
Abstract
Reactions occurring at biogeochemical interfaces such as mineral/water, mineral/microbe, and plant/soil, greatly influence a number of important processes in soil and water environments. These include: sorption, desorption, oxidation-reduction, and precipitation, dissolution. It is useful to study these processes over a range of spatial and temporal scales. The use of molecular scale techniques, especially those that are in-situ and synchrotron-based, have provided a wealth of information on reactivity at biogeochemical interfaces. This review focuses on the use of these techniques, especially X-ray absorption spectroscopy, to speciate and elucidate reaction mechanisms of metal(loids) in soils and plants.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Critical zone
- Multi-scale approach
- Kinetics
- Synchrotron-based techniques
- Molecular environmental science
1 Introduction
The Earth’s Critical Zone is that region of Planet Earth where we live. It extends from the tops of the trees to the water table (Fig. 5.1). There are an array of spatial and temporal scales in the Critical Zone. The spatial scales extend from the atomic to the global and the temporal scales range from milliseconds to eons. Additionally, there are a plethora of physical, chemical, and biological reactions and processes that are coupled and influence some of the major environmental challenges of our time including: water, soil, and air quality, human health, the health of oceans, climate change, and indeed economic vitality and development. There are also a number of significant biogeochemical interfaces such as the mineral/water, microbe/mineral, and plant/soil that play pivotal roles in the fate, transport, toxicity, speciation (form), and bioavailability of nutrients and inorganic and organic contaminants in the environment (Sparks 2005a). It is for all of these reasons that an enhanced understanding of biogeochemical processes and reactivity at Critical Zone interfaces, over a range of spatial and temporal scales, is fundamental to sustaining and enhancing Planet Earth.

Schematic of the Critical Zone. The Critical Zone includes the land surface and its canopy of vegetation, rivers, lakes, and shallow seas, and it extends through the pedosphere, unsaturated vadose zone, and saturated groundwater zone. Interactions at this interface between the solid Earth and its fluid envelopes determine the availability of nearly every life-sustaining resource (Reprinted with permission NRC 2001)
This review will focus on the use of synchrotron radiation to further advance our understanding of the reactivity and mechanisms at important environmental biogeochemical interfaces. It will include past successes and future opportunities. Arguably, the use of synchrotron-based techniques such as X-ray absorption spectroscopy has revolutionized the soil and environmental sciences over the past two decades by greatly advancing discovery research and forging important inter- and multi-disciplinary collaborations that are integral to solving some of the grand, complex challenges we face globally. As a result of these collaborations the interdisciplinary field of molecular environmental science, which involves the study of the chemical and physical forms and distribution of contaminants in soils, sediments, waste materials, natural waters, and the atmosphere at the molecular level (Sparks 2002), is a vibrant interdisciplinary field of research.
There are a number of areas where the application of molecular environmental science is propelling major frontiers. These include speciation of contaminants, which is essential for understanding release mechanisms, spatial resolution, chemical transformations, toxicity, bioavailability, and ultimate impacts on human health; mechanisms of microbial transformations on mineral surfaces; phytoremediation; humic substance structure and chemistry; air and terrestrial emanated particulate reactivity and composition; soil structure; development of predictive models; effective remediation and waste management strategies; and risk assessment (Sparks 2002; Ginder-Vogel and Sparks 2010; Sparks and Ginder-Vogel 2011).
This review will focus on the use of X-ray absorption spectroscopy (XAS), X-ray fluorescence (XRF) spectroscopy, and X-ray computed microtomography to elucidate important information on speciation, binding mechanisms, and contaminant distribution and associations in mineral, soil, and plant systems with emphasis on biogeochemical interfacial processes.
2 Synchrotron Radiation
Intense light is produced at a synchrotron facility. Synchrotron radiation is produced over a wide range of energies from the infrared region with energies <1 eV to the hard X-ray region with energies of 100 keV or more. There are more than 60 synchrotron light sources in over 30 countries (www.lightsources.org). For more information on synchrotron user facilities around the globe and their capabilities in the geosciences, the reader should consult Sutton (2006). It is not the purpose of this review to delve into detail about synchrotron radiation and techniques. The reader can refer to several excellent reviews on the use of synchrotron techniques in the environmental sciences that have been published in the past decade. These reviews focus on the use of synchrotron techniques in low-temperature geochemistry and environmental science (Brown and Parks 2001; Brown and Sturchio 2002; Fenter et al. 2002), clay and soil science (Schulze et al. 1999; Kelly et al. 2008; Ginder-Vogel and Sparks 2010; Sparks and Ginder-Vogel 2011), and the study of heavy metals in the environment (Sparks 2005a). However, some detail is provided on XAS, one of the most widely used synchrotron-based spectroscopic techniques used in the environmental sciences.
2.1 X-Ray Absorption Spectroscopy
XAS was developed in the early 1970s (Sayers et al. 1971). It has a number of desirable features that makes it the technique of choice for studying environmental samples such as minerals, humic materials, soils, biosolids, plants, and particulates. XAS can be used to study most elements in crystalline or non-crystalline solid, liquid, or gaseous states over a concentration range of a few ppm to the pure element. It is also an in-situ technique, which means that one can study reactions in the presence of water. This is a major advantage over many molecular scale techniques, which are ex-situ, and often requiring drying of the sample material, placing it in an ultra-high vacuum (UHV), heating the sample, or employing particle bombardment. Such conditions can alter the sample, creating artifacts, and do not simulate most natural soil conditions (Ginder-Vogel and Sparks 2010; Sparks and Ginder-Vogel 2011).
An XAS experiment, which results in a spectrum (Fig. 5.2), consists of exposing a sample to an incident monochromatic beam of synchrotron X-rays, scanned over a range of energies below and above the absorption edge (K, L, M) of the element of interest (Sparks 2002). The energy region extending from just below to about 50 eV above the absorption edge is the XANES (X-ray absorption near edge structure) portion of the spectrum (Fig. 5.2). Fingerprint information (e.g., oxidation states) can be obtained from this region of the XAS spectrum. The XANES region of the spectrum, while not providing as much quantitative information as the extended X-ray absorption fine structure (EXAFS) region, can provide qualitative or semi-quantitative information on the oxidation state of the measured element (Brown et al. 1995). Such information can be obtained by comparing the features of the XANES spectrum of the sample with features of XANES spectra for well-characterized reference compounds. Some species, such as Cr, yield remarkably different, easily recognizable XANES spectra such as the pre-edge feature that is diagnostic for Cr(VI).

Energy regions of a XAS spectrum
The energy region from 50 to 1,000 eV above the absorption edge is the EXAFS (extended X-ray absorption fine structure) portion of the spectrum (Fig. 5.2). Analysis of an EXAFS spectrum provides information on interatomic distances (R), coordination numbers (CN), and identity of first, second, and more distant shells of neighbors around an absorber (Brown et al. 1995; Sparks 2002; Ginder-Vogel and Sparks 2010; Sparks and Ginder-Vogel 2011).
Additional detail on XAS principles, methodology, sample preparation, and data analyses can be found in a number of excellent sources (Brown 1990; Manceau et al. 1992; Fendorf et al. 1994; Schulze and Bertsch 1995; O’Day 1999; Brown and Parks 2001; Bertsch and Hunter 2001; Fenter et al. 2002; Brown and Sturchio 2002; Kelly et al. 2008; Ginder-Vogel and Sparks 2010; Sparks and Ginder-Vogel 2011).
Other synchrotron-based techniques that allow for investigations of heterogeneous materials, which are characterized by multiple species of metal(loids) and nutrients, at micron and smaller scales include microfocused XAS and XRF and microtomography. Additionally, to study rapid processes in real-time at subsecond time scales and at the molecular level one can employ quick X-ray absorption spectroscopy (Q-XAS).
2.2 Micro-focused XAS and XRF
With micro-focused XAS and XRF one can investigate square micron areas and focus the beam to spatial resolutions of <5 um to determine speciation and elemental distribution and associations. Such ability is critical for understanding heterogeneous systems such as soils and plants. While bulk XAS can and should be used to provide what major species are found in such systems, one often needs to couple these studies with μ-XAS and μ-XRF investigations to more precisely quantify major species and determine associations and distributions of elements over small areas in heterogeneous systems where multiple species exist. Standard bulk XAS techniques explore an area of several square millimeters and provide information on the average local chemical environment. In heterogeneous natural systems where more than one type of surface species is present, bulk XAS will detect only the primary (or average) type of surface product/species in the bulk sample. Soils have particle sizes in the micrometer range, and metal speciation may vary over regions of a few 100 μm2. Consequently, the presence of multiple species in soils results in overlapping atomic shells which makes precise speciation with bulk XAS challenging (McNear et al. 2005b; Ginder-Vogel and Sparks 2010; Sparks and Ginder-Vogel 2011).
Some excellent reviews have appeared on the experimental details and application of μ-XAS and μ-XRF to speciate contaminants in heterogeneous systems (Bertsch and Hunter 1998; Hunter and Bertsch 1998; Bertsch and Seaman 1999; Manceau et al. 2002). A number of investigations have appeared over the past 10 years on the speciation of metals and oxyanions in soils, plants, biosolids, and fly ash (see references in Brown and Sturchio 2002; Ginder-Vogel and Sparks 2010; Sparks and Ginder-Vogel 2011).
2.3 Quick XAS
Many important processes (e.g., adsorption, oxidation-reduction, precipitation) occurring at environmental surfaces are characterized by a rapid initial reaction on time-scales of milliseconds to minutes (Scheidegger and Sparks 1996). In many cases, a significant portion of the entire reaction process has occurred before the first measurements can be made using traditional batch and flow techniques. Thus, a knowledge of these initial reaction rates is critical to determining chemical kinetic rate constants and reaction mechanisms and understanding how the initial reactions impact succeeding processes. Chemical relaxation techniques such as pressure jump (p-jump) and concentration jump (c-jump such as stopped-flow) allow rapid data collection on time scales of milliseconds. However, rate “constants” are calculated from linearized rate equations that include parameters that were determined from equilibrium and modeling studies. Consequently, the rate “constants” are not directly determined (Ginder-Vogel et al. 2009; Ginder-Vogel and Sparks 2010; Sparks and Ginder-Vogel 2011). Direct, in situ, molecular- scale measurement of rapid reactions has, until recently been quite limited. Fendorf et al. (1993) used stop-flow electron paramagnetic resonance (SF-EPR) spectroscopy to measure Mn(II) sorption to birnessite (δ-MnO2) on a time scale of milliseconds. More recently, Parikh et al. (2008) used in situ, Fourier Transform infrared (FTIR) spectroscopy to measure As(III) oxidation rates by hydrous manganese(IV) oxide (HMO) at a time scale of ~2.5 s. However, both of these techniques suffer from limitations. EPR can only be used to measure EPR active nuclei, while FTIR requires both IR active functional groups and relatively high concentrations of the reactants being examined (Parikh et al. 2008).
Quick-scanning X-ray absorption spectroscopy overcomes both of these limitations. Depending on beamline instrumentation and flux, quick-scanning X-ray absorption spectroscopy (Q-XAS) can be used to probe most of the atoms on the periodic table and to relatively low concentrations (Khalid et al. 2010). However, using a unique, cam-operated, continuous-scanning setup at beamline X18B at the National Synchrotron Light Source (NSLS), it is possible to collect XANES and EXAFS spectra as the monochromator travels both up and down in energy (Khalid et al. 2010) on sub-second time scales. Landrot et al. (2010) employed Q-XAS at the NSLS to determine the initial kinetics of Cr(III) oxidation by HMO, and Ginder-Vogel et al. (2009) studied the rates and mechanisms of As(III) oxidation at the HMO mineral surface. In the study of Landrot et al. (2010), Cr(III) oxidation to Cr(VI) was exceedingly rapid as illustrated by the quick appearance of the pre-edge feature (Fig. 5.3), indicative of the appearance of Cr(VI). In both of the Q-XAS studies of Ginder-Vogel et al. (2009) and Landrot et al. (2010) there is strong indication that chemical kinetics are being measured. This is suggested in the study of Landrot et al. (2010), in which initial rate constants were similar as initial concentration of Cr(III) was increased (Sparks 1989).

Cr(III) oxidation kinetics using a Q-XAFS technique, at pH 2.5, [Cr(III)] = 100 mM, [HMO] = 20 g/L, and 0–240 s. Each XANES spectrum shown represents 3 s of the reaction (average of four 0.75 s spectra) (Reprinted with permission Landrot et al. 2010)
2.4 Applications of Synchrotron-Based Techniques to Elucidate Soil Chemical Processes and Reactions
2.4.1 Adsorption of Metals and Oxyanions on Soil Components
A multitude of studies, using bulk XAS to study mineral/water interfacial processes, have appeared in the scientific literature since the first published study by Hayes et al. (1987), who determined the surface complexation products for selenate and selenite on goethite (Sparks 1995, 2005b; Brown and Parks 2001; Brown and Sturchio 2002; Brown et al. 2006). These investigations have provided enlightening information on metal and oxyanion sorption on metal hydr(oxides), clay minerals, humic substances, and other natural materials, including structure, stiochiometry, attachment geometry (inner- vs. outer-sphere, monodentate vs. bidentate or tridentate), the presence of multinuclear complexes and precipitate phases, and the presence of ternary surface complexes when complexing ligands are present in solution (Brown and Parks 2001; Brown and Sturchio 2002; Sparks 2005b). The type of surface complexes on clay minerals and metal-(oxyhydr)oxides that occur with low atomic number elements, such as Al, B, Ca, Mg, S and Si, are not easy to ascertain using XAS under in-situ conditions. However, major advances are being made in the area of soft x-ray XAS spectroscopy that will enable one to directly determine the types of surface complexes that form with these metal(loids) (Ginder-Vogel and Sparks 2010; Sparks and Ginder-Vogel 2011).
Results from XAS studies, as well as from investigations using other in-situ molecular scale techniques such as Fourier transform infrared (FTIR) spectroscopy, allow one to make general conclusions on the predominate types of surface complexes that can occur on soil minerals. Although there are technical difficulties in analyzing alkaline earth cations, i.e., Mg2+, Ca2+, Sr2+, and Ba2+, they primarily form outer-sphere complexes. The divalent first-row transition metal cations Mn2+, Fe2+, Co2+, Ni2+, Cu2+, and Zn2+, and the divalent heavy metal cations such as Cd2+, Hg2+, and Pb2+ primarily form inner-sphere complexes. At higher metal loadings and pHs, sorption of metals such as Co, Cr, Ni, and Zn on phyllosilicates and metal-(oxyhydr) oxides can result in the formation of surface precipitates. The formation of these multinuclear and precipitate phases will be discussed in more detail later.
Experimental limitations are such that it is difficult to directly determine the type of surface complexes that NO –3 , Cl–, and ClO –4 form on mineral surfaces. However, one can propose that they are sorbed as outer-sphere complexes and sorbed on surfaces that exhibit a positive charge. Some researchers have shown that SO 2–4 (Zhang and Sparks 1990) can be sorbed as an outer-sphere complex; however, there is other evidence that SO 2–4 can also be sorbed as an inner-sphere complex (Manceau and Charlet 1994). There is direct spectroscopic evidence to show that selenate can be sorbed as both an outer-sphere and an inner-sphere complex, depending on environmental factors (Hayes et al. 1987; Wijnja and Schulthess 2000).
Most other anions such as molybdate, arsenate, arsenite, selenite, phosphate, and silicate are strongly sorbed as inner-sphere complexes, and sorption occurs through a ligand exchange mechanism. The sorption maximum is often insensitive to ionic strength changes. Sorption of anions via ligand exchange results in a shift in the pzc of the sorbent to a more acidic value (Sparks 2002, 2005b).
However, bulk XAS probes an area of several square millimeters and only provides information on the average local chemical environment of a surface. Thus, if there is more than one type of surface species present, bulk XAS will detect only the primary (or average) type of surface product/species in the bulk sample. Therefore while the data may indicate that the primary surface product is an inner-sphere, this does not mean that outer-sphere complexes are not present. In fact, recent studies, such as those of Catalano et al. (2008), who employed X-ray scattering measurements to study metal(loid) binding on single crystal surfaces, showed that arsenate was sorbed simultaneously as inner- and outer-sphere complexes.
Environmental factors such as pH, surface loading, ionic strength, type of sorbent, and time all affect the type of sorption complex or product. Examples of this include the work of Strawn and Sparks (1999) who investigated Pb sorption on montmorillonite over an ionic strength (I) of 0.006–0.1 and a pH range of 4.48–6.77. At a pH of 4.48 and I of 0.006, outer-sphere complexation on basal planes in the interlayer regions of the montmorillonite predominated. At a pH of 6.77 and I of 0.1, inner-sphere complexation on edge sites of montmorillonite was most prominent, and at pH of 6.76, I of 0.006 and pH of 6.31, I of 0.1, both inner- and outer-sphere complexation occurred. These data are consistent with other findings that inner-sphere complexation is favored at higher pH and ionic strength. Recently, Lafferty et al. (2010), investigating As(III) oxidation kinetics on hydrous manganese oxide(HMO) showed that at shorter times (a few hours) As(V) was bound on HMO as bidentate binuclear and monodentate mononuclear complexes, but at longer times, a third arsenate complex, a bidentate-mononuclear complex appeared. Clearly, there is a continuum of adsorption complexes that can exist in soils (Sparks 2002, 2005b; Ginder-Vogel and Sparks 2010).
2.4.2 Metal(loid) Surface Precipitation/Dissolution
In addition to two-dimensional surface complexes, three dimensional Co2+, Cr3+, Cu2+, Ni2+, and Pb2+ metal hydroxide and mixed metal-Al surface precipitates, can form on metal oxides, phyllosilicates, soil clays, and soils (Charlet and Manceau 1992; Chisholm-Brause et al. 1994; Fendorf et al. 1994; Scheidegger et al. 1996a; Scheidegger et al. 1996b; Scheidegger et al. 1997; Scheidegger et al. 1998; Towle et al. 1997; Elzinga and Sparks 1999; Roberts et al. 1999; Thompson et al. 1999a; Thompson et al. 1999b; Ford and Sparks 2000; Scheckel and Sparks 2001b; Nachtegaal and Sparks 2004; McNear et al. 2006; Peltier et al. 2010). These metal hydroxide phases occur at metal loadings below theoretical monolayer coverage and in a pH range well below the pH where the formation of metal hydroxide precipitates would be expected, according to the thermodynamic solubility product (Scheidegger and Sparks 1996; Sparks 2002, 2005b; Borda and Sparks 2008; Ginder-Vogel and Sparks 2010).
Scheidegger et al. (1997) were the first to show that sorption of metals, such as Ni, on an array of phyllosilicates and Al-oxide, could result in the formation of mixed metal-Al hydroxide surface precipitates. The precipitate phase shares structural features common to the hydrotalcite group of minerals and the layered double hydroxides (LDH) observed in catalyst synthesis. The LDH structure is built of stacked sheets of edge-sharing metal octahedra, containing divalent and trivalent metal ions separated by anions between the interlayer spaces. The LDH structure exhibits a net positive charge x per formula unit which is balanced by an equal negative charge from interlayer anions An–, such as Cl–, Br–, I–, NO –3 , OH–, ClO –4 , and CO 2–3 ; water molecules occupy the remaining interlayer space (Taylor 1984). Recently, Livi et al. (2009), using an array of microscopic techniques including analytical electron microscopy (AEM), high resolution transmission electron microscopy (HRTEM), and powder X-ray diffraction, conducted studies to elucidate the nature of Ni hydroxide precipitates, using the same environmental conditions employed by Scheidegger et al. (1996b, 1997) and reaction times ranging from 1 h to 5 years. While the precipitate phase had a bonding environment similar to Ni-Al LDH, the precipitate was amorphous.
The formation of metal hydroxide surface precipitates can significantly sequester metals (Scheckel and Sparks 2001a). As the surface precipitates age, metal release is greatly reduced (Fig. 5.4). Thus, the metals are less prone to leaching and being taken up by plants and microbes.

Dissolution of Ni from surface precipitates formed on pyrophyllite at residence times of 1 h to 2 years. The figure shows the relative amount of Ni2+ remaining on the pyrophyllite surface following extraction for a 24 h period (each replenishment represents a 24 h extraction) with HNO3 at pH 6.0 (Reprinted with permission Scheckel and Sparks 2001a)
The decrease in metal release and bioavailability is linked to the increasing silication of the interlayer of the LDH phases with increased residence time, resulting in a mineral transformation from a LDH phase to a precursor phyllosilicate surface precipitate (Ford et al. 1999; Ford and Sparks 2000). The mechanism for this transformation is attributed to diffusion of Si, originating from weathering of the sorbent, into the interlayer space of the LDH, replacing the anions such as NO −3 . Peltier et al. (2006), using acid-solution calorimetry, and results from previous calorimetry studies, showed that the enthalpy of formation of LDH phases is more exothermic, indicating great stability, in the order of Cl < NO3 < SO4 < CO3 < Si of interlayer anionic composition, and that LDH phases were much more stable than a Ni(OH)2 phase. The results of these and other studies show that with time, metal sorption on soil minerals involves a continuum of processes, from adsorption to precipitation to solid phase transformation, particularly in the case of certain metals, such as Co2+, Ni2+, and Zn2+. The formation of metal surface precipitates is a way that metals can be naturally attenuated such that they are less mobile and bioavailable. Such products must be considered when modeling the fate and mobility of metals like Co2+, Mn2+, Ni2+, and Zn2+ in soil and water environments (Sparks 2002; Sparks 2005b; Borda and Sparks 2008; Ginder-Vogel and Sparks 2010).
2.5 Speciation of Metal(loids) in Soils
Some examples of studies that have used μ-XAS and μ-XRD to speciate metal(loids) in soils include those by Hunter and Bertsch (1998), Manceau et al. (2000), Isaure et al. (2002), Strawn et al. (2002), Ginder-Vogel et al. (2005), Nachtegaal et al. (2005), Roberts et al. (2005), Arai et al. (2006), McNear et al. (2007), and Gräfe et al. (2008).
Nachtegaal et al. (2005) investigated the speciation of Zn in smelter contaminated soils, employing u-XAS and u-XRF, from a large site in Belgium in which part of the site had been remediated by adding beringite, an aluminosilicate material, compost, and planting metal tolerant plants. The other portion of the site was not treated. The objectives of the study were to determine how Zn speciation differed in the remediated (treated) and non-remediated (non-treated) soils, to investigate if Zn-LDH phases were present in the soils, and to establish the stability of the zinc under different environmental conditions.
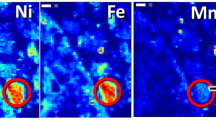
A number of μ-XAS spectra for the treated (Fig. 5.5b) and non-treated (Fig. 5.5d) soils were collected at various regions of interest from the XRF spectra (Fig. 5.5a, treated and Fig. 5.5c, non-treated soils), as well as spectra for reference mineral, sorbed, and solution phases that were probable species in the soils. The μ-XAS spectra of the soils were analyzed via Principle Component Analysis, target transformation, and linear least squares fitting (LLSF) to determine the quantity (%) of each standard species within the individual sample spectra constituting the dataset (Manceau et al. 2002; Nachtegaal et al. 2005). Nachtegaal et al. (2005) found that both mineral (e.g., willemite, hemimorphite, spalerite) and sorbed (Zn-LDH) Zn species predominated in the treated and non-treated soils. The speciation differences in the treated and non-treated soils were minimal, with the major difference being the presence of kerolite, a Zn phyllosilicate phase, that was present in the treated soil (Fig. 5.5b). Significant quantities of Zn-LDH phases were formed in the non-treated soil (Fig. 5.5d). Desorption studies showed that the Zn in both remediated and non-remediated soils was quite stable, reflecting again the role that metal surface precipitates, i.e., Zn-LDH phases, play in sequestering metals such that mobility and bioavailability are diminished.




(a) μ-SXRF tricolor maps for the treated soil samples. The numbers indicate the spots where μ-EXAFS spectra were collected. Red is indicative of the distribution of iron, green of copper and blue of zinc. (b) μ-EXAFS spectra from selected spots on thin sections from the treated soil. (c) μ-SXRF tricolor maps for the non-treated soil samples. (d) μ-EXAFS spectra from selected spots on thin sections from the non-treated soil. The solid line indicates the raw k3c(k) data and the dotted line indicates the best fits obtained with a linear fitting approach (Reprinted with permission Nachtegaal et al. 2005) (Color figure online)
2.6 Speciation of Metals in Hyperaccumulator Plants
A very important biogeochemical interface in the Earth’s Critical Zone is the plant/soil interface. Despite numerous studies, the biochemistry and dynamics of the rhizosphere are still poorly understood. As a way to advance our understanding of the plant/soil interface, scientists have used synchrotron-based XAS, XRF, XRD, and microtomography to investigate plant/soil interfacial processes and reactivity (Sarret et al. 2002, 2006; Küpper et al. 2004; Scheckel et al. 2004; McNear et al. 2005a, b; Tappero et al. 2007). Of particular interest is elucidating the speciation and uptake mechanisms of metals into hyperaccumulator plants. Such an enhanced comprehension could prove invaluable in increasing the effectiveness of phytoremediation in decontaminating metal polluted sites around the world. Phytoremediation is a “green” technology that uses plants to remove contaminants from the environment. Phytoextraction depends on unique plants, capable of accumulating higher than normal metal concentrations (e.g., >1,000 ppm for Ni and Co and >10,000 ppm for Zn) (Baker 1981; McNear et al. 2005a, b). To better understand the mechanisms involved in metal hyperaccumulation and tolerance, it is critical to know whether accumulated metals are bound by strong (specific) ligands or loosely associated with common organic acids (i.e., speciation) as well as where these metals are stored (i.e., localization or compartmentalization). The use of synchrotron-based methods to explore metal speciation and uptake mechanisms in plants is especially attractive because one can use “fresh” hyperaccumulator plant tissues (in vivo) with micrometer resolution and investigate distribution and association of the metals in the plant tissue on two-dimensional (μ-XRF) and three dimensional (microtomography) scales and determine direct metal speciation (μ-XAS). Several studies have employed these techniques to elucidate Ni and Co speciation in Alyssum murale, an important Ni hyperaccumulator plant from the Brassicaceae family that is native to serpentine soils throughout Mediterranean southern Europe (McNear et al. 2005a, b; Tappero et al. 2007; Ginder-Vogel and Sparks 2010; Sparks and Ginder-Vogel 2011).
Tappero et al. (2007) studied metal localization and elemental associations in Alyssum murale plants exposed to metal (Ni, Zn, and Co) co-contaminants using synchrotron-based μ-XRF, μ-XAS, and computed microtomography. Two dimensional XRF images of Alyssum murale leaves showed a marked localization pattern for Co compared to Ni and Zn. The Ni distribution was predominately uniform (Fig. 5.6) which is consistent with previous findings that Ni is compartmentalized in epidermal tissues, i.e., vacuolar sequestration (Krämer et al. 1997, 2000; Broadhurst et al. 2004a, b). Zinc distribution was similar to Ni and was not hyperaccumulated. Cobalt however, was preferentially localized at the tips and margins of A. murale leaves as precipitate phases (Fig. 5.7). Differential absorption computed microtomography (DA-CMT) images (Fig. 5.8) of hydrated A. murale shows leaf Co enrichment in apoplasm tissue. Cobalt near leaf tips was localized primarily on the leaf exterior and the hyperaccumulation mechanism was via exocellular sequestration (Tappero et al. 2007).

Synchrotron X-ray microfluorescence (μ-SXRF) images of the nickel (Ni), cobalt (Co), and zinc (Zn) distributions in a hydrated Alyssum murale leaf from the Ni + Co + Zn treatment. Leaf trichomes are depicted in the Ca channel. The camera image shows the leaf region selected for SXRF imaging (Reprinted with permission Tappero et al. 2007)

Synchrotron X-ray microfluorescence (μ-SXRF) tricolor image (nickel (Ni), cobalt (Co), and calcium (Ca)) of a hydrated Alyssum murale leaf from the Ni + Co + Zn treatment, plus a line profile (fluorescence intensity vs. position) for a segment from the leaf center towards the leaf tip (indicated by a white arrow) (Reprinted with permission Tappero et al. 2007)

Differential absorption (DA-CMT) tomographic projections (5.1 μm slices) of hydrated Alyssum murale leaves depicting (a) cobalt (Co) distribution in the leaf-tip region, (b) Co distribution in the bulk-leaf region, (c) Co distribution in relation to the leaf cell structure (grey), and (d) nickel (Ni) distribution in the leaf-tip and bulk-leaf regions. Leaves were collected from a Co-treated plant (a–c) and from a Ni-treated plant (d). Sinograms recorded above and below the Co or Ni K-edge energy (+30 and −100 eV, respectively) were computationally reconstructed and the resulting projections were subtracted (above – below) to reveal the metal distribution in leaves. Distances are relative to the leaf tissue at the tip as determined from leaf structure images (i.e. below-edge projections) (Reprinted with permission Tappero et al. 2007)
2.7 Future Needs
As our knowledge of environmental systems continues to advance, it is critical that synchrotron-based techniques continue advancing with the field. Currently, a primary limitation of the application of these techniques to environmental systems is the availability of adequate amounts of beamtime. This problem can begin to be resolved by advances in both user support and instrumentation. Several improvements in user administration and services, including standardized data collection software, state-of-the-art data analysis software, increased beamline support staff, and improvements in laboratory support facilities would allow for the more efficient use of limited beamtime. The heterogeneity of environmental samples requires both the application of a wide array of techniques, including X-ray spectroscopic and traditional ones, and more time using each individual technique. The enhanced intensity and flux of third generation synchrotron light-sources allow for the analysis of both lower elemental concentrations and smaller samples; however, many environmental samples also require a broad energy range (5–50 keV), high energy resolution (0.1 eV) and a range of spatial resolutions (bulk, micro, and nano). Additionally, unique endstation capabilities, including flow-through reaction cells and anaerobic gloveboxes, coupled with fast-scanning, high energy resolution fluorescent detection and simultaneous collection of XRD and XAS data, will take advantage of the unique ability of X-ray absorption to analyze samples in situ, allowing a new generation of complex environmental problems to be solved (Ginder-Vogel and Sparks 2010; Sparks and Ginder-Vogel 2011).
References
Arai Y, Lanzirotti A, Sutton SR, Newville M, Dyer J, Sparks DL (2006) Spatial and temporal variability of arsenic solid-state speciation in historically lead arsenate contaminated soils. Environ Sci Technol 40(3):673–679
Baker AJM (1981) Accumulators and excluders – strategies in the response of plants to heavy metals. J Plant Nutr 3:643–654
Bertsch PM, Hunter DB (1998) Elucidating fundamental mechanisms in soil and environmental chemistry: the role of advanced analytical, spectroscopic, and microscopic methods. In: Huang PM (ed) Future prospects for soil chemistry. Soil Science Society of America, Madison, pp 103–122
Bertsch PM, Hunter DB (2001) Applications of synchrotron based X-ray microprobes. Chem Rev 101(6):1809–1842
Bertsch PM, Seaman JC (1999) Characterization of complex mineral assemblages: implications for contaminant transport and environmental remediation. Proc Natl Acad Sci USA 96(7):3350–3357
Borda MJ, Sparks DL (2008) Kinetics and mechanisms of sorption – desorption in soils: a multiscale assessment. In: Violante A, Huang PM, Gadd GM (eds) Biophysico-chemical processes of heavy metals and metalloids in soil environments. Wiley, New York, pp 94–124
Broadhurst CL, Chaney RL, Angle JS, Erbe EF, Maugel TK (2004a) Nickel localization and response to increasing Ni soil levels in leaves of the Ni hyperaccumulator Alyssum murale. Plant Soil 265(1):225–242
Broadhurst CL, Chaney RL, Angle JS, Maugel TK, Erbe EF, Murphy CA (2004b) Simultaneous hyperaccumulation of nickel, manganese, and calcium in Alyssum leaf trichomes. Environ Sci Technol 38(21):5797–5802
Brown GE Jr (1990) Spectroscopic studies of chemisorption reaction mechanisms at oxide-water interfaces. In: Hochella MF, White AF (eds) Mineral-water interface geochemistry. Mineralogical Society of America, Washington, DC, pp 309–353
Brown GE Jr, Parks GA (2001) Sorption of trace elements on mineral surfaces: modern perspectives from spectroscopic studies, and comments on sorption in the marine environment. Int Geogr Rev 43:963–1073
Brown GE Jr, Sturchio NC (2002) An overview of synchrotron radiation applications to low temperature geochemistry and environmental science. In: Fenter PA, Rivers ML, Sturchio NC, Sutton SR (eds) Applications of synchrotron radiation in low-temperature geochemistry and environmental sciences. Mineralogical Society of America, Washington, DC, pp 1–115
Brown GE Jr, Parks GA, O’Day PA (1995) Sorption at mineral-water interfaces: macroscopic and microscopic perspectives. In: Vaughan DJ, Pattrick RAD (eds) Mineral surfaces. Chapman & Hall, London, pp 129–183
Brown GE Jr, Calas G, Hemley RJ (2006) Scientific advances made possible by user facilities. Elements 2(1):23–30
Catalano JG, Park C, Fenter P, Zhang Z (2008) Simultaneous inner- and outer-sphere arsenate adsorption on corundum and hematite. Geochim Cosmochim Acta 72(8):1986–2004
Charlet L, Manceau A (1992) X-ray absorption spectroscopic study of the sorption of Cr(III) at the oxide-water interface. II: adsorption, coprecipitation and surface precipitation on ferric hydrous oxides. J Colloid Interface Sci 148:443–458
Chisholm-Brause CJ, Conradson SD, Buscher CT, Eller PG, Morris DE (1994) Speciation of uranyl sorbed at multiple binding sites on montmorillonite. Geochim Cosmochim Acta 58:3625–3631
Elzinga EJ, Sparks DL (1999) Nickel sorption mechanisms in a pyrophyllite-montmorillonite mixture. J Colloid Interface Sci 213(2):506–512
Fendorf SE, Sparks DL, Franz JA, Camaioni DM (1993) Electron paramagnetic resonance stopped-flow kinetic study of manganese (II) sorption–desorption on birnessite. Soil Sci Soc Am J 57(1):57–62
Fendorf SE, Lamble GM, Stapleton MG, Kelley MJ, Sparks DL (1994) Mechanisms of chromium (III) sorption on silica: 1. Cr(III) surface structure derived by extended x-ray absorption fine structure spectroscopy. Environ Sci Technol 28(2):284–289
Fenter PA, River ML, Sturchio NC, Sutton SR (eds) (2002) Applications of synchrotron radiation in low-temperature geochemistry and environmental science, vol 49, Reviews in mineralogy and geochemistry. The Mineralogical Society of America, Washington, DC
Ford RG, Sparks DL (2000) The nature of Zn precipitates formed in the presence of pyrophyllite. Environ Sci Technol 34(12):2479–2483
Ford RG, Scheinost AC, Scheckel KG, Sparks DL (1999) The link between clay mineral weathering and the stabilization of Ni surface precipitates. Environ Sci Technol 33(18):3140–3144
Ginder-Vogel M, Sparks DL (2010) The impacts of X-ray absorption spectroscopy on understanding soil processes and reaction mechanisms. In: Singh B, Grafe M (eds) Developments in soil science 34: synchrotron-based techniques in soils and sediments. Elsevier, Burlington, pp 1–26
Ginder-Vogel M, Borch T, Mayes MA, Jardine PM, Fendorf S (2005) Chromate reduction and retention processes within arid subsurface environments. Environ Sci Technol 39(20):7833–7839
Ginder-Vogel M, Landrot G, Fischel JS, Sparks DL (2009) Quantification of rapid environmental redox processes with quick-scanning x-ray absorption spectroscopy (Q-XAS). Proc Natl Acad Sci USA 106(38):16124–16128
Gräfe M, Tappero RV, Marcus MA, Sparks DL (2008) Arsenic speciation in multiple metal environments – II. Micro-spectroscopic investigation of a CCA contaminated soil. J Colloid Interface Sci 321(1):1–20
Hayes KF, Roe AL, Brown GE Jr, Hodgson KO, Leckie JO, Parks GA (1987) In situ x-ray absorption study of surface complexes: selenium oxyanions on α-FeOOH. Science 238:783–786
Hunter DB, Bertsch PM (1998) In situ examination of uranium contaminated soil particles by micro-X-ray absorption and micro-fluorescence spectroscopies. J Radioanal Nucl Chem 234(1–2):237–242
Isaure MP, Laboudigue A, Manceau M et al (2002) Quantitative Zn speciation in a contaminated dredged sediment by μ-PIXE, μ-SXRF, EXAFS spectroscopy and principal component analysis. Geochim Cosmochim Acta 66(9):1549–1567
Kelly SD, Hesterberg D, Ravel B (2008) Analysis of soils and minerals using X-ray absorption spectroscopy. In: Ulery AL, Drees LR (eds) Methods of soil analysis part 5 – mineralogical methods. Soil Science Society of America, Madison, pp 387–464
Khalid S, Caliebe WA, Siddons P et al (2010) Quick extended x-ray absorption fine structure instrument with millisecond time scale, optimized for in-situ applications. Rev Sci Instrum 81(1):015105
Krämer U, Smith RD, Wenzel WW, Raskin I, Salt DE (1997) The role of metal transport and tolerance in nickel hyperaccumulation by Thlaspi goesingense Halacsy. Plant Physiol 115(4):1641–1650
Krämer U, Pickering IJ, Prince RC, Raskin I, Salt DE (2000) Subcellular localization and speciation of nickel in hyperaccumulator and non-accumulator Thlaspi species. Plant Physiol 122(4):1343–1353
Küpper H, Mijovilovich A, Meyer-Klaucke W, Kroneck PMH (2004) Tissue- and age-dependent differences in the complexation of cadmium and zinc in the cadmium/zinc hyperaccumulator Thlaspi caerulescens (Ganges ecotype) revealed by X-ray absorption spectroscopy. Plant Physiol 134(2):748–757
Lafferty B, Ginder-Vogel M, Sparks DL (2010) Arsenite oxidation by a poorly crystalline manganese oxide. 1. Stirred-flow experiments. Environ Sci Technol 44:8460–8466
Landrot G, Ginder-Vogel M, Sparks DL (2010) Kinetics of chromium(III) oxidation by manganese(IV) oxides using quick scanning X-ray absorption fine structure spectroscopy (Q-XAFS). Environ Sci Technol 44:143–149
Livi KJT, Senesi GS, Scheinost AC, Sparks DL (2009) Microscopic examination of nanosized mixed Ni-Al hydroxide surface precipitates on pyrophyllite. Environ Sci Technol 43(5):1299–1304
Manceau A, Charlet L (1994) The mechanism of selenate adsorption on goethite and hydrous ferric oxide. J Colloid Interface Sci 168:87–93
Manceau A, Charlet L, Boisset MC, Didier B, Spadini L (1992) Sorption and speciation of heavy metals on hydrous Fe and Mn oxides. From microscopic to macroscopic. Appl Clay Sci 7:201–223
Manceau A, Lanson B, Schlegel ML et al (2000) Quantitative Zn speciation in smelter-contaminated soils by EXAFS spectroscopy. Am J Sci 300(4):289–343
Manceau A, Marcus MA, Tamura N (2002) Quantitative speciation of heavy metals in soils and sediments by synchrotron X-ray techniques. In: Fenter P, Sturchio NC (eds) Applications of synchrotron radiation in low-temperature geochemistry and environmental science. Mineralogical Society of America, Washington, DC, pp 341–428
McNear DH, Peltier E, Everhart J et al (2005a) Application of quantitative fluorescence and absorption-edge computed microtomography to image metal compartmentalization in Alyssum murale. Environ Sci Technol 39(7):2210–2218
McNear DH Jr, Tappero R, Sparks DL (2005b) Shining light on metals in the environment. Elements 1:211–216
McNear DH, Tappero R, Sparks DL (2006) Elemental analysis of roots and fungi. In: Luster J, Finlay R (eds) Handbook of methods used in rhizosphere research. Swiss Federal Research Institute, Birmensdorf, pp 204–205
McNear DH, Chaney RL, Sparks DL (2007) The effects of soil type and chemical treatment on nickel speciation in refinery enriched soils: a multi-technique investigation. Geochim Cosmochim Acta 71(9):2190–2208
Nachtegaal M, Sparks DL (2004) Effect of iron oxide coatings on zinc sorption mechanisms at the clay-mineral/water interface. J Colloid Interface Sci 276:13–23
Nachtegaal M, Marcus MA, Sonke JE et al (2005) Effects of in situ remediation on the speciation and bioavailability of zinc in a smelter contaminated soil. Geochim Cosmochim Acta 69(19):4649–4664
NRC (2001) Basic research opportunities in earth science. National Academy Press, Washington, DC
O’Day PA (1999) Molecular environmental geochemistry. Rev Geophys 37(2):249–274
Parikh SJ, Lafferty BJ, Sparks DL (2008) An ATR-FTIR spectroscopic approach for measuring rapid kinetics at the mineral/water interface. J Colloid Interface Sci 320(1):177–185
Peltier E, Allada R, Navrotsky A, Sparks DL (2006) Nickel solubility and precipitation in soils: a thermodynamic study. Clays Clay Miner 54(2):153–164
Peltier E, van der Lelie D, Sparks DL (2010) Formation and stability of Ni-Al hydroxide phases in Delaware soils. Environ Sci Technol 44:302–308
Roberts DR, Scheidegger AM, Sparks DL (1999) Kinetics of mixed Ni-Al precipitate formation on a soil clay fraction. Environ Sci Technol 33(21):3749–3754
Roberts D, Nachtegaal M, Sparks DL (2005) Speciation of metals in soils. In: Tabatabai MA, Sparks DL (eds) Chemical processes in soils. Soil Science Society of America, Madison, pp 619–654
Sarret G, Saumitou-Laprade P, Bert V et al (2002) Forms of zinc accumulated in the hyperaccumulator Arabidopsis halleri. Plant Physiol 130(4):1815–1826
Sarret G, Harada E, Choi YE et al (2006) Trichomes of tobacco excrete zinc as zinc-substituted calcium carbonate and other zinc-containing compounds. Plant Physiol 141(3):1021–1034
Sayers DE, Stern EA, Lytle FW (1971) New technique for investigating noncrystalline structures: fourier analysis of extended X-ray – absorption fine structure. Phys Rev Lett 27(18):1204–1207
Scheckel KG, Sparks DL (2001a) Dissolution kinetics of nickel surface precipitates on clay mineral and oxide surfaces. Soil Sci Soc Am J 65(3):685–694
Scheckel KG, Sparks DL (2001b) Temperature effects on nickel sorption kinetics at the mineral-water interface. Soil Sci Soc Am J 65(3):719–728
Scheckel KG, Lombi E, Rock SA, McLaughlin NJ (2004) In vivo synchrotron study of thallium speciation and compartmentation in lberis intermedia. Environ Sci Technol 38(19):5095–5100
Scheidegger AM, Sparks DL (1996) A critical assessment of sorption – desorption mechanisms at the soil mineral/water interface. Soil Sci 161(12):813–831
Scheidegger AM, Fendorf M, Sparks DL (1996a) Mechanisms of nickel sorption on pyrophyllite: macroscopic and microscopic approaches. Soil Sci Soc Am J 60(6):1763–1772
Scheidegger AM, Lamble GM, Sparks DL (1996b) Investigation of Ni sorption on pyrophyllite: an XAFS study. Environ Sci Technol 30(2):548–554
Scheidegger AM, Lamble GM, Sparks DL (1997) Spectroscopic evidence for the formation of mixed-cation hydroxide phases upon metal sorption on clays and aluminum oxides. J Colloid Interface Sci 186(1):118–128
Scheidegger AM, Strawn DG, Lamble GM, Sparks DL (1998) The kinetics of mixed Ni-Al hydroxide formation on clay and aluminum oxide minerals: a time-resolved XAFS study. Geochim Cosmochim Acta 62(13):2233–2245
Schulze DG, Bertsch PM (1995) Synchrotron x-ray techniques in soil, plant, and environmental research. Adv Agron 55:1–66
Schulze DG, Stucki JW, Bertsch PM (1999) Synchrotron x-ray methods in clay science, vol 98, CMS Workshop Lectures. Clay Minerals Society, Boulder
Sparks DL (1989) Kinetics of soil chemical processes. Academic, New York
Sparks DL (1995) Environmental soil chemistry. Academic, San Diego
Sparks DL (2002) Environmental soil chemistry, 2nd edn. Academic, San Diego
Sparks DL (2005a) Toxic metals in the environment: the role of surfaces. Elements 1:193–197
Sparks DL (2005b) Metal and oxyanion sorption on naturally occurring oxide and clay mineral surfaces. In: Grassian VH (ed) Environmental catalyis. Taylor & Francis, Boca Raton, pp33–36
Sparks DL, Ginder-Vogel M (2011) The role of synchrotron radiation in elucidating the biogeochemistry of metal(loids) and nutrients at critical zone interfaces. In: Sumner ME, Li Y (eds) Handbook of soil sci. Taylor & Francis, Boca Raton
Strawn DG, Sparks DL (1999) The use of XAFS to distinguish between inner- and outer-sphere lead adsorption complexes on montmorillonite. J Colloid Interface Sci 216:257–269
Strawn D, Doner H, Zavarin M, McHugo S (2002) Microscale investigation into the geochemistry of arsenic, selenium, and iron in soil developed in pyritic shale materials. Geoderma 108:237–257
Sutton SR, Caffee MW, Dove MT (2006) Synchrotron radiation, neutron, and mass spectrometry techniques at user facilities. Elements 2:15–21
Tappero R, Peltier E, Grafe M et al (2007) Hyperaccumulator Alyssum murale relies on a different metal storage mechanism for cobalt than for nickel. New Phytol 175(4):641–654
Taylor RM (1984) The rapid formation of crystalline double hydroxy salts and other compounds by controlled hydrolysis. Clay Miner 19(4):591–603
Thompson HA, Parks GA, Brown GE Jr (1999a) Ambient-temperature synthesis, evolution, and characterization of cobalt-aluminum hydrotalcite-like solids. Clays Clay Miner 47(4):425–438
Thompson HA, Parks GA, Brown GE Jr (1999b) Dynamic interactions of dissolution, surface adsorption, and precipitation in an aging cobalt(II)-clay-water system. Geochim Cosmochim Acta 63(11–12):1767–1779
Towle SN, Bargar JR, Brown GE, Parks GA (1997) Surface precipitation of Co(II)(aq) on Al2O3. J Colloid Interface Sci 187(1):62–82
Wijnja H, Schulthess CP (2000) Vibrational spectroscopic study of selenate and sulfate adsorption mechanisms on Fe and A1 (hydr)oxides surfaces. J Colloid Interface Sci 229:286–297
Zhang PC, Sparks DL (1990) Kinetics and mechanisms of sulfate adsorption desorption on goethite using pressure-jump relaxation. Soil Sci Soc Am J 54(5):1266–1273
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht.
About this chapter
Cite this chapter
Sparks, D.L. (2013). Advances in the Use of Synchrotron Radiation to Elucidate Environmental Interfacial Reaction Processes and Mechanisms in the Earth’s Critical Zone. In: Xu, J., Sparks, D. (eds) Molecular Environmental Soil Science. Progress in Soil Science. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-4177-5_5
Download citation
DOI: https://doi.org/10.1007/978-94-007-4177-5_5
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-4176-8
Online ISBN: 978-94-007-4177-5
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)