
Abstract
Sphingolipid de novo biosynthesis is related with metabolic diseases. However, the mechanism is still not quite clear. Sphingolipids are ubiquitous and critical components of biological membranes. Their biosynthesis starts with soluble precursors in the endoplasmic reticulum and culminates in the Golgi complex and plasma membrane. The interaction of sphingomyelin, cholesterol, and glycosphingolipid drives the formation of plasma membrane rafts. Lipid rafts have been shown to be involved in cell signaling, lipid and protein sorting, and membrane trafficking. It is well known that toll-like receptors, class A and B scavenger receptors, and insulin receptor are located in lipid rafts. Sphingomyelin is also a reservoir for other sphingolipids. So, sphingomyelin has important impact in cell signaling through its structural role in lipid rafts or its catabolic inter-mediators, such as ceramide and glycoceramide. In this chapter, we will discuss both aspects.
This work was supported by NIH grants HL093419, HL69817, VA Merit 000900–01 to XCJ.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Sphingomyelin
- Ceramide
- Diacylglycerol
- Sphingolipids
- Sphingolipid biosynthesis
- Lipid rafts
- Cholesterol homeostasis
1.1 Introduction
Although the discovery of sphingomyelin (SM) was reported more than a century ago, its role as a significant ‘bioactive lipid’ along with other members of the sphingolipid family have been recognized just couple of decades ago. Technological advances in lipid detection, analysis, and quantitation have played a key role in promoting the development of the sphingolipid research field. There have been numerous studies establishing sphingolipids’ multifunctional roles in the regulation of various cellular processes such as cell growth, death, senescence, adhesion, migration, inflammation, angiogenesis and intracellular trafficking [1, 2].
However, the concept that SM is involved in cellular signaling is relatively new. We believe that SM mediated cellular signaling can be broadly manifested in two ways:
-
(i)
SM metabolism resulting in the production of various interconvertible bioactive sphingolipids or derivatives such as ceramide, diacylglyceride, and sphingosine-1-phosphate. These bioactive lipids act on their specific targets within the cell and regulate various signal transduction pathways, thereby affecting cellular functions.
-
(ii)
SM-enriched lipid raft mediated cell signaling. The interaction of SM with cholesterol and glycosphingolipid is known to drive the formation of plasma membrane microdomains called lipid rafts [3]. As much as 70 % of all cellular SM is found in these rafts [4, 5] and they have proven to be involved in cell signaling, lipid, and protein sorting, and membrane trafficking [3, 6, 7].
This review specifically aims at deciphering the role of SM as a bioactive lipid in cellular signaling through its metabolism and its contribution to lipid rafts.
1.2 Structure, Sub-cellular Localization, and Measurement of Sphingomyelin Levels
The SM molecule consists of two regions: a phosphorylcholine head group attached to a ceramide molecule (Fig. 1.1). The latter in turn is made up of a sphingosine backbone and a fatty acid (acyl chain). SM usually contains 16:0, 18:0, 22:0, 24:0, and 24:1 acyl chains but the most abundant SM species found in mammalian tissues are 16:0 [8]. Whether or not the differing acyl chain lengths in SMs dictate unique functions or important biophysical distinctions has not yet been established.

Molecular structure of sphingomyelin
SM is the most abundant sphingolipid in mammalian cells and the majority of the cellular SM is located in the outer leaflet of plasma membranes [5, 9]. SM is indispensable for mammalian cell viability, as evidenced by the inability of mammalian cells to survive in culture, when they are unable to produce SM [10].
SM levels can be measured by the following methods: (i) enzyme-based assay: tissue homogenates can be incubated with bacterial sphingomyelinase, alkaline phosphatase, choline oxidase, peroxidase, N-ethyl-N-(2-hydroxy-3-sulfopropyl)-3,5-dimethoxyaniline, and 4-aminoantipyrine for 45 min. This results in a product with blue color, whose intensity is proportional to the SM present in the tissue sample, and can be measured at an optimal absorption of 595 nm [11]; (ii) liquid chromatography tandem mass spectrometry (LC/MS/MS); and, (iii) lysenin-mediated cell lysis assay. Lysenin is a SM-specific cytotoxin, which recognizes SM only when it forms aggregates or microdomains and eventually leads to cell lysis [12]. Based on the lysenin-mediated cell lysis intensity, plasma membrane SM levels can be indirectly evaluated. More SM on the plasma membrane can cause high cell mortality [12, 13].
1.3 Sphingomyelin Metabolism-Mediated Cell Signaling
1.3.1 De Novo Sphingomyelin Synthesis
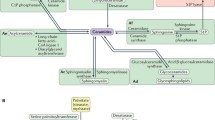
SM biosynthesis initiates in the endoplasmic reticulum (ER), utilizing non-sphingolipid hydrophilic precursor molecules, serine, and palmitoyl-CoA (Fig. 1.2). The condensation of L-serine and palmitoyl-CoA to form 3-ketodihydrosphingosine is facilitated by ER membrane associated serine palmitoyltransferase (SPT). The next step in the sphingolipid biosynthesis is the reduction of 3-ketodihydrosphingosine to dihydrosphingosine by a reductase. N-acylation of the dihydrosphingosine gives rise to dihydroceramide, a product that is still relatively hydrophilic. Conversion of dihydroceramide to ceramide is facilitated by ceramide synthases and involves a desaturation step. Ceramides are hydrophobic and therefore become membrane associated. The majority of ceramides are transported from ER to the Golgi by ceramide transport protein (CERT), and the rest are converted to ceramide phosphoethanolamine (CPE). In the Golgi apparatus, ceramides are further converted to sphingomyelin by the sphingomyelin synthase (SMS) [14, 15], to glucosylceramide by the glucosylceramide synthase and, then, to more complex sphingolipids such as glucosylceramide and hematoside (GM3) by their respective synthases (Fig. 1.2). These products are then transported to plasma membrane, the major cellular reservoir for these lipids. SM and other sphingolipids may reach to the blood circulation through lipoprotein secretion or lipid efflux (Fig. 1.2).

Scheme of sphingomyelin biosynthesis. SMS 1 and 2, sphingomyelin synthase 1 and 2; SMSr, sphingomyelin synthase related protein; GCS glucosylceramide synthase, CPE ceramide phosphoethanolamine synthase, CERT ceramide trafficking protein, GM3 hematoside, ER endoplasmic reticulum, PM plasma membrane
1.3.2 Sphingomyelin and Its Related Bioactive Lipids
Sphingomyelin synthase (SMS), utilizing ceramide and phosphatidylcholine as its two substrates to produce SM and diacylglyceride, sits at the crossroads of bioactive lipid synthesis (Fig. 1.3). SM can also be hydrolyzed by sphingomyelinase (SMase) to yield ceramide and choline phosphate. The resulting ceramide can be further converted into sphingosine and sphingosine-1-phosphate (Fig. 1.3). Potentially, manipulating SMS and SMase could influence these bioactive lipid levels, thus influencing cell biological functions.

SMS and SMase-related bioactive lipid productions. SMS sphingomyelin synthase, SMase sphingomyelinase
1.3.2.1 Sphingomyelinase-Mediated Ceramide Production
Twenty years ago, it had been disclosed that SMase-mediated SM hydrolysis (SM cycle) is a novel pathway of transmembrane signal transduction. In response to extracellular agonists, membrane SM can be hydrolyzed by SMase to yield ceramide and choline phosphate [16–20].
So far, five type of SMases have been reported and they are classified based on their optimal pH and metal ion dependence activity [21]. They are lysosomal acid SMase, secreted zinc-dependent acid SMase, magnesium-dependent neutral SMase, magnesium-independent neutral SMase, and alkaline SMase. Multiple reviews have summarized the current knowledge about these SMases, from an overview of structure and catalysis to specific properties, roles, and regulation of these enzymes in physiological and pathological contexts [22–24].
Ceramide is a product of SMase reaction (Fig. 1.3) and has been identified as a second messenger, mediating the effects of cell growth, cell differentiation, and apoptosis. Hannun and Obeid [25] have recently summarized a large body of information with regards to metabolism, structure, and function of ceramides.
1.3.2.2 Sphingomyelin Synthase-Mediated Diacylglycerol Production
There are three different pathways that can produce diacylglycerol [26] (Fig. 1.4). Many studies have clearly established the significant role of diacylglycerol in the regulation of fundamental cellular functions such as proliferation and apoptosis through the activation of protein kinase C [27–29]. However, we still do not know the importance of the diacylglycerol produced by the reaction catalyzed by SMS. This is because hydrolysis of membrane inositol phospholipids by phospholipase C, or hydrolysis of other membrane phospholipids, particularly choline phospholipids, by phospholipase D and phospholipase A2 can produce diacylglycerol that links extracellular signals to intracellular events through activation of protein kinase C [30]. However, it is conceivable that SMS activity-mediated diacylglycerol can potentially play an important role in maintaining cellular diacylglycerol pools [26].

Three pathways for diacylglyceride production. (1) phosphatidylinositol phospholipase C; (2) phosphatidylcholine phospholipase D; (3) phosphatidic acid phosphatase; (4) sphingomyelin synthase
1.4 Sphingomyelin as a Critical Component of Lipid Rafts in Mediating Signal Transduction
1.4.1 Sphingomyelin-Enriched Cell Membrane Lipid Rafts
Sphingolipids, including SM and glycosphingolipids, together with cholesterol, have been implicated in lateral microdomain or ‘lipid raft’ formation in biological membranes. These microdomains serve as signaling platforms and are involved in cellular processes, such as signal transduction, membrane trafficking, and protein sorting [31, 32]. Other lipids found in raft structures include phosphatidylethanolamine, glycerophospholipids, phosphatidylserine, arachidonic acid, phosphatidylglucoside, ceramide, and lactosylceramide [33, 34].
The formation of lipid rafts in biological membranes is driven by lipid–lipid interactions, which are largely dependent on the structure and biophysical properties of the lipid components. It is favored by the presence of long-chain saturated sphingolipids and phospholipids as well as by physiological proportions of cholesterol [35, 36]. There is strong evidence suggesting a preferential interaction between SM and cholesterol, stabilized by hydrogen bonding [37–39]. Infrared spectroscopic studies have also confirmed the presence of intermolecular hydrogen bonding between the amide group of SM and the 3-hydroxyl group of cholesterol [40]. The levels of cholesterol and SM in the plasma membrane are also tightly controlled [41, 42]. Greater lateral packing density in SM-containing membranes is known to be responsible for lowering the rate of spontaneous cholesterol transfer from SM-containing membranes [43]. Highly saturated glycosphingolipids are also capable of forming extensive hydrogen-bonding network with cholesterol and are therefore found in lipid rafts. However, in the presence of both SM and glycosphingolipids, cholesterol preferentially interacts with SM [44]. The rafts co-existing with the fluid matrix of the plasma membrane exist in the liquid-ordered phase [45, 46] due to its cholesterol content. Cholesterol promotes phase separation of saturated SMs [47] and SM needs cholesterol to be detergent-insoluble [45].
1.4.2 Lipid Rafts and Cell Signaling
Lipid rafts act as organizing centers for processes such as membrane trafficking and signal transduction [48, 49]. Cytoplasmic proteins that are covalently modified by saturated fatty acids (palmitoyl or myristoyl moieties) and cell surface proteins that are attached via a glycosyl phosphatidylinositol (GPI) anchor are highly concentrated within lipid rafts. Many proteins involved in signal transduction, such as Src family kinases, G proteins, growth factor receptors, mitogen-activated protein kinase (MAPK), and protein kinase C are predominantly found in lipid rafts [32]. In addition, lipid rafts are dynamic in nature, which tends to scaffold certain signaling molecules, while excluding others. By such spatial segregation, lipid rafts not only provide a favorable environment for intra-molecular cross talk but also aid to expedite the signal relay.
Due to their insolubility in nonionic detergents such as Triton X-100, lipid rafts have been frequently termed as ‘detergent resistant membranes’ (DRMs). In fact, subpopulations of rafts have been proposed, in part based on their size, constituents, and functional properties [50, 51]. Caveolae are a subset of rafts and are considered to be 50–100 nm flask-like invaginations of the plasma membrane. Rafts and caveolae are dynamic entities, forming and dissipating in response to various external stimuli [52]. Upon stimulation, they internalize and serve a clathrin (coated pit)-independent mechanism of endocytosis of plasma membrane constituents. Raft/caveolae-mediated endocytosis is reported to facilitate transportation of entities to other cellular regions and across the cell (transcytosis) [53–55].
Membrane rafts and caveolae usually express specific proteins like flotillins and caveolins (Cavs) within their structure [56]. Cavs are structural proteins that provide an important, defining feature of caveolae and can be secreted into the extracellular space [57, 58]. Cavs are highly conserved among species and the three different isoforms of Cavs (Cav-1, -2, and -3) are differentially expressed in cells: Cav-3 is restricted to skeletal, cardiac, and smooth muscle, Cav-1 is more ubiquitously expressed, while expression of Cav-2 generally parallels that of Cav-1 [59]. Cavs also undergo covalent modifications like palmitoylation and phosphorylation [57–61]. It is known that insulin receptor (IR) is located in caveolae [62] and insulin receptor can interact with Cav1 [63]. In caveolae, the mobility of IR is increased by dissociation of the IR–Cav1 interaction [63]. It has been reported that SMS2 is able to regulate the dynamic structure of SM-rich lipid microdomains on the plasma membrane [64, 65] and could modify protein function, such as that of CD36 or Cav 1 located in the lipid microdomains [64]. SMS2 gene knockout (KO) mice exhibited disrupted regulation of the lipid microdomains function, leading to a prevention of lipid droplet formations, fatty liver, obesity, and insulin resistance [64, 65].
1.4.3 Role of the Lipid Rafts in Inflammatory Signaling
Toll like receptors (TLRs) are critically involved in inflammatory responses [66, 67]. Lipid rafts appear to provide a platform for the interaction of TLRs with their ligands in cells [68–71].
Each one of TLRs has a unique extracellular domain that allows specific ligand recognition. The intracellular toll/interleukin-1 (IL-1) receptor (TIR) domain of TLRs shares high degree of homology, but there are enough differences to cause diversified functions mediated by different TLRs [66, 67, 72, 73]. Upon ligand-induced stimulation, the TIR domain of TLRs associates with the TIR domain of their respective adaptor molecules to initiate intracellular signaling. Myeloid differentiation primary response gene 88 (MyD88) is a common TLR adaptor used by all TLRs, except for TLR3 [73]. Upon stimulation with a specific ligand, the membrane-associated TLRs (such as TLR2 and TLR4, and other components of the TLR complex) are recruited into the lipid rafts [74, 75]. These rafts aid in the interaction of TLRs with their ligands in macrophages [68–71], initiating nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) and MAP kinase activation and proinflammatory cytokine production, thus resulting into inflammatory responses.
Tumor necrosis factor alpha (TNFα) is one of the cytokines involved in systemic inflammation. TNFα can specifically bind to TNF receptors (TNFRs). It is known that lipid rafts play an essential role in TNFR1 clustering [76]. Upon contact with TNFα, TNF receptors form trimers and this binding causes a conformational change to occur in the receptor, leading to the dissociation of the inhibitory protein silencer of death domain (SODD) from the intracellular death domain. This dissociation enables the adaptor protein TNFR type 1-associated DEATH domain protein (TRADD) to bind to the death domain, serving as a platform for subsequent protein binding. Following TRADD binding, three pathways can be initiated [77, 78]: (1) activation of NFκB, (2) activation of MAPK pathways, and, (3) induction of cell death signaling.
Luberto et al. [79] found that D609, a nonspecific SMS inhibitor, blocks TNFα and phorbol ester-mediated NFκB activation that was concomitant with decreased levels of SM and diacylglyceride. Moreover, this did not affect the generation of ceramide, suggesting SM and diacylglycerol, derived from SM synthesis, are involved in NFκB activation. However, D609 is widely used to inhibit PC-phospholipase C (PLC) (Fig. 1.4), a well-known regulator of NFκB activation via diacylglyceride-mediated signaling [80]. Thus, it remains unclear what pathway is inhibited by D609 in particular that causes a diminished NFκB activation.
1.4.4 Role of Lipid Raft Sphingomyelin in Inflammatory Signaling
Studies from our laboratory [81] indicate that SMS2 knockdown in macrophages results in blockage of ligand-induced internalization as well as recruitment of TNFR1 to lipid rafts, suggesting a mechanism for the modulation of NFκB activity by SMS2. On similar lines, lipopolysaccharide (LPS)-induced plasma membrane recruitment of TLR4-MD-2 (TLR4 co-receptor) complex is also diminished in SMS2-knockout macrophages. As a result, SMS2 deficiency attenuates both NFκB and MAP kinase pathways, both of which are signaled via raft-associated TNFR1 and TLR4 along with their adaptor proteins. These findings strongly suggest the critical role of SMS2-synthesized SM for the normal function of TNFR1 and TLR4 on the plasma membrane following stimulation by their respective ligands (TNFα and LPS) [81].
We also created SMS1 knockout mice and found that SMS1 deficiency significantly decreased SM in plasma, liver, and macrophages but had only a marginal effect on ceramide levels [82]. Surprisingly, we found that SMS1 deficiency dramatically increased glucosylceramide and hematoside (GM3) levels in plasma, liver, and macrophages (4- to 12-fold), while SMS2 deficiency had no such effect. We evaluated total SMS activity in tissues and found that SMS1 deficiency causes 77 % reduction of SMS activity in macrophages [82], while SMS2 deficiency causes 70 % reduction of SMS activity in the liver [13], indicating SMS1 is the major SMS in macrophages, whereas SMS2 is predominant in the liver. We also found that SMS1 deficiency significantly attenuated TLR4-mediated NFκB and MAP kinase activation after LPS treatment.
The content of SM in the plasma membrane can also be modulated by SPT, the first and rate-limiting enzyme of the sphingolipid biosynthetic pathway [83]. SPT deficiency in macrophages also results in lower plasma membrane SM content as evidenced by lysenin-sensitivity assays, making the cells more resistant to lysis when treated with lysenin [81, 84]. LPS treatment of SPT deficient macrophages results in lesser recruitment of TLR4-MD2 complex, thereby attenuating both NFκB and MAP kinase activation. SPT deficient macrophages produce less TNFα and IL-6 in vitro when treated with LPS. SM supplementation experiments further prove that exogenous SM can enrich plasma membrane SM levels and can eventually restore the wild-type inflammatory phenotype in SPT deficient macrophages [128]. In general, SMS2 deficiency and SPT partial deficiency yield similar phenotypes, in terms of membrane SM levels, NFκB and MAP kinase activation. Unlike SMS2 deficiency, SPT partial deficiency does not change ceramide at the intracellular level or either in the plasma membrane or its lipid rafts. Thus, ceramide levels may have negligible role in mediating inflammatory signaling [128]. A reduction of plasma membrane SM levels are closely related to inflammation [81, 82].
1.4.5 Role of Lipid Raft Sphingomyelin Content in Cholesterol Homeostasis
Reverse cholesterol transport (RCT) is a multi-step process resulting in the net movement of cholesterol from peripheral tissues back to the liver via the plasma [85] and it plays a major role in cholesterol homeostasis. The first and most crucial step of RCT is cholesterol efflux from peripheral tissues, such as macrophages [85].
Foam cell formation due to excessive accumulation of cholesterol by macrophages is a pathological hallmark of atherosclerosis [86]. Macrophage scavenger receptor class A is implicated in the deposition of cholesterol in arterial walls during atherogenesis, through receptor-mediated endocytosis of modified low-density lipoproteins [87]. A member of scavenger receptor class B, CD36, is also involved in macrophage foam cell formation [88]. However, macrophages cannot limit the uptake of cholesterol, and therefore depend on cholesterol efflux pathways for preventing their transformation into foam cells. Several ATP-binding cassette (ABC) transporters, including ABCA1 [89] and ABCG1 [90], as well as scavenger receptor class B1 (SR-B1) [90], facilitate the efflux of cholesterol from macrophages.
In macrophages, ABCA1 exports cholesterol and phospholipids to lipid-free apolipoproteins, while ABCG1 and SR-BI export cholesterol to phospholipid-containing acceptors [90]. ABCA1-dependent cholesterol efflux requires aid from membrane lipid rafts [91, 92], while ABCG1 is mainly found intracellularly in the basal state, with little cell surface presentation. Under stimulation, for example by liver X receptor agonist treatment, ABCG1 redistributes itself to the plasma membrane, and increases cholesterol mass efflux to HDL [93]. ABCA1 and ABCG1 are known to cooperate in cholesterol efflux [90]. SR-BI also facilitates cholesterol efflux from macrophages [94]. ABCA1, ABCG1, and SR-BI are located in the plasma membrane, and exist either in rafts (SR-BI) [95, 96], or associated with the redistribution of lipids in the plasma membrane (ABCA1 and ABCG1) [90, 97]. It is, therefore, conceivable that fundamental changes in SM and glycosphingolipid levels of the plasma membrane can influence the functions of these proteins and alter cholesterol efflux [98, 99].
SM is also known as a cholesterol-binding molecule and there by plays an important role in cholesterol efflux. There are two possible SM-mediated cholesterol efflux mechanisms. Firstly, SM is involved in the recruitment of efflux-related transporters to the plasma membrane [94]. Indeed, SM-deficient cells enhance apoA-I-dependent cholesterol efflux by ABCA1 [98, 99]. This is further supported by SMS2 deficient and SPT partial deficient macrophage studies, where decrease of SM levels in macrophage plasma membrane increases both ABCA1 and ABCG1 protein levels on macrophage surfaces, thereby increasing cholesterol efflux in vitro and in vivo [100, 128]. Although ABCA1 is known to be located in a non-raft region, its levels influence lipid raft composition [101]. Overexpression of ABCA1 [97] and treatment of cells with high- density lipoprotein (HDL) or apoA-I [102, 103] disrupts or depletes raft domains, inhibiting raft-dependent signaling. This indicates a possible interaction between ABCA1 and raft-containing lipids.
Secondly, SM is also critical for cholesterol sequestration in the plasma membrane. It is known that lysosomal SMase is involved in cholesterol transport from lysosomes to the plasma membrane [98]. Because SM avidly binds cholesterol [104], SMase deficiency inhibits macrophage cholesterol efflux through promoting cholesterol sequestration by SM. Thus, SPT deficiency, leading to reduced plasma membrane SM levels, produces the inverse effect of SMase deficiency with reference to macrophage cholesterol efflux. SPT deficiency, therefore, aids in cholesterol efflux by inducing less cholesterol sequestration in the macrophage plasma membrane [128]. This is further supported by the finding that exogenously added SM significantly diminishes cholesterol efflux mediated by ABCA1 [99], suggesting that the increase of SM content in the plasma membrane prevents cholesterol efflux.
1.4.6 The Effect of Macrophage Lipid Raft Sphingomyelin Levels on Cholesterol Efflux and Inflammation
It is known that macrophage cholesterol efflux and inflammation are inversely related to each other. Yvan-Charvet et al. reported that macrophage ABCA1 and ABCG1 deficiencies increase free cholesterol accumulation and increase cell signaling via TLRs [105]. Zhu et al. reported that macrophage ABCA1 reduces MyD88-dependent TLR trafficking to lipid rafts by reduction of lipid raft cholesterol [106]. In addition, ABCA1 expression decreases cellular plasma membrane rigidity by reducing formation of tightly packed lipid rafts [97]. Therefore, more cholesterol efflux is related to less inflammation in macrophages. A recent report indicated that IL-6 markedly induced ABCA1 expression and enhanced ABCA1-mediated cholesterol efflux from human macrophages to apoA-I [107]. We found that SPT partial deficient macrophages have significantly lower SM levels in plasma membrane lipid rafts. This reduction not only impaired inflammatory responses triggered by TRL4 and its downstream NFκB and MAPK pathways, but also enhanced reverse cholesterol transport mediated by ABC transporters [128]. Our findings in this study clearly provided the evidence that plasma membrane SM levels are also critical for the inverse relationship between macrophage cholesterol efflux and inflammation.
1.4.7 Significance of Lipid Raft Sphingomyelin in Insulin Sensitivity
1.4.7.1 A Question from SPT Inhibition Studies
A previous study has indicated that treatment with myriocin, a potent SPT inhibitor and an immune suppressor, effectively ameliorates glucocorticoid-, saturated fat-, and obesity-induced insulin resistance [108]. Insulin resistance is a pathological condition where the insulin becomes less effective at lowering blood sugars. The authors attributed that effect to the reduction of ceramide in tissues, but they did not evaluate SM levels, especially in the plasma membrane. We and others have noted that myriocin treatment not only reduces ceramide, but also SM and glycosphingolipid levels [109–111]. This arises a fundamental question from the SPT inhibition studies: which sphingolipid in particular, ceramide, or SM, is responsible for causing the insulin resistance?
1.4.7.2 Ceramide and Insulin Resistance
There are two considerations linking ceramide and insulin resistance. Firstly, ceramide blocks the translocation of Akt/Protein Kinase B (PKB) to the plasma membrane [112]. It has also been reported that ceramide inactivation of Akt/PKB requires the atypical PKC isoform PKCζ [113, 114]. Secondly, ceramide may impair the action of insulin by facilitating signaling pathways initiated by inflammatory cytokines, such as TNFα and IL-6, which are known to impair insulin signaling [115, 116]. However, it has also been reported that various glycosphingolipid synthase inhibitors augment insulin-stimulated phosphorylation of the insulin receptor, as well as Akt/PKB and/or mammalian target of rapamycin phosphorylation, in the skeletal muscle [117] and liver [117, 118] of obese rodents, without altering ceramide levels. While ceramide accumulates in some insulin-resistant models [119, 120], it fails to do so in lipid-infused animals. In fact, the relative increase of ceramide in obese rodents and humans is rather quite small [121, 122]. Moreover, it is not known whether muscle ceramide content is a major factor in muscle insulin sensitivity. Adams et al. demonstrated that ceramide content is increased in skeletal muscle from obese, insulin-resistant humans [121], while Skovbro et al. found that human skeletal muscle ceramide content is not a major factor in muscle insulin sensitivity [123]. In general, the role of cellular ceramide in insulin resistance is controversial. Therefore more studies needs to be done in this field to establish this relationship.
1.4.7.3 Lipid Rafts and Insulin Resistance
Insulin resistance, abdominal obesity, dyslipidemia coupled with high blood pressure, and a proinflammatory state are common disorders associated with type 2 diabetes and atherosclerosis [124]. An important question is how these interrelated risk factors could be mechanistically coupled in a physiological situation. Considering a simple scenario, lipid raft disruption could affect insulin signaling. It has been suggested that lipid rafts play an important role in the pathogenesis of insulin resistance [125]. Indeed, disruption of caveolae in cultured cells by cholesterol extraction with β-cyclodextrin results in progressive inhibition of tyrosine phosphorylation of insulin receptor substrate 1, as well as reduced activation of glucose transport in response to insulin [126].
Glycosphingolipids are also known to be structurally and functionally important components in the lipid rafts [127]. Pharmacological inhibition of glycosphingolipid synthesis markedly improves insulin sensitivity in rodent models of insulin resistance [118]. Deficiency of GM3 ganglioside (a key glycosphingolipid in the rafts) is also known to enhance insulin receptor tyrosine phosphorylation [62]. Moreover, GM3 could dissociate insulin receptor/Cav-1 complex, thus causing insulin receptor dysfunctionality [63]. Since SM is also one of the major components within lipid rafts, it is conceivable that diminishing SM in the plasma membrane could have a similar impact on insulin signaling.
1.4.7.4 Reducing Plasma Membrane Sphingomyelin Increases Insulin Sensitivity
We utilized two models: SPT partial deficient mice and SMS2 knockout mice for the insulin sensitivity study [65]. We found that: (i) both SPT partial and SMS2 complete deficiency enhances insulin sensitivity; (ii) both deficiencies decrease plasma membrane SM levels, which contribute to the enhancement of insulin sensitivity; (iii) SPT deficiency decreases ceramide, while SMS2 deficiency increases it, therefore, ceramide is probably not the regulator of insulin sensitivity; (iv) there, they were no significant changes of glucosylceramide and GM3 levels in tissues or even cell plasma membrane, so they might not play a significant role in insulin sensitivity in the above models; and finally, (v) this leads us to conclude that SPT or SMS2 inhibition is a promising pharmacological approach for the treatment of insulin resistance and metabolic syndrome.
1.5 Conclusions
The role of SM as a signaling molecule other than its membrane structural properties is recently being recognized in greater depth. And only a few of these important functions have been highlighted in this chapter, however, the scope of SM as a bioactive lipid mediator is enormous. Therefore, it is indeed essential to study and understand how SM is synthesized and degraded via its complex metabolic network. This would further shed light on this enigmatic molecule and its noteworthy roles in physiological processes. On the other hand, accumulating evidence showed that dynamic modification of SM in lipid rafts on cell plasma membrane controls development of obesity, insulin resistance, fatty liver, inflammation, and atherosclerosis. It is important to investigate the relationship between SM and other lipids, and between SM and functional proteins, such as insulin receptor, CD36, TLRs, and so on, in the lipid rafts.
References
Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9:139–150
Kleger A, Liebau S, Lin Q, von Wichert G, Seufferlein T (2011) The impact of bioactive lipids on cardiovascular development. Stem Cells Int 2011:916180
Simons K, Ikonen E (1997) Functional rafts in cell membranes. Nature 387:569–572
Li Z, Hailemariam TK, Zhou H, Li Y, Duckworth DC et al (2007) Inhibition of sphingomyelin synthase (SMS) affects intracellular sphingomyelin accumulation and plasma membrane lipid organization. Biochim Biophys Acta 1771:1186–1194
Shaul PW, Anderson RG (1998) Role of plasmalemmal caveolae in signal transduction. Am J Physiol 275:L843–L851
Futerman AH, Hannun YA (2004) The complex life of simple sphingolipids. EMBO Rep 5:777–782
Holthuis JC, van Meer G, Huitema K (2003) Lipid microdomains, lipid translocation and the organization of intracellular membrane transport (Review). Mol Membr Biol 20:231–241
Calhoun WI, Shipley GG (1979) Fatty acid composition and thermal behavior of natural sphingomyelins. Biochim Biophys Acta 555:436–441
Kolesnick RN (1991) Sphingomyelin and derivatives as cellular signals. Prog Lipid Res 30:1–38
Tafesse FG, Ternes P, Holthuis JC (2006) The multigenic sphingomyelin synthase family. J Biol Chem 281:29421–29425
Hojjati MR, Jiang XC (2006) Rapid, specific, and sensitive measurements of plasma sphingomyelin and phosphatidylcholine. J Lipid Res 47:673–676
Ishitsuka R, Yamaji-Hasegawa A, Makino A, Hirabayashi Y, Kobayashi T (2004) A lipid-specific toxin reveals heterogeneity of sphingomyelin-containing membranes. Biophys J 86:296–307
Liu J, Zhang H, Li Z, Hailemariam TK, Chakraborty M et al (2009) Sphingomyelin synthase 2 is one of the determinants for plasma and liver sphingomyelin levels in mice. Arterioscler Thromb Vasc Biol 29:850–856
Huitema K, van den Dikkenberg J, Brouwers JF, Holthuis JC (2004) Identification of a family of animal sphingomyelin synthases. EMBO J 23:33–44
Yamaoka S, Miyaji M, Kitano T, Umehara H, Okazaki T (2004) Expression cloning of a human cDNA restoring sphingomyelin synthesis and cell growth in sphingomyelin synthase-defective lymphoid cells. J Biol Chem 279:18688–18693
Okazaki T, Bell RM, Hannun YA (1989) Sphingomyelin turnover induced by vitamin D3 in HL-60 cells. Role in cell differentiation. J Biol Chem 264:19076–19080
Kim MY, Linardic C, Obeid L, Hannun Y (1991) Identification of sphingomyelin turnover as an effector mechanism for the action of tumor necrosis factor alpha and gamma-interferon. Specific role in cell differentiation. J Biol Chem 266:484–489
Dressler KA, Mathias S, Kolesnick RN (1992) Tumor necrosis factor-alpha activates the sphingomyelin signal transduction pathway in a cell-free system. Science 255:1715–1718
Ballou LR, Chao CP, Holness MA, Barker SC, Raghow R (1992) Interleukin-1-mediated PGE2 production and sphingomyelin metabolism. Evidence for the regulation of cyclooxygenase gene expression by sphingosine and ceramide. J Biol Chem 267:20044–20050
Mathias S, Younes A, Kan CC, Orlow I, Joseph C et al (1993) Activation of the sphingomyelin signaling pathway in intact EL4 cells and in a cell-free system by IL-1 beta. Science 259:519–522
Marchesini N, Hannun YA (2004) Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochem Cell Biol 82:27–44
Zeidan YH, Hannun YA (2010) The acid sphingomyelinase/ceramide pathway: biomedical significance and mechanisms of regulation. Curr Mol Med 10:454–466
Clarke CJ, Wu BX, Hannun YA (2011) The neutral sphingomyelinase family: identifying biochemical connections. Adv Enzyme Regul 51:51–58
Duan RD (2006) Alkaline sphingomyelinase: an old enzyme with novel implications. Biochim Biophys Acta 1761:281–291
Hannun YA, Obeid LM (2011) Many ceramides. J Biol Chem 286:27855–27862
Becker KP, Hannun YA (2005) Protein kinase C and phospholipase D: intimate interactions in intracellular signaling. Cell Mol Life Sci 62:1448–1461
Goni FM, Alonso A (1999) Structure and functional properties of diacylglycerols in membranes. Prog Lipid Res 38:1–48
Liu WS, Heckman CA (1998) The sevenfold way of PKC regulation. Cell Signal 10:529–542
Quest AF, Ghosh S, Xie WQ, Bell RM (1997) DAG second messengers: molecular switches and growth control. Adv Exp Med Biol 400A:297–303
Nishizuka Y (1992) Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 258:607–614
Brown DA, London E (2000) Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem 275:17221–17224
Simons K, Toomre D (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1:31–39
Sonnino S, Prinetti A, Nakayama H, Yangida M, Ogawa H et al (2009) Role of very long fatty acid-containing glycosphingolipids in membrane organization and cell signaling: the model of lactosylceramide in neutrophils. Glycoconj J 26:615–621
Sonnino S, Prinetti A (2010) Lipids and membrane lateral organization. Front Physiol 1:153
Brown RE (1998) Sphingolipid organization in biomembranes: what physical studies of model membranes reveal. J Cell Sci 111(Pt 1):1–9
London E, Brown DA (2000) Insolubility of lipids in triton X-100: physical origin and relationship to sphingolipid/cholesterol membrane domains (rafts). Biochim Biophys Acta 1508:182–195
Li XM, Momsen MM, Smaby JM, Brockman HL, Brown RE (2001) Cholesterol decreases the interfacial elasticity and detergent solubility of sphingomyelins. Biochemistry 40:5954–5963
Sankaram MB, Thompson TE (1990) Modulation of phospholipid acyl chain order by cholesterol. A solid-state 2H nuclear magnetic resonance study. Biochemistry 29:10676–10684
Bittman R, Kasireddy CR, Mattjus P, Slotte JP (1994) Interaction of cholesterol with sphingomyelin in monolayers and vesicles. Biochemistry 33:11776–11781
Veiga MP, Arrondo JL, Goni FM, Alonso A, Marsh D (2001) Interaction of cholesterol with sphingomyelin in mixed membranes containing phosphatidylcholine, studied by spin-label ESR and IR spectroscopies. A possible stabilization of gel-phase sphingolipid domains by cholesterol. Biochemistry 40:2614–2622
Slotte JP (1997) Cholesterol-sphingomyelin interactions in cells–effects on lipid metabolism. Subcell Biochem 28:277–293
Ridgway ND (2000) Interactions between metabolism and intracellular distribution of cholesterol and sphingomyelin. Biochim Biophys Acta 1484:129–141
Kan CC, Ruan ZS, Bittman R (1991) Interaction of cholesterol with sphingomyelin in bilayer membranes: evidence that the hydroxy group of sphingomyelin does not modulate the rate of cholesterol exchange between vesicles. Biochemistry 30:7759–7766
Masserini M, Ravasi D (2001) Role of sphingolipids in the biogenesis of membrane domains. Biochim Biophys Acta 1532:149–161
Schroeder R, London E, Brown D (1994) Interactions between saturated acyl chains confer detergent resistance on lipids and glycosylphosphatidylinositol (GPI)-anchored proteins: GPI-anchored proteins in liposomes and cells show similar behavior. Proc Natl Acad Sci U S A 91:12130–12134
Simons K, Ikonen E (2000) How cells handle cholesterol. Science 290:1721–1726
Wolf C, Koumanov K, Tenchov B, Quinn PJ (2001) Cholesterol favors phase separation of sphingomyelin. Biophys Chem 89:163–172
Galbiati F, Razani B, Lisanti MP (2001) Emerging themes in lipid rafts and caveolae. Cell 106:403–411
Pike LJ (2005) Growth factor receptors, lipid rafts and caveolae: an evolving story. Biochim Biophys Acta 1746:260–273
Mishra S, Joshi PG (2007) Lipid raft heterogeneity: an enigma. J Neurochem 103(Suppl 1):135–142
Patra SK (2008) Dissecting lipid raft facilitated cell signaling pathways in cancer. Biochim Biophys Acta 1785:182–206
Tsutsumi YM, Horikawa YT, Jennings MM, Kidd MW, Niesman IR et al (2008) Cardiac-specific overexpression of caveolin-3 induces endogenous cardiac protection by mimicking ischemic preconditioning. Circulation 118:1979–1988
Minshall RD, Malik AB (2006) Transport across the endothelium: regulation of endothelial permeability. Handb Exp Pharmacol 176:107–144
Mukherjee S, Tessema M, Wandinger-Ness A (2006) Vesicular trafficking of tyrosine kinase receptors and associated proteins in the regulation of signaling and vascular function. Circ Res 98:743–756
Nichols B (2003) Caveosomes and endocytosis of lipid rafts. J Cell Sci 116:4707–4714
Frick M, Bright NA, Riento K, Bray A, Merrified C et al (2007) Coassembly of flotillins induces formation of membrane microdomains, membrane curvature, and vesicle budding. Curr Biol 17:1151–1156
Tahir SA, Yang G, Ebara S, Timme TL, Satoh T et al (2001) Secreted caveolin-1 stimulates cell survival/clonal growth and contributes to metastasis in androgen-insensitive prostate cancer. Cancer Res 61:3882–3885
Tahir SA, Yang G, Goltsov AA, Watanabe M, Tabata K et al (2008) Tumor cell-secreted caveolin-1 has proangiogenic activities in prostate cancer. Cancer Res 68:731–739
Williams TM, Lisanti MP (2004) The caveolin proteins. Genome Biol 5:214
Dietzen DJ, Hastings WR, Lublin DM (1995) Caveolin is palmitoylated on multiple cysteine residues. Palmitoylation is not necessary for localization of caveolin to caveolae. J Biol Chem 270:6838–6842
Li S, Seitz R, Lisanti MP (1996) Phosphorylation of caveolin by src tyrosine kinases. The alpha-isoform of caveolin is selectively phosphorylated by v-Src in vivo. J Biol Chem 271:3863–3868
Yamashita T, Hashiramoto A, Haluzik M, Mizukami H, Beck S et al (2003) Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc Natl Acad Sci U S A 100:3445–3449
Kabayama K, Sato T, Saito K, Loberto N, Prinetti A et al (2007) Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proc Natl Acad Sci U S A 104:13678–13683
Mitsutake S, Zama K, Yokota H, Yoshida T, Tanaka M et al (2011) Dynamic modification of sphingomyelin in lipid microdomains controls development of obesity, fatty liver, and type 2 diabetes. J Biol Chem 286:28544–28555
Li Z, Zhang H, Liu J, Liang CP, Li Y et al (2011) Reducing plasma membrane sphingomyelin increases insulin sensitivity. Mol Cell Biol 31:4205–4218
Beutler B, Jiang Z, Georgel P, Crozat K, Croker B et al (2006) Genetic analysis of host resistance: toll-like receptor signaling and immunity at large. Annu Rev Immunol 24:353–389
Takeda K, Akira S (2005) Toll-like receptors in innate immunity. Int Immunol 17:1–14
Lee HK, Dunzendorfer S, Soldau K, Tobias PS (2006) Double-stranded RNA-mediated TLR3 activation is enhanced by CD14. Immunity 24:153–163
Wang R, Town T, Gokarn V, Flavell RA, Chandawarkar RY (2006) HSP70 enhances macrophage phagocytosis by interaction with lipid raft-associated TLR-7 and upregulating p38 MAPK and PI3K pathways. J Surg Res 136:58–69
Nakahira K, Kim HP, Geng XH, Nakao A, Wang X et al (2006) Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med 203:2377–2389
Szabo G, Dolganiuc A, Dai Q, Pruett SB (2007) TLR4, ethanol, and lipid rafts: a new mechanism of ethanol action with implications for other receptor-mediated effects. J Immunol 178:1243–1249
Miggin SM, O’Neill LA (2006) New insights into the regulation of TLR signaling. J Leukoc Biol 80:220–226
Miyake K (2006) Roles for accessory molecules in microbial recognition by Toll-like receptors. J Endotoxin Res 12:195–204
Triantafilou M, Brandenburg K, Kusumoto S, Fukase K, Mackie A et al (2004) Combinational clustering of receptors following stimulation by bacterial products determines lipopolysaccharide responses. Biochem J 381:527–536
Triantafilou M, Morath S, Mackie A, Hartung T, Triantafilou K (2004) Lateral diffusion of Toll-like receptors reveals that they are transiently confined within lipid rafts on the plasma membrane. J Cell Sci 117:4007–4014
Legler DF, Micheau O, Doucey MA, Tschopp J, Bron C (2003) Recruitment of TNF receptor 1 to lipid rafts is essential for TNFalpha-mediated NF-kappaB activation. Immunity 18:655–664
Wajant H, Pfizenmaier K, Scheurich P (2003) Tumor necrosis factor signaling. Cell Death Differ 10:45–65
Chen G, Goeddel DV (2002) TNF-R1 signaling: a beautiful pathway. Science 296:1634–1635
Luberto C, Yoo DS, Suidan HS, Bartoli GM, Hannun YA (2000) Differential effects of sphingomyelin hydrolysis and resynthesis on the activation of NF-kappa B in normal and SV40-transformed human fibroblasts. J Biol Chem 275:14760–14766
Schutze S, Potthoff K, Machleidt T, Berkovic D, Wiegmann K et al (1992) TNF activates NF-kappa B by phosphatidylcholine-specific phospholipase C-induced “acidic” sphingomyelin breakdown. Cell 71:765–776
Hailemariam TK, Huan C, Liu J, Li Z, Roman C et al (2008) Sphingomyelin synthase 2 deficiency attenuates NFkappaB activation. Arterioscler Thromb Vasc Biol 28:1519–1526
Li Z, Fan Y, Liu J, Li Y, Huan C et al (2012) Impact of sphingomyelin synthase 1 deficiency on sphingolipid metabolism and atherosclerosis in mice. Arterioscler Thromb Vasc Biol 32:1577–1584
Merrill AH Jr, Jones DD (1990) An update of the enzymology and regulation of sphingomyelin metabolism. Biochim Biophys Acta 1044:1–12
Yamaji-Hasegawa A, Makino A, Baba T, Senoh Y, Kimura-Suda H et al (2003) Oligomerization and pore formation of a sphingomyelin-specific toxin, lysenin. J Biol Chem 278:22762–22770
Tall AR (1998) An overview of reverse cholesterol transport. Eur Heart J 19(Suppl A):A31–A35
Ross R (1993) The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 362:801–809
Freeman M, Ashkenas J, Rees DJ, Kingsley DM, Copeland NG et al (1990) An ancient, highly conserved family of cysteine-rich protein domains revealed by cloning type I and type II murine macrophage scavenger receptors. Proc Natl Acad Sci U S A 87:8810–8814
Kodama T, Freeman M, Rohrer L, Zabrecky J, Matsudaira P et al (1990) Type I macrophage scavenger receptor contains alpha-helical and collagen-like coiled coils. Nature 343:531–535
Cavelier C, Lorenzi I, Rohrer L, von Eckardstein A (2006) Lipid efflux by the ATP-binding cassette transporters ABCA1 and ABCG1. Biochim Biophys Acta 1761:655–666
Jessup W, Gelissen IC, Gaus K, Kritharides L (2006) Roles of ATP binding cassette transporters A1 and G1, scavenger receptor BI and membrane lipid domains in cholesterol export from macrophages. Curr Opin Lipidol 17:247–257
Gaus K, Kritharides L, Schmitz G, Boettcher A, Drobnik W et al (2004) Apolipoprotein A-1 interaction with plasma membrane lipid rafts controls cholesterol export from macrophages. FASEB J 18:574–576
Mendez AJ, Lin G, Wade DP, Lawn RM, Oram JF (2001) Membrane lipid domains distinct from cholesterol/sphingomyelin-rich rafts are involved in the ABCA1-mediated lipid secretory pathway. J Biol Chem 276:3158–3166
Wang N, Ranalletta M, Matsuura F, Peng F, Tall AR (2006) LXR-induced redistribution of ABCG1 to plasma membrane in macrophages enhances cholesterol mass efflux to HDL. Arterioscler Thromb Vasc Biol 26:1310–1316
Huang ZH, Gu D, Lange Y, Mazzone T (2003) Expression of scavenger receptor BI facilitates sterol movement between the plasma membrane and the endoplasmic reticulum in macrophages. Biochemistry 42:3949–3955
Graf GA, Connell PM, van der Westhuyzen DR, Smart EJ (1999) The class B, type I scavenger receptor promotes the selective uptake of high density lipoprotein cholesterol ethers into caveolae. J Biol Chem 274:12043–12048
Babitt J, Trigatti B, Rigotti A, Smart EJ, Anderson RG et al (1997) Murine SR-BI, a high density lipoprotein receptor that mediates selective lipid uptake, is N-glycosylated and fatty acylated and colocalizes with plasma membrane caveolae. J Biol Chem 272:13242–13249
Landry YD, Denis M, Nandi S, Bell S, Vaughan AM et al (2006) ATP-binding cassette transporter A1 expression disrupts raft membrane microdomains through its ATPase-related functions. J Biol Chem 281:36091–36101
Leventhal AR, Chen W, Tall AR, Tabas I (2001) Acid sphingomyelinase-deficient macrophages have defective cholesterol trafficking and efflux. J Biol Chem 276:44976–44983
Nagao K, Takahashi K, Hanada K, Kioka N, Matsuo M et al (2007) Enhanced apoA-I-dependent cholesterol efflux by ABCA1 from sphingomyelin-deficient Chinese hamster ovary cells. J Biol Chem 282:14868–14874
Liu J, Huan C, Chakraborty M, Zhang H, Lu D et al (2009) Macrophage sphingomyelin synthase 2 deficiency decreases atherosclerosis in mice. Circ Res 105:295–303
Fessler MB, Parks JS (2011) Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. J Immunol 187:1529–1535
Peshavariya H, Dusting GJ, Di Bartolo B, Rye KA, Barter PJ et al (2009) Reconstituted high-density lipoprotein suppresses leukocyte NADPH oxidase activation by disrupting lipid rafts. Free Radic Res 43:772–782
Fielding PE, Russel JS, Spencer TA, Hakamata H, Nagao K et al (2002) Sterol efflux to apolipoprotein A-I originates from caveolin-rich microdomains and potentiates PDGF-dependent protein kinase activity. Biochemistry 41:4929–4937
Slotte JP (1999) Sphingomyelin-cholesterol interactions in biological and model membranes. Chem Phys Lipids 102:13–27
Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M et al (2008) Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation 118:1837–1847
Zhu X, Owen JS, Wilson MD, Li H, Griffiths GL et al (2010) Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res 51:3196–3206
Frisdal E, Lesnik P, Olivier M, Robillard P, Chapman MJ et al (2011) Interleukin-6 protects human macrophages from cellular cholesterol accumulation and attenuates the pro-inflammatory response. J Biol Chem 286:30926–30936
Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM et al (2007) Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 5:167–179
Park TS, Panek RL, Mueller SB, Hanselman JC, Rosebury WS et al (2004) Inhibition of sphingomyelin synthesis reduces atherogenesis in apolipoprotein E-knockout mice. Circulation 110:3465–3471
Hojjati MR, Li Z, Zhou H, Tang S, Huan C et al (2005) Effect of myriocin on plasma sphingolipid metabolism and atherosclerosis in apoE-deficient mice. J Biol Chem 280:10284–10289
Glaros EN, Kim WS, Wu BJ, Suarna C, Quinn CM et al (2007) Inhibition of atherosclerosis by the serine palmitoyl transferase inhibitor myriocin is associated with reduced plasma glycosphingolipid concentration. Biochem Pharmacol 73:1340–1346
Stratford S, DeWald DB, Summers SA (2001) Ceramide dissociates 3′-phosphoinositide production from pleckstrin homology domain translocation. Biochem J 354:359–368
Powell DJ, Hajduch E, Kular G, Hundal HS (2003) Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCzeta-dependent mechanism. Mol Cell Biol 23:7794–7808
Blouin CM, Prado C, Takane KK, Lasnier F, Garcia-Ocana A et al (2010) Plasma membrane subdomain compartmentalization contributes to distinct mechanisms of ceramide action on insulin signaling. Diabetes 59:600–610
Holland WL, Summers SA (2008) Sphingolipids, insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocr Rev 29:381–402
de Mello VD, Lankinen M, Schwab U, Kolehmainen M, Lehto S et al (2009) Link between plasma ceramides, inflammation and insulin resistance: association with serum IL-6 concentration in patients with coronary heart disease. Diabetologia 52:2612–2615
Zhao H, Przybylska M, Wu IH, Zhang J, Siegel C et al (2007) Inhibiting glycosphingolipid synthesis improves glycemic control and insulin sensitivity in animal models of type 2 diabetes. Diabetes 56:1210–1218
Aerts JM, Ottenhoff R, Powlson AS, Grefhorst A, van Eijk M et al (2007) Pharmacological inhibition of glucosylceramide synthase enhances insulin sensitivity. Diabetes 56:1341–1349
Gorska M, Dobrzyn A, Zendzian-Piotrowska M, Gorski J (2004) Effect of streptozotocin-diabetes on the functioning of the sphingomyelin-signalling pathway in skeletal muscles of the rat. Horm Metab Res 36:14–21
Straczkowski M, Kowalska I, Nikolajuk A, Dzienis-Straczkowska S, Kinalska I et al (2004) Relationship between insulin sensitivity and sphingomyelin signaling pathway in human skeletal muscle. Diabetes 53:1215–1221
Adams JM 2nd, Pratipanawatr T, Berria R, Wang E, DeFronzo RA et al (2004) Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 53:25–31
Turinsky J, O’Sullivan DM, Bayly BP (1990) 1,2-Diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. J Biol Chem 265:16880–16885
Skovbro M, Baranowski M, Skov-Jensen C, Flint A, Dela F et al (2008) Human skeletal muscle ceramide content is not a major factor in muscle insulin sensitivity. Diabetologia 51:1253–1260
Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414:799–806
Virkamaki A, Ueki K, Kahn CR (1999) Protein-protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J Clin Invest 103:931–943
Parpal S, Karlsson M, Thorn H, Stralfors P (2001) Cholesterol depletion disrupts caveolae and insulin receptor signaling for metabolic control via insulin receptor substrate-1, but not for mitogen-activated protein kinase control. J Biol Chem 276:9670–9678
Langeveld M, Aerts JM (2009) Glycosphingolipids and insulin resistance. Prog Lipid Res 48:196–205
Chakraborty M, Lou C, Huan C, Kuo M, Park, T, Cao, G, Jiang XC (2013) Myeloid cell-specific serine palmitoyltransferase subunit 2 haploinsufficiency reduces mouse atherosclerosis. J Clin Invest. doi:10.1172/JC16041S
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Chakraborty, M., Jiang, XC. (2013). Sphingomyelin and Its Role in Cellular Signaling. In: Capelluto, D. (eds) Lipid-mediated Protein Signaling. Advances in Experimental Medicine and Biology, vol 991. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6331-9_1
Download citation
DOI: https://doi.org/10.1007/978-94-007-6331-9_1
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6330-2
Online ISBN: 978-94-007-6331-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)