
Abstract
Cadherins and catenins are the central cell–cell adhesion molecules in adherens junctions (AJs). This chapter reviews the knowledge concerning the role of cadherins and catenins in epithelial cancer and examines the published literature demonstrating the changes in the expression and function of these proteins in human cancer and the association of these changes with patient outcomes. The chapter also covers the mechanistic studies aiming at uncovering the significance of changes in cadherin and catenin expression in cancer and potential molecular mechanisms responsible for the causal role of AJs in cancer initiation and progression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The ability to adhere to each other is one of the most fundamental cellular functions necessary for the formation of metazoan organisms. Cell–cell adhesion of properly polarized cells is directly responsible for complex architecture of all organs and tissues. The mechanisms of intercellular adhesion are very complex and differ significantly between cells in different organs and tissues. The cell–cell adhesion is especially strong in epithelial tissues, where the cells can tightly seal their membranes to prevent the formation of gaps between neighboring cells. This is important, because the primary function of epithelia is formation of a barrier that separates organism from both external and internal environments (epidermis, intestines, epithelial tubes in various glandular organs) and also separates different tissues from each other (blood vessels).
Tumors originating in epithelial tissues are known as carcinomas and these cancers account for the majority of human malignancies. The most significant manifestation of carcinoma is the focal loss of normal tissue architecture and aberrant accumulation of epithelial cells. Since maintenance of normal tissue organization is one of the primary functions of cell–cell adhesion, it is perhaps not surprising that cell–cell adhesion structures are often abnormal in epithelial tumors. Interestingly, weakening of intercellular adhesion in carcinomas has been noted by pathologists many decades ago (Coman 1944). While gaps and separations between the neighboring cells are often visible on histological sections of low-grade early tumors, they are especially noticeable in more lethal high-grade carcinomas, which are more likely to spread to distant organs and kill the patient. Only recently, with the advancements of biochemistry and molecular biology it became possible to decipher the molecular mechanisms responsible for cell–cell adhesion in normal tissues and the loss or weakening of cell–cell adhesion in epithelial tumors. Interestingly, while cells can form different types of cell–cell adhesion structures, the adherens junctions (AJs) generated by classical cadherins are especially important for normal epithelial self organization and are also frequently affected in epithelial tumors. This chapter will summarize and review the literature concerning the role and mechanisms of AJs in tumor initiation and progression.
2 Core Proteins Involved in the Formation and Maintenance of the AJs
Many proteins localize to the AJs, however, only few groups of proteins are essential for AJ formation. These proteins belong to the cadherin and catenin families (Fig. 16.1). Cadherins are a large family of proteins that contains several calcium -binding cadherin domains in the extracellular portion of the molecule. Cadherin domains are directly responsible for the homophilic and calcium -dependent adhesive interactions between cadherin molecules on the neighboring cells. According to the overall domain structure and the number of cadherin domains, the cadherin family can be divided into classical, atypical and protocadherin cadherins. The classical cadherins include the best-studied E(epithelial)-, N(neural)-, P(placental)-, and VE(vasculo-endothelial)-cadherins. The cytoplasmic portion of these transmembrane proteins can bind to catenins and this confers strong adhesive interactions between the membranes of adjacent cells (Cavey and Lecuit 2009; Meng and Takeichi 2009). Since the role of nonclassical and atypical cadherins in mammalian cancer is still poorly understood, this chapter will focus on the role and significance of classical cadherins in cancer initiation and progression.

Major proteins involved in the formation of the AJs. Classical cadherins are the transmembrane proteins containing multiple extracellular cadherin domains, which are directly involved in cis- and trans-adhesive interactions. Cytoplasmic domains of cadherins interact with p120- and β-catenins. β-catenin binds to α-catenin, which provides a functional link to the actin cytoskeleton. Actin filaments help to cluster the cadherin–catenin complexes at the membrane and strengthen the adhesive interactions between the neighboring cells
Catenins are the proteins that interact with cytoplasmic domain of classical cadherins and are required for strengthening of cell–cell contacts and proper AJ formation (Fig. 16.1). p120-catenin directly interacts with cadherins and plays an important role in delivery and stabilization of cadherins at the plasma membrane (Chen et al. 2003b; Davis et al. 2003; Ishiyama et al. 2010). In addition, p120-catenin is also a prominent negative regulator of RhoA and a positive regulator of Rac1 small GTPases, which orchestrate the dynamics of the actin cytoskeleton at the AJs (Anastasiadis and Reynolds 2001; Noren et al. 2000). β-catenin or a similar protein γ-catenin (plakoglobin) also directly interacts with the cytoplasmic domain of cadherin. Importantly, both β-catenin and plakoglobin bind to α-catenin and the principal function of these proteins in the AJs is to bring α-catenin to the cadherin–catenin complex at the cell membrane. In addition to its critical role in the AJs, β-catenin is also a crucial transcriptional co-activator in the canonical Wnt signal transduction pathway, which orchestrates normal development and, when abnormally activated, can cause cancer. β-catenin translocates to the nucleus , binds to TCF family of transcription factors and activates transcription of genes that regulate morphogenetic, as well as, self-renewal and differentiation programs (Clevers 2006; Wend et al. 2010).
At the AJs, β-catenin links cadherin with α-catenin. α-catenin is the only catenin containing a well-defined actin-binding domain, and it is believed, that it functionally links cadherin–catenin proteins at the membrane with the actin cytoskeleton. The mechanisms of this linkage are poorly understood because purified cadherin-β-catenin-α-catenin protein complexes cannot bind to actin filaments (Drees et al. 2005; Yamada et al. 2005). α-catenin may be involved in regulation of actin polymerization at the AJs (Kobielak et al. 2004; Kovacs et al. 2002; Vasioukhin et al. 2000) and, additionally, it may use other actin-binding proteins Eplin or Vinculin to link to the actin filaments indirectly (Abe and Takeichi 2008; Yonemura et al. 2010). Mammalian genomes contain three α-catenin genes, αE(epithelial), αN-(neural) and αT(testicular), which differ primarily by their tissue-specific pattern of expression (Janssens et al. 2001). In this chapter, we will concentrate primarily on epithelial αE-catenin.
In addition to cadherin–catenin protein complexes, the Nectin -afadin cell–cell adhesion system is also present at the AJs (Ogita et al. 2010). Nectins and Nectin-like molecules are transmembrane adhesion proteins containing immunoglobulin-like domains, which interact in trans with each other and are responsible for weak calcium -independent cell–cell adhesion. Cytoplasmic domains of Nectins and Nectin-like proteins bind to afadin, which contains actin-binding domain and links Nectin–afadin molecules at the membrane to the actin cytoskeleton. Interestingly, afadin also binds to α-catenin and this interaction can link Nectin and cadherin adhesion systems at the AJs. The role of the Nectin–afadin adhesion system is discussed in detail in chap. 7 and it will not be covered in this chapter.
3 Decrease in Adherens Junction Function and Weakening of Intercellular Adhesion in Human Epithelial Cancer
In normal tissues epithelial cells tightly adhere to each other; however, in many epithelial tumors and especially in high-grade advanced cancers the tumor cells display decreased cell–cell adhesion and increased invasion into surrounding stromal tissues. Generally, tumors displaying invasion and local dissemination of epithelial tumor cells are considered to be more likely to result in metastatic progression and death of the patient. Therefore, knowledge about the molecular mechanisms responsible for the differences between well-circumscribed tumors containing tightly adhering cells and tumors containing loosely adhering cells is very important for understanding the biology of cancer progression and metastasis . Since AJs play an important role in normal cell–cell adhesion, many investigators analyzed the AJs in human tumors. Immunostainings of various tumor types with antibodies recognizing specific proteins of the AJs resulted in numerous papers describing the changes in these proteins expression and the correlations between these changes and patient outcomes.
3.1 Changes in Cadherin Expression. Decrease in E-Cadherin and Upregulation of Mesenchymal Cadherins in High-Grade Invasive Carcinoma
E-cadherin is the most prominently expressed classical cadherin in normal epithelial cells. Immunostaining with anti-E-cadherin antibodies revealed significant decreases in the expression levels of this protein in the variety of human epithelial tumors (Berx and van Roy 2009). Interestingly, not all the tumors are exactly the same, as there are significant differences between different organs and even different tumor types within the organ. In breast carcinoma, loss of E-cadherin expression is an early and highly penetrant event (up to 85%) in lobular tumors (Berx et al. 1995; Berx and Van Roy 2001; Cowin et al. 2005). Conversely, E-cadherin expression in ductal breast carcinoma is decreased only in advanced high-grade tumors (Jeschke et al. 2007; Nagae et al. 2002; Oka et al. 1993; Park et al. 2007; Pedersen et al. 2002; Rakha et al. 2005).
Similar to lobular breast carcinoma, the diffuse-type gastric cancer also displays the early onset loss of E-cadherin expression (92%) (Mayer et al. 1993). In contrast, intestinal-type gastric cancer shows loss of E-cadherin expression primarily in high-grade invasive tumors (Shun et al. 1998). In squamous cell carcinoma, prostate cancer , non-small cell lung carcinoma and colon cancer the expression of E-cadherin is usually maintained in early low-grade tumors and is downregulated in a subset of late high-grade tumors (Bohm et al. 1994; Schipper et al. 1991; Schuhmacher et al. 1999; Umbas et al. 1992). Loss or decrease in expression of E-cadherin usually correlates with tumor invasiveness, distant metastasis and unfavorable patient outcome (Asgeirsson et al. 2000; Dolled-Filhart et al. 2006; Heimann et al. 2000; Nakopoulou et al. 2002; Park et al. 2007; Pedersen et al. 2002; Rakha et al. 2005).
E-cadherin is not the only classical cadherin expressed in normal epithelial tissues. In addition to E-cadherin, epithelial cells often express other cadherins. For example, skin keratinocytes express both E-cadherin and P-cadherin and loss of expression of both of these genes is required for disruption of the AJs (Tinkle et al. 2008b). In contrast to E-cadherin, which is expressed at relatively similar levels by all epithelial cells within the tissue, P-cadherin often displays much more differential levels of expression. For example, in skin epidermis only basal and hair germ cells express P-cadherin. Similarly, in breast epithelium, P-cadherin is expressed primarily by myoepithelial cells, Thus, perhaps not surprisingly, the results of the analyses of P-cadherin expression in epithelial tumors were very different from what was found for E-cadherin. In breast cancer , P-cadherin is often overexpressed and its expression correlates with poor patient prognosis (Paredes et al. 2007; Turashvili et al. 2011). P-cadherin is not expressed in normal adult colon, but its expression is activated in colon cancer (Milicic et al. 2008). In contrast, in oral squamous cell carcinoma loss of P-cadherin expression is a hallmark of aggressive tumors (Lo Muzio et al. 2004; Munoz-Guerra et al. 2005).
While expression of E-cadherin in high-grade epithelial tumors is downregulated, tumor cells often show increased expression of N-cadherin , which is normally present only in mesenchymal cells and neurons (Cavallaro et al. 2002; Wheelock et al. 2008). This phenomenon is called “cadherin switching” and it is considered to be one of the hallmarks of epithelial to mesenchymal transition (EMT ). EMT is a normal process of a dramatic change in cellular morphology which occurs during specific stages of normal embryonic development, when highly adhesive epithelial cells downregulate expression of epithelial cadherins, upregulate expression of mesenchymal cadherins and become highly motile and invasive (Shook and Keller 2003). Notable examples of these developmental events include gastrulation, formation of neural crest cells , heart valve formation and delamination of muscle precursor cells from the dermomyotome during muscle morphogenesis (Micalizzi et al. 2010). This ability of epithelial cells to drastically change their phenotype and become highly invasive mesenchymal cells is maintained in epithelial tumors and EMT is likely to play an important role during tumor dissemination and metastasis (Hugo et al. 2007). While downregulation of epithelial cadherins is clearly the causal event responsible for the transition from epithelial to mesenchymal phenotype, the role of upregulation of N-cadherin is not completely clear. Early experiments with established cancer cell lines indicated that upregulation of N-cadherin in epithelial cells can cause a prominent increase in cell migration and invasion (Nieman et al. 1999; Zahir and Weaver 2004). N-cadherin can activate cell migration in tissue culture by interacting with fibroblast growth factor receptor (FGFR ) and stimulating FGF signaling, as well as by activating Rac1 and Cdc42 small GTPases, which regulate the actin cytoskeleton (Wheelock et al. 2008). In vivo experiments revealed a more complex picture. Overexpression of N-cadherin in breast tumors, which were induced by upregulation of ErbB2, did not produce a significant phenotype; however, overexpression of N-cadherin in breast carcinoma, which was induced by expression of polyoma virus middle T antigen, resulted in increased metastasis (Hulit et al. 2007; Knudsen et al. 2005). Thus, it is likely that the role of N-cadherin upregulation during EMT in epithelial tumors depends on the nature of the signaling pathways responsible for tumor initiation and progression.
3.2 Changes in Expression of Catenins
In addition to E-cadherin, AJs in epithelial cells also require expression of catenins, which were also analyzed in a range of epithelial tumors. In general, immunostaining with anti-α-catenin antibodies revealed changes that are very similar to the changes in expression of E-cadherin. Tumors showing loss or decrease in expression of E-cadherin, often also show the downregulation of α-catenin (Aaltomaa et al. 1999; Kimura et al. 2000; Richmond et al. 1997; Toyoyama et al. 1999; Umbas et al. 1992; Zhou et al. 2005). 81% of breast carcinomas display loss or decrease in staining for α-catenin (Rimm et al. 1995). α-catenin is absent in 50% of squamous cell carcinomas of esophagus (Setoyama et al. 2007) and in 41% of gastric carcinomas (Zhou et al. 2005). Decrease or loss of expression of α-catenin often correlate with invasive phenotype, lymph node metastasis , recurrence of the disease and poor patient outcome (Aaltomaa et al. 1999, 2005; Kadowaki et al. 1994; Nakanishi et al. 1997; Richmond et al. 1997; Setoyama et al. 2007; van Oort et al. 2007).
In addition to its important role in AJs, β-catenin is also a transcriptional co-activator, which can cause tumor formation, when it is constitutively stabilized and localized to the nucleus . The important role of β-catenin in cell transformation is the primary reason for the extensive analyses of this protein in variety of human malignancies. The amount of information about β-catenin in human cancer is massive and it has been extensively covered in excellent recent reviews (Clevers 2006; Heuberger and Birchmeier 2010; Klaus and Birchmeier 2008; Wend et al. 2010). Overall, β-catenin is prominently upregulated in colorectal cancer , in subsets of medulloblastomas and basal cell carcinomas, and type of hair follicle cancer called pilomatricoma (Clevers 2006; El-Bahrawy et al. 2003; Gat et al. 1998; Gibson et al. 2010; Saldanha et al. 2004; Yamazaki et al. 2001). While there are many studies that report the presence of nuclear β-catenin in various additional tumor types, these findings are not always corroborated by other laboratories. For example, the presence of significant levels of nuclear β-catenin was reported in breast cancer tumors in one report (Lin et al. 2000), but this finding was not confirmed in the follow up papers (Dolled-Filhart et al. 2006; Gillett et al. 2001; Pedersen et al. 2002; Wong et al. 2002). In many epithelial tumors, the levels of junctional β-catenin parallels the expression of E-cadherin and it is downregulated in the advanced high-grade tumors (Aaltomaa et al. 2005; Carico et al. 2001; Chung et al. 2007; Dolled-Filhart et al. 2006; Fukumaru et al. 2007; Ishizaki et al. 2004; Jaggi et al. 2005; Nakanishi et al. 1997).
4 Molecular Mechanisms Responsible for the Decrease of Cadherin-Catenin-Mediated Adhesion in Human Epithelial Tumors
As described earlier, extensive literature indicates that epithelial cadherins and associated catenins are frequently downregulated in advanced human epithelial tumors. The molecular mechanisms responsible for this downregulation are very diverse and include regulation at the level of genes, gene transcription , and multiple post-transcriptional stages.
4.1 Mutation and Deletion of Cadherin and Catenin Genes in Human Cancer
The most direct effects on gene function are gene mutations, deletions or amplifications. While “next generation” sequencing approaches currently make it possible to analyze tumor cell mutations in all human genes, earlier efforts concentrated on sequencing of specific genes in DNA isolated from human tumors. Cancer genome re-sequencing of E-cadherin (CDH1) revealed frequent inactivating mutations in E-cadherin in lobular breast and diffuse-type gastric cancers, where up to 50% of primary tumors contain mutations in E-cadherin (Becker et al. 1993, 1994;. Berx et al. 1995, 1996). In breast cancer , the wild-type allele of E-cadherin is often lost and the overall loss of heterozygosity of E-cadherin is a frequent event in breast cancer (Berx et al. 1996; Cleton-Jansen et al. 2001). Besides the lobular breast and diffuse-type gastric cancer , mutations in E-cadherin are not frequent in other carcinomas (Endo et al. 2001; Risinger et al. 1994; Soares et al. 1997; Taddei et al. 2000; Wijnhoven et al. 1999).
The homozygous loss-of-function mutations in E-cadherin, β-catenin(Ctnnb1) and αE-catenin (Ctnna1) genes are embryonic lethal in mice and it is likely that they result in lethality in humans (Haegel et al. 1995; Huelsken et al. 2000; Larue et al. 1994; Torres et al. 1997). While mice with heterozygous mutations in E-cadherin, β-catenin and αE-catenin genes appear normal, germ line mutation in one allele of E-cadherin in humans strongly predisposes to development of diffuse-type gastric cancer (Guilford et al. 1998). Interestingly, the probability of development of lobular breast cancer in females carrying the germ line mutation in E-cadherin is only slightly higher than in general population (Pharoah et al. 2001).
Loss-of-function mutations in α- and β-catenin genes have been described in cell lines derived from various human epithelial tumors; however, there is no knowledge about the prevalence of these mutations in primary tumors. Cancer genome re-sequencing projects identified mutations and deletions in α E- and αN-catenin genes; however, since only few tumors were sequenced, it is impossible to conclude the overall prevalence of these mutations (Ding et al. 2010; Wood et al. 2007).
In contrast to the loss-of-function mutations, protein stabilizing mutations in β-catenin are frequently found in primary epithelial tumors (Polakis 2000). These mutations concentrate in the amino-terminal part of the protein and often affect serine and threonine amino acids, which need to be phosphorylated for protein degradation (see below). Thus, the functional outcome of these mutations is stabilization of the protein and constitutive activation of the canonical Wnt signaling pathway, which is causally involved in cancer initiation. Mutations in β-catenin are found in 27% of intestinal type gastric cancers (Park et al. 1999), in up to 20% of hepatocellular carcinomas (de La Coste et al. 1998; Miyoshi et al. 1998), in 15% of pediatric kidney cancers (Koesters et al. 1999), in 61% of anaplastic thyroid carcinoma (Garcia-Rostan et al. 1999), and in 75% of hair follicle tumors called pilomatricoma (Chan et al. 1999).
4.2 Transcriptional Downregulation of Cadherin and Catenin Gene Expression
While decreases in the expression of epithelial cadherins and loss of cell–cell adhesion occur in the majority of advanced epithelial tumors, E-cadherin is frequently mutated or lost in only a few specific tumor types. Indeed, the most frequent mechanisms responsible for the loss of E-cadherin function is transcriptional downregulation of its expression (Berx and van Roy 2009). Expression of E-cadherin is downregulated by either promoter hypermethylation or an increase in expression of transcriptional factors that down-regulate E-cadherin promoter activity.
Promoter hypermethylation is likely the most frequent event responsible for the loss of E-cadherin expression in advanced tumors. Even in gastric diffuse-type tumors containing an inactivating mutation of E-cadherin, the second nonmutant allele of E-cadherin is silenced via methylation of a CpG island in the promoter of the E-cadherin gene (Grady et al. 2000). Abnormally increased methylation of the E-cadherin promoter is found in primary gastric, breast, prostate, non-small cell lung, thyroid, bladder, cervical, esophageal, renal, colorectal, hepatocellular and other types of cancer (Chen et al. 2003a; Garinis et al. 2002; Graff et al. 1995, 1998; Kim et al. 2007; Matsumura et al. 2001; Nojima et al. 2001; Ribeiro-Filho et al. 2002; Si et al. 2001; Tamura et al. 2000; Yoshiura et al. 1995). Increased methylation of the E-cadherin promoter enhances the binding of methyl-CpG-interacting proteins MeCP2 and MBP2, which recruit HDACs that cause histone 3 (H3) deacetylation and shut down transcription (Koizume et al. 2002). Moreover, proteins that bind to methylated H3 at the E-cadherin promoter also play an important role in regulation of E-cadherin expression. Methyl-H3K9-binding protein MPP8 localizes to the E-cadherin promoter and mediates E-cadherin silencing by directing DNA methylation via interaction with DNA methyltransferase 3A (Kokura et al. 2010). Interestingly, in some cell types transcription factors critical for proper epithelial differentiation may neutralize the effect of E-cadherin promoter hypermethylation. For example, the members of the forkhead transcription factor family FOXA1/2 are downregulated in pancreatic ductal adenocarcinoma resulting in the loss of E-cadherin expression. Re-expression of FOXA1/2 in pancreatic cancer cells with extensive E-cadherin promoter hypermethylation results in re-activation of E-cadherin expression (Song et al. 2010).
In addition to promoter methylation, several transcription repressors can mediate downregulation of E-cadherin expression through direct interaction with E-cadherin promoter. Earlier somatic cell hybrid experiments indicated the presence of factors that utilize E-box sequences within the E-cadherin promoter to downregulate its activity (Giroldi et al. 1997). Later studies identified multiple E-box-binding transcriptional repressors that can bind to the E-cadherin promoter, downregulate its transcriptional activity and promote EMT , cell migration and invasion . Specifically, the Zinc-finger protein SNAIL represses E-cadherin promoter by the recruitment of the H3K27 histone methyltransferases polycomb repressive complex 2 (PRC2) and Sin3 A/HDAC1/2 (Batlle et al. 2000; Cano et al. 2000; Herranz et al. 2008; Peinado et al. 2004). E-box binding proteins ZEB1 /2 downregulate E-cadherin expression by interacting with the transcription co-repressor CtBP-1 (Comijn et al. 2001; Grooteclaes and Frisch 2000; Shi et al. 2003). In addition, ZEB1 can regulate E-cadherin independently of CtBP, by interacting with the SWI/SNF chromatin-remodeling protein BRG1 (Sanchez-Tillo et al. 2010). In addition to Snail and ZEB1/2, basic helix-loop-helix proteins E12/E47 and Twist repress the transcription of E-cadherin through direct interaction with its promoter (Perez-Moreno et al. 2001; Yang et al. 2004). The forkhead domain transcription factor FOXQ1 is overexpressed in high-grade basal-type breast cancers. It downregulates expression of E-cadherin and promotes EMT, gain of stem cell-like properties, and acquisition of resistance to chemotherapy-induced apoptosis (Qiao et al. 2011). EZH2 is a histone-lysine-methylase and the member of the polycomb group of proteins involved in transcriptional repression of target genes. EZH2 is upregulated in prostate, breast and bladder cancers and it mediates tumor aggressiveness by transcriptional silencing of E-cadherin (Cao et al. 2008). Bmi1 is another polycomb-group protein frequently overexpressed in human cancers. Bmi1 is expressed in stem cells, it maintains self-renewal, and it cooperates with Twist1 to repress E-cadherin and p16INK4a and to promote epithelial-mesenchymal transition and tumor initiating capacity (Yang et al. 2010). Zeppo1 (zinc finger elbow-related proline domain protein 1) is amplified and overexpressed in breast cancer . It binds to Groucho, represses E-cadherin expression and promotes breast cancer progression in mouse models of metastatic breast cancer (Slorach et al. 2011). Overall, various transcriptional repressors are utilized during normal development to quickly downregulate E-cadherin expression and promote EMT. During progression of epithelial tumors, these factors are also upregulated and this causes tumor cell EMT and promotes tumor invasion and metastasis (Peinado et al. 2007; Yang et al. 2004).
4.3 Posttranscriptional Inactivation of Cadherin–Catenin Adhesion System by MicroRNAs
While transcriptional regulation of E-cadherin has been extensively studied, it is clear that cells can employ multiple posttranscriptional mechanisms to control the levels of epithelial cadherins. A number of microRNAs exercise a potent control over E-cadherin protein production at the level of translation. MicroRNAs usually recognize the 3’ UTR region of their target mRNAs and negatively regulate protein translation. The Weinberg laboratory recently demonstrated that a microRNA upregulated in breast cancer cells, miR-9, directly targets E-cadherin mRNA (Ma et al. 2010). Overexpression of miR-9 in a nonmetastatic cell line downregulated E-cadherin and enabled the cells to form metastases. Conversely, downregulation of miR-9 levels in highly malignant cells inhibited metastasis . Interestingly, transcription of miR-9 itself was controlled by MYC and MYCN proteins, which are strongly implicated in cancer . Similar to miR-9, a microRNA overexpressed in esophageal squamous cell carcinoma, miR-92a, also directly targets E-cadherin 3’UTR and promotes cell migration and invasion (Chen et al. 2011). Highly expressed in breast cancer stem cells miR-495 also directly targets E-cadherin and promotes tumorigenesis in mice. Interestingly, the transcription factor E12/E47, previously implicated in the direct regulation of E-cadherin, was also shown to control the expression of miR-495 (Hwang-Verslues et al. 2011).
While the majority of microRNAs target the 3’UTR of mRNAs and negatively regulate protein translation, some microRNAs target promoter regions and activate transcription . For example, miR-373 has a binding site in the E-cadherin promoter and it activates E-cadherin transcription (Place et al. 2008). Besides this noncanonical regulation, miR-373 can target LATS2 and CD44 mRNAs and promote tumor invasion and metastasis (Huang et al. 2008; Voorhoeve et al. 2006).
While miR-9, mir-92a and miR-495 directly regulate E-cadherin mRNA, other microRNAs act on transcriptional factors that can control E-cadherin expression. For example, miR-141, mir-200 and mir-205 miRNAs directly repress ZEB1 and ZEB2 (Burk et al. 2008; Gregory et al. 2008; Korpal et al. 2008; Olson et al. 2009; Park et al. 2008). In addition, the miR-200 family directly targets Suz12, a subunit of a polycomb repressor complex (PRC2), which negatively regulates E-cadherin expression and promotes formation of breast cancer stem cells (Iliopoulos et al. 2010). miR-708 is downregulated in human renal cell carcinoma, and in normal cells, this microRNA targets E-cadherin regulators ZEB2 and BMI1 (Saini et al. 2011). In some cases, the microRNA regulation of epithelial cadherin expression is even more complex. miR-221 and miR-222 are expressed in basal-type breast cancer , where they target the 3’UTR of TRPS1 (trichorhinophalangeal syndrome type 1), which is a member of the GATA family of transcriptional repressors directly repressing expression of ZEB2 (Stinson et al. 2011).
Overall, microRNAs elicit a powerful control of E-cadherin expression in normal epithelial cells and deregulation of these microRNAs can have profound consequences for the loss of epithelial phenotype and increased invasion and metastasis in high-grade epithelial tumors.
4.4 Posttranslational Effects on the Cadherin–Catenin Adhesion System
During development epithelial cells constantly remodel their cell–cell adhesion structures to allow for normal tissue morphogenesis. Moreover, in the majority of adult organs, homeostasis is maintained through constant tissue renewal via a well-orchestrated process of stem cell-mediated self-renewal and differentiation. These processes also involve constant epithelial cell movements within the tissue, which implies the need for quick cell–cell junction breakdown and re-formation that often takes place without the overall loss of the epithelial phenotype. To accomplish this task, cells evolved several mechanisms eliciting posttranslational control of AJ function. Cadherin–catenin complexes can be quickly regulated by phosphorylation-mediated disruption of cadherin–catenin protein complexes that is often coupled with endocytosis of cadherin and its degradation.
Protein phosphorylation is a powerful mechanism that can be rapidly employed to change protein function. Stability of the AJs depends on the efficient formation of cadherin–catenin protein complexes. Phosphorylation of cadherins and catenins influences their binding affinities and overall AJ complex stability. E-cadherin at the membrane associates with multiple receptor-type tyrosine kinases (RTKs) including IGF1R, ErbB2, Met and EGFR (Canonici et al. 2008; Hiscox and Jiang 1999; Ochiai et al. 1994; Reshetnikova et al. 2007). Activation of RTKs results in decreases in the affinity between cadherin and catenins and disassembly of the cadherin–catenin complexes (Behrens et al. 1993; Hamaguchi et al. 1993). RTKs and/or Src -family kinases phosphorylate E-cadherin and this creates a binding site for Hakai , which mono-ubiquitinates E-cadherin and promotes its interaction with mono-ubiquitin-binding protein HRS, internalization and subsequent lysosome -mediated degradation (Fujita et al. 2002; Palacios et al. 2005; Shen et al. 2008; Toyoshima et al. 2007). The AJ protein Shrew-1 plays an important role in EGF -induced endocytosis of E-cadherin; however, the mechanism of Shrew-1 function is not well understood (Gross et al. 2009). Endocytosed E-cadherin can also be ubiquitinated by MDM 2, which is overexpressed in breast cancer , and this is followed by E-cadherin degradation (Yang et al. 2006). Similar to E-cadherin, β-catenin is a prominent substrate for tyrosine phosphorylation by Src-family, BCL-Abl, MET and RET tyrosine kinases and this phosphorylation decreases the affinities between β-catenin and both E-cadherin and α-catenin, and in addition, causes an activation of β-catenin-mediated transcription (Brembeck et al. 2004; Coluccia et al. 2006, 2007; Gujral et al. 2008; Lilien and Balsamo 2005; Roura et al. 1999; Zeng et al. 2006).
p120-catenin was first discovered as a prominent substrate for Src -family tyrosine kinases (Reynolds et al. 1994). Tyrosine phosphorylation of p120-catenin modulates its binding to and inhibition of RhoA and this can have a significant impact on the stability of the AJs (Castano et al. 2007). Src can also disrupt AJs and promote EMT by phosphorylation and targeted degradation of the Rac1 activator Tiam1, which is required for AJ formation and maintenance (Woodcock et al. 2009).
In addition to tyrosine kinases , the stability of the AJs is also regulated by serine-threonine phosphorylation. The cadherin cytoplasmic tail contains a serine-rich domain and its phosphorylation by casein kinase I results in the disruption of cadherin–catenin complexes (Ochiai et al. 1994). Serine-threonine phosphorylation of β-catenin by GSK3 β plays a critical role in the destruction of cytoplasmic β-catenin and the negative regulation of its transcriptional activity (Aberle et al. 1997; Clevers 2006). In contrast, phosphorylation by AKT and cyclic AMP-dependent protein kinases causes activation of β-catenin transcriptional activity (Fang et al. 2007; Hino et al. 2005).
AJs link to the actin cytoskeleton and changes in the actin cytoskeleton play crucial roles in both AJs formation and disruption. Cadherin–catenin clustering is regulated by the actin cytoskeleton (Angres et al. 1996; Hirano et al. 1992). Rho-family GTPases are the general regulators of the actin cytoskeleton, and they play a crucial role in the regulation of cell–cell adhesion, cell migration and invasion (Ellenbroek and Collard 2007). While α-catenin is the only catenin with a well-defined f-actin binding domain, p120-catenin is the principal regulator of Rho family GTPases. P120-catenin binds and inactivates RhoA and activates Rac1 , and this promotes AJ stabilization (Reynolds 2007). While some dynamics of the actin cytoskeleton are necessary for AJ formation, sustained activation of either RhoA or Rac1, which is frequently induced by oncogenes, is likely to destabilize cadherin–catenin contacts and stimulate cell migration and invasion (Gimond et al. 1999; Lozano et al. 2008; Zhong et al. 1997). For example, Abl and Arg intracellular tyrosine kinases impact AJs assembly through regulation of Rho-ROCK-myosin signaling pathway (Zandy et al. 2007). Inhibition of these kinases results in activation of Rho-ROCK pathway and disruption of the AJs, which is mediated by actomyosin contraction.
Cell surface levels of cadherin can be rapidly altered by activation of cadherin endocytosis. For example, during normal embryonic development a dramatic EMT takes place during gastrulation. In gastrulation, activin/nodal members of the TGF-β superfamily induce expression of Fibronectin Leucine-rich Repeat Transmembrane 3 (FLRT3), a transmembrane protein containing extracellular leucine-rich repeats, and the small GTPase Rnd1 (Ogata et al. 2007). These two proteins interact physically and decrease cell adhesion by sequestering cadherin through a dynamin-dependent endocytosis pathway.
We previously discussed the important role of p120-catenin in cadherin stabilization at the plasma membrane (Kowalczyk and Reynolds 2004). Interestingly, the interaction site between p120-catenin and cadherin extends over the cadherin residues required for clathrin-mediated endocytosis and Hakai -dependent ubiquitination of this protein (Ishiyama et al. 2010). Thus, interaction between cadherin and p120-catenin should inhibit cadherin internalization and stabilize it at the cell surface. Upon endocytosis, E-cadherin can associate with the MDM 2 E3 ubiquitin ligase, which ubiquitinates cadherin and targets it for degradation (Yang et al. 2006). Interestingly, expression levels of MDM2 negatively correlate with the levels of E-cadherin in clinical samples of breast cancer , and overexpression of MDM 2 promotes E-cadherin internalization and degradation in breast cancer cell lines (Yang et al. 2006).
The TGF-β signal transduction pathway is a critical regulator of EMT in various epithelial tumors. Activated by TGF-β treatment SMAD3/4 transcription factors interact with Snail1 at the promoters of E-cadherin and tight-junctional genes and co-repress their transcription to induce EMT (Vincent et al. 2009). Another mechanism responsible for TGF-β-induced EMT involves phosphorylation of Ser 43 of hnRNP E1 by protein kinase Akt2, which results in the release of the hnRNP from the 3’ UTR of Disabled-2 (Dab2) and interleukin-like EMT inducer (ILEI) transcripts and translational activation of Dab2 and ILEI (Chaudhury et al. 2010; Hussey et al. 2011). Dab2 mediates directional trafficking and polarized distribution of several cell surface proteins including E-cadherin (Yang et al. 2007). Moreover, Dab2 is a critical protein responsible for EMT in TGF-β-treated cells (Prunier and Howe 2005).
Progression of epithelial tumors is frequently associated with activation of cell surface proteases, which promote tumor invasion and metastasis. Interestingly, many of these enzymes directly cleave cadherin molecules. Cleavage of cadherins by metalloproteinases (MMPs), disintegrin and metalloproteinases (ADAM ) or γ-secretase results in the disruption of cadherin-mediated adhesion (Ferber et al. 2008; Lochter et al. 1997; Marambaud et al. 2002; Maretzky et al. 2005; Solanas et al. 2011). MMPs can be responsible for EMT induced by potent oncogenes. For example, in lung epithelial cells, oncogenic K-Ras can promote disruption of the AJs and EMT via ERK-mediated induction of MMP-9 and cleavage of E-cadherin at two sites (Wang et al. 2009). Interestingly, both extracellular and intracellular fragments of cadherin generated after cleavage can have an important function in regulation of cell adhesion and signaling. The extracellular fragments have dominant negative activity, binding to cadherin on the cell surface and interfering with its function in cell–cell adhesion (Damsky et al. 1983). The cytoplasmic domain of E-cadherin can translocate to the nucleus and regulate p120-catenin -Kaiso transcription activity (Ferber et al. 2008). Similarly, upon translocation to the nucleus , the cytoplasmic domain of N-cadherin enhances transcription activity of β-catenin and represses CBP/CREB-mediated transcription (Marambaud et al. 2003; Shoval et al. 2007).
To summarize, the advanced epithelial tumors may employ a variety of mechanisms to down-regulate the function of epithelial cadherins and decrease epithelial cell adhesion. We will now discuss the potential clinical significance of these events.
5 Causal Role of AJ Proteins in Cancer Initiation and Progression
Analyses of human tumors clearly demonstrated a decrease of the epithelial cadherin–catenin cell–cell adhesion system in advanced malignancies. This, however, does not necessarily indicate that loss of epithelial-type AJs is causally involved in tumor initiation or progression. One of the potential causal proofs frequently used in the clinical literature is the evidence by association. Indeed, primary tumors demonstrating loss of E-cadherin expression statistically are more likely to present with metastasis and result in poor patient outcome. However, one has to be careful in making conclusions based on associations, because there is a potential caveat in this simple explanation. Since decrease in E-cadherin expression is more frequently seen in histologically advanced tumors, the association between decreased E-cadherin expression and tumor metastasis may be circumstantial and not causal. An experimental approach is usually necessary to establish causality. We will now discuss the potential causal connection between the cadherin–catenin adhesion system and cancer initiation and progression.
Many cell lines generated from human tumors display decreased expression of E-cadherin and/or catenins. Re-expression of missing cell–cell adhesion molecules and the analysis of resulting cell lines provided a powerful tool for the investigation of the role of AJ proteins in human cancer . Many groups also used a complementary loss-of-function approach to decrease the expression of cadherins or catenins in normal epithelial cells or in cancer cell lines with the cadherin–catenin adhesion system. Overall, studies with E-cadherin demonstrated that it plays a critical role in the establishment and maintenance of epithelial cell phenotypes and the attenuation of EMT , cell migration , invasion and metastasis (Behrens et al. 1989; Chen and Obrink 1991; Frixen et al. 1991; Vleminckx et al. 1991). Re-expression of α-catenin in cell lines missing α-catenin resulted in even more dramatic phenotypes and caused not only reversion of EMT and attenuation of cell invasion , but also decreased rates of cell proliferation and attenuated primary tumor formation in immunocompromised mice (Bullions et al. 1997; Ewing et al. 1995; Watabe et al. 1994). The role of p120-catenin in human cancer cell lines is even more complex and it appears to depend on the expression of E-cadherin. In cells expressing E-cadherin, p120-catenin inhibits Ras and attenuates cell proliferation; however, in cell lines missing E-cadherin, p120-catenin does not attenuate Ras and, instead, it activates Rac1 -MAPK signaling and promotes cell proliferation (Dohn et al. 2009; Soto et al. 2008).
Cell lines are a powerful experimental model, which can quickly assess the tumorigenic potential and significance of specific genetic and epigenetic changes that take place in human tumors. However, experiments with cell lines are unable to capture the complexity of tumor initiation and progression in human patients, because of the important role of three-dimensional tissue organization and overall cellular diversity that is missing in these models (Bissell and Hines 2011; Lee and Vasioukhin 2008). Genetic experiments with mice are usually necessary to capture all the details of normal tissue microenvironment and three-dimentional tissue organization. The Christofori laboratory generated the first genetic evidence of an important role of E-cadherin in tumor progression . The progression from well-differentiated adenoma to invasive carcinoma in the mouse model of pancreatic β-cell carcinoma (Rip1Tag2 mice) is marked by the decrease in E-cadherin expression (Perl et al. 1998). Re-expression of E-cadherin in these tumors in vivo resulted in the arrest of tumor progression at the adenoma stage and, conversely, expression of a dominant-negative E-cadherin caused enhanced tumor invasion and metastasis (Perl et al. 1998). In a different genetic cancer model, the growth and metastatic progression of non-small-cell lung cancer driven by C-Raf overexpression was enhanced by simultaneous deletion of E-cadherin (Ceteci et al. 2007).
Unlike the majority of human epithelial tumors, E-cadherin is lost early in lobular breast carcinoma. These tumors also display frequent inactivation of the p53 tumor suppressor gene. Tissue-specific deletion of tumor suppressor p53 in mice is sufficient to cause development of breast cancer . While deletion of E-cadherin in the same tissues was not sufficient to cause cancer , simultaneous deletion of E-cadherin and p53 resulted in earlier development of invasive and metastatic lobular breast carcinoma (Derksen et al. 2006). Interestingly, deletion of E-cadherin in breast epithelial cells in this model caused resistance to anoikis, an apoptotic cell death caused by the loss of attachment to the extracellular matrix.
While the experiments with genetic inactivation of E-cadherin clearly demonstrated that the loss of E-cadherin expression is playing a causal role in tumor progression , it was also very clear that E-cadherin is not a canonical tumor suppressor, because deletion of E-cadherin was not sufficient to cause tumor formation (Derksen et al. 2006). Somewhat different results were obtained in the experiments with genetic ablation of p120- and α-catenins. Deletion of α-catenin in hair follicle stem and progenitor cells caused the formation of prominent inflammatory skin lesions and development of skin squamous cell carcinoma tumors (Silvis et al. 2011). Moreover, transplantation of α-catenin -/-keratinocytes on the skin of nude mice resulted in wound-like microenvironment and formation of skin lesions resembling squamous cell carcinoma (Kobielak and Fuchs 2006). Similarly, transplantation of p120-catenin -/- keratinocytes on the skin of nude mice caused formation of prominent inflammatory lesions and tumor-like growth (Perez-Moreno et al. 2008). Conditional deletion of p120-catenin in the mouse oral cavity, esophagus, and forestomach results in inflammation and invasive squamous cell cancer (Stairs et al. 2011). Similarly, ablation of p120-catenin in intestinal epithelial cells results in chronic inflammation and formation of tumors (Smalley-Freed et al. 2011). These genetic in vivo experiments demonstrate that both p120- and α-catenins can function as tumor suppressors.
Unlike p120 and α-catenins, β-catenin is not critical for AJ formation in epithelial cells, because loss of β-catenin can be compensated by plakoglobin, which can provide a link between cadherins and α-catenin. In contrast to p120- and α-catenins, β-catenin is a very potent proto-oncogene playing a central role in the canonical Wnt signal transduction pathway (Clevers 2006). Constitutive activation of Wnt signaling is oncogenic in many organs and tissues. β-catenin levels are negatively regulated by a destruction complex containing APC, Axin, GSK3 β and Casein kinase I (CKI). GSK3β and CKI phosphorylate β-catenin and target it for degradation. Deletion or mutation of the phosphorylation sites stabilizes β-catenin and prominently increases its transcriptional activity. Expression of a stabilized form of β-catenin in the variety of mouse organs and tissues resulted in development of cancer , essentially proving that stabilization of β-catenin is sufficient for tumor development (Clevers 2006). Inactivation of β-catenin destruction complex also results in increase in β-catenin levels and signaling activities. Mutation of one allele of APC in mice results in upregulation of β-catenin and development of multiple polyps and eventually intestinal cancer (Clevers 2004). Similarly, mutation of APC in humans results in Familial adenomatous polyposis (FAP), which is an inherited disorder characterized by development of multiple intestinal polyps and predisposition to cancer of the large intestine , as well as fibromas, osteomas and medulloblastomas. In general, human and mouse phenotypes vary significantly, depending on the nature of the APC gene mutation. Interestingly, while loss of one allele of APC should be theoretically sufficient to activate β-catenin in all cell types, the most prevalent phenotype in APC mutants is the development of intestinal tumors (Clevers 2004). The reasons for such specificity are not known, but it may indicate an exquisite sensitivity of the intestinal stem cells to changes in β-catenin signaling. Since transcriptional activity of β-catenin is required for the maintenance of stem and progenitor cells in many organs and tissues, conditional deletion of β-catenin usually results in the depletion of these cell populations and failure of normal development or adult organ homeostasis (Fevr et al. 2007; Machon et al. 2003; Zechner et al. 2003). This provides a significant opportunity for therapeutic intervention, as tumors are likely to be very sensitive to the loss of β-catenin signaling activity (Malanchi et al. 2008).
Overall, loss-of-function and gain-of-function experiments using cell lines and genetically engineered mice demonstrated that AJ proteins play an important role in epithelial tumor initiation and progression. E-cadherin downregulation in epithelial tumors is not sufficient for tumor initiation, but it promotes tumor invasion and metastasis . P120- and α-catenins can function as tumor suppressors and cause tumor development in some organs and tissues. Finally, β-catenin is a proto-oncogene, since upregulation of β-catenin protein levels is sufficient for tumor development. We will now discuss potential molecular mechanisms responsible for tumor- and metastasis suppression by AJs.
6 Mechanisms of AJ Proteins in Preventing Cancer Initiation and Progression
6.1 Maintenance of the Epithelial Phenotype and Adhesion-Mediated Attenuation of Tumor Invasion
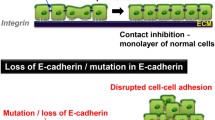
One of the most important functions of the AJs is mediation of strong intercellular adhesion between epithelial cells, which is necessary for the three-dimensional architecture of organs and tissues. This function is mediated by the connection of AJs to the actin cytoskeleton, which helps to generate the forces necessary to drive the membranes of neighboring cells together and promote cell–cell adhesion (Cavey and Lecuit 2009; Meng and Takeichi 2009). During epithelial tumor development, properly functioning AJs continue to mediate strong cell–cell adhesion between the tumor cells and this should attenuate primary tumor invasion , which is necessary for tumor progression and dissemination. Thus, the purely adhesive function of AJ proteins should be able to suppress tumor progression and metastasis by simply preventing the tumor cells from separating from the bulk of the tumor and invading the stromal cell compartment (Fig. 16.2a). It has been assumed that the decrease in expression of E-cadherin and the increase in expression of N-cadherin in tumor cells would reduce the affinity of tumor cells for each other and promote their affinity for mesenchymal cells, expressing N-cadherin. Theoretically, this should promote tumor cell invasion . The recent development of novel experimental techniques that include conditional gene knockouts and gene replacements technologies made it possible to test this hypothesis experimentally in live animals. The experimental results demonstrated that this explanation is probably too simplistic. Ablation of classical cadherins (both E- and P-cadherin) or α-catenin in epidermal keratinocytes in mice causes prominent cell–cell adhesion defects, but does not result in the loss of epithelial cell phenotypes and intermingling of mutant epithelial cells with stromal fibroblasts (Tinkle et al. 2008b; Vasioukhin et al. 2001). Moreover, genetic replacement of E-cadherin with N-cadherin in intestinal epithelial cells does not result in disruption of tissue morphology and intermingling of N-cadherin-expressing epithelial cells with the stromal cells (Libusova et al. 2010). It appears that simple ablation of the AJs in normal epithelial cells in vivo does not result in a complete loss of the epithelial cell phenotype. Since this does happen in many tumor cells, it is likely that AJs do play an important role in the maintenance of epithelial phenotype, but this function is reinforced by some unknown additional mechanisms in the normal cells, and these mechanisms may be lost in epithelial tumors.

Mechanisms of Adherens Junction function in cancer. a AJs in epithelial cells prevent tumor cell dispersion and local tissue invasion, which attenuates tumor progression and metastasis. b Epithelial cadherins bind to the variety of tyrosine kinase receptor proteins and negatively regulate their signaling outputs. c In the tumor cells with attenuated cytoplasmic β-catenin destruction machinery, cadherin sequesters β-catenin from the nucleus and negatively regulates β-catenin signaling activity. d AJ proteins support Hippo pathway signaling by negatively regulating nuclear localization of Yap1
6.2 Modulation of Growth Factor Receptor Signaling
Cell surface growth factor receptors play a critical role in regulation of cell proliferation and many of these proteins function as proto-oncogenes or tumor-suppressors. Cadherin–catenin protein complexes at the membrane interact with multiple growth factor receptors and this profoundly influences their signaling outputs (Fig. 16.2b). For example, E-cadherin interacts with and negatively regulates signaling of multiple receptor type tyrosine kinases (Qian et al. 2004; Takahashi and Suzuki 1996). Furthermore, some of the mutations of E-cadherin that are found in primary tumors result in decreased binding between E-cadherin and epithelial growth factor receptor (EGFR ) and this enhances EGFR activity (Bremm et al. 2008). Interestingly, E-cadherin-mediated adhesion impacts only some, but not all EGFR downstream signaling pathways. For example, clustering of E-cadherin at the cell surface with E-cadherin-coated beads specifically impacts EGFR-mediated activation of STAT5 (Perrais et al. 2007). E-cadherin can also regulate signaling of growth factor receptors indirectly through neuronal cell adhesion molecule (NCAM) (Lehembre et al. 2008). Loss of E-cadherin expression results in upregulation of NCAM, its translocation to the lipid rafts, where it activates non-receptor tyrosine kinase Fyn, leading to the phosphorylation and activation of the focal adhesion kinase and the assembly of integrin-mediated focal adhesions , cell spreading and EMT (Lehembre et al. 2008).
In contrast to the relationship between EGFR and E-cadherin, fibroblast growth factor receptor (FGFR ) interaction with N-cadherin potentiates FGFR signaling and causes increased activation of MAPK pathway and upregulation of MMP9, which promotes cellular invasion (Suyama et al. 2002). Similarly, interaction between N-cadherin and platelet-derived growth factor-receptor β (PDGFR β) promotes PDGF signaling-induced cell migration (Theisen et al. 2007).
Cadherin-mediated adhesion can also potentiate specific signaling branches downstream from the cell surface receptor-type tyrosine kinases . For example, VE-cadherin is necessary for proper vascular endothelial growth factor receptor-mediated activation of phosphoinositide 3 (PI3)-kinase (Carmeliet et al. 1999; Kang et al. 2007). Similarly, when Ewing sarcoma cells are grown in anchorage-independent conditions in soft agar, they upregulate E-cadherin, which increases ErbB4-phosphatidylinositol 3-kinase signaling and promotes cell survival (Kang et al. 2007). This PI3-kinase pathway-associated pro-survival function of AJs may play an important role during tumor cell dissemination and formation of metastatic lesions. Indeed, while primary epithelial tumors frequently show the decrease in expression of E-cadherin, the metastatic lesions in the same patients often display the reemergence of E-cadherin expression (Bukholm et al. 2000; Hung et al. 2006; Imai et al. 2004). During later stages of metastasis , cancer cells have to survive in foreign microenvironments, which are often lacking the proper extracellular matrix proteins, that otherwise facilitate formation of integrin-based adhesion structures and associated activation of PI3-kinase signaling. In these conditions, AJs may promote activation of PI3-kinase signaling necessary for cell survival and, thus, promote metastasis .
In addition to cadherins, catenins can also profoundly change the growth factor receptor signaling pathways. p120-catenin -mediated inhibition of RhoA -ROCK pathway is necessary for anchorage-independent growth of MDCK tumor cells overexpressing activated Src or Rac1 proteins (Dohn et al. 2009). Quite surprisingly, in lobular breast cancer developing upon ablation of E-cadherin and p53, p120-catenin promotes anoikis resistance by indirect upregulation of Rho-Rock signaling (Schackmann et al. 2011). Although it is confusing, p120-catenin is known to have different and some times opposite functions depending on whether the cells express E-cadherin (Soto et al. 2008). Similar to p120-catenin, α-catenin also impacts the signaling by the growth factor receptors. While the mechanisms are still not well understood, the ablation of α-catenin in skin keratinocytes results in increased insulin-like growth factor (IGFR)–Ras -MAPK signaling (Vasioukhin et al. 2001). IGF signaling can activate both Akt and MAPK kinase pathways, but only the MAPK pathway was affected in αE-catenin -/- keratinocytes .
6.3 Negative Regulation of Oncogenic β-Catenin Signaling
β-catenin is a potent proto-oncogene and upregulation of its levels in vivo results in the development of cancer in a variety of organs and tissues (Clevers 2006; Polakis 2000). Since AJs also utilize β-catenin, upregulation of the levels of epithelial cadherins may sequester β-catenin to the AJs and attenuate its transcriptional activity in the nucleus (Fig. 16.2c). This type of relationship has been demonstrated in variety of model systems. Overexpression of E-cadherin and α-catenin in Xenopus and Drosophila embryos and cancer cell lines results in attenuation of β-catenin-mediated signaling (Giannini et al. 2000; Gottardi et al. 2001; Heasman et al. 1994; Merdek et al. 2004; Onder et al. 2008; Orsulic et al. 1999; Sanson et al. 1996; Sehgal et al. 1997; Simcha et al. 1998). Similarly, inactivation of epithelial cadherins in many cancer cell lines with an activated β-catenin signaling pathway results in increased β-catenin signaling (Kuphal and Behrens 2006; Onder et al. 2008). β-catenin is also hyperactive in E-cadherin -/- embryonic stem cells (Orsulic et al. 1999). In contrast, inactivation of cadherins in other cell lines does not cause increase in β-catenin-mediated signaling (Kuphal and Behrens 2006; van de Wetering et al. 2001). Similarly, loss-of-function experiments involving epithelial cadherins and α-catenin in live organisms for the most part do not show impacts on β-catenin signaling. Tissue-specific inactivation of E-cadherin and α-catenin in keratinocytes , as well as the knockout of α-catenin in the developing brain do not cause increases in β-catenin signaling (Lien et al. 2006; Vasioukhin et al. 2001; Young et al. 2003). Moreover, genetic inactivation of epithelial cadherins in mouse models of epithelial cancer often promotes tumor progression , but rarely causes concomitant increase in β-catenin signaling. For example, loss of E-cadherin promotes pancreatic cancer progression, but it causes no changes in β-catenin signaling (Herzig et al. 2007). Similarly, deletion of E-cadherin in the mouse model of breast and skin cancer accelerates tumor development and promotes metastasis , but it does not impact β-catenin signaling (Derksen et al. 2006). Conditional deletion of αE-catenin in skin hair follicle stem cells results in the development of inflammatory lesions and squamous cell carcinoma, but β-catenin signaling is not activated in αE-catenin -/- cells (Silvis et al. 2011). In contrast, inactivation of cadherin function by overexpression of dominant-negative E-cadherin in a mouse model of Raf-driven lung cancer results in increased β-catenin signaling (Ceteci et al. 2007).
Overall, it appears that sequestration of β-catenin to the AJs can attenuate β-catenin nuclear signaling, but this is especially evident in cells where the β-catenin destruction machinery is inactivated and the excess of β-catenin is not efficiently cleared by degradation (Heuberger and Birchmeier 2010).
6.4 Positive Regulation of the Tumor-Suppressive Hippo Signaling
Initially discovered in Drosophila , the Hippo pathway in mammalian organisms regulates the size of the organs, and protects them from tumor development (Pan 2010; Zhao et al. 2011). The canonical Hippo pathway consists of a kinase cascade that culminates in phosphorylation of transcriptional co-activators Yap1 and Taz (WWTR1) (Fig. 16.2d). Activation of Hippo signaling results in phosphorylation and degradation of Yap and Taz. Decreased Hippo pathway activity results in nuclear translocation of Yap and Taz, interaction with TEADs, as well as several other transcription factors, and transcriptional regulation of genes involved in proliferation, differentiation and apoptotic cell death. Constitutive activation of Yap1 or Taz signaling in mammary epithelial cells results in cell transformation (Chan et al. 2008; Dong et al. 2007; Overholtzer et al. 2006). Moreover, tissue specific activation of Yap1 in liver and skin progenitors results in development of liver cancer and skin squamous cell carcinoma (Dong et al. 2007; Schlegelmilch et al. 2011). Interestingly, Hippo signaling is upregulated by increased cell density and it appears to be a critical pathway regulating contact inhibition of cell proliferation (Zhao et al. 2007). This is intriguing because contact inhibition of cell proliferation is regulated by cadherins and catenins (Takahashi and Suzuki 1996; Vasioukhin et al. 2001).
Several recent studies discovered a functional connection between the Hippo pathway and the AJ protein α-catenin (Schlegelmilch et al. 2011; Silvis et al. 2011). Mass Spectrometry analysis identified α-catenin as a prominent Yap1 interacting partner in keratinocytes and the loss of α-catenin expression resulted in constitutive nuclear localization and activation of Yap1 (Schlegelmilch et al. 2011). Moreover, constitutively nuclear Yap1 was necessary for tumor formation associated with loss of α-catenin in keratinocytes (Silvis et al. 2011). Interestingly, in keratinocytes α-catenin did not regulate Mst1/2 and Lats1/2, the canonical kinases of the Hippo pathway. Instead, it interacted with Yap1 and this interaction attenuated its nuclear translocation (Schlegelmilch et al. 2011; Silvis et al. 2011). In addition, this functional impact on Yap1 localization was specific for α-catenin, because the knockdowns of epithelial cadherins did not affect Yap1 localization (Schlegelmilch et al. 2011). This is consistant with the prominent phenotypic differences between α-catenin-null and E-/P-cadherin-null epidermises. While both show disruption of the AJs, only ablation of α-catenin results in epidermal hyperplasia (Tinkle et al. 2008a; Vasioukhin et al. 2001). In contrast to keratinocytes , disruption of AJs is sufficient for nuclear translocation of Yap1 in breast epithelial MCF10 A cells (Kim et al. 2011). In these cells, AJ activation of the canonical Hippo kinase cascade is responsible for the contact inhibition of cell proliferation. The mechanisms responsible for the connection between AJs and the Hippo kinases in MCF10 A cells are not well understood. It is quite intriguing that the tumor suppressor NF2 is implicated in regulation of the Hippo signaling (Zhang et al. 2010), and it is also a direct interactor of α-catenin (Gladden et al. 2010). Future research will help to determine whether NF2-α-catenin interaction is an important mechanistic link between the AJs and the Hippo signaling pathway.
7 Summary and Future Perspectives
AJs play a critical role in human epithelial tumors. Loss of epithelial cadherins and disruption of AJs is an early and causal event in lobular breast carcinoma and diffuse type gastric cancer. In other tumor types, disruption of the AJs usually happens during the transition from low-grade well-differentiated tumors to high-grade poorly differentiated invasive cancer. Loss of epithelial cadherin usually correlates with poor patient outcome, and mechanistic experiments in cell lines and model organisms demonstrated a causal role of AJs in tumor initiation and progression. While significant progress has been achieved in elucidation of the signaling events responsible for the tumor and metastasis suppressor function of cadherins and catenins, the available knowledge is still fragmented and quite rudimentary. This direction represents an exciting avenue for the future research.
References
Aaltomaa S, Lipponen P, Ala-Opas M, Eskelinen M, Kosma VM (1999) Alpha-catenin expression has prognostic value in local and locally advanced prostate cancer. Br J Cancer 80:477–482
Aaltomaa S, Karja V, Lipponen P, Isotalo T, Kankkunen JP, Talja M, Mokka R (2005) Reduced alpha- and beta-catenin expression predicts shortened survival in local prostate cancer. Anticancer Res 25:4707–4712
Abe K, Takeichi M (2008) EPLIN mediates linkage of the cadherin catenin complex to F-actin and stabilizes the circumferential actin belt. Proc Natl Acad Sci U S A 105:13–19
Aberle H, Bauer A, Stappert J, Kispert A, Kemler R (1997) Beta-catenin is a target for the ubiquitin-proteasome pathway. Embo J 16:3797–3804
Anastasiadis PZ, Reynolds AB (2001) Regulation of Rho GTPases by p120-catenin. Curr Opin Cell Biol 13:604–610
Angres B, Barth A, Nelson WJ (1996) Mechanism for transition from initial to stable cell-cell adhesion: kinetic analysis of E-cadherin-mediated adhesion using a quantitative adhesion assay. J Cell Biol 134:549–557
Asgeirsson KS, Jonasson JG, Tryggvadottir L, Olafsdottir K, Sigurgeirsdottir JR, Ingvarsson S, Ogmundsdottir HM (2000) Altered expression of E-cadherin in breast cancer. patterns, mechanisms and clinical significance. Eur J Cancer 36:1098–1106
Batlle E, Sancho E, Franci C Dominguez D, Monfar M, Baulida J, Garcia De Herreros A (2000) The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2:84–89
Becker KF, Atkinson MJ, Reich U Huang HH, Nekarda H, Siewert JR, Hofler H (1993) Exon skipping in the E-cadherin gene transcript in metastatic human gastric carcinomas. Hum Mol Genet 2:803–804
Becker KF, Atkinson MJ, Reich U, Becker I, Nekarda H, Siewert JR, Hofler H (1994) E-cadherin gene mutations provide clues to diffuse type gastric carcinomas. Cancer Res 54:3845–3852
Behrens J, Mareel MM, Van Roy FM, Birchmeier W (1989) Dissecting tumor cell invasion: epithelial cells acquire invasive properties after the loss of uvomorulin-mediated cell-cell adhesion. J Cell Biol 108:2435–2447
Behrens J, Vakaet L, Friis R, Winterhager E, Van Roy F, Mareel MM, Birchmeier W (1993) Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J Cell Biol 120:757–766
Berx G, Van Roy F (2001) The E-cadherin/catenin complex: an important gatekeeper in breast cancer tumorigenesis and malignant progression. Breast Cancer Res 3:289–293
Berx G, van Roy F (2009) Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol 1:a003129
Berx G, Cleton-Jansen AM, Nollet F, de Leeuw WJ, van de Vijver M, Cornelisse C, van Roy F (1995) E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. Embo J 14:6107–6115
Berx G, Cleton-Jansen AM, Strumane K, de Leeuw WJ, Nollet F,van Roy F, Cornelisse C(1996) E-cadherin is inactivated in a majority of invasive human lobular breast cancers by truncation mutations throughout its extracellular domain. Oncogene 13:1919–1925
Bissell MJ, Hines WC (2011) Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med 17:320–329
Bohm M, Totzeck B, Birchmeier W, Wieland I (1994) Differences of E-cadherin expression levels and patterns in primary and metastatic human lung cancer. Clin Exp Metastasis 12:55–62
Brembeck FH, Schwarz-Romond T, Bakkers J, Wilhelm S, Hammerschmidt M, Birchmeier W (2004) Essential role of BCL9–2 in the switch between beta-catenin’s adhesive and transcriptional functions. Genes Dev 18:2225–2230
Bremm A, Walch A, Fuchs M, Mages J, Duyster J, Keller G, Hermannstadter C, Becker KF, Rauser S, Langer R, von Weyhern CH, Hofler H, Luber B (2008) Enhanced activation of epidermal growth factor receptor caused by tumor-derived E-cadherin mutations. Cancer Res 68:707–714
Bukholm IK, Nesland JM, Borresen-Dale AL (2000) Re-expression of E-cadherin, alpha-catenin and beta-catenin, but not of gamma-catenin, in metastatic tissue from breast cancer patients (seecomments). J Pathol 190:15–19
Bullions LC, Notterman DA, Chung LS, Levine AJ (1997) Expression of wild-type alpha-catenin protein in cells with a mutant alpha-catenin gene restores both growth regulation and tumor suppressor activities. Mol Cell Biol 17:4501–4508
Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T (2008) A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 9:582–589
Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2:76–83
Canonici A, Steelant W, Rigot V, Khomitch-Baud A, Boutaghou-Cherid H, Bruyneel E, Van Roy F, Garrouste F, Pommier G, Andre F (2008) Insulin-like growth factor-I receptor, E-cadherin and alpha v integrin form a dynamic complex under the control of alpha-catenin. Int J Canc 122:572–582
Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, Mehra R, Laxman B, Cao X, Kleer CG, Varambally S, Chinnaiyan AM (2008) Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 27:7274–7284
Carico E, Atlante M, Bucci B, Nofroni I, Vecchione A (2001) E-cadherin and alpha-catenin expression during tumor progression of cervical carcinoma. Gynecol Oncol 80:156–161
Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oostuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert JM, Collen D, Dejana E (1999) Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 98:147–157
Castano J, Solanas G, Casagolda D, Raurell I, Villagrasa P, Bustelo XR, Garcia de Herreros A, Dunach M (2007) Specific phosphorylation of p120-catenin regulatory domain differently modulates its binding to RhoA. Mol Cell Biol 27:1745–1757
Cavallaro U, Schaffhauser B, Christofori G (2002) Cadherins and the tumour progression: is it all in a switch? Cancer Lett 176:123–128
Cavey M, Lecuit T (2009) Molecular bases of cell-cell junctions stability and dynamics. Cold Spring Harb Perspect Biol 1:a002998
Ceteci F, Ceteci S, Karreman C, Kramer BW, Asan E, Gotz R, Rapp UR (2007) Disruption of tumor cell adhesion promotes angiogenic switch and progression to micrometastasis in RAF-driven murine lung cancer. Cancer Cell 12:145–159
Chan EF, Gat U, McNiff JM, Fuchs E (1999) A common human skin tumour is caused by activating mutations in beta-catenin. Nat Genet 21:410–413
Chan SW, Lim CJ, Guo K, Ng CP, Lee I, Hunziker W, Zeng Q, Hong W (2008) A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res 68:2592–2598
Chaudhury A, Hussey GS, Ray PS, Jin G, Fox PL, Howe PH (2010) TGF-beta-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nat Cell Biol 12:286–293
Chen CL, Liu SS, Ip SM, Wong LC, Ng TY, Ngan HY (2003a) E-cadherin expression is silenced by DNA methylation in cervical cancer cell lines and tumours. Eur J Cancer 39:517–523
Chen WC, Obrink B (1991) Cell-cell contacts mediated by E-cadherin (uvomorulin) restrict invasive behavior of L-cells. J Cell Biol 114:319–327
Chen X, Kojima S, Borisy GG, Green KJ (2003b) p120 catenin associates with kinesin and facilitates the transport of cadherin-catenin complexes to intercellular junctions. J Cell Biol 163:547–557
Chen ZL, Zhao XH, Wang JW, Li BZ, Wang Z, Sun J, Tan FW, Ding DP, Xu XH, Zhou F, Tan XG, Hang J, Shi SS, Feng XL, He J (2011) microRNA-92a promotes lymph node metastasis of human esophageal squamous cell carcinoma via E-cadherin. J Biol Chem 286:10725–10734
Chung Y, Lam AK, Luk JM, Law S, Chan KW, Lee PY, Wong J (2007) Altered E-cadherin expression and p120 catenin localization in esophageal squamous cell carcinoma. Ann Surg Oncol 14:3260–3267
Cleton-Jansen AM, Callen DF, Seshadri R, Goldup S, McCallum B, Crawford J, Powell JA, Settasatian C, van Beerendonk H, Moerland EW, Smit VT, Harris WH, Millis R, Morgan NV, Barnes D, Mathew CG, Cornelisse CJ (2001) Loss of heterozygosity mapping at chromosome arm 16q in 712 breast tumors reveals factors that influence delineation of candidate regions. Cancer Res 61:1171–1177
Clevers H (2004) Wnt breakers in colon cancer. Cancer Cell 5:5–6
Clevers H (2006) Wnt/beta-catenin signaling in development and disease. Cell 127:469–480
Coluccia AM, Benati D, Dekhil H, De Filippo A, Lan C, Gambacorti-Passerini C (2006) SKI-606 decreases growth and motility of colorectal cancer cells by preventing pp60(c-Src)-dependent tyrosine phosphorylation of beta-catenin and its nuclear signaling. Cancer Res 66:2279–2286
Coluccia AM, Vacca A, Dunach M, Mologni L, Redaelli S, Bustos VH, Benati D, Pinna LA, Gambacorti-Passerini C (2007) Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia through its tyrosine phosphorylation. Embo J 26:1456–1466
Coman D (1944) Decreased mutual adhesiveness, a property of cells from squamous cell carcinomas. Cancer Res 4:625–629
Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F (2001) The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 7:1267–1278
Cowin P, Rowlands TM, Hatsell SJ (2005) Cadherins and catenins in breast cancer. Curr Opin Cell Biol 17:499–508
Damsky CH, Richa J, Solter D, Knudsen K, Buck CA (1983) Identification and purification of a cell surface glycoprotein mediating intercellular adhesion in embryonic and adult tissue. Cell 34:455–466
Davis MA, Ireton RC, Reynolds AB (2003) A core function for p120-catenin in cadherin turnover. J Cell Biol 163:525–534
de La Coste A, Romagnolo B Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C, Kahn A, Perret C (1998) Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A 95:8847–8851
Derksen PW, Liu X, Saridin F, Van Der Gulden H, Zevenhoven J, Evers B, van Beijnum JR, Griffioen AW, Vink J, Krimpenfort P, Peterse JL, Cardiff RD, Berns A, Jonkers J (2006) Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell 10:437–449
Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, Abbott RM, Hoog J, Dooling DJ, Koboldt DC, Schmidt H, Kalicki J, Zhang Q, Chen L, Lin L, Wendl MC, McMichael JF, Magrini VJ, Cook L, McGrath SD, Vickery TL, Appelbaum E, Deschryver K, Davies S, Guintoli T, Crowder R, Tao Y, Snider JE, Smith SM, Dukes AF, Sanderson GE, Pohl CS, Delehaunty KD, Fronick CC, Pape KA, Reed JS, Robinson JS, Hodges JS, Schierding W, Dees ND, Shen D, Locke DP, Wiechert ME, Eldred JM, Peck JB, Oberkfell BJ, Lolofie JT, Du F, Hawkins AE, O’Laughlin MD, Bernard KE, Cunningham M, Elliott G, Mason MD, Thompson DM Jr, Ivanovich JL, Goodfellow PJ, Perou CM, Weinstock GM, Aft R, Watson M, Ley TJ, Wilson RK, Mardis ER (2010) Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 464:999–1005
Dohn MR, Brown MV, Reynolds AB (2009) An essential role for p120-catenin in Src- and Rac1-mediated anchorage-independent cell growth. J Cell Biol 184:437–450
Dolled-Filhart M, McCabe A, Giltnane J, Cregger M, Camp RL, Rimm DL (2006) Quantitative in situ analysis of beta-catenin expression in breast cancer shows decreased expression is associated with poor outcome. Cancer Res 66:5487–5494
Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D (2007) Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130:1120–1133
Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI (2005) Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell 123:903–915
El-Bahrawy M, El-Masry N, Alison M, Poulsom R,Fallowfield M (2003) Expression of beta-catenin in basal cell carcinoma. Br J Dermatol Syph 148:964–970
Ellenbroek SI, Collard JG (2007) Rho GTPases: functions and association with cancer. Clin Exp Metastasis 24:657–672
Endo K, Ashida K, Miyake N, Terada T (2001) E-cadherin gene mutations in human intrahepatic cholangiocarcinoma. J Pathol 193:310–317
Ewing CM, Ru N, Morton RA, Robinson JC, Wheelock MJ, Johnson KR, Barrett JC, Isaacs WB (1995) Chromosome 5 suppresses tumorigenicity of PC3 prostate cancer cells: correlation with re-expression of alpha-catenin and restoration of E-cadherin function. Cancer Res 55:4813–4817
Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu Z (2007) Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem 282:11221–11229
Ferber EC, Kajita M, Wadlow A, Tobiansky L, Niessen C, Ariga H, Daniel J, Fujita Y (2008) A role for the cleaved cytoplasmic domain of e-cadherin in the nucleus. J Biol Chem 283:12691–12700
Fevr T, Robine S, Louvard D, Huelsken J (2007) Wnt/beta-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol Cell Biol 27:7551–7559
Frixen UH, Behrens J, Sachs M, Eberle G, Voss B, Warda A, Lochner D, Birchmeier W (1991) E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol 113:173–185
Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, Sommer T, Birchmeier W (2002) Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol 4:222–231
Fukumaru K, Yoshii N, Kanzaki T, Kanekura T (2007) Immunohistochemical comparison of beta-catenin expression by human normal epidermis and epidermal tumors. J Dermatol 34:746–753
Garcia-Rostan G, Tallini G, Herrero A, D’Aquila TG, Carcangiu ML, Rimm DL (1999) Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res 59:1811–1815
Garinis GA, Menounos PG, Spanakis NE, Papadopoulos K, Karavitis G, Parassi I, Christeli E, Patrinos GP, Manolis EN, Peros G (2002) Hypermethylation-associated transcriptional silencing of E-cadherin in primary sporadic colorectal carcinomas. J Pathol 198:442–449
Gat U, DasGupta R, Degenstein L, Fuchs E (1998) De Novo hair follicle morphogenesis and hair tumors in mice expressing a truncated beta-catenin in skin. Cell 95:605–614
Giannini AL, Vivanco M, Kypta RM (2000) alpha-catenin inhibits beta-catenin signaling by preventing formation of a beta-catenin*T-cell factor*DNA complex. J Biol Chem 275:21883–21888
Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, Kranenburg TA, Hogg T, Poppleton H, Martin J, Finkelstein D, Pounds S, Weiss A, Patay Z, Scoggins M, Ogg R, Pei Y, Yang ZJ, Brun S, Lee Y, Zindy F, Lindsey JC, Taketo MM, Boop FA, Sanford RA, Gajjar A, Clifford SC, Roussel MF, McKinnon PJ, Gutmann DH, Ellison DW, Wechsler-Reya R, Gilbertson RJ (2010) Subtypes of medulloblastoma have distinct developmental origins. Nature 468:1095–1099
Gillett CE, Miles DW, Ryder K, Skilton D, Liebman RD, Springall RJ, Barnes DM, Hanby AM (2001) Retention of the expression of E-cadherin and catenins is associated with shorter survival in grade III ductal carcinoma of the breast. J Pathol Bacteriol 193:433–441
Gimond C, Van Der Flier A, van Delft S, Brakebusch C, Kuikman I, Collard JG, Fassler R, Sonnenberg A (1999) Induction of cell scattering by expression of beta1 integrins in beta1-deficient epithelial cells requires activation of members of the rho family of GTPases and downregulation of cadherin and catenin function. J Cell Biol 147:1325–1340
Giroldi LA, Bringuier PP, de Weijert M, Jansen C, van Bokhoven A, Schalken JA (1997) Role of E boxes in the repression of E-cadherin expression. Biochem Biophys Res Commun 241:453–458
Gladden AB, Hebert AM, Schneeberger EE, McClatchey AI (2010) The NF2 tumor suppressor, Merlin, regulates epidermal development through the establishment of a junctional polarity complex. Dev Cell 19:727–739
Gottardi CJ, Wong E, Gumbiner BM (2001) E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J Cell Biol 153:1049–1060
Grady WM, Willis J, Guilford PJ, Dunbier AK, Toro TT, Lynch H, Wiesner G, Ferguson K, Eng C, Park JG, Kim SJ, Markowitz S (2000) Methylation of the CDH1 promoter as the second genetic hit in hereditary diffuse gastric cancer. Nat Genet 26:16–17
Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, Jarrard DF, Isaacs WB, Pitha PM, Davidson NE, Baylin SB (1995) E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 55:5195–5199
Graff JR, Greenberg VE, Herman JG, Westra WH, Boghaert ER, Ain KB, Saji M, Zeiger MA, Zimmer SG, Baylin SB (1998) Distinct patterns of E-cadherin CpG island methylation in papillary, follicular, Hurthle’s cell, and poorly differentiated human thyroid carcinoma. Cancer Res 58:2063–2066
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10:593–601
Grooteclaes ML, Frisch SM (2000) Evidence for a function of CtBP in epithelial gene regulation and anoikis. Oncogene 19:3823–3828
Gross JC, Schreiner A, Engels K, Starzinski-Powitz A (2009) E-cadherin surface levels in epithelial growth factor-stimulated cells depend on adherens junction protein shrew-1. Mol Biol Cell 20:3598–3607
Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, Taite H, Scoular R, Miller A, Reeve AE (1998) E-cadherin germline mutations in familial gastric cancer. Nature 392:402–405
Gujral TS, van Veelen W, Richardson DS, Myers SM, Meens JA, Acton DS, Dunach M, Elliott BE, Hoppener JW, Mulligan LM (2008) A novel RET kinase-beta-catenin signaling pathway contributes to tumorigenesis in thyroid carcinoma. Cancer Res 68:1338–1346
Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R (1995) Lack of beta-catenin affects mouse development at gastrulation. Development 121:3529–3537
Hamaguchi M, Matsuyoshi N, Ohnishi Y, Gotoh B, Takeichi M, Nagai Y (1993) p60v-src causes tyrosine phosphorylation and inactivation of the N-cadherin-catenin cell adhesion system. Embo J 12:307–314
Heasman J, Crawford A, Goldstone K, Garner-Hamrick P, Gumbiner B, McCrea P, Kintner C, Noro CY, Wylie C (1994) Overexpression of cadherins and underexpression of beta-catenin inhibit dorsal mesoderm induction in early Xenopus embryos. Cell 79:791–803
Heimann R, Lan F, McBride R, Hellman S (2000) Separating favorable from unfavorable prognostic markers in breast cancer: the role of E-cadherin. Cancer Res 60:298–304
Herranz N, Pasini D, Diaz VM, Franci C, Gutierrez A, Dave N, Escriva M, Hernandez-Munoz I, Di Croce L, Helin K, Garcia de Herreros A, Peiro S (2008) Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol Cell Biol 28:4772–4781
Herzig M, Savarese F, Novatchkova M, Semb H, Christofori G (2007) Tumor progression induced by the loss of E-cadherin independent of beta-catenin/Tcf-mediated Wnt signaling. Oncogene 26:2290–2298
Heuberger J, Birchmeier W (2010) Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb Perspect Biol 2:a002915
Hino S, Tanji C, Nakayama KI, Kikuchi A (2005) Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase stabilizes beta-catenin through inhibition of its ubiquitination. Mol Cell Biol 25:9063–9072
Hirano S, Kimoto N, Shimoyama Y, Hirohashi S, Takeichi M (1992) Identification of a neural alpha-catenin as a key regulator of cadherin function and multicellular organization. Cell 70:293–301
Hiscox S, Jiang WG (1999) Association of the HGF/SF receptor, c-met, with the cell-surface adhesion molecule, E-cadherin, and catenins in human tumor cells. Biochem Biophys Res Commun 261:406–411
Huang Q, Gumireddy K, Schrier M, le Sage C, Nagel R, Nair S, Egan DA, Li A, Huang G, Klein-Szanto AJ, Gimotty PA, Katsaros D, Coukos G, Zhang L, Pure E, Agami R (2008) The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat Cell Biol 10:202–210
Huelsken J, Vogel R, Brinkmann V, Erdmann B, Birchmeier C, Birchmeier W (2000) Requirement for beta-catenin in anterior-posterior axis formation in mice. J Cell Biol 148:567–578
Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW (2007) Epithelial--mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J Cell Physiol 213:374–383
Hulit J, Suyama K, Chung S, Keren R, Agiostratidou G, Shan W, Dong X, Williams TM, Lisanti MP, Knudsen K, Hazan RB (2007) N-cadherin signaling potentiates mammary tumor metastasis via enhanced extracellular signal-regulated kinase activation. Cancer Res 67:3106–3116
Hung KF, Chang CS, Liu CJ, Lui MT, Cheng CY, Kao SY (2006) Differential expression of E-cadherin in metastatic lesions comparing to primary oral squamous cell carcinoma. J Oral Pathol Med 35:589–594
Hussey GS, Chaudhury A, Dawson AE, Lindner DJ, Knudsen CR, Wilce MC, Merrick WC, Howe PH (2011) Identification of an mRNP complex regulating tumorigenesis at the translational elongation step. Mol Cell 41:419–431
Hwang-Verslues WW, Chang PH, Wei PC, Yang CY, Huang CK, Kuo WH, Shew JY, Chang KJ, Lee EY, Lee WH (2011) miR-495 is upregulated by E12/E47 in breast cancer stem cells, and promotes oncogenesis and hypoxia resistance via downregulation of E-cadherin and REDD1. Oncogene 30:2463–2474
Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K (2010) Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell 39:761–772
Imai T, Horiuchi A, Shiozawa T, Osada R, Kikuchi N, Ohira S, Oka K, Konishi I (2004) Elevated expression of E-cadherin and alpha-, beta-, and gamma-catenins in metastatic lesions compared with primary epithelial ovarian carcinomas. Hum Pathol 35:1469–1476
Ishiyama N, Lee SH, Liu S, Li GY, Smith MJ, Reichardt LF, Ikura M (2010) Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell 141:117–128
Ishizaki Y, Omori Y, Momiyama M, Nishikawa Y, Tokairin T, Manabe M, Enomoto K (2004) Reduced expression and aberrant localization of p120catenin in human squamous cell carcinoma of the skin. J Dermatol Sci. 34:99–108
Jaggi M, Johansson SL, Baker JJ, Smith LM, Galich A, Balaji KC (2005) Aberrant expression of E-cadherin and beta-catenin in human prostate cancer. Urol Oncol 23:402–406
Janssens B, Goossens S, Staes K, Gilbert B, van Hengel J, Colpaert C, Bruyneel E, Mareel M, van Roy F (2001) alphaT-catenin: a novel tissue-specific beta-catenin-binding protein mediating strong cell-cell adhesion. J Cell Sci 114:3177–3188
Jeschke U, Mylonas I, Kuhn C, Shabani N, Kunert-Keil C, Schindlbeck C, Gerber B, Friese K (2007) Expression of E-cadherin in human ductal breast cancer carcinoma in situ, invasive carcinomas, their lymph node metastases, their distant metastases, carcinomas with recurrence and in recurrence. Anticancer Res 27:1969–1974
Kadowaki T, Shiozaki H, Inoue M, Tamura S, Oka H, Doki Y, Iihara K, Matsui S, Iwazawa T, Nagafuchi A, et al (1994) E-cadherin and alpha-catenin expression in human esophageal cancer. Cancer Res 54:291–296
Kang HG, Jenabi JM, Zhang J, Keshelava N, Shimada H, May WA, Ng T, Reynolds CP, Triche TJ, Sorensen PH (2007) E-cadherin cell-cell adhesion in ewing tumor cells mediates suppression of anoikis through activation of the ErbB4 tyrosine kinase. Cancer Res 67:3094–3105
Kim DS, Kim MJ, Lee JY, Kim YZ, Kim EJ, Park JY (2007) Aberrant methylation of E-cadherin and H-cadherin genes in nonsmall cell lung cancer and its relation to clinicopathologic features. Cancer 110:2785–2792
Kim NG, Koh E, Chen X, Gumbiner BM (2011) E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc Natl Acad Sci U S A 108:11930–11935
Kimura K, Endo Y, Yonemura Y, Heizmann CW, Schafer BW, Watanabe Y, Sasaki T (2000) Clinical significance of S100A4 and E-cadherin-related adhesion molecules in non-small cell lung cancer. Int J Oncol 16:1125–1131
Klaus A, Birchmeier W (2008) Wnt signalling and its impact on development and cancer. Nature reviews. Cancer 8:387–398
Knudsen KA, Sauer C, Johnson KR, Wheelock MJ (2005) Effect of N-cadherin misexpression by the mammary epithelium in mice. J Cell Biochem 95:1093–1107
Kobielak A, Fuchs E (2006) Links between alpha-catenin, NF-kappaB, and squamous cell carcinoma in skin. Proc Natl Acad Sci U S A 103:2322–2327
Kobielak A, Pasolli HA, Fuchs E (2004) Mammalian formin-1 participates in adherens junctions and polymerization of linear actin cables. Nat Cell Biol 6:21–30
Koesters R, Ridder R, Kopp-Schneider A, Betts D, Adams V, Niggli F, Briner J, von Knebel Doeberitz M (1999) Mutational activation of the beta-catenin proto-oncogene is a common event in the development of Wilms’ tumors. Cancer Res 59:3880–3882
Koizume S, Tachibana K, Sekiya T, Hirohashi S, Shiraishi M (2002) Heterogeneity in the modification and involvement of chromatin components of the CpG island of the silenced human CDH1 gene in cancer cells. Nucleic Acids Res 30:4770–4780
Kokura K, Sun L, Bedford MT, Fang J (2010) Methyl-H3K9-binding protein MPP8 mediates E-cadherin gene silencing and promotes tumour cell motility and invasion. EMBO J 29:3673–3687
Korpal M, Lee ES, Hu G, Kang Y (2008) The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 283:14910–14914
Kovacs EM, Goodwin M, Ali RG, Paterson AD, Yap AS (2002) Cadherin-directed actin assembly: E-cadherin physically associates with the Arp2/3 complex to direct actin assembly in nascent adhesive contacts. Curr Biol 12:379–382
Kowalczyk AP, Reynolds AB (2004) Protecting your tail: regulation of cadherin degradation by p120-catenin. Curr Opin Cell Biol 16:522–527
Kuphal F, Behrens J (2006) E-cadherin modulates Wnt-dependent transcription in colorectal cancer cells but does not alter Wnt-independent gene expression in fibroblasts. Exp Cell Res 312:457–467
Larue L, Ohsugi M, Hirchenhain J, Kemler R (1994) E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc Natl Acad Sci U S A 91:8263–8267
Lee M, Vasioukhin V (2008) Cell polarity and cancer--cell and tissue polarity as a non-canonical tumor suppressor. J Cell Sci 121:1141–1150
Lehembre F, Yilmaz M, Wicki A, Schomber T, Strittmatter K, Ziegler D, Kren A, Went P, Derksen PW, Berns A, Jonkers J, Christofori G (2008) NCAM-induced focal adhesion assembly: a functional switch upon loss of E-cadherin. EMBO J 27:2603–2615
Libusova L, Stemmler MP, Hierholzer A, Schwarz H, Kemler R (2010) N-cadherin can structurally substitute for E-cadherin during intestinal development but leads to polyp formation. Development 137:2297–2305
Lien WH, Klezovitch O, Fernandez TE, Delrow J, Vasioukhin V (2006) alphaE-catenin controls cerebral cortical size by regulating the hedgehog signaling pathway. Science 311:1609–1612
Lilien J, Balsamo J (2005) The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr Opin Cell Biol 17:459–465
Lin SY, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y, Pestell RG, Hung MC (2000) Beta-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci U S A 97:4262–4266
Lo Muzio L, Pannone G, Mignogna MD, Staibano S, Mariggio MA, Rubini C, Procaccini M, Dolci M, Bufo P, De Rosa G, Piattelli A (2004) P-cadherin expression predicts clinical outcome in oral squamous cell carcinomas. Histol Histopathol 19:1089–1099
Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ (1997) Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol 139:1861–1872
Lozano E, Frasa MA, Smolarczyk K, Knaus UG, Braga VM (2008) PAK is required for the disruption of E-cadherin adhesion by the small GTPase Rac. J Cell Sci 121:933–938
Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S, Westermann F, Speleman F, Vandesompele J, Weinberg RA (2010) miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol 12:247–256
Machon O, Van Den Bout CJ, Backman M, Kemler R, Krauss S (2003) Role of beta-catenin in the developing cortical and hippocampal neuroepithelium. Neuroscience 122:129–143
Malanchi I, Peinado H, Kassen D, Hussenet T, Metzger D, Chambon P, Huber M, Hohl D, Cano A, Birchmeier W, Huelsken J (2008) Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature 452:650–653
Marambaud P, Shioi J Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK (2002) A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. Embo J 21:1948–1956
Marambaud P, Wen PH Dutt A, Shioi J, Takashima A, Siman R, Robakis NK (2003) A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell 114:635–645
Maretzky T, Reiss K, Ludwig A, Buchholz J, Scholz F, Proksch E, de Strooper B, Hartmann D, Saftig P (2005) ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc Natl Acad Sci U S A 102:9182–9187
Matsumura T, Makino R, Mitamura K (2001) Frequent down-regulation of E-cadherin by genetic and epigenetic changes in the malignant progression of hepatocellular carcinomas. Clin Cancer Res 7:594–599
Mayer B, Johnson JP, Leitl F, Jauch KW, Heiss MM, Schildberg FW, Birchmeier W, Funke I (1993) E-cadherin expression in primary and metastatic gastric cancer: down-regulation correlates with cellular dedifferentiation and glandular disintegration. Cancer Res 53:1690–1695
Meng W, Takeichi M (2009) Adherens junction: molecular architecture and regulation. Cold Spring Harb Perspect Biol 1:a002899
Merdek KD, Nguyen NT, Toksoz D (2004) Distinct activities of the alpha-catenin family, alpha-catulin and alpha-catenin, on beta-catenin-mediated signaling. Mol Cell Biol 24:2410–2422
Micalizzi DS, Farabaugh SM, Ford HL (2010) Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia 15:117–134
Milicic A, Harrison LA, Goodlad RA, Hardy RG, Nicholson AM, Presz M, Sieber O, Santander S, Pringle JH, Mandir N, East P, Obszynska J, Sanders S, Piazuelo E, Shaw J, Harrison R, Tomlinson IP, McDonald SA, Wright NA, Jankowski JA (2008) Ectopic expression of P-cadherin correlates with promoter hypomethylation early in colorectal carcinogenesis and enhanced intestinal crypt fission in vivo. Cancer Res 68:7760–7768
Miyoshi Y, Iwao K, Nagasawa Y, Aihara T, Sasaki Y, Imaoka S, Murata M, Shimano T, Nakamura Y (1998) Activation of the beta-catenin gene in primary hepatocellular carcinomas by somatic alterations involving exon 3. Cancer Res 58:2524–2527
Munoz-Guerra MF, Marazuela EG, Fernandez-Contreras ME, Gamallo C (2005) P-cadherin expression reduced in squamous cell carcinoma of the oral cavity: an indicatior of poor prognosis. Cancer 103:960–969
Nagae Y, Kameyama K, Yokoyama M, Naito Z, Yamada N, Maeda S, Asano G, Sugisaki Y, Tanaka S (2002) Expression of E-cadherin catenin and C-erbB-2 gene products in invasive ductal-type breast carcinomas. Journal of Nippon Medical School = Nihon Ika Daigaku zasshi 69:165–171
Nakanishi Y, Ochiai A, Akimoto S, Kato H, Watanabe H, Tachimori Y, Yamamoto S, Hirohashi S (1997) Expression of E-cadherin, alpha-catenin, beta-catenin and plakoglobin in esophageal carcinomas and its prognostic significance: immunohistochemical analysis of 96 lesions. Oncology 54:158–165
Nakopoulou L, Gakiopoulou-Givalou H, Karayiannakis AJ, Giannopoulou I, Keramopoulos A, Davaris P, Pignatelli M (2002) Abnormal alpha-catenin expression in invasive breast cancer correlates with poor patient survival. Histopathology 40:536–546
Nieman MT, Prudoff RS, Johnson KR, Wheelock MJ (1999) N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J Cell Biol 147:631–644
Nojima D, Nakajima K, Li LC, Franks J, Ribeiro-Filho L, Ishii N, Dahiya R (2001) CpG methylation of promoter region inactivates E-cadherin gene in renal cell carcinoma. Mol Carcinog 32:19–27
Noren NK, Liu BP, Burridge K, Kreft B (2000) p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J Cell Biol 150:567–580
Ochiai A, Akimoto S, Kanai Y, Shibata T, Oyama T, Hirohashi S (1994) c-erbB-2 gene product associates with catenins in human cancer cells. Biochem Biophys Res Commun 205:73–78
Ogata S, Morokuma J, Hayata T, Kolle G, Niehrs C, Ueno N, Cho KW (2007) TGF-beta signaling-mediated morphogenesis: modulation of cell adhesion via cadherin endocytosis. Genes Dev 21:1817–1831
Ogita H, Rikitake Y, Miyoshi J, Takai Y (2010) Cell adhesion molecules nectins and associating proteins: Implications for physiology and pathology. Proc Jpn Acad Ser B Phys Biol Sci 86:621–629
Oka H, Shiozaki H, Kobayashi K, Inoue M, Tahara H, Kobayashi T, Takatsuka Y, Matsuyoshi N, Hirano S, Takeichi M et al (1993) Expression of E-cadherin cell adhesion molecules in human breast cancer tissues and its relationship to metastasis. Cancer Res 53:1696–1701
Olson P, Lu J, Zhang H, Shai A, Chun MG, Wang Y, Libutti SK, Nakakura EK, Golub TR, Hanahan D (2009) MicroRNA dynamics in the stages of tumorigenesis correlate with hallmark capabilities of cancer. Genes Dev. 23:2152–2165
Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA (2008) Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res 68:3645–3654
Orsulic S, Huber O, Aberle H, Arnold S, Kemler R (1999) E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J Cell Sci 112(Pt 8):1237–1245
Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng CX, Brugge JS, Haber DA (2006) Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A 103:12405–12410
Palacios F, Tushir JS, Fujita Y, D’Souza-Schorey C (2005) Lysosomal targeting of E-cadherin: a unique mechanism for the down-regulation of cell-cell adhesion during epithelial to mesenchymal transitions. Mol Cell Biol 25:389–402
Pan D (2010) The hippo signaling pathway in development and cancer. Dev Cell 19:491–505
Paredes J, Correia AL, Ribeiro AS, Albergaria A, Milanezi F, Schmitt FC (2007) P-cadherin expression in breast cancer: a review. Breast Cancer Res 9:214
Park D, Karesen R, Axcrona U, Noren T, Sauer T (2007) Expression pattern of adhesion molecules (E-cadherin, alpha-, beta-, gamma-catenin and claudin-7), their influence on survival in primary breast carcinoma, and their corresponding axillary lymph node metastasis. Apmis 115:52–65
Park SM, Gaur AB, Lengyel E, Peter ME (2008) The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 22:894–907
Park WS, Oh RR, Park JY, Lee SH, Shin MS, Kim YS, Kim SY, Lee HK, Kim PJ, Oh ST, Yoo NJ, Lee JY (1999) Frequent somatic mutations of the beta-catenin gene in intestinal-type gastric cancer. Cancer Res 59:4257–4260
Pedersen KB, Nesland JM, Fodstad O, Maelandsmo GM (2002) Expression of S100A4, E-cadherin, alpha- and beta-catenin in breast cancer biopsies. Br J Cancer 87:1281–1286
Peinado H, Ballestar E, Esteller M, Cano A (2004) Snail mediates E-cadherin repression by the recruitment of the Sin3 A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol 24:306–319
Peinado H, Olmeda D, Cano A (2007) Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev 7:415–428
Perez-Moreno M, Song W, Pasolli HA, Williams SE, Fuchs E (2008) Loss of p120 catenin and links to mitotic alterations, inflammation, and skin cancer. Proc Natl Acad Sci U S A 105:15399–15404
Perez-Moreno MA, Locascio A, Rodrigo I, Dhondt G, Portillo F, Nieto MA, Cano A (2001) A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem 276:27424–27431
Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G (1998) A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 392:190–193
Perrais M, Chen X, Perez-Moreno M, Gumbiner BM (2007) E-Cadherin Homophilic Ligation Inhibits Cell Growth and Epidermal Growth Factor Receptor Signaling Independently of Other Cell Interactions. Mol Biol Cell 18:2013–2025
Pharoah PD, Guilford P, Caldas C (2001) Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 121:1348–1353
Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R (2008) MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci U S A 105:1608–1613
Polakis P (2000) Wnt signaling and cancer. Genes Dev 14:1837–1851
Prunier C, Howe PH (2005) Disabled-2 (Dab2) is required for transforming growth factor beta-induced epithelial to mesenchymal transition (EMT). J Biol Chem 280:17540–17548
Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR (2004) E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. Embo J 23:1739–1748
Qiao Y, Jiang X, Lee ST, Karuturi RK, Hooi SC, Yu Q (2011) FOXQ1 regulates epithelial-mesenchymal transition in human cancers. Cancer Res 71:3076–3086
Rakha EA, Abd El Rehim D, Pinder SE, Lewis SA, Ellis IO (2005) E-cadherin expression in invasive non-lobular carcinoma of the breast and its prognostic significance. Histopathology 46:685–693
Reshetnikova G, Troyanovsky S, Rimm DL (2007) Definition of a direct extracellular interaction between Met and E-cadherin. Cell Biol Int 31:366–373
Reynolds AB (2007) p120-catenin: Past and present. Biochim Biophys Acta 1773:2–7
Reynolds AB, Daniel J, McCrea PD, Wheelock MJ, Wu J, Zhang Z (1994) Identification of a new catenin: the tyrosine kinase substrate p120cas associates with E-cadherin complexes. Mol Cell Biol 14:8333–8342
Ribeiro-Filho LA, Franks J, Sasaki M, Shiina H, Li LC, Nojima D, Arap S, Carroll P, Enokida H, Nakagawa M, Yonezawa S, Dahiya R (2002) CpG hypermethylation of promoter region and inactivation of E-cadherin gene in human bladder cancer. Mol Carcinog 34:187–198
Richmond PJ, Karayiannakis AJ, Nagafuchi A, Kaisary AV, Pignatelli M (1997) Aberrant E-cadherin and alpha-catenin expression in prostate cancer: correlation with patient survival. Cancer Res 57:3189–3193
Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS (1995) Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci U S A 92:8813–8817
Risinger JI, Berchuck A, Kohler MF, Boyd J (1994) Mutations of the E-cadherin gene in human gynecologic cancers. Nat Genet 7:98–102
Roura S, Miravet S, Piedra J, Garcia de Herreros A, Dunach M (1999) Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J Biol Chem 274:36734–36740
Saini S, Yamamura S, Majid S, Shahryari V, Hirata H, Tanaka Y, Dahiya R (2011) MicroRNA-708 induces apoptosis and suppresses tumorigenicity in renal cancer cells. Cancer Res
Saldanha G, Ghura V, Potter L, Fletcher A (2004) Nuclear beta-catenin in basal cell carcinoma correlates with increased proliferation. Br J Dermatol 151:157–164
Sanchez-Tillo E, Lazaro A, Torrent R, Cuatrecasas M, Vaquero EC, Castells A, Engel P, Postigo A (2010) ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 29:3490–3500
Sanson B, White P, Vincent JP (1996) Uncoupling cadherin-based adhesion from wingless signalling in Drosophila. Nature 383:627–630
Schackmann RC, van Amersfoort M, Haarhuis JH, Vlug EJ, Halim VA, Roodhart JM, Vermaat JS, Voest EE, Van Der Groep P, van Diest PJ, Jonkers J, Derksen PW (2011) Cytosolic p120-catenin regulates growth of metastatic lobular carcinoma through Rock1-mediated anoikis resistance. J Clin Invest 121:3176–3188
Schipper JH, Frixen UH, Behrens J, Unger A, Jahnke K, Birchmeier W (1991) E-cadherin expression in squamous cell carcinomas of head and neck: inverse correlation with tumor dedifferentiation and lymph node metastasis. Cancer Res 51:6328–6337
Schlegelmilch K, Mohseni M, Kirak O, Pruszak J, Rodriguez JR, Zhou D, Kreger BT, Vasioukhin V, Avruch J, Brummelkamp TR, Camargo FD (2011) Yap1 Acts Downstream of alpha-Catenin to Control Epidermal Proliferation. Cell 144:782–795
Schuhmacher C, Becker I, Oswald S, Atkinson MJ, Nekarda H, Becker KF, Mueller J, Siewert JR, Hofler H (1999) Loss of immunohistochemical E-cadherin expression in colon cancer is not due to structural gene alterations. Virchows Arch 434:489–495
Sehgal RN, Gumbiner BM, Reichardt LF (1997) Antagonism of cell adhesion by an alpha-catenin mutant, and of the Wnt-signaling pathway by alpha-catenin in Xenopus embryos. J Cell Biol 139:1033–1046
Setoyama T, Natsugoe S, Okumura H, Matsumoto M, Uchikado Y, Yokomakura N, Ishigami S, Aikou T (2007) alpha-catenin is a significant prognostic factor than E-cadherin in esophageal squamous cell carcinoma. J Surg Oncol 95:148–155
Shen Y, Hirsch DS, Sasiela CA, Wu WJ (2008) Cdc42 regulates E-cadherin ubiquitination and degradation through an epidermal growth factor receptor to Src-mediated pathway. J Biol Chem 283:5127–5137
Shi Y, Sawada J, Sui G, Affarel B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y (2003) Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422:735–738
Shook D, Keller R (2003) Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech Dev 120:1351–1383
Shoval I, Ludwig A, Kalcheim C (2007) Antagonistic roles of full-length N-cadherin and its soluble BMP cleavage product in neural crest delamination. Development 134:491–501
Shun CT, Wu MS, Lin JT, Wang HP, Houng RL, Lee WJ, Wang TH, Chuang SM (1998) An immunohistochemical study of E-cadherin expression with correlations to clinicopathological features in gastric cancer. Hepatogastroenterology 45:944–949
Si HX, Tsao SW, Lam KY, Srivastava G, Liu Y, Wong YC, Shen ZY, Cheung AL (2001) E-cadherin expression is commonly downregulated by CpG island hypermethylation in esophageal carcinoma cells. Cancer Lett 173:71–78
Silvis MR, Kreger BT, Lien WH, Klezovitch O, Rudakova GM, Camargo FD, Lantz DM, Seykora JT, Vasioukhin V (2011) {alpha}-Catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Sci Signal 4:ra33
Simcha I, Shtutman M, Salomon D, Zhurinsky J, Sadot E, Geiger B, Ben-Ze’ev A (1998) Differential nuclear translocation and transactivation potential of beta-catenin and plakoglobin. J Cell Biol 141:1433–1448
Slorach EM, Chou J, Werb Z (2011) Zeppo1 is a novel metastasis promoter that represses E-cadherin expression and regulates p120-catenin isoform expression and localization. Genes Dev 25:471–484
Smalley-Freed WG, Efimov A, Short SP, Jia P, Zhao Z, Washington MK, Robine S, Coffey RJ, Reynolds AB (2011) Adenoma formation following limited ablation of p120-catenin in the mouse intestine. PLoS ONE 6(5):e19880. doi:10.1371/journal.pone.0019880
Soares P, Berx G, van Roy F, Sobrinho-Simoes M (1997) E-cadherin gene alterations are rare events in thyroid tumors. International journal of cancer. Int J Cancer 70:32–38
Solanas G, Cortina C, Sevillano M, Batlle E (2011) Cleavage of E-cadherin by ADAM10 mediates epithelial cell sorting downstream of EphB signalling. Nat Cell Biol 13:1100–1107
Song Y, Washington MK, Crawford HC (2010) Loss of FOXA1/2 is essential for the epithelial-to-mesenchymal transition in pancreatic cancer. Cancer Res 70:2115–2125
Soto E, Yanagisawa M, Marlow LA, Copland JA, Perez EA, Anastasiadis PZ (2008) p120 catenin induces opposing effects on tumor cell growth depending on E-cadherin expression. J Cell Biol 183:737–749
Stairs DB, Bayne LJ, Rhoades B, Vega ME, Waldron TJ, Kalabis J, Klein-Szanto A, Lee JS, Katz JP, Diehl JA, Reynolds AB, Vonderheide RH, Rustgi AK (2011) Deletion of p120-catenin results in a tumor microenvironment with inflammation and cancer that establishes it as a tumor suppressor gene. Cancer Cell 19:470–483
Stinson S, Lackner MR, Adai AT, Yu N, Kim HJ, O’Brien C, Spoerke J, Jhunjhunwala S, Boyd Z, Januario T, Newman RJ, Yue P, Bourgon R, Modrusan Z, Stern HM, Warming S, de Sauvage FJ, Amler L, Yeh RF, Dornan D (2011) miR-221/222 Targeting of Trichorhinophalangeal 1 (TRPS1) promotes epithelial-to-mesenchymal transition in breast cancer. Sci Signal 4:pt5
Suyama K, Shapiro I, Guttman M, Hazan RB (2002) A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell 2:301–314
Taddei I, Piazzini M, Bartoletti R, Dal Canto M, Sardi I (2000) Molecular alterations of E-cadherin gene: possible role in human bladder carcinogenesis. Int J Mol Med 6:201–208
Takahashi K, Suzuki K (1996) Density-dependent inhibition of growth involves prevention of EGF receptor activation by E-cadherin-mediated cell-cell adhesion. Exp Cell Res 226:214–222
Tamura G, Yin J, Wang S, Fleisher AS, Zou T, Abraham JM, Kong D, Smolinski KN, Wilson KT, James SP, Silverberg SG, Nishizuka S, Terashima M, Motoyama T, Meltzer SJ (2000) E-Cadherin gene promoter hypermethylation in primary human gastric carcinomas. J Natl Cancer Inst 92:569–573
Theisen CS, Wahl JK, 3rd, Johnson KR, Wheelock MJ (2007) NHERF links the N-cadherin/catenin complex to the platelet-derived growth factor receptor to modulate the actin cytoskeleton and regulate cell motility. Mol Biol Cell 18:1220–1232
Tinkle CL, Pasoli HA, Stokes N, Fuchs E (2008a) New insights into cadherin function in epidermal sheet formation and maintenance of tissue integrity. Proc Natl Acad Sci U S A 105(40):15225-15226, doi:10.1073/pnas.0808458105
Tinkle CL, Pasolli HA, Stokes N, Fuchs E (2008b) New insights into cadherin function in epidermal sheet formation and maintenance of tissue integrity. Proc Natl Acad Sci U S A 105:15405–15410
Torres M, Stoykova A, Huber O, Chowdhury K, Bonaldo P, Mansouri A, Butz S, Kemler R, Gruss P (1997) An alpha-E-catenin gene trap mutation defines its function in preimplantation development. Proc Natl Acad Sci U S A 94:901–906
Toyoshima M, Tanaka N, Aoki J, Tanaka Y, Murata K, Kyuuma M, Kobayashi H, Ishii N, Yaegashi N, Sugamura K (2007) Inhibition of tumor growth and metastasis by depletion of vesicular sorting protein Hrs: its regulatory role on E-cadherin and beta-catenin. Cancer Res 67:5162–5171
Toyoyama H, Nuruki K, Ogawa H, Yanagi M, Matsumoto H, Nishijima H, Shimotakahara T, Aikou T, Ozawa M (1999) The reduced expression of e-cadherin, alpha-catenin and gamma-catenin but not beta-catenin in human lung cancer. Oncol Rep 6:81–85
Turashvili G, McKinney SE, Goktepe O, Leung SC, Huntsman DG, Gelmon KA, Los G, Rejto PA, Aparicio SA (2011) P-cadherin expression as a prognostic biomarker in a 3992 case tissue microarray series of breast cancer. Mod Pathol 24:64–81
Umbas R, Schalken JA, Aalders TW, Carter BS, Karthaus HF, Schaafsma HE, Debruyne FM, Isaacs WB (1992) Expression of the cellular adhesion molecule E-cadherin is reduced or absent in high-grade prostate cancer. Cancer Res 52:5104–5109
van de Wetering M, Barker N, Harkes IC, Van Der Heyden M, Dijk NJ, Hollestelle A, Klijn JG, Clevers H, Schutte M (2001) Mutant E-cadherin breast cancer cells do not display constitutive Wnt signaling. Cancer Res 61:278–284
van Oort IM, Tomita K, van Bokhoven A, Bussemakers MJ, Kiemeney LA, Karthaus HF, Witjes JA, Schalken JA (2007) The prognostic value of E-cadherin and the cadherin-associated molecules alpha-, beta-, gamma-catenin and p120ctn in prostate cancer specific survival: a long-term follow-up study. Prostate 67:1432–1438
Vasioukhin V, Bauer C, Yin M, Fuchs E (2000) Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell 100:209–219
Vasioukhin V, Bauer C, Degenstein L, Wise B, Fuchs E (2001) Hyperproliferation and defects in epithelial polarity upon conditional ablation of alpha-catenin in skin. Cell 104:605–617
Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL, Crystal RG, de Herreros AG, Moustakas A, Pettersson RF, Fuxe J (2009) A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol 11:943–950
Vleminckx K, Vakaet L Jr, Mareel M, Fiers W, van Roy F (1991) Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 66:107–119
Voorhoeve PM, le Sage C, Schrier M, Gillis AJ, Stoop H, Nagel R, Liu YP, van Duijse J, Drost J, Griekspoor A, Zlotorynski E, Yabuta N, De Vita G, Nojima H, Looijenga LH, Agami R (2006) A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 124:1169–1181
Wang XQ, Li H, Van Putten V, Winn RA, Heasley LE, Nemenoff RA (2009) Oncogenic K-Ras regulates proliferation and cell junctions in lung epithelial cells through induction of cyclooxygenase-2 and activation of metalloproteinase-9. Mol Biol Cell 20:791–800
Watabe M, Nagafuchi A, Tsukita S, Takeichi M (1994) Induction of polarized cell-cell association and retardation of growth by activation of the E-cadherin-catenin adhesion system in a dispersed carcinoma line. J Cell Biol 127:247–256
Wend P, Holland JD, Ziebold U, Birchmeier W (2010) Wnt signaling in stem and cancer stem cells. Semin Cell Dev Biol 21:855–863
Wheelock MJ, Shintani Y, Maeda M, Fukumoto Y, Johnson KR (2008) Cadherin switching. J Cell Sci 121:727–735
Wijnhoven BP, de Both NJ, van Dekken H, Tilanus HW, Dinjens WN (1999) E-cadherin gene mutations are rare in adenocarcinomas of the oesophagus. Br J Cancer 80:1652–1657
Wong SC, Lo SF, Lee KC, Yam JW, Chan JK, Wendy Hsiao WL (2002) Expression of frizzled-related protein and Wnt-signalling molecules in invasive human breast tumours. J Pathol 196:145–153
Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B (2007) The genomic landscapes of human breast and colorectal cancers. Science 318 (5853):1108–1113
Woodcock SA, Rooney C, Liontos M, Connolly Y, Zoumpourlis V, Whetton AD, Gorgoulis VG, Malliri A (2009) SRC-induced disassembly of adherens junctions requires localized phosphorylation and degradation of the rac activator tiam1. Mol Cell 33:639–653
Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ (2005) Deconstructing the cadherin-catenin-actin complex. Cell 123:889–901
Yamazaki F, Aragane Y, Kawada A, Tezuka T (2001) Immunohistochemical detection for nuclear beta-catenin in sporadic basal cell carcinoma. Br J Dermatol 145:771–777
Yang DH, Cai KQ, Roland IH, Smith ER, Xu XX (2007) Disabled-2 is an epithelial surface positioning gene. J Biol Chem 282:13114–13122
Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117:927–939
Yang JY, Zong CS, Xia W, Wei Y, Ali-Seyed M, Li Z, Broglio K, Berry DA, Hung MC (2006) MDM 2 promotes cell motility and invasiveness by regulating E-cadherin degradation. Mol Cell Biol 26:7269–7282
Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY, Yang WH, Huang CH, Kao SY, Tzeng CH, Tai SK, Chang SY, Lee OK, Wu KJ (2010) Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol 12:982–992
Yonemura S, Wada Y, Watanabe T, Nagafuchi A, Shibata M (2010) Alpha-Catenin as a tension transducer that induces adherens junction development. Nat Cell Biol 12:533–542
Yoshiura K, Kanai Y, Ochiai A, Shimoyama Y, Sugimura T, Hirohashi S (1995) Silencing of the E-cadherin invasion-suppressor gene by CpG methylation in human carcinomas. Proc Natl Acad Sci U S A 92:7416–7419
Young P, Boussadia O, Halfter H, Grose R, Berger P, Leone DP, Robenek H, Charnay P, Kemler R, Suter U (2003) E-cadherin controls adherens junctions in the epidermis and the renewal of hair follicles. Embo J 22:5723–5733
Zahir N, Weaver VM (2004) Death in the third dimension: apoptosis regulation and tissue architecture. Curr Opin Genet Dev 14:71–80
Zandy NL, Playford M, Pendergast AM (2007) Abl tyrosine kinases regulate cell-cell adhesion through Rho GTPases. Proc Natl Acad Sci U S A 104:17686–17691
Zechner D, Fujita Y, Hulsken J, Muller T, Walther I, Taketo MM, Crenshaw EB 3rd, Birchmeier W, Birchmeier C (2003) Beta-Catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev Biol 258:406–418
Zeng G, Apte U, Micsenyi A, Bell A, Monga SP (2006) Tyrosine residues 654 and 670 in beta-catenin are crucial in regulation of Met-beta-catenin interactions. Exp Cell Res 312:3620–3630
Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, Giovannini M, Liu P, Anders RA, Pan D (2010) The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell 19:27–38
Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, Halder G, Lai ZC, Guan KL (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21:2747–2761
Zhao B, Tumaneng K, Guan KL (2011) The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol 13:877–883
Zhong C, Kinch MS, Burridge K (1997) Rho-stimulated contractility contributes to the fibroblastic phenotype of Ras-transformed epithelial cells. Mol Biol Cell 8:2329–2344
Zhou YN, Xu CP, Chen Y, Han B, Yang SM, Fang DC (2005) Alpha-catenin expression is decreased in patients with gastric carcinoma. World J Gastroenterol 11:3468–3472
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Vasioukhin, V. (2012). Adherens Junctions and Cancer. In: Harris, T. (eds) Adherens Junctions: from Molecular Mechanisms to Tissue Development and Disease. Subcellular Biochemistry, vol 60. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-4186-7_16
Download citation
DOI: https://doi.org/10.1007/978-94-007-4186-7_16
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-4185-0
Online ISBN: 978-94-007-4186-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)