin analogues of Archaea. Biochim Biophys Acta 1788:2101–2106
Fry M, Blondin GA, Green DE (1980) The localization of tightly bound cardiolipin in cytochrome oxidase. J Biol Chem 255:9967–9970
Fry M, Green DE (1980) Cardiolipin requirement by cytochrome oxidase and the catalytic role of phospholipid. Biochem Biophys Res Commun 93:1238–1246
Fry M, Green DE (1981) Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J Biol Chem 256:1874–1880
Lange C, Nett JH, Trumpower BL, Hunte C (2001) Specific roles of protein-phospholipid interactions in the yeast cytochrome bc1 complex structure. EMBO J 20:6591–6600
Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schagger H (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem 278:52873–52880
Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9:139–150
Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG (2005) Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1:223–232
Schug ZT, Gottlieb E (2009) Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim Biophys Acta 1788:2022–2031
Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD (2002) Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111:331–342
Cao J, Shen W, Chang Z, Shi Y (2009) ALCAT1 is a polyglycerophospholipid acyltransferase potently regulated by adenine nucleotide and thyroid status. Am J Physiol Endocrinol Metab 296:E647–E653
Li J, Liu X, Wang H, Zhang W, Chan DC, Shi Y (2012) Lysocardiolipin acyltransferase 1 (ALCAT1) controls mitochondrial DNA fidelity and biogenesis through modulation of MFN2 expression. Proc Natl Acad Sci U S A 109:6975–6980
Li J, Romestaing C, Han X, Li Y, Hao X, Wu Y, Sun C, Liu X, Jefferson LS, Xiong J, Lanoue KF, Chang Z, Lynch CJ, Wang H, Shi Y (2010) Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab 12:154–165
Lee HJ, Mayette J, Rapoport SI, Bazinet RP (2006) Selective remodeling of cardiolipin fatty acids in the aged rat heart. Lipids Health Dis 5:2
Paradies G, Petrosillo G, Paradies V, Ruggiero FM (2010) Oxidative stress, mitochondrial bioenergetics, and cardiolipin in aging. Free Radic Biol Med 48:1286–1295
Chen S, He Q, Greenberg ML (2008) Loss of tafazzin in yeast leads to increased oxidative stress during respiratory growth. Mol Microbiol 68:1061–1072
Zhong Q, Gvozdenovic-Jeremic J, Webster P, Zhou J, Greenberg ML (2005) Loss of function of KRE5 suppresses temperature sensitivity of mutants lacking mitochondrial anionic lipids. Mol Biol Cell 16:665–675
Zhong Q, Li G, Gvozdenovic-Jeremic J, Greenberg ML (2007) Up-regulation of the cell integrity pathway in saccharomyces cerevisiae suppresses temperature sensitivity of the pgs1Delta mutant. J Biol Chem 282:15946–15953
Zhou J, Zhong Q, Li G, Greenberg ML (2009) Loss of cardiolipin leads to longevity defects that are alleviated by alterations in stress response signaling. J Biol Chem 284:18106–18114
Chen S, Tarsio M, Kane PM, Greenberg ML (2008) Cardiolipin mediates cross-talk between mitochondria and the vacuole. Mol Biol Cell 19:5047–5058
Chang SC, Heacock PN, Clancey CJ, Dowhan W (1998) The PEL1 gene (renamed PGS1) encodes the phosphatidylglycero-phosphate synthase of Saccharomyces cerevisiae. J Biol Chem 273:9829–9836
Osman C, Haag M, Wieland FT, Brugger B, Langer T (2010) A mitochondrial phosphatase required for cardiolipin biosynthesis: the PGP phosphatase Gep4. EMBO J 29:1976–1987
Chang SC, Heacock PN, Mileykovskaya E, Voelker DR, Dowhan W (1998) Isolation and characterization of the gene (CLS1) encoding cardiolipin synthase in Saccharomyces cerevisiae. J Biol Chem 273:14933–14941
Tamai KT, Greenberg ML (1990) Biochemical characterization and regulation of cardiolipin synthase in Saccharomyces cerevisiae. Biochim Biophys Acta 1046:214–222
Tuller G, Hrastnik C, Achleitner G, Schiefthaler U, Klein F, Daum G (1998) YDL142c encodes cardiolipin synthase (Cls1p) and is non-essential for aerobic growth of Saccharomyces cerevisiae. FEBS Lett 421:15–18
Jiang F, Rizavi HS, Greenberg ML (1997) Cardiolipin is not essential for the growth of Saccharomyces cerevisiae on fermentable or non-fermentable carbon sources. Mol Microbiol 26:481–491
Beranek A, Rechberger G, Knauer H, Wolinski H, Kohlwein SD, Leber R (2009) Identification of a cardiolipin-specific phospholipase encoded by the gene CLD1 (YGR110W) in yeast. J Biol Chem 284:11572–11578
Gu Z, Valianpour F, Chen S, Vaz FM, Hakkaart GA, Wanders RJ, Greenberg ML (2004) Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome. Mol Microbiol 51:149–158
Kawasaki K, Kuge O, Chang SC, Heacock PN, Rho M, Suzuki K, Nishijima M, Dowhan W (1999) Isolation of a chinese hamster ovary (CHO) cDNA encoding phosphatidylglycerophosphate (PGP) synthase, expression of which corrects the mitochondrial abnormalities of a PGP synthase-defective mutant of CHO-K1 cells. J Biol Chem 274:1828–1834
Zhang J, Guan Z, Murphy AN, Wiley SE, Perkins GA, Worby CA, Engel JL, Heacock P, Nguyen OK, Wang JH, Raetz CR, Dowhan W, Dixon JE (2011) Mitochondrial phosphatase PTPMT1 is essential for cardiolipin biosynthesis. Cell Metab 13:690–700
Hostetler KY, Van den Bosch H, Van Deenen LL (1971) Biosynthesis of cardiolipin in liver mitochondria. Biochim Biophys Acta 239:113–119
Hostetler KY, van den Bosch H, van Deenen LL (1972) The mechanism of cardiolipin biosynthesis in liver mitochondria. Biochim Biophys Acta 260:507–513
Chen D, Zhang XY, Shi Y (2006) Identification and functional characterization of hCLS1, a human cardiolipin synthase localized in mitochondria. Biochem J 398:169–176
Houtkooper RH, Akbari H, van Lenthe H, Kulik W, Wanders RJ, Frentzen M, Vaz FM (2006) Identification and characterization of human cardiolipin synthase. FEBS Lett 580:3059–3064
Hostetler KY, Galesloot JM, Boer P, Van Den Bosch H (1975) Further studies on the formation of cardiolipin and phosphatidylglycerol in rat liver mitochondria. Effect of divalent cations and the fatty acid composition of CDP-diglyceride. Biochim Biophys Acta 380:382–389
Nowicki M, Muller F, Frentzen M (2005) Cardiolipin synthase of Arabidopsis thaliana. FEBS Lett 579:2161–2165
Mancuso DJ, Kotzbauer P, Wozniak DF, Sims HF, Jenkins CM, Guan S, Han X, Yang K, Sun G, Malik I, Conyers S, Green KG, Schmidt RE, Gross RW (2009) Genetic ablation of calcium-independent phospholipase A2{gamma} leads to alterations in hippocampal cardiolipin content and molecular species distribution, mitochondrial degeneration, autophagy, and cognitive dysfunction. J Biol Chem 284:35632–35644
Mancuso DJ, Sims HF, Han X, Jenkins CM, Guan SP, Yang K, Moon SH, Pietka T, Abumrad NA, Schlesinger PH, Gross RW (2007) Genetic ablation of calcium-independent phospholipase A2gamma leads to alterations in mitochondrial lipid metabolism and function resulting in a deficient mitochondrial bioenergetic phenotype. J Biol Chem 282:34611–34622
Xu Y, Malhotra A, Ren M, Schlame M (2006) The enzymatic function of tafazzin. J Biol Chem 281:39217–39224
Xu Y, Kelley RI, Blanck TJ, Schlame M (2003) Remodeling of cardiolipin by phospholipid transacylation. J Biol Chem 278:51380–51385
Malhotra A, Xu Y, Ren M, Schlame M (2009) Formation of molecular species of mitochondrial cardiolipin. 1. A novel transacylation mechanism to shuttle fatty acids between sn-1 and sn-2 positions of multiple phospholipid species. Biochim Biophys Acta 1791:314–320
Schlame M, Acehan D, Berno B, Xu Y, Valvo S, Ren M, Stokes DL, Epand RM (2012) The physical state of lipid substrates provides transacylation specificity for tafazzin. Nat Chem Biol 8:862–869
Schlame M, Ren M, Xu Y, Greenberg ML, Haller I (2005) Molecular symmetry in mitochondrial cardiolipins. Chem Phys Lipids 138:38–49
Taylor WA, Hatch GM (2003) Purification and characterization of monolysocardiolipin acyltransferase from pig liver mitochondria. J Biol Chem 278:12716–12721
Taylor WA, Hatch GM (2009) Identification of the human mitochondrial linoleoyl-coenzyme A monolysocardiolipin acyltransferase (MLCL AT-1). J Biol Chem 284:30360–30371
Cao J, Liu Y, Lockwood J, Burn P, Shi Y (2004) A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. J Biol Chem 279:31727–31734
Ng Y, Barhoumi R, Tjalkens RB, Fan YY, Kolar S, Wang N, Lupton JR, Chapkin RS (2005) The role of docosahexaenoic acid in mediating mitochondrial membrane lipid oxidation and apoptosis in colonocytes. Carcinogenesis 26:1914–1921
Hong MY, Chapkin RS, Barhoumi R, Burghardt RC, Turner ND, Henderson CE, Sanders LM, Fan YY, Davidson LA, Murphy ME, Spinka CM, Carroll RJ, Lupton JR (2002) Fish oil increases mitochondrial phospholipid unsaturation, upregulating reactive oxygen species and apoptosis in rat colonocytes. Carcinogenesis 23:1919–1925
Watkins SM, Carter LC, German JB (1998) Docosahexaenoic acid accumulates in cardiolipin and enhances HT-29 cell oxidant production. J Lipid Res 39:1583–1588
Sparagna GC, Lesnefsky EJ (2009) Cardiolipin remodeling in the heart. J Cardiovasc Pharmacol 53:290–301
Christodoulou J, McInnes RR, Jay V, Wilson G, Becker LE, Lehotay DC, Platt BA, Bridge PJ, Robinson BH, Clarke JT (1994) Barth syndrome: clinical observations and genetic linkage studies. Am J Med Genet 50:255–264
Barth PG, Wanders RJ, Vreken P (1999) X-linked cardioskeletal myopathy and neutropenia (Barth syndrome)-MIM 302060. J Pediatr 135:273–276
Bione S, D’Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D (1996) A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet 12:385–389
Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJ, Barth PG (2000) Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Biophys Res Commun 279:378–382
Schlame M, Towbin JA, Heerdt PM, Jehle R, DiMauro S, Blanck TJ (2002) Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann Neurol 51:634–637
Sparagna GC, Chicco AJ, Murphy RC, Bristow MR, Johnson CA, Rees ML, Maxey ML, McCune SA, Moore RL (2007) Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J Lipid Res 48:1559–1570
Saini-Chohan HK, Holmes MG, Chicco AJ, Taylor WA, Moore RL, McCune SA, Hickson-Bick DL, Hatch GM, Sparagna GC (2009) Cardiolipin biosynthesis and remodeling enzymes are altered during development of heart failure. J Lipid Res 50:1600–1608
Garcia MJ, McNamara PM, Gordon T, Kannel WB (1974) Morbidity and mortality in diabetics in the Framingham population. Sixteen year follow-up study. Diabetes 23:105–111
Nichols GA, Hillier TA, Erbey JR, Brown JB (2001) Congestive heart failure in type 2 diabetes: prevalence, incidence, and risk factors. Diabetes Care 24:1614–1619
Poornima IG, Parikh P, Shannon RP (2006) Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res 98:596–605
Wang J, Song Y, Wang Q, Kralik PM, Epstein PN (2006) Causes and characteristics of diabetic cardiomyopathy. Rev Diabet Stud (RDS) 3:108–117
Fang ZY, Prins JB, Marwick TH (2004) Diabetic cardiomyopathy: evidence, mechanisms, and therapeutic implications. Endocr Rev 25:543–567
Han X, Yang J, Yang K, Zhao Z, Abendschein DR, Gross RW (2007) Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: a shotgun lipidomics study. Biochemistry 46:6417–6428
Han X, Yang J, Cheng H, Yang K, Abendschein DR, Gross RW (2005) Shotgun lipidomics identifies cardiolipin depletion in diabetic myocardium linking altered substrate utilization with mitochondrial dysfunction. Biochemistry 44:16684–16694
Gorbenko GP (1999) Structure of cytochrome c complexes with phospholipids as revealed by resonance energy transfer. Biochim Biophys Acta 1420:1–13
Iverson SL, Orrenius S (2004) The cardiolipin-cytochrome c interaction and the mitochondrial regulation of apoptosis. Arch Biochem Biophys 423:37–46
Gonzalvez F, Pariselli F, Jalmar O, Dupaigne P, Sureau F, Dellinger M, Hendrickson EA, Bernard S, Petit PX (2010) Mechanistic issues of the interaction of the hairpin-forming domain of tBid with mitochondrial cardiolipin. PLoS One 5:e9342
Gonzalvez F, Schug ZT, Houtkooper RH, MacKenzie ED, Brooks DG, Wanders RJ, Petit PX, Vaz FM, Gottlieb E (2008) Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol 183:681–696
Li H, Zhu H, Xu CJ, Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94:491–501
Spooner PJ, Watts A (1992) Cytochrome c interactions with cardiolipin in bilayers: a multinuclear magic-angle spinning NMR study. Biochemistry 31:10129–10138
Choi SY, Gonzalvez F, Jenkins GM, Slomianny C, Chretien D, Arnoult D, Petit PX, Frohman MA (2007) Cardiolipin deficiency releases cytochrome c from the inner mitochondrial membrane and accelerates stimuli-elicited apoptosis. Cell Death Differ 14:597–606
Belikova NA, Vladimirov YA, Osipov AN, Kapralov AA, Tyurin VA, Potapovich MV, Basova LV, Peterson J, Kurnikov IV, Kagan VE (2006) Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin-containing membranes. Biochemistry 45:4998–5009
Aguilar L, Ortega-Pierres G, Campos B, Fonseca R, Ibanez M, Wong C, Farfan N, Naciff JM, Kaetzel MA, Dedman JR, Baeza I (1999) Phospholipid membranes form specific nonbilayer molecular arrangements that are antigenic. J Biol Chem 274:25193–25196
Lutter M, Perkins GA, Wang X (2001) The pro-apoptotic Bcl-2 family member tBid localizes to mitochondrial contact sites. BMC Cell Biol 2:22
Tuominen EK, Wallace CJ, Kinnunen PK (2002) Phospholipid-cytochrome c interaction: evidence for the extended lipid anchorage. J Biol Chem 277:8822–8826
Petrosillo G, Casanova G, Matera M, Ruggiero FM, Paradies G (2006) Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: induction of permeability transition and cytochrome c release. FEBS Lett 580:6311–6316
Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S (2002) Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci U S A 99:1259–1263
Petrosillo G, Ruggiero FM, Paradies G (2003) Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. FASEB J: Off Publ Fed Am Soc Exp Biol 17:2202–2208
Garcia Fernandez M, Troiano L, Moretti L, Nasi M, Pinti M, Salvioli S, Dobrucki J, Cossarizza A (2002) Early changes in intramitochondrial cardiolipin distribution during apoptosis. Cell Growth Differ Mol Biol J Am Assoc Cancer Res 13:449–455
Thiagarajan P, Tait JF (1990) Binding of annexin V/placental anticoagulant protein I to platelets. Evidence for phosphatidylserine exposure in the procoagulant response of activated platelets. J Biol Chem 265:17420–17423
Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X (2000) Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol 2:754–761
Gonzalvez F, Pariselli F, Dupaigne P, Budihardjo I, Lutter M, Antonsson B, Diolez P, Manon S, Martinou JC, Goubern M, Wang X, Bernard S, Petit PX (2005) tBid interaction with cardiolipin primarily orchestrates mitochondrial dysfunctions and subsequently activates Bax and Bak. Cell Death Differ 12:614–626
Heit B, Yeung T, Grinstein S (2011) Changes in mitochondrial surface charge mediate recruitment of signaling molecules during apoptosis. Am J Physiol Cell Physiol 300:C33–C41
Liu J, Weiss A, Durrant D, Chi NW, Lee RM (2004) The cardiolipin-binding domain of Bid affects mitochondrial respiration and enhances cytochrome c release. Apoptosis: Int J Program Cell Death 9:533–541
Sani MA, Dufourc EJ, Grobner G (2009) How does the Bax-alpha1 targeting sequence interact with mitochondrial membranes? The role of cardiolipin. Biochim Biophys Acta 1788:623–631
Schlame M, Augustin W (1985) Association of creatine kinase with rat heart mitochondria: high and low affinity binding sites and the involvement of phospholipids. Biomed Biochim Acta 44:1083–1088
Schlattner U, Gehring F, Vernoux N, Tokarska-Schlattner M, Neumann D, Marcillat O, Vial C, Wallimann T (2004) C-terminal lysines determine phospholipid interaction of sarcomeric mitochondrial creatine kinase. J Biol Chem 279:24334–24342
Maniti O, Lecompte MF, Marcillat O, Desbat B, Buchet R, Vial C, Granjon T (2009) Mitochondrial creatine kinase binding to phospholipid monolayers induces cardiolipin segregation. Biophys J 96:2428–2438
Epand RF, Tokarska-Schlattner M, Schlattner U, Wallimann T, Epand RM (2007) Cardiolipin clusters and membrane domain formation induced by mitochondrial proteins. J Mol Biol 365:968–980
Epand RF, Schlattner U, Wallimann T, Lacombe ML, Epand RM (2007) Novel lipid transfer property of two mitochondrial proteins that bridge the inner and outer membranes. Biophys J 92:126–137
Speer O, Back N, Buerklen T, Brdiczka D, Koretsky A, Wallimann T, Eriksson O (2005) Octameric mitochondrial creatine kinase induces and stabilizes contact sites between the inner and outer membrane. Biochem J 385:445–450
Liu J, Chen J, Dai Q, Lee RM (2003) Phospholipid scramblase 3 is the mitochondrial target of protein kinase C delta-induced apoptosis. Cancer Res 63:1153–1156
Liu J, Dai Q, Chen J, Durrant D, Freeman A, Liu T, Grossman D, Lee RM (2003) Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol Cancer Res: MCR 1:892–902
Van Q, Liu J, Lu B, Feingold KR, Shi Y, Lee RM, Hatch GM (2007) Phospholipid scramblase-3 regulates cardiolipin de novo biosynthesis and its resynthesis in growing HeLa cells. Biochem J 401:103–109
Kim TH, Zhao Y, Ding WX, Shin JN, He X, Seo YW, Chen J, Rabinowich H, Amoscato AA, Yin XM (2004) Bid-cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome C release. Mol Biol Cell 15:3061–3072
Sorice M, Circella A, Cristea IM, Garofalo T, Di Renzo L, Alessandri C, Valesini G, Esposti MD (2004) Cardiolipin and its metabolites move from mitochondria to other cellular membranes during death receptor-mediated apoptosis. Cell Death Differ 11:1133–1145
Esposti MD, Erler JT, Hickman JA, Dive C (2001) Bid, a widely expressed proapoptotic protein of the Bcl-2 family, displays lipid transfer activity. Mol Cell Biol 21:7268–7276
Beyer K, Klingenberg M (1985) ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by 31P nuclear magnetic resonance. Biochemistry 24:3821–3826
Beyer K, Nuscher B (1996) Specific cardiolipin binding interferes with labeling of sulfhydryl residues in the adenosine diphosphate/adenosine triphosphate carrier protein from beef heart mitochondria. Biochemistry 35:15784–15790
Sedlak E, Robinson NC (1999) Phospholipase A(2) digestion of cardiolipin bound to bovine cytochrome c oxidase alters both activity and quaternary structure. Biochemistry 38:14966–14972
Joshi AS, Zhou J, Gohil VM, Chen S, Greenberg ML (2009) Cellular functions of cardiolipin in yeast. Biochim Biophys Acta 1793:212–218
Claypool SM (2009) Cardiolipin, a critical determinant of mitochondrial carrier protein assembly and function. Biochim Biophys Acta 1788:2059–2068
Pitkanen S, Robinson BH (1996) Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of superoxide dismutase. J Clin Invest 98:345–351
Grant CM, MacIver FH, Dawes IW (1997) Mitochondrial function is required for resistance to oxidative stress in the yeast Saccharomyces cerevisiae. FEBS Lett 410:219–222
Barros MH, Netto LE, Kowaltowski AJ (2003) H(2)O(2) generation in Saccharomyces cerevisiae respiratory pet mutants: effect of cytochrome c. Free Radic Biol Med 35:179–188
Schagger H, Pfeiffer K (2000) Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J 19:1777–1783
Zhang M, Mileykovskaya E, Dowhan W (2002) Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem 277:43553–43556
Zhang M, Mileykovskaya E, Dowhan W (2005) Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J Biol Chem 280:29403–29408
McKenzie M, Lazarou M, Thorburn DR, Ryan MT (2006) Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol 361:462–469
Claypool SM, Oktay Y, Boontheung P, Loo JA, Koehler CM (2008) Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J Cell Biol 182:937–950
Haines TH, Dencher NA (2002) Cardiolipin: a proton trap for oxidative phosphorylation. FEBS Lett 528:35–39
Hoch FL (1998) Cardiolipins and mitochondrial proton-selective leakage. J Bioenerg Biomembr 30:511–532
Jiang F, Ryan MT, Schlame M, Zhao M, Gu Z, Klingenberg M, Pfanner N, Greenberg ML (2000) Absence of cardiolipin in the crd1 null mutant results in decreased mitochondrial membrane potential and reduced mitochondrial function. J Biol Chem 275:22387–22394
Chen YC, Taylor EB, Dephoure N, Heo JM, Tonhato A, Papandreou I, Nath N, Denko NC, Gygi SP, Rutter J (2012) Identification of a protein mediating respiratory supercomplex stability. Cell Metab 15:348–360
Turrens JF, Alexandre A, Lehninger AL (1985) Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys 237:408–414
Barja G (1999) Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J Bioenerg Biomembr 31:347–366
Grivennikova VG, Vinogradov AD (2006) Generation of superoxide by the mitochondrial Complex I. Biochim Biophys Acta 1757:553–561
Kushnareva Y, Murphy AN, Andreyev A (2002) Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P) + oxidation-reduction state. Biochem J 368:545–553
Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E (1998) Peroxidative damage to cardiac mitochondria: cytochrome oxidase and cardiolipin alterations. FEBS Lett 424:155–158
Paradies G, Petrosillo G, Pistolese M, Ruggiero FM (2000) The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles. FEBS Lett 466:323–326
Paradies G, Petrosillo G, Pistolese M, Ruggiero FM (2001) Reactive oxygen species generated by the mitochondrial respiratory chain affect the complex III activity via cardiolipin peroxidation in beef-heart submitochondrial particles. Mitochondrion 1:151–159
Robinson NC, Strey F, Talbert L (1980) Investigation of the essential boundary layer phospholipids of cytochrome c oxidase using Triton X-100 delipidation. Biochemistry 19:3656–3661
Powell GL, Knowles PF, Marsh D (1987) Spin-label studies on the specificity of interaction of cardiolipin with beef heart cytochrome oxidase. Biochemistry 26:8138–8145
Abramovitch DA, Marsh D, Powell GL (1990) Activation of beef-heart cytochrome c oxidase by cardiolipin and analogues of cardiolipin. Biochim Biophys Acta 1020:34–42
Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Serena D, Ruggiero FM (1999) Lipid peroxidation and alterations to oxidative metabolism in mitochondria isolated from rat heart subjected to ischemia and reperfusion. Free Radic Biol Med 27:42–50
Petrosillo G, Portincasa P, Grattagliano I, Casanova G, Matera M, Ruggiero FM, Ferri D, Paradies G (2007) Mitochondrial dysfunction in rat with nonalcoholic fatty liver Involvement of complex I, reactive oxygen species and cardiolipin. Biochim Biophys Acta 1767:1260–1267
Ferlini C, De Angelis C, Biselli R, Distefano M, Scambia G, Fattorossi A (1999) Sequence of metabolic changes during X-ray-induced apoptosis. Exp Cell Res 247:160–167
Ushmorov A, Ratter F, Lehmann V, Droge W, Schirrmacher V, Umansky V (1999) Nitric-oxide-induced apoptosis in human leukemic lines requires mitochondrial lipid degradation and cytochrome C release. Blood 93:2342–2352
Liu X, Ye B, Miller S, Yuan H, Zhang H, Tian L, Nie J, Imae R, Arai H, Li Y, Cheng Z, Shi Y (2012) Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Mol Cell Biol 32:4493–4504
Maftah A, Ratinaud MH, Dumas M, Bonte F, Meybeck A, Julien R (1994) Human epidermal cells progressively lose their cardiolipins during ageing without change in mitochondrial transmembrane potential. Mech Ageing Dev 77:83–96
Lewin MB, Timiras PS (1984) Lipid changes with aging in cardiac mitochondrial membranes. Mech Ageing Dev 24:343–351
Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E (1993) Age-dependent decrease in the cytochrome c oxidase activity and changes in phospholipids in rat-heart mitochondria. Arch Gerontol Geriatr 16:263–272
Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E (1997) Age-dependent decline in the cytochrome c oxidase activity in rat heart mitochondria: role of cardiolipin. FEBS Lett 406:136–138
Lenaz G, Bovina C, Castelluccio C, Fato R, Formiggini G, Genova ML, Marchetti M, Pich MM, Pallotti F, Parenti Castelli G, Biagini G (1997) Mitochondrial complex I defects in aging. Mol Cell Biochem 174:329–333
Paradies G, Ruggiero FM (1990) Age-related changes in the activity of the pyruvate carrier and in the lipid composition in rat-heart mitochondria. Biochim Biophys Acta 1016:207–212
Hoch FL (1992) Cardiolipins and biomembrane function. Biochim Biophys Acta 1113:71–133
Hagen TM, Ingersoll RT, Wehr CM, Lykkesfeldt J, Vinarsky V, Bartholomew JC, Song MH, Ames BN (1998) Acetyl-L-carnitine fed to old rats partially restores mitochondrial function and ambulatory activity. Proc Natl Acad Sci U S A 95:9562–9566
Sen T, Sen N, Tripathi G, Chatterjee U, Chakrabarti S (2006) Lipid peroxidation associated cardiolipin loss and membrane depolarization in rat brain mitochondria. Neurochem Int 49:20–27
Sen T, Sen N, Jana S, Khan FH, Chatterjee U, Chakrabarti S (2007) Depolarization and cardiolipin depletion in aged rat brain mitochondria: relationship with oxidative stress and electron transport chain activity. Neurochem Int 50:719–725
Wisneski JA, Gertz EW, Neese RA, Gruenke LD, Morris DL, Craig JC (1985) Metabolic fate of extracted glucose in normal human myocardium. J Clin Invest 76:1819–1827
Christe ME, Rodgers RL (1994) Altered glucose and fatty acid oxidation in hearts of the spontaneously hypertensive rat. J Mol Cell Cardiol 26:1371–1375
Davila-Roman VG, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP, Gropler RJ (2002) Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol 40:271–277
Nalecz KA, Bolli R, Wojtczak L, Azzi A (1986) The monocarboxylate carrier from bovine heart mitochondria: partial purification and its substrate-transporting properties in a reconstituted system. Biochim Biophys Acta 851:29–37
Noel H, Pande SV (1986) An essential requirement of cardiolipin for mitochondrial carnitine acylcarnitine translocase activity. Lipid requirement of carnitine acylcarnitine translocase. Eur J Biochem 155:99–102
Paradies G, Petrosillo G, Gadaleta MN, Ruggiero FM (1999) The effect of aging and acetyl-L-carnitine on the pyruvate transport and oxidation in rat heart mitochondria. FEBS Lett 454:207–209
Paradies G, Ruggiero FM, Petrosillo G, Gadaleta MN, Quagliariello E (1995) Carnitine-acylcarnitine translocase activity in cardiac mitochondria from aged rats: the effect of acetyl-L-carnitine. Mech Ageing Dev 84:103–112
Hagen TM, Wehr CM, Ames BN (1998) Mitochondrial decay in aging. Reversal through supplementation of acetyl-L-carnitine and N-tert-butyl-alpha-phenyl-nitrone. Ann N Y Acad Sci 854:214–223
Maccari F, Arseni A, Chiodi P, Ramacci MT, Angelucci L (1990) Levels of carnitines in brain and other tissues of rats of different ages: effect of acetyl-L-carnitine administration. Exp Gerontol 25:127–134
Dzugasova V, Obernauerova M, Horvathova K, Vachova M, Zakova M, Subik J (1998) Phospha-tidylglycerolphosphate synthase encoded by the PEL1/PGS1 gene in Saccharomyces cerevisiae is localized in mitochondria and its expression is regulated by phospholipid precursors. Curr Genet 34:297–302
Jiang F, Gu Z, Granger JM, Greenberg ML (1999) Cardiolipin synthase expression is essential for growth at elevated temperature and is regulated by factors affecting mitochondrial development. Mol Microbiol 31:373–379
Popolo L, Vai M, Gatti E, Porello S, Bonfante P, Balestrini R, Alberghina L (1993) Physiological analysis of mutants indicates involvement of the Saccharomyces cerevisiae GPI-anchored protein gp115 in morphogenesis and cell separation. J Bacteriol 175:1879–1885
de Nobel H, Ruiz C, Martin H, Morris W, Brul S, Molina M, Klis FM (2000) Cell wall perturbation in yeast results in dual phosphorylation of the Slt2/Mpk1 MAP kinase and in an Slt2-mediated increase in FKS2-lacZ expression, glucanase resistance and thermotolerance. Microbiology 146(Pt 9):2121–2132
Zhong Q, Greenberg ML (2005) Deficiency in mitochondrial anionic phospholipid synthesis impairs cell wall biogenesis. Biochem Soc Trans 33:1158–1161
Lussier M, White AM, Sheraton J, di Paolo T, Treadwell J, Southard SB, Horenstein CI, Chen-Weiner J, Ram AF, Kapteyn JC, Roemer TW, Vo DH, Bondoc DC, Hall J, Zhong WW, Sdicu AM, Davies J, Klis FM, Robbins PW, Bussey H (1997) Large scale identification of genes involved in cell surface biosynthesis and architecture in Saccharomyces cerevisiae. Genetics 147:435–450
Heinisch JJ, Lorberg A, Schmitz HP, Jacoby JJ (1999) The protein kinase C-mediated MAP kinase pathway involved in the maintenance of cellular integrity in Saccharomyces cerevisiae. Mol Microbiol 32:671–680
Levin DE (2005) Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol Mol Biol Rev: MMBR 69:262–291
Terashima H, Yabuki N, Arisawa M, Hamada K, Kitada K (2000) Up-regulation of genes encoding glycosylphosphatidylinositol (GPI)-attached proteins in response to cell wall damage caused by disruption of FKS1 in Saccharomyces cerevisiae. Mol Gen Genet 264:64–74
Jung US, Levin DE (1999) Genome-wide analysis of gene expression regulated by the yeast cell wall integrity signalling pathway. Mol Microbiol 34:1049–1057
Conde R, Pablo G, Cueva R, Larriba G (2003) Screening for new yeast mutants affected in mannosylphosphorylation of cell wall mannoproteins. Yeast 20:1189–1211
Page N, Gerard-Vincent M, Menard P, Beaulieu M, Azuma M, Dijkgraaf GJ, Li H, Marcoux J, Nguyen T, Dowse T, Sdicu AM, Bussey H (2003) A Saccharomyces cerevisiae genome-wide mutant screen for altered sensitivity to K1 killer toxin. Genetics 163:875–894
Velours G, Boucheron C, Manon S, Camougrand N (2002) Dual cell wall/mitochondria localization of the ‘SUN’ family proteins. FEMS Microbiol Lett 207:165–172
Green R, Lesage G, Sdicu AM, Menard P, Bussey H (2003) A synthetic analysis of the Saccharomyces cerevisiae stress sensor Mid2p, and identification of a Mid2p-interacting protein, Zeo1p, that modulates the PKC1-MPK1 cell integrity pathway. Microbiology 149:2487–2499
Sickmann A, Reinders J, Wagner Y, Joppich C, Zahedi R, Meyer HE, Schonfisch B, Perschil I, Chacinska A, Guiard B, Rehling P, Pfanner N, Meisinger C (2003) The proteome of Saccharomyces cerevisiae mitochondria. Proc Natl Acad Sci U S A 100:13207–13212
Zahedi RP, Sickmann A, Boehm AM, Winkler C, Zufall N, Schonfisch B, Guiard B, Pfanner N, Meisinger C (2006) Proteomic analysis of the yeast mitochondrial outer membrane reveals accumulation of a subclass of preproteins. Mol Biol Cell 17:1436–1450
Kaeberlein M, Guarente L (2002) Saccharomyces cerevisiae MPT5 and SSD1 function in parallel pathways to promote cell wall integrity. Genetics 160:83–95
Hayashi M, Maeda T (2006) Activation of the HOG pathway upon cold stress in Saccharomyces cerevisiae. J Biochem 139:797–803
Winkler A, Arkind C, Mattison CP, Burkholder A, Knoche K, Ota I (2002) Heat stress activates the yeast high-osmolarity glycerol mitogen-activated protein kinase pathway, and protein tyrosine phosphatases are essential under heat stress. Eukaryot Cell 1:163–173
Hahn JS, Thiele DJ (2002) Regulation of the Saccharomyces cerevisiae Slt2 kinase pathway by the stress-inducible Sdp1 dual specificity phosphatase. J Biol Chem 277:21278–21284
Davenport KR, Sohaskey M, Kamada Y, Levin DE, Gustin MC (1995) A second osmosensing signal transduction pathway in yeast. Hypotonic shock activates the PKC1 protein kinase-regulated cell integrity pathway. J Biol Chem 270:30157–30161
Garcia-Rodriguez LJ, Valle R, Duran A, Roncero C (2005) Cell integrity signaling activation in response to hyperosmotic shock in yeast. FEBS Lett 579:6186–6190
Hohmann S (2002) Osmotic stress signaling and osmoadaptation in yeasts. Microbiol Mol Biol Rev: MMBR 66:300–372
Rep M, Krantz M, Thevelein JM, Hohmann S (2000) The transcriptional response of Saccharomyces cerevisiae to osmotic shock. Hot1p and Msn2p/Msn4p are required for the induction of subsets of high osmolarity glycerol pathway-dependent genes. J Biol Chem 275:8290–8300
Kojima K, Bahn YS, Heitman J (2006) Calcineurin, Mpk1 and Hog1 MAPK pathways independently control fludioxonil antifungal sensitivity in Cryptococcus neoformans. Microbiology 152:591–604
Klionsky DJ, Herman PK, Emr SD (1990) The fungal vacuole: composition, function, and biogenesis. Microbiol Rev 54:266–292
Latterich M, Watson MD (1993) Evidence for a dual osmoregulatory mechanism in the yeast Saccharomyces cerevisiae. Biochem Biophys Res Commun 191:1111–1117
Butow RA, Avadhani NG (2004) Mitochondrial signaling: the retrograde response. Mol Cell 14:1–15
Liu Z, Butow RA (2006) Mitochondrial retrograde signaling. Annu Rev Genet 40:159–185
Brett CL, Tukaye DN, Mukherjee S, Rao R (2005) The yeast endosomal Na + K+/H + exchanger Nhx1 regulates cellular pH to control vesicle trafficking. Mol Biol Cell 16:1396–1405
Ali R, Brett CL, Mukherjee S, Rao R (2004) Inhibition of sodium/proton exchange by a Rab-GTPase-activating protein regulates endosomal traffic in yeast. J Biol Chem 279:4498–4506
Zinser E, Sperka-Gottlieb CD, Fasch EV, Kohlwein SD, Paltauf F, Daum G (1991) Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J Bacteriol 173:2026–2034
Lemasters JJ (2005) Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 8:3–5
Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ (2009) Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 17:98–109
Tal R, Winter G, Ecker N, Klionsky DJ, Abeliovich H (2007) Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. J Biol Chem 282:5617–5624
Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y (1992) Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol 119:301–311
Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169:425–434
Shintani T, Huang WP, Stromhaug PE, Klionsky plasm to vacuole targeting pathway. Dev Cell 3:825–837
Hughes AL, Gottschling DE (2012) An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature 492:261–265
McCord JM (1988) Free radicals and myocardial ischemia: overview and outlook. Free Radic Biol Med 4:9–14
Ambrosio G, Zweier JL, Flaherty JT (1991) The relationship between oxygen radical generation and impairment of myocardial energy metabolism following post-ischemic reperfusion. J Mol Cell Cardiol 23:1359–1374
Lucas DT, Szweda LI (1998) Cardiac reperfusion injury: aging, lipid peroxidation, and mitochondrial dysfunction. Proc Natl Acad Sci U S A 95:510–514
Acehan D, Vaz F, Houtkooper RH, James J, Moore V, Tokunaga C, Kulik W, Wansapura J, Toth MJ, Strauss A, Khuchua Z (2011) Cardiac and skeletal muscle defects in a mouse model of human Barth syndrome. J Biol Chem 286:899–908
Sato H, Tanaka T, Kasai K, Kita T, Tanaka N (2007) Role of p38 mitogen-activated protein kinase on cardiac dysfunction after hemorrhagic shock in rats. Shock 28:291–299
Peter PS, Brady JE, Yan L, Chen W, Engelhardt S, Wang Y, Sadoshima J, Vatner SF, Vatner DE (2007) Inhibition of p38 alpha MAPK rescues cardiomyopathy induced by overexpressed beta 2-adrenergic receptor, but not beta 1-adrenergic receptor. J Clin Invest 117:1335–1343
Allo SN, Carl LL, Morgan HE (1992) Acceleration of growth of cultured cardiomyocytes and translocation of protein kinase C. Am J Physiol 263:C319–C325
Palaniyandi SS, Sun L, Ferreira JC, Mochly-Rosen D (2009) Protein kinase C in heart failure: a therapeutic target? Cardiovasc Res 82:229–239
Paoletti R, Maffei A, Madaro L, Notte A, Stanganello E, Cifelli G, Carullo P, Molinaro M, Lembo G, Bouche M (2010) Protein kinase Ctheta is required for cardiomyocyte survival and cardiac remodeling. Cell Death Dis 1:e45
Ping P, Zhang J, Qiu Y, Tang XL, Manchikalapudi S, Cao X, Bolli R (1997) Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circ Res 81:404–414
Liu GS, Cohen MV, Mochly-Rosen D, Downey JM (1999) Protein kinase C-epsilon is responsible for the protection of preconditioning in rabbit cardiomyocytes. J Mol Cell Cardiol 31:1937–1948
Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM, Ping P (2003) Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ Res 92:873–880
Hambleton M, York A, Sargent MA, Kaiser RA, Lorenz JN, Robbins J, Molkentin JD (2007) Inducible and myocyte-specific inhibition of PKCalpha enhances cardiac contractility and protects against infarction-induced heart failure. Am J Physiol Heart Circ Physiol 293:H3768–H3771
Belin RJ, Sumandea MP, Allen EJ, Schoenfelt K, Wang H, Solaro RJ, de Tombe PP (2007) Augmented protein kinase C-alpha-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ Res 101:195–204
Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD (2004) PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med 10:248–254
Acknowledgements
The Greenberg laboratory acknowledges support from the National Institutes of Health (R21 HL 084218) and the Barth Syndrome Foundation (BSF) to M.L.G., and Wayne State University Graduate Enhancement Research Funds to V.A.P.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Patil, V.A., Greenberg, M.L. (2013). Cardiolipin-Mediated Cellular Signaling. In: Capelluto, D. (eds) Lipid-mediated Protein Signaling. Advances in Experimental Medicine and Biology, vol 991. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6331-9_11
Download citation
DOI: https://doi.org/10.1007/978-94-007-6331-9_11
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6330-2
Online ISBN: 978-94-007-6331-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)