
Abstract
Phosphoinositides, especially phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2] are required for the activity of many different ion channels. This chapter will highlight various aspects of this paradigm, by discussing current knowledge on four different ion channel families: inwardly rectifying K+ (Kir) channels, KCNQ voltage gated K+ channels, voltage gated Ca2+ (VGCC) channels and Transient Receptor Potential (TRP) channels. Our main focus is to discuss functional aspects of this regulation, i.e. how changes in the concentration of PtdIns(4,5)P2 in the plasma membrane upon phospholipase C activation may modulate the activity of ion channels, and what are the major determinants of this regulation. We also discuss how channels act as coincidence detectors sensing phosphoinositide levels and other signalling molecules. We also briefly discuss the available methods to study phosphoinositide regulation of ion channels, and structural aspects of interaction of ion channel proteins with these phospholipids. Finally, in several cases the effect of PtdIns(4,5)P2 is more complex than a simple dependence of ion channel activity on the lipid, and we will discuss some these complexities.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
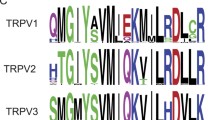


Keywords
10.1 Introduction
Membrane phosphoinositides play a multitude of roles in a variety of biological processes. Phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2], generally referred to as PIP2, is the substrate for phospholipase C (PLC) (Fig. 10.1) and constitutes up to 1% of the phospholipids in the plasma membrane, where it is localized in the inner, cytoplasmic leaflet. Its immediate precursor PtdIns(4)P is found at comparable quantities, whereas phosphatidylinositol is more abundant, but it is usually not efficient in regulating ion channels. Other phosphoinositides, such as PtdIns(3,4)P2 and PtdIns(3,4,5)P3, the products of PI3 Kinases are found in much smaller quantities even in stimulated cells than PtdIns(4,5)P2. PtdIns(4,5)P2 is the physiologically most important regulator of most studied ion channels, because it is more abundant, and/or more active than other phosphoinositides.

Phosphoinositide metabolism. PtdIns(4,5)P2 is generated from its precursor PtdIns by two consecutive phosphorylation steps by phosphatidylinositol 4-kinases (PI4K) and phosphatidylinositol 4-phosphate 5 kinases (PIP5K). The reversibility of these processes is ensured by various phosphatases (Ptase). Phosphoinositide 3 kinases (PI3K) generate PtdIns(3,4)P2 and PtdInse(3,4,5)P3. Various PLC isoforms, activated by G Protein Coupled Receptors (GPCR), receptor tyrosine kinases (RTK) and other factors, hydrolyse PtdIns(4,5)P2, and generate inositol 1,4,5 trisphopshate (IP3) and diacylglycerol (DAG). IP3 liberates Ca2+ from intracellular stores, and DAG activates protein kinase C (PKC). (From Rohacs (2007) with permission)
Tremendous progress has been made in the last one and a half decades in understanding the regulation of transporters and ion channels by these lipids. Several sporadic early studies reported the modulation of various membrane transporters by phosphoinositides, such as the plasma membrane Ca2+ ATP-ase; these studies have been thoroughly reviewed recently (Huang 2007). It was, however, Hilgeman’s seminal paper in 1996 (Hilgemann and Ball 1996) reporting PIP2 dependence of two cardiac ion transport proteins in excised patches, that sparked a new era of research on this field. Shortly after this discovery, a surprising number and variety of ion channels have been reported to be regulated by PIP2. It currently seems that dependence of activity on PIP2 is a property of a large number, if not the majority, of mammalian plasma membrane ion channel proteins (Gamper and Shapiro 2007a; Suh and Hille 2008). This chapter will discuss this topic using the example of four different groups of ion channels, all of which share the property of being regulated by membrane phosphoinositides (chiefly PIP2) and illustrate various features of this paradigm. Our first two examples are the inwardly-rectifying K+ (Kir) channels and the M-type K+ channels (Kv7 or KCNQ) for which the regulation by phosphoinositides is widely studied and relatively well understood. It is clear that all members of both families require the presence of PIP2 for activity and depletion of the lipid in the plasma membrane inhibits them; there is also a consensus for the physiological role of the apparent affinity for PIP2 in the regulation of these channels. The next example is the family of voltage-gated Ca2+ channels (VGCC) which also require PIP2 for their activity although whether PIP2 is a physiological regulator of VGCC activity in vivo remains under discussion. Finally our last example is the Transient Receptor Potential (TRP) channel family; these channels are in the focus of intense research, however, their phosphoinositide regulation is complex in many cases, not very well understood, and even controversial.
10.2 Tools to Study Phosphoinositide Regulation of Ion Channels
A powerful toolkit for studying regulation of ion channels by phosphoinositides has been developed over the recent years. Since a similar combination of methods and approaches has been applied to investigation of regulation by phosphoinositides of all channel families discussed here, we will briefly summarize them in this section.
-
(i)
Excised patch recordings. In this experimental paradigm an inside-out excised patch configuration of the patch clamp technique is used. After the excision of the membrane patch, the activity of PIP2 dependent ion channels decreases, a phenomenon referred to as run-down. For many PIP2-sensitive channels the mechanism underlying run-down is the decrease in PIP2 concentration in the patch due to the action of membrane-bound lipid phosphatases present in the patch membrane. Mg2+ applied to the intracellular surface of the patch accelerates channel run-down by providing a cofactor for lipid phosphatases (Huang et al. 1998) and also by the direct screening of the negative charges of the phosphoinositides (Suh and Hille 2007; Piron et al. 2010). MgATP can prevent current run-down, or re-activate the channels after run-down (Hilgemann 1997; Sui et al. 1998) by providing substrate for lipid kinases that regenerate PtdIns(4,5)P2 from its precursors. Run-down can be slowed down with an inhibitory cocktail against lipid phosphatases (Hilgemann and Ball 1996; Zhang et al. 1999). This implies that most major kinases and phosphatases remain associated with the patch membrane even after excision. The velocity of the run-down of the activity of a given channel generally correlates with its apparent affinity for PIP2; channels with higher PIP2 affinity run down slower than channels with lower affinity. One can also apply PIP2 chelating agents, such as PIP2 antibody (Huang et al. 1998) or poly-Lysine (Lopes et al. 2002) to excised patches to accelerate run-down. Poly-Lys is less selective than the antibody but it works more reliably.
Perhaps the most direct way to study the effects of PIP2 on ion channels is to apply phsophoinositides directly to the cytoplasmic surface of excised inside-out patches after current rundown. Phosphoinositides with various lipid side chains are available for these experiments; PIP2 from natural sources has mainly arachidonyl-stearyl side chains, while synthetic PIP2 usually contains two palmitoyl side chains. These long acyl chain lipids accumulate in the patch membrane, thus it is difficult to control their effective concentration. After activation with these PIP2 analogues most ion channels run down quite slowly upon cessation of the application of the lipid, making repeated application of the these analogues impractical (Rohacs et al. 2002). Short acyl chain, water soluble (e.g. DiC8) analogues of PIP2 on the other hand activate most ion channels quickly and reversibly, presumably because they diffuse out the membrane easily upon washout (Rohacs et al. 1999). DiC8 phosphoinositides are water soluble, whereas long acyl chain phosphoinositides are found in micelles in aqueous solutions; these solutions often need to be sonicated in order to prevent aggregation of the lipids. It is important to keep in mind that the patch membrane contains significant amount of PIP2 in the cell-attached configuration.
-
(ii)
Biochemical approaches to study phosphoinositide binding to ion channels. Several techniques have been used to measure direct biochemical binding of phosphoinositides to ion channels (Huang et al. 1998; Soom et al. 2001; MacGregor et al. 2002). Most of these studies were performed with truncated cytoplasmic segments of ion channels. The advantage of this approach is that it measures direct association of phosphoinositides with ion channels. On the other hand, it is possible that the binding to these isolated channel fragments does not correspond to the biologically important interactions. In several cases mutations that affected PIP2 binding were reintroduced into the full length channel and functional effects were shown on phosphoinositide sensitivity (Huang et al. 1998). This is a strong argument for direct activation of a channel by PIP2. A perhaps even stronger evidence for direct activation is the demonstration of the effect of the lipid on a purified channel reconstituted in the artificial membranes of known composition. Recently activation by PIP2 of purified TRPM8 channels in lipid bilayers (Zakharian et al. 2009, 2010) and Kir channels in liposomes (D’Avanzo et al. 2010) have been demonstrated.
-
(iii)
Manipulations of phosphoinositide levels in living cells. Only limited pharmacological tools are available to inhibit various enzymes involved in PIP2 metabolism, and they are not very specific. At relatively low concentrations wortmannin (10–100 nM) and LY294002 (10 mM) are widely used as PI3K inhibitors. At higher concentrations (> 5 mM for wortmannin and > 100 mM for LY294002) they also inhibit PI4K isoforms (Balla 2001), thus slowly depleting PIP2 and also preventing the recovery of PIP2 levels after the PLC-mediated hydrolysis. PLC can be inhibited by U73122 and edelfosine, but these drugs have a number of side effects (Horowitz et al. 2005). In intact cells PIP2 levels can be modified using a variety of tools. PIP2 levels can be decreased by activating PLC via G-protein coupled receptors (PLCβ), receptor tyrosine kinases (PLCγ) or Ca2+ influx (probably PLCd). However, PLCs not only hydrolyze PIP2 but concurrently release IP3 and DAG which, in turn, trigger their own signalling cascades (e.g. release of Ca2+ from the IP3-sensitive stores, activation of PKC, arachidonic acid release etc.; Fig. 10.1) which often complicates interpretation of results. An alternative approach to modify PIP2 levels in living cells is by over-expression of various lipid kinases and phosphatases. The first generation of such tools contained constitutively active enzymes; for instance a widely used approach to tonically deplete PIP2 in cells is to overexpress construct contained PIP2-specific 5’ phosphotases of INP family from yeast (Stolz et al. 1998) tagged with GFP and a membrane localization sequence from tyrosine kinase Lyn. When overexpressed in cells such constructs localize to the plasma membrane and tonically deplete PIP2 by converting it into PIP. Similarly, overexpression of PI4- and PIP5-kinases is used to tonically increase membrane PIP2 levels. In a new generation of such probes, a phenomenon of chemically-induced dimeryzation (CID) has been utilized to make the lipid 5’ phosphatase or kinase activity to become acutely inducible in living cell. These constructs were independently developed in the labs of Tobias Meyer (Suh et al. 2006) and Tamas Balla (Varnai et al. 2006). The PIP2 depleting CID system uses two different proteins with high affinity to immunosuppressant rapamycin: the FRB domain of the mammalian target of rapamycin (mTOR) and the FK506 binding protein FKBP12. In a study by Suh and colleagues the rapamycin-binding domain of FRB was fused to the membrane-localisation tag of Lyn kinase while the rapamycin-binding domain of FKBP was attached to the 5’ phosphatase Inp54p and CFP. When these constructs were co-transfected together with Kv7.2/Kv7.3 channels into ts-A cells, acute addition of the rapamycin analogue induced rapid recruitment of Inp54p to plasma membrane, dephosphorylation of PIP2 and virtually complete inhibition of M channel activity (Suh et al. 2006). Similarly, TRPM8 channel activity was almost completely inhibited by rapamycin-induced PIP2 depletion (Varnai et al. 2006). Another type of inducible lipid phosphatase which is used to study PIP2-sensitivity of ion channels is voltage-sensitive phosphatase VSP which contains a voltage sensor domain homologous to those of voltage-gated ion channels and a 5’-phosphatase domain homologous to PTEN (Iwasaki et al. 2008). The phosphatase domain of VSP is inactive at potentials below 0 mV but can be activated by strong depolarization pulses (e.g. to above + 40 mV) (Iwasaki et al. 2008), thus strong depolarizing pulses to above + 40 mV induce rapid, reversible depletion of PIP2 in cells overexpressing VSP; this, in turn, was shown to inhibit Kir and Kv7 channels (Murata and Okamura 2007; Falkenburger et al. 2010c).
-
(iv)
Optical probes for phosphoinositide metabolism. Attachment of fluorescent proteins to different phosphoinositide-binding domains has been used for live monitoring of phosphoinositide levels in living cells. Thus, the pleckstrin homology (PH) domain of PLCd1 fused with GFP (PLC-PH-GFP) has been widely used to monitor PIP2 hydrolysis by PLC or dephosphorylation by Inp phosphatases. At basal conditions this probe localizes to the inner leaflet of the plasma membrane where it binds to PIP2; when PIP2 concentration in the membrane decreases (e.g. due to its hydrolysis to IP3 and DAG by PLC), the probe translocates to the cytoplasm, which can be easily monitored using confocal microscope. Similar fluorescence resonance energy transfer (FRET)-based variants of this probe have also been developed (van der Wal et al. 2001). These probes have been widely used for correlating kinetics of PIP2 hydrolysis with the activity of ion channel of interest (Mitchell et al. 1996; Hsuan et al. 1998). The disadvantage of the PLC-PH-GFP probe is that it has higher affinity to IP3 in vitro as compared to PIP2 (Hirose et al. 1999; Varnai and Balla 1998) thus, the interpretation of its translocation in terms of PIP2 levels is not straightforward (Varnai and Balla 2006; Liu et al. 2010). Recently a new PIP2 probe that does not bind IP3 has been developed (Quinn et al. 2008), it is based on the PIP2 affinity of the transcription factor tubby. The probe is probably a better reporter of the membrane PIP2 levels, although it is less sensitive an indicator of PLC activity compared to PLC-PH-GFP (Szentpetery et al. 2009; Liu et al. 2010). In addition to PIP2, similar optical probes have been developed for other lipids, such as PtdIns(3,4)P2, PtdIns(3,4,5)P3, PtdIns(4)P, DAG and others (Balla and Varnai 2009; Balla 2009).
10.3 Inwardly Rectifying K+ (Kir) Channels
Kir channels are K+ selective ion channels that conduct more current in the inward than in the outward direction if measured through a range of voltages in patch clamp experiments. They have two transmembrane domains per subunit, four of which form the functional homo or heterotetrameric channels. Most of them are open near the resting membrane potential and they conduct outward currents in most cases, but they close down upon major depolarizations, thus they allow the development of the action potentials. The mammalian family has 15 members, divided into various subfamilies, numbered 1–7 (Hibino et al. 2010). Most Kir channels are constitutively active, with two exceptions. G-protein activated inwardly rectifying K+ (GIRK) channels are members of the Kir3.x subfamily, while ATP inhibited K+ (KATP) channels are members of the Kir6.x family. GIRK channels are activated by the βγ subunits of heterotrimeric G-proteins and play roles in processes such as the regulation of heart rate by muscarinic stimulation and in the analgesic effects of opioids. KATP channels are inhibited by cytoplasmic ATP, and they are best known for their role in glucose-induced stimulation of insulin secretion. It has been shown that all members of the mammalian Kir channel family require PIP2 for activity (Rohacs et al. 2003; Du et al. 2004). Mutations in Kir channel genes may cause a variety of disease, such as diabetes, hyperinsulinemia, Andersen’s syndrome, Bartter’s syndrome, and vitreoretinal degeneration (Hibino et al. 2010).
10.3.1 How Does PIP2 Interact with Ion Channels?
This question is most thoroughly studied in Kir channels, and our understanding of how PIP2 interacts with channels is probably the most comprehensive here. The generally accepted view is that the negatively charged head group of PIP2 interacts with positively charged amino acid residues in the cytoplasmic domains of ion channels (Fig. 10.2b, d). Early work identified residues in the proximal C-terminus, close to the pore- forming second transmembrane domain that are involved in PIP2 interactions (Fan and Makielski 1997; Hilgemann and Ball 1996). Later several studies systematically mutated conserved positively charged residues in Kir channels to identify additional PIP2 interacting residues. As a general rule neutralizing a PIP2 interacting residue decreases Kir channel apparent PIP2 affinity (Fig. 10.2c), which is manifested in the decrease of channel open probability, decreased macroscopic current amplitude, increased speed of run down in excised patches, and increased inhibition by depletion of PIP2 (see below). The putative PIP2 interacting residues identified this way were located in various places in the linear sequence, including more distal regions in the C-terminus, and residues in the N-terminus (Lopes et al. 2002).

Phosphoinositide regulation of Kir channels. a Kir2.1 activity in an excised macropatch from a Xenopus oocyte at −80 mV. Current activity runs down after establishment of the inside out configuration (i/o). The channels are re-activated by the application of PIP2. (Modified from Rohacs et al. (1999) with permission). b Simplified cartoon showing that PIP2 interacts with a Kir channel through binding of the negatively charged head-group of the lipid to positively charged residues in the C- and N-termini of the channel protein. c The effect of the apparent affinity of the channel for PIP2. (From Rohacs (2007) with permission). Hypothetical concentration dependence of the effect of PIP2 on a channel with high (solid line), and low (dashed line) apparent affinity for PIP2. d A 3D model of Kir2.1 channels with putative PIP2 interacting residues, based on partial crystal structures of various Kir channels. (From Logothetis et al. (2007a) with permission)
When the partial crystal structures of various Kir channels were published, most of the putative PIP2 interacting residues identified earlier by mutagenesis (Lopes et al. 2002) lined up on the interface of the channel with the membrane, compatible with the idea that they are part of the PIP2 binding site. This shows that the mutagenesis approach is useful in finding putative PIP2 interacting residues. A homology model based on partial crystal structures of various Kir channels has been proposed to depict PIP2 interacting residues in Kir channels (Logothetis et al. 2007b) (Fig. 10.2d). Based on this model it is likely that positively charged residues from different parts of the channel come close together in 3 dimensions to form a PIP2 binding pocket.
Even though this relatively simple model is quite widely accepted, the real picture may be somewhat more complex, thus, several points of caution need to be made. First, the phosphoinositide specificity profile of Kir channels is variable, some channels, such as KATP are activated equally well by PtdIns(4,5)P2, PtdIns(3,4)P2 and PtdIns(3,4,5,)P3, whereas others such as Kir2.1, are activated specifically by PtdIns (4,5)P2, but not by the other two lipids (Rohacs et al. 2003). This stereospecific activation is difficult to explain with a purely charge mediated binding. Second, crystal structures of known phosphoinositide interacting soluble proteins show that in addition to positively charged amino acids, non-charged residues also invariably contribute to phosphoinositide binding (Rosenhouse-Dantsker and Logothetis 2007). Virtually no effort has been made so far to identify non-charged residues that interact with phosphoinositides in ion channels. Third, even when a positively charged residue is identified, mutation of which alters channel activation by PIP2, it is very difficult to tell with certainty, based on mutagenesis data whether this residue interacts directly with PIP2, or its mutation alters PIP2 interactions indirectly (Colquhoun 1998). Even when the crystal structure of the channel is solved without the interacting lipid, it is not trivial to dock PIP2 and tell which residue it is in contact with.
10.3.2 The Role of Apparent Affinity for PIP2 and Relationship to Other Regulators
Under resting conditions the inner leaflet of the plasma membrane contains significant amounts of PIP2. Whether this is enough to keep a particular PIP2 sensitive channel maximally open, depends on the apparent affinity of the channel for the lipid (Fig. 10.2c). Channels with high affinity for PIP2 cannot be further activated by excess PIP2 because resting PIP2 levels are saturating them. On the other hand, channels with lower PIP2 affinity can be theoretically further activated by increased PIP2 levels. Conversely, and more importantly, channels with lower PIP2 affinity can be easily inhibited by depletion of PIP2, whereas channels with high PIP2 affinity are not inhibited significantly by moderate (physiological) PIP2 depletions (Fig. 10.2c). However, even high affinity channels can be inhibited by complete depletion of PIP2, such as by applying a PIP2 chelator in excised patches.
Mutation of PIP2 interacting residues may convert a high affinity channel to a low affinity one, shifting its PIP2 dose-response to the right and rendering it more sensitive to inhibition by PIP2 depletion. This phenomenon is utilized in mutation studies aiming at locating putative PIP2 interacting residues (Lopes et al. 2002). In several Kir channels mutations are also found that strengthen PIP2 interactions, leading to decreased sensitivity for PIP2 depletion (Zhang et al. 1999; Du et al. 2004).
Even in the absence of mutations, the apparent affinity for PIP2 is not necessarily static. A classic example of PIP2 affinity modulated by a channel ligand is GIRK channels, where it was proposed that both Gβγ and Na+ open the channels by stabilizing its interaction with PIP2 (Huang et al. 1998; Zhang et al. 1999). This would manifest as a left shift in the PIP2 dose-response, and increased channel activity at a constant PIP2 concentration. Consistently with this, GIRK channel currents are inhibited less by PLC induced PIP2 depletion in the presence of excess Gβγ (Keselman et al. 2007). Intracellular Na+, another activator of several GIRK channels has a similar effect (Zhang et al. 1999). For Na+, a compelling mechanistic model was proposed recently to explain how it increases PIP2 sensitivity of Kir3.4 channels: Na+ binds to an Asp and His that triggers a structural switch that frees a crucial Arg enabling it to interact with PIP2 (Rosenhouse-Dantsker et al. 2008). A similar model was proposed later for Kir3.2, based on crystallographic studies (Inanobe et al. 2010). Many other modulators of Kir channels, such as protein kinase C, intracellular Mg2+ and pH have also been reported to affect channel PIP2 interactions (Du et al. 2004; Keselman et al. 2007).
10.3.3 Metabolic Regulation and Phosphoinositides—KATP Channels
Kir6.x channels are the pore-forming subunits of KATP channels. They are considered to be metabolic sensors directly inhibited by cytoplasmic ATP and they open in conditions when cytoplasmic ATP concentrations decrease. Functional KATP channels have an auxiliary subunit, the sulfonylurea receptor (SUR). ATP inhibits the channel through direct binding to the pore-forming Kir6.2 subunit and the SUR subunit modifies this effect. ADP on the other hand activates the channels through the SUR subunit, and the KATP channels are considered to be sensors of cellular ATP/ADP ratio. The two best characterized combinations are Kir6.2–SUR1, the KATP channel in insulin secreting pancreatic beta cells and the cardiac Kir6.2–SUR2A, found in ventricular cardiomyocytes. The physiological function of the pancreatic KATP channel is very well established, these cells respond to physiological changes in extracellular glucose concentrations by changes in intracellular ATP levels. An increase in extracellular glucose increases the ATP/ADP ratio inside these cells, leading to the closing of KATP channels, depolarizing the membrane potential and the consequential opening of voltage gated Ca2+ channels which, in turn, stimulates insulin secretion. In other cell types such as cardiomyocytes, physiological changes in cellular metabolism are not expected to change cellular ATP levels. KATP channels there are likely to act as brakes on cellular metabolism under severe metabolic conditions, such as ischemia, when they open, hyperpolarize the cell, and thus limit further activity.
KATP channels require PIP2 for activity, and their phosphoinositide regulation is intimately related to their metabolic regulation. In excised patches ATP sensitivity of these channels show a marked reduction after application of phosphoinositides (Shyng and Nichols 1998; Baukrowitz et al. 1998) and it was proposed that different phosphoinositide levels among different cells may underlie the well known variability of ATP sensitivity in excised patches. Both ATP and the head-group of PIP2 are highly negatively charged, and binding of both molecules to KATP channels is thought to involve positively charged residues. Mechanistically, it is possible that PIP2 and ATP bind to overlapping binding sites, and binding of ATP displaces the activating lipids head-group. Another important activator of KATP channels is long acyl chain coenzyme-A (LC-CoA) (Tucker and Baukrowitz 2008; Shumilina et al. 2006). Even though originally it was proposed that PIP2 and LC-CoA activates KATP channels via different mechanisms (Gribble et al. 1998), there has been a growing consensus that the negatively charged LC-CoA acts through the phosphoinositide binding site of Kir6.2 (Tucker and Baukrowitz 2008), based mainly on the following data. Most Kir channels show some level of isomer specificity among various phosphoinositides, with PtdIns(4,5)P2 being the most active, and are inhibited by LC-CoA (Rohacs et al. 2003; Rapedius et al. 2005). Kir6.2 channels on the other hand show no isomer selectivity among phosphoinositides, and are activated by LC-CoA (Rohacs et al. 2003). When Kir2.1 and Kir7.1 channels were engineered to be less selective among various isomers of PIP2, they were activated by LC-CoA (Rohacs et al. 2003). Furthermore, it was demonstrated that PIP2 binding to the C-terminus of Kir6.2 and Kir2.1 is antagonized by LC-CoA (Rapedius et al. 2005).
10.4 M-type (Kv7, KCNQ) Channels
Following Kir channels, the Kv7 K+ channel family gives another example of ‘classical’ PIP2-sensitive channels for which phosphoinositide binding site has been suggested and physiological role of the channel-PIP2 interaction has been confirmed. The direct PIP2-dependency of Kv7 open probability even allowed some researchers to use Kv7 channels as biosensors of plasma membrane PIP2 levels (much like for the case of Kir channels; see e.g. (Suh et al. 2006; Murata and Okamura 2007)).
In mammals there are five KCNQ genes (KCNQ1-5) coding for five Kv7 α-subunits (Kv7.1–Kv7.5) which give rise to several physiologically important potassium currents. In the mammalian central and peripheral nervous systems Kv7.2, Kv7.3 and Kv7.5 form the so-called ‘M-type channels’ underlying neuronal M current, an important cellular instrument for stabilizing neuronal resting membrane potential, setting the threshold for action potential firing and controlling firing frequency (Wang et al. 1998; Shapiro et al. 2000; Selyanko et al. 2002); reviewed in (Delmas and Brown 2005; Brown and Passmore 2009). The M current was discovered some 30 years ago by David Brown and colleagues (Brown and Adams 1980) as a specific K+ current fraction in sympathetic neurons which is characterized by slow kinetics of activation and inactivation, very negative (negative to −60 mV) threshold for activation and no inactivation under physiological conditions (Fig. 10.3a). In the original study (Brown and Adams 1980) this current fraction was eliminated by stimulation of muscarinic acetylcholine receptors (mAchR), hence, it received the name ‘M current’. Distinctive biophysical properties of neuronal M channels bestow them a strong control over neuronal excitability. Thus, the negative threshold for activation and no inactivation allows a fraction of M channels to be open near the resting membrane potential of a neuron whilst slow kinetics of activation confers a role in the accommodation (wearing-off) within the bursts of action potentials (Delmas and Brown 2005; Brown and Passmore 2009). The importance of M currents in mammalian CNS is exemplified by the fact that loss-of-function mutations within principal M channel genes KCNQ2 and KCNQ3 often result in a form of epilepsy, benign familial neonatal convulsions (BFNC) and even mutations causing as little as 25% of the M current reduction are sufficient to cause a disease (Maljevic et al. 2010); genetic deletion of KCNQ2 in mice is lethal (Watanabe et al. 2000). The general ‘rule of thumb’ is that neurons expressing high levels of M-current-forming Kv7 channels are ‘phasic’ neurons with high threshold for action potential firing (Jia et al. 2008); acute pharmacological or receptor-induced inhibition of M current (Jia et al. 2008; Liu et al. 2010) or genetic downregulation of KCNQ expression (Mucha et al. 2010) can switch these neurons into highly excitable, constantly firing (‘tonic’) phenotype which explains KCNQ-associated seizures as well as recently reported role of M channels in pain (Linley et al. 2008; Liu et al. 2010; Mucha et al. 2010).

PIP2 sensitivity of the Kv7 channels. a Whole-cell current traces elicited in CHO cells overexpressing Kv7.5 by the train of voltage pulses depicted underneath. Activation curve (normalized tail current amplitudes plotted against voltage) is given on the right. b Inhibition of Kv7.2/Kv7.3 current in CHO cells by the PIP2 depletion with Inp54p phosphatase. Cells were transfected with plasmids coding for KCNQ2, KCNQ3 with or without membrane-targeted Inp54p (Inp54p). Bars represent Kv7.2/7.3 current densities recorded in the whole-cell patch clamp mode from the cells transfected with KCNQ2, KCNQ3 only (Control) or with KCNQ2, KCNQ3 and Inp54p (Inp54p) or from KCNQ2, KCNQ3, Inp54p transfected cells with 100 mM DiC8-PIP2 added to the pipette solution. Trace on the left represent the time course of Kv7.2/7.3 current recovery in Inp54p-overexpressing cell upon breaking into whole cell with a pipette solution containing 100 mM DiC8-PIP2. (Modified from Linley et al. (2008) with permission). c PIP2 sensitivity of the Kv7 channels studied using singe-channel recordings. Individual traces recorded in the cell-attached (‘on-cell’) or inside-out configurations (as indicated) from patches of CHO cell membrane containing single Kv7.2 (left), Kv7.3 (middle) or Kv7.4 (right) channels in the presence of the indicated concentrations of DiC8-PIP2. (From Li et al. (2005) with permission). d. A dependency of the Po of Kv7.2 (squares), Kv7.3 (triangles), Kv7.4 (inverted triangles) and Kv7.2/Kv7.3 (circles) from the DiC8-PIP2 concentration in inside-out patches (from (Li et al. 2005) with permission). e Alignment of the putative PIP2-interacive domains of Kv7.2, Kv7.3 and Kv7.4 channels. The large and small boxes enclose a cluster of positively charged residues and a critical conserved basic residue, respectively. (Based on the data from Hernandez et al. (2008a))
Another important K+ current conducted by a member of Kv7 family, Kv7.1, can be found in the heart. In cardiomyocytes Kv7.1 multimerizes with its auxiliary subunit, KCNE1 to produce the slow component of the cardiac delayed rectifier current (IK), IKs (Barhanin et al. 1996; Sanguinetti et al. 1996; Wang et al. 1996). The IKs is responsible for the repolarization of the cardiac action potential and for the control of action potential duration (reviewed in Charpentier et al. (2010)). The loss-of-function mutations within KCNQ1 gene often result in the group of cardiac arrhythmias called inherited long QT syndrome form 1 (e.g. the autosomal dominant Romano-Ward syndrome and the autosomal recessive Jervel and Lange-Nielsen syndrome); the gain-of-function KCNQ1 mutations have also been reported and these result in familial atrial fibrillation and another form of arrhythmia—short QT syndrome (reviewed in Charpentier et al. (2010)).
Kv7.4 is a Kv7 subunit which is predominantly expressed in the auditory pathways and loss-of-function mutations within the KCNQ4 result in DFNA2 nonsyndromic hearing loss (Kubisch et al. 1999). KCNQ4 is abundantly expressed in the inner ear, particularly in the outer hair cells (OHCs) of the organ of Corti (Kubisch et al. 1999) as well as in several nuclei and tracts of the auditory pathways in the brainstem (Kharkovets et al. 2000). In OHCs Kv7.4 localizes to the basal membrane and might provide a pathway for the extrusion of potassium entering OHCs through the mechanosensitive channels at the apical membrane (Kharkovets et al. 2000, 2006).
In addition to the neuronal and cardiac roles, several Kv7 subunits are expressed in smooth (Greenwood and Ohya 2009) and skeletal (Iannotti et al. 2010) muscles and in epithelia (Vallon et al. 2005) where they may contribute to the control of contractility (Greenwood and Ohya 2009), skeletal muscle proliferation (Iannotti et al. 2010), transepithelial transport (Vallon et al. 2005) and cell volume regulation (Piron et al. 2010).
As discussed above, a number of important physiological roles for Kv7 channels have been identified, accordingly, mutations or other impairments of Kv7 channels often result in severe disorders (e.g. seizures, pain, arrhythmias and deafness), therefore Kv7 channel regulation (to which PIP2 plays one of the key roles) has attracted some high-profile research.
10.4.1 M Channel Modulation: Focus on PIP2
The M current has been discovered as a neuronal K+ current fraction inhibited by mAchRs. Later it became apparent that not only mAchRs (more precisely, M1, M3 and M5 mAchR isoforms), but also receptors for bradykinin, angiotensin II, histamine, protease activated receptor-2 (PAR-2), P2Y receptors and potentially any other GPCR that is coupled to the Gq/11 subtype of G proteins can inhibit M channels (reviewed in (Delmas and Brown 2005; Gamper and Shapiro 2007a; Linley et al. 2010)). Deciphering the signalling pathways linking GPCR and M channels took some time though.
Before the KCNQ genes have been cloned, most of the M current research has been performed on sympathetic neurons which express robust M currents. It was soon discovered that in these neurons the muscarinic inhibition of M current is mediated by M1 mAchR (Marrion et al. 1989) and require Gq or G11 type of Gα subunits (Haley et al. 1998, 2000) and their usual downstream effector, PLCβ. An important experiment by A. Selyanko in David Brown’s group demonstrated that external application of muscarinic agonist inhibited M channels isolated in cell-attached patches from the superior cervical ganglion (SCG) sympathetic neurons. Based on the distinction first made by Soejima and Noma (1984) between “membrane-delimited” and “diffusible messenger” signalling (Soejima and Noma 1984), it was concluded that muscarinic M current inhibition must be mediated by a diffusible intracellular second messenger (Selyanko et al. 1992). It required an additional 10 years of concentrated effort before this elusive “mystery” messenger has been identified and, as it often happens, it turned out to be not quite what everyone has been looking for.
After all ‘usual suspects’ of the PLC signalling cascade (Fig. 10.1) have been exhaustively probed and failed to satisfy the experimental data (see e.g. (Robbins et al. 1993; Hille 1994; Marrion 1997)), complementary studies by the Hille and Logothetis groups came to a suggestion that the actual mediator of PLC-induced M current inhibition may not be a downstream product of PIP2 hydrolysis but the PIP2 hydrolysis itself. Indeed, the recovery of M current amplitude from the mAchR-mediated inhibition was shown to be prevented by blocking PIP2 resynthesis with a PI4-kinase inhibitor (Suh and Hille 2002; Zhang et al. 2003). Furthermore, the application of standard phosphoinositide research toolkit soon revealed that Kv7/M channels (both in the expression systems and in SCG neurons) are indeed highly PIP2 sensitive. Thus, currents from cloned Kv7.2/Kv7.3 channels expressed in Xenopus oocytes ran-down upon patch excision and this run-down was successfully prevented or attenuated by addition of PIP2 (or an analog) to the inner leaflet of the plasma membrane; Kv7 current in excised patches was promptly inhibited by PIP2 scavengers such as anti-PIP2 antibody and polylysine (Zhang et al. 2003). Overexpression of Inp54p phosphotase tonically inhibited Kv7.2/7.3 currents in CHO cells while perfusion of DiC8-PIP2 through the patch pipette rapidly recovered current amplitude in the Inp54p-overexpressing cells (Fig. 10.3b; (Li et al. 2005; Linley et al. 2008)). Similar experiments were later repeated for Kv7.1 and IKs channels (Loussouarn et al. 2003; Piron et al. 2010) and a similar PIP2 requirement for channel activity has been seen (with the exception of voltage dependency of the PIP2 effect seen with Kv7.1 channels, see below).
Development of new optical and biochemical tools for monitoring and manipulating PIP2 levels in living cells allowed researchers to further probe the relationships between receptor-mediated PIP2 hydrolysis and Kv7 channel activity. Thus the PLCd–PH probes have been extensively used to correlate the PIP2 hydrolysis by PLC with the Kv7/M current inhibition in the expression systems and neurons (e.g. Horowitz et al. 2005; Winks et al. 2005; Falkenburger et al. 2010c); indeed, these experiments found a good correlation between the kinetics of both processes (especially in the expression systems) and allowed to compose a detailed kinetic models taking into account affinities, abundances and kinetics of interaction for different key players of Gq/11 signalling (such as receptors, Gα, Gβ γ, PLC, etc.), PIP2 and Kv7 subunits (Hernandez et al. 2009; Falkenburger et al. 2010b).
A significant complication of PLC signalling, which for some time casted a cloud of doubt over the ‘PIP2 hypothesis’, is the fact that the PIP2 hydrolysis is always accompanied by the concurrent release of several second messengers (Fig. 10.1) and some of them were also shown to cause M current inhibition (e.g. Ca2+ (Selyanko and Brown 1996; Cruzblanca et al. 1998; Gamper and Shapiro 2003; Gamper et al. 2005) and DAG/PKC (Hoshi et al. 2003)). Thus, while PIP2 sensitivity of Kv7 channels per se was convincingly demonstrated, it was not clear whether receptor-mediated Kv7/M current inhibition in native cells can be solely mediated by the PIP2 depletion. To a certain degree this question has been clarified with the use of inducible 5’-phosphatases (VSPs and rapamycin-inducible CID system). Unlike PLC, these phosphatases convert PtdIns(4,5)P2 to PtdIns(4)P without the release of any relevant second messengers; nevertheless, both type of inducible phosphatases were shown to be able to inhibit Kv7/M channel activity almost completely (Suh et al. 2006; Murata and Okamura 2007; Falkenburger et al. 2010c). There were also other types of experiments which solidified the ‘PIP2 hypothesis’. Thus, application of highly-basic palmitoilated PIP2-binding peptides reduced M current in SCG neurons and sensitized the current to depression by muscarinic stimulation (Robbins et al. 2006). In contrast, when tonic membrane PIP2 levels were elevated by over-expression of PIP5-kinase, the tonic amplitude of overexpressed Kv7.2/7.3 channels dramatically increased (Li et al. 2005). A similar maneuver reduced the extent of muscarinic suppression of M current in sympathetic neurons (Winks et al. 2005).
10.4.2 Structure-functional Aspects of Kv7 Channel Sensitivity to PIP2
Single channel recordings from the cells expressing cloned Kv7 channels revealed that homomeric channels assembled from the individual Kv7 subunits have distinct and highly-variable maximal open probability (Po) in cell attached patches (Selyanko et al. 2001; Li et al. 2004). Thus, the tonic Po (at saturating voltages) of Kv7.2 and Kv7.4 is very low (~ 0.1), the Po of Kv7.3 is near unity and the Po of Kv7.2/7.3 heteromultimers is in the range of 0.3 (Selyanko et al. 2001; Li et al. 2004, 2005). In a series of inside-out single channel experiments Li et al. (2005) found out that the Po of Kv7 channels tested (Kv7.2, Kv7.3, heteromeric Kv7.2/Kv7.3 and Kv7.4) can be interpreted as a Hill function of the DiC8-PIP2 concentration (with Hill coefficients between 1 and 1.7; Fig. 10.3c, d). Interestingly, these experiments also revealed that Kv7 channels with low tonic Pomax (Kv7.2 and Kv7.4) had approximately 100 times lower apparent PIP2 affinity as compared to Kv7.3 which tonic Pomax » 1 (DiC8-PIP2 EC50 ~ 200 mM vs.~ 2 mM); Kv7.2/7.3 heteromultimers had an intermediate values for both the PIP2 affinity (EC50 ~ 40 mM) and Pomax (~ 0.3), in accord with them being heteromeric channels containing subunits with both high and low PIP2 affinity. Thus, it has been suggested that the tonic activity of Kv7 subunits depends directly on their apparent PIP2 affinity and on the tonic concentration of PIP2 in the plasma membrane. Furthermore, it was hypothesized that the Po of Kv7 channels is directly governed by membrane PIP2 abundance.
The different intrinsic affinity for PIP2 of different Kv7 channels implies that M channels assembled from different Kv7 subunits should respond to muscarinic stimulation with different sensitivities, and indeed, this is what has been observed: the concentration-dependency of the inhibition of homomeric Kv7.3 and Kv7.4 channels by M1 mAchR agonist Oxotremorin-M had IC50 of 1 mM and 66 nM respectively (Hernandez et al. 2009).
The single channel analysis of Kv7 channel PIP2 dependence has been extended in the further work by the Shapiro group which used chimeras between high- and low-PIP2-affinity Kv7 isoforms (Kv7.3 and Kv7.4) to pin-point a site of PIP2 binding within the Kv7 channels. This chimeric approach in combination with point-mutations, homology modeling and energy minimization analysis revealed a cluster of positively-charged amino acids within the linker between the first two (out of four) helical domains of Kv7 carboxy-termini (helixes A and B) as such PIP2 binding site (Fig. 10.3e (Hernandez et al. 2008a)). The motif identified in Kv7 channels contained conserved K/R residues at the positions (in Kv7.2) 425, 452, 459, 461, 463 and 467 (Fig. 10.3e) which were suggested to play a key role in the channel interaction with PIP2. Homology modelling based on the solved structure of the PIP2 binding sites of Kir2.1 (Pegan et al. 2005) implied that Kv7 channels may have PIP2-binding modules which structurally are similar to Kir2.1. In another study (Zhang et al. 2003) a more proximal positively charged residue of the C-terminus (H328 of Kv7.2) has been suggested to participate in the channel interaction with PIP2. It is thus conceivable that while chimeric approach did identify some core PIP2 binding residues within Kv7 channel, there may be some other regions within Kv7 channel proteins that participate in the interactions with PIP2; in addition a caution needs to be taken regarding the modelling of Kv7 channels on the basis Kir channel structure as the homology between these two channel families is not close.
Interestingly, while swapping the A-B linker between Kv7.4 and Kv7.3 does invert PIP2 sensitivity of the channels, the suggested PIP2-binding K/R residues are conserved among the Kv7.2-Kv7.5 channels (but not in Kv7.1, see below). Thus, it is still unclear if the strikingly different apparent PIP2 affinities of individual Kv7 subunits (e.g. ~ 100-fold difference in DiC8-PIP2 EC50 between Kv7.4 and Kv7.3) arise from the different biochemical binding affinities of individual PIP2 binding sites or from the divergent coupling efficiencies between the PIP2-binding domains and the gating machinery of the channel (Hernandez et al. 2008a).
Kv7.1 is a member of Kv7 family which in many structural and functional aspects stands apart from the rest of the family (e.g. it is the only Kv7 channel that inactivates, it is not inhibited by Ca2+ etc.); the part of the Kv7.1 C-terminus which is homologous to the putative PIP2 binding motif of Kv7.2–Kv7.5 carries much less similarity with the rest of the family. Accordingly, the putative PIP2-interacting residues that were identified within the Kv7.1 are distributed more diffusely. Among three putative PIP2-interacting residues identified within the Kv7.1 two belong to the helix B (R539 and R555) of the C-terminus, another putative site was identified as an arginine within the S4-S5 linker (R243) (Park et al. 2005).
Interestingly, PIP2 dependency of the Kv7.1 channel gating shows a noticeable dissimilarity from that of other Kv7s: the action of PIP2 on Kv7.2–Kv7.4 comprises of the voltage-independent increase in channel Po while voltage-dependence and kinetics of channel activation and deactivation is not affected (Li et al. 2005; Delmas et al. 2005); in contrast, binding of PIP2 to Kv7.1 induces negative shift in voltage-dependence and slows deactivation (Loussouarn et al. 2003; Piron et al. 2010). While it is accepted that for all Kv7s PIP2 acts to stabilise the open state of the channel, the difference in the effect of PIP2 on channel gating further highlights likely structural dissimilarity in PIP2 action on KV7.1 and the rest of Kv7 family.
10.4.3 PIP2 Depletion by GPCR Activation in Neurons—Is It Really Happening?
While experiments with inducible phosphatases did unambiguously prove that Kv7 channels can be acutely inhibited by PIP2 depletion in living cells, what these experiments did not prove is whether physiological M channel inhibition by the PLC-coupled GPCR in vivo is indeed mediated by PIP2 depletion or, to put it differently, whether GPCR activation in neurons can produce enough PIP2 depletion to inhibit native M current without the need for other second messengers. For the muscarinic suppression of M current in sympathetic SCG neurons the answer is most likely ‘yes’ as the other second messengers produced by the PLC hydrolysis were ruled out by exhaustive experimentation (reviewed in Delmas et al. (2005); Gamper and Shapiro (2007b)), moreover, M1 receptors in SCG do not couple to IP3-sensitive Ca2+ stores and do not release Ca2+, which is another potent inhibitor of M channels (Selyanko and Brown 1996; Cruzblanca et al. 1998; Gamper and Shapiro 2003; Zaika et al. 2007). However, in more general terms the answer is probably “no” or “not quite” as even the pioneers of ‘PIP2 hypothesis’, Hillgeman (Hilgemann et al. 2001) and Hille (Falkenburger et al. 2010a) acknowledge that it is very difficult to envision a PLC-mediated PIP2 depletion as a specific signalling mechanism on its own as strong PIP2 depletion would simultaneously ‘shut down’ too many membrane proteins. Accordingly, a closer look at the modulation of native M currents by other Gq/11-PLC-coupled receptors revealed more complex nature of this signalling cascade. Thus, endogenous B2 (bradykinin) and P2Y (purinergic) receptors in SCG neurons do induce IP3-mediated rises in cytosolic Ca2+ and only weakly suppress M current if intracellular Ca2+ is held constant, IP3 receptors are blocked, Ca2+ stores are depleted or when an IP3 phosphatase or an IP3 sponge is over-expressed (Shapiro et al. 1994; Cruzblanca et al. 1998; Delmas et al. 2002; Gamper and Shapiro 2003; Zaika et al. 2006, 2007). Similarly, in sensory neurons from dorsal root ganglia (DRG), bradykinin B2 (Liu et al. 2010) and PAR-2 receptors (Linley et al. 2008) robustly inhibit native M current but mostly via Ca2+-mediated mechanism while saturation of the plasma membrane with the excess of DiC8-PIP2 by the intracellular dialysis only marginally reduces such inhibition (Linley et al. 2008). Moreover, study by Liu and colleagues suggested that the degree of membrane PIP2 depletion estimated with the optical probes based on the PH domain of PLCd (hitherto a major PIP2 probe used by many labs) is likely to be overestimated as this probe has higher affinity for IP3 than for PIP2 (Hirose et al. 1999). Indeed, translocation of the probe from membrane to the cytosol may not necessarily indicate a significant drop in membrane PIP2 levels, as IP3, the hydrolysis product of PIP2, may also cause the probe to translocate (Gamper et al. 2004; Liu et al. 2010). Accordingly, in DRG neurons PLCd-PH probe robustly translocated to cytosol in response to bradykinin stimulation but another PIP2 probe, YFP-tubby, which does not bind IP3 (Quinn et al. 2008), did not translocate unless exogenous B2 receptors are overexpressed (Liu et al. 2010). These observations suggest that in DRG neurons bradykinin induces enough PIP2 hydrolysis to produce IP3 necessary for Ca2+ release from the stores and to cause PLCd-PH probe to translocate but the overall drop in the membrane PIP2 level is not sufficient to cause YFP-tubby probe translocation or to significantly inhibit M current. Thus it is likely that for the many PLC-mediated signalling pathways PIP2 depletion is a contributing factor but not a sole mediator of M current inhibition. In a most likely scenario, activation of PLC by a GPCR concomitantly triggers three different signals that modulate M channel activity in a cumulative way: (i) some drop in membrane PIP2 (probably localized, although see (Gamper and Shapiro 2007b) for discussion of problems with local PIP2 depletion); (ii) release of Ca2+ from intracellular stores, Ca2+-bound calmodulin then inhibits M channels (Gamper and Shapiro 2003; Gamper et al. 2005); (iii) activation of PKC and AKAP-dependent phosphorylation of M channel protein (Hoshi et al. 2003; Bal et al. 2010). These concurrent pathways are ultimately interrelated as phosphorylation of Kv7.2 by PKC increases the sensitivity of this M channel to muscarinic inhibition (presumably by decreasing channel affinity to PIP2) (Hoshi et al. 2003; Bal et al. 2010), likewise, since the suggested calmodulin- and PIP2 binding sites are in close proximity or overlap (Yus-Najera et al. 2002; Gamper and Shapiro 2003; Hernandez et al. 2008a), calmodulin binding to M channel could compete PIP2 off the channel and thus reduce channel PIP2 affinity (the opposite should also hold true: PIP2 depletion should increase the affinity for calmodulin binding) (Gamper and Shapiro 2007a). This putative ‘coincidence detection’ mechanism may insure the fidelity and specificity of PLC-mediated regulation of M channels (see more on this issue in our recent reviews (Gamper and Shapiro 2007a, b)).
10.4.4 Physiological Significance of PLC-mediated M Channel Inhibition
This topic has been discussed at length in many recent reviews (e.g. Delmas and Brown 2005; Gamper and Shapiro 2007b; Hernandez et al. 2008b; Brown and Passmore 2009) therefore here we will just briefly outline the major concepts: (i) PLC-mediated M-type channel inhibition underlies the excitatory action of neurotransmitters (acetylcholine) and neuropeptides (e.g. bradykinin and angiotensin II; reviewed in Delmas and Brown (2005)); (ii) muscarinic inhibition of presinaptic M currents has been suggested to facilitate neurotransmitter release (Hernandez et al. 2008b; Kubista et al. 2009); (iii) in the PNS inhibition of M channels in nociceptive sensory fibers by the inflammatory mediators bradykinin and proteases mediates acute inflammatory pain (Linley et al. 2008, 2010; Liu et al. 2010).
10.5 Voltage-gated Ca2+ Channels
Although PIP2 sensitivity of voltage-gated Ca2+ channels is less understood than that of Kv7s, the research in both fields historically paralleled each other in many ways as both VGCC and M channels are inhibited by M1 AchR stimulation in SCG neurons and a common second (‘mystery’) messenger has been suggested (Bernheim et al. 1991; Mathie et al. 1992; Hille 1994).
VGCC form a large family of voltage gated ion channels which are selectively permeable to Ca2+. VGCC are expressed in all types of excitable cells where they mediate release of neurotransmitters from synaptic terminals, secretion of neuromediators and hormones by neurons and neuroendocrine cells, excitation-contraction coupling and gene expression (see (Catterall 2000) for review). The family contains ‘high-voltage-activated’ channels (L-, N-, P/Q- and R-type) which are activated by strong depolarizations (above ~− 30 mV), and ‘low-voltage-activated’ T-type channels which are activated at more negative voltages (threshold voltage ~− 50 mV). The assembly of a VGCC is quite complex, it contains a pore-forming subunit α1, which has 24 transmembrane domains (TMD) organized in four 6-TMD repeats (with each 6-TMD repeat being analogous to a single α subunit of a voltage-gated K+ channel). One α1 subunit is sufficient to provide a pore-forming channel core, however, functional VGCC are usually assembled with auxiliary subunits: β, α2d and, in some cases, γ (with the exception of the T-type channels α1 subunit, which is sufficient to form functional channel). There are three groups of α1 subunits: Cav1.1–Cav1.4 are pore-forming subunits of L-type channels; Cav2.1 form P/Q channels, Cav2.2 form N-type channels, Cav2.3 form R-type channels and Cav3.1–Cav3.3 form T-type channels.
VGCC are very important mediators of Ca2+ influx and, thus, these channels are targeted by multiple and complex regulatory and modulatory signalling cascades. Particularly well researched is the regulation of VGCC by GPCR. There are two major pathways of such regulation. (i) The ‘fast’ pathway is voltage-dependent, membrane delimited and is mediated by the Gβγ subunits; this pathway is mediated by the Pertussis Toxin-sensitive, Go/i-coupled GPCR and is understood as direct voltage-dependent interaction of the channel with Gβγ subunits (Bean 1989; Lipscombe et al. 1989; Herlitze et al. 1996; Zamponi and Snutch 1998). (ii) The ‘slow’ pathway encompass a group of mechanisms which share some common features such as voltage independence, often lack of sensitivity to PTX, and much slower (10 s seconds) kinetics as compared to direct Gβγ inhibition (100 s ms). One of such slow pathways is initiated by the Gq-coupled receptors (Bernheim et al. 1991; Delmas and Brown 2005; Michailidis et al. 2007; Roberts-Crowley et al. 2009) and it has been suggested that this Gq-mediated slow pathway may require the same second messenger as the M channel inhibition (Bernheim et al. 1991; Mathie et al. 1992; Hille 1994). A growing body of evidence suggest that indeed, as in the case of Kv7/M channels, many types of VGCC are PIP2 sensitive and receptor-mediated PIP2 depletion is, again, a plausible candidate for a mediator of VGCC inhibition by some Gq/11 receptor agonists.
10.5.1 Experimental Evidence for PIP2 Sensitivity of N-, P/Q- and L-type Channels
The first indications that some VGCC may require PIP2 for their activity were published around the same time as that for the M channels: Wu et al discovered that the run-down of cloned P/Q-type Ca2+ channels in inside-out macropatches can be reversed by application of PIP2 to the inner leaflet of the plasma membrane (Wu et al. 2002). Later, these findings were extended to N- (Gamper et al. 2004) and L-type (Michailidis et al. 2007) channels (Fig. 10.4a). The same set of techniques used to study PIP2 sensitivity of Kir and Kv7 channels has since been applied to VGCC. Thus, anti-PIP2 antibodies accelerated the run-down of P/Q-type channels, an effect reversed by direct application of PIP2 (Wu et al. 2002). Chelation of membrane PIP2 by the overexpression of PLCd-PH construct (Gamper et al. 2004; Suh et al. 2010) or tonic depletion of PIP2 by overexpression of Inp54p 5’ phosphatase reduced current density of native VGCC (mostly N-type) in SCG neurons and reduced the amount of VGCC inhibition by M1 AchR triggering (Gamper et al. 2004). Furthermore, it has been found that recovery of N-type current from the muscarinic inhibition was abolished by PI4 kinase blockade with wortmannin (Fig. 10.4b) whereas the inhibition itself was attenuated by dialysis of DiC8-PIP2 via the patch pipette (Gamper et al. 2004). Hille’s group used the inducible phosphatase approach to probe if PLC-independent PIP2 depletion can inhibit heterologousely expressed VGCC and also to screen for PIP2-sensitive VGCC isoforms (Suh et al. 2010). This study has confirmed major original findings of Jiang’s and Shapiro’s groups (Wu et al. 2002; Gamper et al. 2004) and brought several important conclusions in support of the ‘PIP2 hypothesis’ for VGCC: (i) inducible enzymatic depletion of membrane PIP2 without any GPCR or PLC activation and without co-release of any relevant signalling molecules can inhibit N-, P/Q- and L-type (Cav1.2 and Cav1.3) channels (Fig. 10.4c); (ii) inducible enzymatic PIP2 depletion prevented (Cav1.3) or dramatically reduced (Cav2.2) subsequent muscarinic inhibition of Ca2+ currents; (iii) kinetics of PIP2-sensitive VGCC inhibition and recovery follows the kinetics of enzymatic PIP2 depletion and recovery closely (especially true for Cav1.3 channels although not so true for Cav2.2; see below).

PIP2 sensitivity of VGCC. a Reactivation of N-type Ca2+ channels by PIP2 in inside-out macropatches. Shown on the right is a current trace evoked by the voltage step from −80 to +100 mV (voltage protocol is given above the trace) in the macropatch from Xenopus laevis oocyte overexpressing N-type Ca2+ channels. Shown on the right is a time course of rundown and reactivation by PIP2 application of the tail current induced by stepping from +100 to −40 mV. (From Gamper et al. (2004) with permission). b Inhibition of PIP2 resynthesis with wortmannin in cultured sympathetic neurons prevents recovery of VGCC from muscarinic modulation. Plotted are the amplitudes of inward Ca2+ currents evoked by 15 ms depolarizing voltage pulses given every 3 s from a holding potential of −80 to +5 mV recorded in the perforated patch configuration of the patch-clamp technique. Top panel shows a control experiment in which the neuron was treated with vehicle (0.1% DMSO). DMSO, Oxotremorine (Oxo, 10 mM), and CdCl2 (Cd2+; 100 mM) were applied during the periods indicated by the shaded areas. Insets on the right depict current traces recorded at times indicated. Lower panel depicts similar experiment but wortmannin (Wort; 50 mM) was applied instead of DMSO. (From Gamper et al. (2004) with permission). c Inhibition of CaV1.3 L-type Ca2+ channels in tsA cells by voltage-sensitive phosphatase. Typical current traces before and after activation of Dr-VSP by depolarizations to +120 mV. Cells without Dr-VSP (Control), cells transfected with Dr-VSP, or cells transfected with Dr-VSP and PI5-K received a 10 ms test pulse to −10 mV and then were depolarized to +120 mV for zero or 0.5 s followed by a second test pulse (voltage protocol is depicted above). The currents before (A) and after (B) the + 120 mV-depolarizing pulse are superimposed. (From Suh et al. (2010) with permission). d Putative PIP2-interactive residues within VGCC. Shown are amino acid sequences of the S6 TMDs in the four repeats of Cav2.1 α subunit and the S6 segment in the third repeat of Cav2.2 and Cav1.2 α subunits. I1520 in Cav2.1 and homologous residues in Cav2.2 and Cav1.2 are shown in grey. Cartoon depicting transmembrane topology of the α1 subunit of voltage-gated Ca2+ channels is shown above; grey dot at the intracellular end of S6 within the third repeat indicates the location of I1520. (From Zhen et al. (2006) with permission)
Not all experiments on PIP2 dependency of VGCC are coherent, thus, cloned P/Q- (Wu et al. 2002) and N-type (Gamper et al. 2004) VGCC expressed in oocytes appear to display a bi-modal sensitivity to PIP2: low concentrations of PIP2 produced a voltage-independent stabilizing effect, whereas higher concentrations induced a positive shift of channel voltage-dependence reminiscent of the transition from ‘willing’ to ‘reluctant’ (terms used to denote either free or Gβγ-bound channels in a Go/i-coupled receptor modulation paradigm (Bean 1989; Ikeda 1996; Herlitze et al. 1997)) states of VGCC. Accordingly, a model has been proposed in which P/Q- and N-type channels have two distinct PIP2 binding sites: a higher-affinity site that binds PIP2 to maintain channel activity and a lower affinity site which, when PIP2 is bound, shifts the channel into the ‘reluctant’ mode (Wu et al. 2002; Michailidis et al. 2007). However, the voltage-dependent action of PIP2 on these channels was not observed in the whole cell experiments in SCG neurons (Gamper et al. 2004), likewise, little evidence for voltage dependence of PIP2 effect was found in the whole cell experiments on the L- and N-type channels overexpressed in ts-A cells (Suh et al. 2010). It has been hypothesized that channel phosphorylation or possibly some cytosolic factor that modifies VGCC sensitivity to PIP2 can be lost in excised-patch experiments (Suh et al. 2010). Despite of this slight discrepancy, the excised-patch and the whole-cell experiments do suggest that the ‘slow’ pathway of Gq/11-coupled receptor-induced VGCC inhibition in neurons can be mediated (at least in part) by a high-affinity, voltage independent action of PIP2.
PIP2-binding site(s) within VGCC remain elusive. Low-specificity interactions between the Cav2.1 (P/Q-type) subunit C-terminus and several phosphoinositide species have been reported (Rousset et al. 2004). In addition, a substitution of single isoleucin (I1520) by histidine or aspartate in the cytosolic loop after S6 in the third 6-TMD repeat significantly attenuated the run-down of recombinant P/Q channels in inside-out patches and prevented channel inhibition by PIP2-scavenging MARCKS peptide (Fig. 10.4d); similar effects were seen after substitution of homologous residues in N- and L-type channels (Zhen et al. 2006). These effects were attributed to the changes in channel-PIP2 interaction. Mutagenesis experiments described above are suggestive but further work is needed to characterize PIP2 binding sites within VGCC.
10.5.2 PIP2 vs. Arachidonic Acid
There is a competing hypothesis for the ‘slow’ Gq/11-mediated inhibition of L-, N- and P/Q channels according to which the main second messenger is the arachidonic acid (AA). AA is a frequent constituent of phospholipids, including PIP2 as it is covalently attached to the C2 (sn-2) carbon atom of the glycerol backbone of phospholipids; estimated 80% of PIP2 has AA in the sn-2 position (Wenk et al. 2003; Roberts-Crowley et al. 2009). Phospholipase A2 group IVa (cPLA2) selectivity cleaves AA at the sn-2 position of phospholipids (Leslie 2004). cPLA2 can bind to PIP2 via its C2 domain and Gq/11 receptor stimulation can acutely activate cPLA2 via the ERK1/2-dependent phosphorylation (Roberts-Crowley et al. 2009). Thus, the same receptors that trigger PIP2 hydrolysis can cause concurrent release of the AA which, according to the ‘AA hypothesis’, is the main signal mediating VGCC inhibition. In support of this hypothesis, exogenously applied AA inhibits currents of native and recombinant VGCC of major subtypes with IC50 in the range of 1–10 mM, which is considered as a physiologically relevant range (Xiao et al. 1997; Vellani et al. 2000; Zhang et al. 2000; Liu et al. 2001; Talavera et al. 2004; Liu 2007). In contrast to PIP2 which stabilise the open state of the channels, AA was suggested to stabilize the closed state (Roberts-Crowley et al. 2009). Several other experiments, mostly by the Rittenhouse group, suggested involvement of the cPLA2 in the Gq-mediated inhibition of N- and L-type VGCC in SCG neurons. Thus, muscarinic stimulation of SCG neurons was shown to induce phosphorylation of the cPLA2 protein (Liu et al. 2006), moreover, pharmacological inhibition of PLA reduced N-type Ca2+ current inhibition by Oxo-M (Liu and Rittenhouse 2003). Likewise, L-type channel inhibition by Oxo-M was lost in neurons from cPLA2 − / − mice (no change in M current inhibition by Oxo-M in such neurons was noticed) (Liu et al. 2006) (for further discussion of regulation of VGCC by AA see the excellent recent review (Roberts-Crowley et al. 2009)).
As for the case of PIP2, the evidence for the sensitivity of VGCC to the exogenously applied AA is sound but whether AA is a second messenger of the receptor-mediated physiological signals regulating the VGCC in native neurons is much harder to prove due to the plethora of second messengers released by GPCR. In addition, some labs were unable to find evidence in support for the requirement of PLA for the Gq/11-mdiated inhibition of N- and L-type channels (Bannister et al. 2002; Gamper et al. 2004; Lechner et al. 2005). An experimental design which would allow enzymatic release of AA without concurrent production of other second messengers (similar to inducible phosphotases developed to probe the PIP2 sensitivity of channels) would help to further support the ‘AA hypothesis’.
As in the case for M channels, attempts to unify the ‘PIP2’ and the ‘AA’ hypotheses into a ‘coincidence detection’ mechanism has been made (e.g. Gamper and Shapiro 2007a; Roberts-Crowley et al. 2009) with the most comprehensive model proposed by the Rittenhouse group. In this hypothesis it is suggested that PIP2 is docked within a VGCC channel complex in such a way that its inositol head group binds to one site within the channel while its AA tail interacts with another binding site. Stimulation of a Gq/11-coupled receptor simultaneously (or in a rapid succession) activates PLC, PLA2 and DAG lipase which, in turn, comprehensively degrade PIP2 molecule into IP3, glycerol and free fatty acids. This full degradation is needed for maximal destabilization of the open state of the channel (Roberts-Crowley et al. 2009). The hypothesis is very attractive as it accounts for many conflicting evidence from both ‘PIP2’ and ‘AA’ hypotheses and also provides some way of specificity for the Gq/11 signalling towards the VGCC channels as it ensures that simple PIP2 hydrolysis is not enough to produce maximal inhibition of VGCC. However, new data from the inducible phosphatase experiments (Suh et al. 2010), which suggest that L-, N- and P/Q-type VGCC can be significantly inhibited by the conversion of PtdIns(4,5)P2 into PtdIns(4)P (without AA release) pose some difficulty here. Nevertheless, at least for the N-type channels, there is an additional small component of inhibition induced by Oxo-M which is not prevented by PIP2 dephosphorylation (Suh et al. 2010). This may indicate a need for a cofactor such as AA. Clearly further research is needed to develop inclusive model for the slow pathway of VGCC inhibition.
10.5.3 Possible Physiological Implications
The physiological significance of VGCC regulation is hard to overestimate since the activity of these channels control synaptic transmission, muscle contraction and gene expression. Accordingly, dysfunctions of VGCC cause severe human conditions ranging from movement disorders, arrhythmias and hypertension to neurological disorders, epilepsy and migraine (Gribkoff 2006). Emerging evidence suggest that PIP2 stabilizes activity of P/Q-, N- and L-type VGCC and receptor-mediated PIP2 depletion underlies (or at least contributes to) inhibition of these channels. Therefore, it is straightforward to suggest that PIP2 sensitivity of VGCC may provide one of the core mechanisms for control over the physiological processes which are regulated through VGCC.
10.6 TRP Channels
Transient Receptor Potential (TRP) channels are distant relatives of the voltage gated ion channel superfamily (Yu and Catterall 2004). They have six transmembrane domains per subunit and four subunits form the functional channel (Ramsey et al. 2006). Most TRP channels are non-selective, Ca2+ permeable cation channels, and display outward rectification. Based on sequence homology, mammalian TRP channels are subdivided into six groups: TRPC (Classical or Canonical), TRPV (Vanilloid), TRPM (Melastatin), TRPP (Polycystin), TRPML (Mucolipin) and TRPA (Ankyrin). They play essential roles in a wide variety of physiological processes, such as thermosensation, mechanosensation, nociception, taste, vision, fertilization, intra- and extracellular Ca2+ and Mg2+ homeostasis (Clapham et al. 2001; Montell et al. 2002; Minke and Cook 2002). Mutations in TRP channels and TRP related proteins cause various diseases such as hypomagnesemia (Walder et al. 2002), polycystic kidney disease (Wilson 2004), familial focal segmental glomerulosclerosis (Winn et al. 2005) and mucolipidosis (Raychowdhury et al. 2004), reviewed in Nilius et al. (2005); Nilius and Owsianik (2010). In congruence with the variety of functions they are involved in, their activation mechanisms are also quite diverse; these include temperature, mechanical stimuli, pH, and various signalling pathways and chemical ligands.
Despite the high diversity of activation mechanisms and physiological functions, most, if not all TRP channels are regulated by phosphoinositides (Rohacs 2007, 2009; Nilius et al. 2008). However, their regulation by phosphoinositides is quite complex. All ion channel families discussed so far were activated by PIP2, in other words their activity depended on the presence of the lipid. Dependence of activity on phosphoinositides have been described for many TRP family members as well; it is possible that this is a conserved feature of this ion channel family, but inhibition by phosphoinositides have also been described for many of them. Table 10.1 summarizes current knowledge based on the primary literature on phosphoinositide effects on TRP channels. Here we discuss the literature on a selected few channels. Two of our examples (TRPM8 and TRPV5/6) are similar to the channels discussed so far, their activity depends on PIP2, whereas our other two examples, TRPCs and TRPV1 are channels where the regulation by PIP2 is quite complex and controversial, and our understanding is limited.
10.6.1 TRPM Channels
TRPMs are the functionally most diverse group in the TRP channel superfamily with eight mammalian members. Most TRPMs are non-selective Ca2+ permeable cation channels, similar to other TRP-s; exceptions are TRPM4 and TRPM5, which conduct monovalent cations, but not Ca2+. PIP2 regulation has been reported for 4 members of this group, in all cases PIP2 activated the respective channel (Table 10.1) and thus PIP2 dependence is probably a common feature of TRPM channels. Here we discuss the literature on TRPM8, which, with respect to phosphoinositide regulation, is the most thoroughly studied member of this family.
TRPM8 is an ion channel activated by cold temperatures and cooling agents such as menthol or icilin in sensory neurons (McKemy et al. 2002; Peier et al. 2002). Genetic deletion of TRPM8 in mice convincingly demonstrated the involvement of this channel in sensing cold temperatures (Dhaka et al. 2007; Colburn et al. 2007; Bautista et al. 2007). TRPM8 has also been proposed to be involved in mediating the analgesic effects of moderate cold and menthol (Proudfoot et al. 2006).
TRPM8 clearly requires PIP2 for activity. Its activity runs down in excised patches, and application of PIP2 reactivates the channel (Liu and Qin 2005; Rohacs et al. 2005). The activating effect is isomer specific; PtdIns(4,5)P2, is more effective than PtdIns(3,4)P2, PtdIns(3,4,5)P3 or PtdIns(4)P (Rohacs et al. 2005). PIP2 chelating agents, such as PIP2 antibody, or poly-Lysine also inhibit TRPM8 in excised patches (Liu and Qin 2005; Rohacs et al. 2005). The activity of the purified TRPM8 reconstituted into lipid bilayers depends on the presence of PIP2 with a similar phosphoinositide specificity profile as in excised patches, providing a strong evidence for direct activation of the channel by PIP2 (Zakharian et al. 2009, 2010), see also Fig. 10.5a, b. Activation of PLC via cell surface receptors (Liu and Qin 2005; Rohacs et al. 2005), by Ca2+ influx through TRPM8 (Rohacs et al. 2005; Daniels et al. 2008) or pharmacologically with m-3M3FBS (Daniels et al. 2008) inhibits TRPM8. PLC independent depletion of PIP2 using a rapamycin-inducible phosphatase (Varnai et al. 2006; Wang et al. 2008; Daniels et al. 2008) or high concentrations of wortmannin (Liu and Qin 2005; Rohacs et al. 2005) also inhibits TRPM8 further supporting its dependence on PIP2.

a PIP2 regulation of TRP channels. Activation of TRPM8 currents in an excised inside out macropatch in Xenopus oocytes. Currents are show after run-down, and the effects of various diC8 phosphoinositides are shown. (From Rohacs et al. (2005) with permission). b Activation of the purified TRPM8 by various diC8 phosphoinositides in planar lipid bilayers. (From Zakharian et al. (2010) with permission). c A model for Ca2+ induced inactivation/desensitization for various TRP channels. (Modified from Rohacs (2009) with permission)
In addition to being important for channel activity, PIP2 is also likely to be involved in desensitization of TRPM8. TRPM8 currents activated by menthol (McKemy et al. 2002; Rohacs et al. 2005; Daniels et al. 2008) or cold (Reid et al. 2002; Daniels et al. 2008) gradually diminish in the presence of extracellular Ca2+, a process called desensitization or adaptation. This effect has been reported both in expression systems (McKemy et al. 2002; Rohacs et al. 2005) and in native sensory neurons (Reid et al. 2002). Similarly, physiological responses to cold (Darian-Smith et al. 1973) and menthol (Eccles 1994) have been shown to desensitize. It was proposed that the mechanism of desensitization is the Ca2+-induced activation of PLC and the ensuing depletion of PIP2 (Rohacs et al. 2005) (Fig. 10.5c). This idea is based on the following findings. As mentioned earlier, PIP2 activates TRPM8 in excised patches and depletion of the lipid inhibits the channel in intact cells. Ca2+ influx through TRPM8 leads to activation of PLC and the depletion of PIP2, (Rohacs et al. 2005; Daniels et al. 2008). TRPM8 desensitization is slowed down by co-expressing PIP5K that synthesizes PIP2, and accelerated by co-expressing the highly Ca2+ sensitive PLC isoform PLCd1 (Rohacs et al. 2005).
How does PIP2 activate TRPM8? Two questions will be discussed here briefly: what is the relationship of PIP2 to other regulators of TRPM8, and where are the PIP2 interacting residues? TRPM8 is activated by cold and cooling agents, such as menthol. Cooling agents shift the activation threshold of the channel to warmer temperatures (McKemy et al. 2002). It was shown that both cold and menthol increase sensitivity of TRPM8 to PIP2, i.e. shift PIP2 dose-response curves to the left. Concurrently, the channel becomes less sensitive to PIP2 depletion in the presence of menthol (Rohacs et al. 2005). This is similar to the effect of Gβγ on Kir channels, as discussed earlier.
TRP channels are also thought to be directly activated by PIP2 through binding to positively charged residues, but only limited efforts have been made to identify those residues in TRP channels so far. Mutation of positively charged residues in the highly conserved TRP domain of TRPM8 substantially decreased the apparent affinity of the channel for PIP2 (Rohacs et al. 2005). The same mutations rendered the channel more sensitive to inhibition by depletion of PIP2. This is compatible with the idea that these residues are part of a PIP2 binding site. However, the R1008Q mutation that had the most dramatic effect on PIP2 sensitivity also affected menthol and cold sensitivity. PIP2 sensitivity of this mutant was however still much less than that of the wild-type channel when examined at lower temperatures and higher menthol concentrations arguing for a primary effect on PIP2 sensitivity. Nevertheless, as discussed earlier, it cannot be excluded that these mutations affect PIP2 sensitivity indirectly. Two of the three TRP domain mutants only moderately affected PIP2 sensitivity, thus it is unlikely that this domain is solely responsible for PIP2 sensitivity of TRPM8. It is likely that other parts of the channel also contribute to PIP2 binding, a notion further supported by the fact that mutation of equivalent TRP domain residues did not affect PIP2 sensitivity of TRPM4 (Nilius et al. 2006).
10.6.2 TRPV Channels
The TRPV family has 6 mammalian members. They can be separated into 2 groups. TRPV1-4 are sensory channels, all are activated by heat with various thresholds. Most of these channels are expressed in sensory neurons, or keratinocytes in the skin. TRPV4, in addition to being activated by heat, is also a mechanosensitive channel. TRPV5 and 6 on the other hand are epithelial Ca2+ channels involved in organism level Ca2+ homeostasis. PIP2 regulation was reported for three members of this family: TRPV1, TRPV5 and TRPV6 (Table 10.1). All three of these channels are activated by PIP2 in excised patches but for TRPV1 an additional indirect inhibitory effect of the lipid in intact cells may complicate the picture.
10.6.2.1 TRPV5 and TRPV6
TRPV5 and TRPV6 are Ca2+ selective channels, located on the apical membrane of epithelial cells that are responsible for active transcellular Ca2+ transport (Hoenderop et al. 2005). They share high homology to each other, but much less to the other members of the TRPV family. Unlike all other TRP channels, TRPV5 and 6 show inward rectification and are selective for calcium and other divalent cations (Hoenderop et al. 2005). TRPV5 is expressed in the kidney, in the late distal convoluted and the connecting tubules, whereas TRPV6 is mainly expressed in the duodenum. TRPV6 is regulated at the transcriptional level by active vitamin D3 (calcitriol). Genetic deletion of either of these channels results in disturbances in calcium homeostasis in mice (Bianco et al. 2006; Hoenderop et al. 2003). The rate of TRPV6 protein evolution was shown to be accelerated in the human lineage (Akey et al. 2006) and its ancestral overactive variant was shown to be associated with increased prevalence of kidney stones in humans, presumably by increased intestinal Ca2+ absorption and compensatory hypercalciuria (Suzuki et al. 2008). Both TRPV5 and TRPV6 undergo Ca2+-induced inactivation, which presumably protects the cells form toxic Ca2+ levels and limits epithelial Ca2+ transport.
Both TRPV5 and TRPV6 require PIP2 for activity; their activity runs down in excised patches, which is accelerated by poly-Lysine (Rohacs et al. 2005) and they are reactivated by application of PIP2 (Lee et al. 2005; Thyagarajan et al. 2008). We have proposed that Ca2+-induced inactivation of TRPV6 proceeds through PLC activation and the resulting depletion of PIP2 (Thyagarajan et al. 2008, 2009), similarly to TRPM8 (Fig. 10.5b). This model is based on the following findings. TRPV6 is activated in excised patches by PIP2 but not PIP. Ca2+-induced inactivation is inhibited by dialyzing PtdIns(4,5)P2, but not PtdIns(4)P through the patch pipette in whole-cell patch clamp experiments. Ca2+ influx through TRPV6 leads to depletion of PIP2 and formation of IP3, indicating activation of PLC. PLC independent depletion of PIP2 with the rapamycin-inducible PIP2 phosphatase, or high concentrations of wortmannin inhibited TRPV6 (Thyagarajan et al. 2008). Both PIP2 depletion and Ca2+-induced inactivation of TRPV6 were inhibited by PLC inhibitors (Thyagarajan et al. 2009).
The calcium sensor calmodulin has also been proposed to play a role in Ca2+-induced inactivation of TRPV6 (Derler et al. 2006; Niemeyer et al. 2001). Again, just like in other cases, it is possible that both mechanisms contribute to Ca2+-induced inactivation. Competition of CaM with PIP2, as proposed for other TRP channels (Kwon et al. 2007) and Kv7 channels (see above) is a feasible mechanism that would integrate CaM and PIP2 regulation, but it has not been experimentally tested on TRPV6.
10.6.2.2 TRPV1
TRPV1 was the first non-canonical mammalian TRP channel to be cloned (Caterina et al. 1997). Its major activators are heat, capsaicin (the pungent compound in hot peppers), and tissue acidosis. This channel is involved in nociception and there are many other factors that activate or regulate it (Pingle et al. 2007). TRPV1 was also the first mammalian TRP channel that was reported to be regulated by PIP2. It was proposed that PIP2 tonically inhibits TRPV1, and depletion of this lipid by pro-inflammatory agents, such as bradykinin, relieves this inhibition, and potentiates TRPV1 activity at low stimulation levels (Chuang et al. 2001). This potentiation is thought to underlie thermal hyperalgesia, the increased sensitivity of inflamed areas to heat. Later however, several laboratories reported that in contradiction to this model, PIP2 and other phosphoinositides activate the channel in excised patches (Stein et al. 2006; Lukacs et al. 2007; Klein et al. 2008; Kim et al. 2008c). Agents that chelate PIP2 (such as poly-Lysine) inhibit TRPV1 in excised patches, thus supporting the activating effect of the lipid (Stein et al. 2006; Lukacs et al. 2007). This apparent controversy is similar to that seen with TRPC5, see later.
What is the functional role of the activating effect of PIP2? It is likely that depletion of the lipid plays a role in the Ca2+-dependent desensitization of TRPV1, similarly to several other TRP channels, such as TRPM8 (Rohacs et al. 2005), TRPM4 (Nilius et al. 2006) and TRPV6 (Thyagarajan et al. 2008). The model is simple: when Ca2+ enters a cell through TRPV1, it activates a Ca2+ sensitive PLC, and the resulting PIP2 depletion leads/contributes to decreased channel activity (Fig. 10.5c). This model is based on the following data. (i) As already mentioned, TRPV1 requires PIP2 for activity in excised patches. (ii) Application of capsaicin in the presence of extracellular Ca2+ leads to hydrolysis of PIP2 (Liu et al. 2005; Lukacs et al. 2007; Akopian et al. 2007; Yao and Qin 2009). (iii) Recovery from desensitization depends on the ability of the cell to resynthesize PIP2 (Liu et al. 2005). (iv) PLC inhibitors reduce desensitization (Lukacs et al. 2007; Lishko et al. 2007). (v) Supplying excess PtdIns(4,5)P2 or PtdIns(4)P through the patch pipette in whole-cell patch clamp experiments reduces desensitization (Lukacs et al. 2007; Lishko et al. 2007). PtdIns(4)P also activates TRPV1 in excised patches, and it is also depleted upon PLC activation (Lukacs et al. 2007). As the concentration of PtdIns(4)P is thought to be comparable to that of PIP2, it may also play a role, together with PIP2, in keeping TRPV1 open.
PIP2 depletion is unlikely to be the mechanism solely responsible for desensitization of TRPV1, as both PLC inhibition and supplying excess PIP2 only partially inhibited desensitization. Also in one study supplying PIP2 through the patch pipette in whole-cell experiments only moderately reduced capsaicin-induced desensitization (Akopian et al. 2007). The ubiquitous Ca2+ sensor calmodulin has also been proposed to play a role in desensitization, both acting on the channel directly (Numazaki et al. 2003; Rosenbaum et al. 2004; Lishko et al. 2007), and by activating calcineurin (Docherty et al. 1996; Mohapatra and Nau 2005), and thus inducing dephosphorylation of the channel.
There seems to be a general agreement on the role of PIP2 in activating TRPV1, and the originally proposed tonic inhibitory effect of PIP2 is somewhat debated. Is it possible that PIP2 has both inhibitory effects and is required for channel activity, similar to what was proposed for VGCC (Wu et al. 2002). It was found that depletion of the lipid with the rapamycin-inducible PIP2 phosphatase system (Varnai et al. 2006) leads to further activation when the channel is only moderately stimulated by capsaicin or heat (Lukacs et al. 2007). This finding suggests a partial inhibition by PIP2 in intact cells, in addition to its activating effect. Importantly, potentiation by PIP2 depletion was only seen when the channel was stimulated by low concentration of capsaicin, or moderate heating, conditions where PLC mediated sensitization also occurs. When the channel was maximally stimulated by high capsaicin concentrations, neither activation, nor inhibition by the inducible phosphatase was observed (Lukacs et al. 2007). The lack of inhibition at high capsaicin concentrations was explained with PtdIns(4)P keeping the channel open under such conditions. PtdIns(4)P is not depleted by the phosphatase, indeed it is expected that its level increases when PIP2 is converted to PtdIns(4)P. Conversely, when we over-expressed the PIP5K enzyme, generating excess of PIP2, TRPV1 activity was inhibited at low, but not at high capsaicin concentrations (Lukacs et al. 2007). This finding is also compatible with a partial inhibitory effect of PIP2 at moderate stimulation levels. This inhibitory effect, however, is likely to be indirect, because it is not detectable in excised patches.
Another article, on the other hand found that the rapamycin-inducible PIP2 phosphatase inhibited TRPV1 both high and low concentrations of capsaicin (Klein et al. 2008). This is compatible with the activating effect of PIP2 in excised patches, and argues against an inhibitory effect of the lipid. It is hard to tell what causes the discrepancies between the two studies (Lukacs et al. 2007; Klein et al. 2008). There are a number of differences in the experimental conditions including, the cell-type, the rapamycin analogue, the concentrations of capsaicin used, and the origin of the rapamycin-phosphatase system (Suh et al. (2006) vs. Varnai et al. (2006)). Some of these differences may explain the opposing findings of the two studies. It is worth noting however, that the same two articles reached very similar conclusions on the effects of the phosphoinositides PtdIns(4,5)P2, PtdIns(4)P and PtdIns(3,4,5)P3 in excised patches, despite several differences in experimental conditions (Lukacs et al. 2007; Klein et al. 2008).
A recent addition to the complexity of phosphoinositide regulation of TRPV1 is the discovery of Pirt (Kim et al. 2008a). Pirt is a two transmembrane domain protein, specifically expressed in DRG neurons and it interacts both with TRPV1 and phosphoinositides. It was proposed that phosphoinositides activate TRPV1 through binding to Pirt. PIP2 however activates TRPV1 in excised patches in expression systems (Lukacs et al. 2007), where Pirt is unlikely to be present. It is unlikely that Pirt is an obligatory subunit for TRPV1 modulation by phosphoinositides, but it is present in the native environment of TRPV1; it interacts with the channel and modulates its function. Thus it is probably an important modulator of native TRPV1 channels, but clarifying its exact role in phosphoinositide regulation of these channels will require further experimental work.
In conclusion, TRPV1 clearly requires phosphoinositides for activity; PIP2 reproducibly activates the channel in excised patches. There also seems to be an agreement that depletion of the lipid contributes to Ca2+-induced desensitization. If there is a partial inhibition by PIP2 in intact cells, it is likely to depend on a factor lost upon patch excision (indirect effect) because several laboratories found no evidence of it in excised patches using a variety of tools (Lukacs et al. 2007; Klein et al. 2008). PIP2 regulation of TRPV1 has recently been reviewed with a discussion of ideas to integrate the activating and the possible inhibitory effects of PIP2 in the PLC mediated regulation of TRPV1 (Rohacs et al. 2008).
10.6.3 TRPC Channels
TRPC channels are activated downstream of PLC, and mediate Ca2+ influx and presumably depolarization. The exact mechanism by which they are activated by PLC is not clear in most cases (Trebak et al. 2007). TRPC3, 6 and 7 has been shown to be activated by DAG, the downstream product of PLC activation (Hofmann et al. 1999), but the other TRPC isoforms are generally thought to be insensitive to DAG. Many TRPC isoforms have been shown to be inhibited by PIP2 (table), and relief from tonic inhibition by PIP2 upon PLC activation has been proposed as a mechanism for TRPC channel activation. As we will see, this mechanism may play a role in certain cases, but it is unlikely to be a general paradigm among TRPCs.
TRPCs are the closest mammalian homologues of the drosophila TRP and TRPL channels. In the drosophila eye the TRP/TRPL complex is activated by light in a PLC dependent manner, thus generating the receptor potential (Hardie and Raghu 2001). The TRPL channel was shown to be inhibited by PIP2 in excised patches in an expression system (Estacion et al. 2001). The same channel was also activated by DAG analogues. Later studies showed that activation of the TRP/TRPL complex by PIP2 depletion is unlikely to be the major mechanism to generate the receptor potential in the drosophila eye, even though it may play some auxiliary role (Hardie 2007). Confounding these observations, a recent report, found activation of heterologously expressed TRPL by PIP2 in excised patches. The two precursors, PtdIns and PtdIns(4)P, on the other hand inhibited TRPL in excised patches (Huang et al. 2010).
The mammalian TRPC4 splice variant TRPC4α, but not TRPC4β is inhibited when PIP2 is dialyzed through the patch pipette in whole-cell patch clamp experiments (Otsuguro et al. 2008). PIP2 was shown to bind to the C-terminus of TRPC4α, but not TRPC4β. The inhibition by PIP2 could be disrupted with the cytoskeletal inhibitor cytochalasin D or by deleting the C-terminal PDZ binding motif from TRPC4α. PIP2 depletion, however, was not sufficient in itself to open the channels. The effects of PIP2 in excised patches were not examined in this study (Otsuguro et al. 2008).
Another article showed that TRPC5 can be moderately activated by depleting PIP2 using two inhibitors of PI4K, wortmannin and LY294002 (Trebak et al. 2008). Activation by wortmannin was inhibited by dialyzing PIP2 through the patch pipette. Interestingly, depletion of PIP2 using a rapamycin-inducible PIP2 phosphatase inhibited TRPC5 when the channel was activated by a low concentration of carbachol (Trebak et al. 2008). When PIP2 was tested in excised patches, however, it activated TRPC5 (Trebak et al. 2008), similarly to TRPC3, 6, and 7 (Lemonnier et al. 2007). These data suggest that PIP2 has both activating and inhibitory effects on TRPC5. The inhibitory effect of PIP2 is likely to be an indirect effect, because it is not detected in excised patches.
Presently there is a controversy whether TRPC6 and TRPC7 channels are activated or inhibited by PIP2. One study showed that in an expression system, PIP2 activates TRPC6 and TRPC7 in inside-out patches (Lemonnier et al. 2007). Another study found that in vascular smooth muscle cells PIP2 inhibits native TRPC6 channels in excised patches. (Albert et al. 2008; Ju et al. 2010). The same study also showed that dialyzing PIP2 through the whole-cell patch pipette inhibited activation of TRPC6 by angiotensin II and DAG. Collectively, the regulation of TRPC channels by PIP2 is likely to be quite complex and not yet fully understood (Table 10.1).
In conclusion, the activity of many TRP channels depends on the presence of PIP2 in the plasma membrane; in this respect, these channels are similar to Kir and KCNQ channels. Some TRP channels, such as TRPM8 and TRPV5 and 6, behave very similar to classical PIP2 sensitive channels. Some other TRP channels however, are also reported to be both activated and inhibited by PIP2. The difference between whether PIP2 activates or inhibits was either the experimental setting, i.e. intact cells versus excised patches or endogenous versus heterologously expressed channels. Given the sheer prevalence of this “dual regulation”, it is hard to dismiss it as an artifact, or unreliable data. Differences between native vs. expressed channels can be explained by different cellular components expressed in these cell in addition to the channel, whereas difference between excised patch and whole-cell measurements can be explained with lost cellular components in the latter. Altogether, regulation of many TRP channels by PIP2 is quite complex and its understanding requires further investigation.
10.7 Conclusions
A large number and variety of ion channels are modulated by plasma membrane phosphoinositides. In most cases, the activity of the channels depend on the presence of PtdIns(4,5)P2, and the depletion of the lipid inhibits them. In the last 10–15 years we have seen an explosion in the number of PIP2-sensitive ion channels and transporters; in addition to the ones discussed here the list of PIP2-sensitive ion channels now includes K2P, HERG, CNG, ENaC, CFTR, P2X—to name a few, but also many others. Importantly, we have also seen a tremendous progress in the development of tools and approaches to study this phenomenon; this progress gives hope that in the near future we will see further insights into the mechanisms and significance of ion channel interaction with phosphoinositides. Indeed, there are many intriguing yet unanswered questions ahead. One of such questions is why so many ion channel proteins display sensitivity to phosphoinositides? One hypothesis suggests that for many plasma membrane ion channels requirement for PIP2 provides a mechanism for silencing these channels until they reach plasma membrane (Hilgemann et al. 2001). Indeed, during their life cycle, plasma membrane ion channels travel trough the various membranous organelles (ER, Golgi, endosomes etc.) but in most cases it is only plasma membrane where their activity is needed. Accordingly, in contrast to the plasma membrane, intracellular membranes usually contain very little PIP2 and for the majority of PIP2-sensitive ion channels the requirement for PIP2 is permissive. Thus, at least for the channels with high PIP2 affinity, the PIP2-dependence may serve to ensure that their activity is ‘turned off’ until they reach their designated cellular localization. Ion channels with moderate and low PIP2 affinity are however likely to be modulated by physiological fluctuations in plasma membrane PIP2 abundance. The next ‘hot’ question therefore is how the specificity of PIP2 signalling is achieved? One possible mechanism for such specificity is a local PIP2 depletion which would affect only those PIP2-sensitive membrane proteins that are in close spatial juxtaposition to a PIP2-depleting activity (e.g. GPCR coupled to PLC). The idea of local PIP2 depletion is attractive but is not easily reconcilable with the suggested fast lateral diffusion of PIP2 in the biological membranes (Yaradanakul and Hilgemann 2007) nor with the experimental data in neurons demonstrating that extracellular application of GPCR agonists can inhibit PIP2-sensitive ion channels (e.g. Kv7) within the isolated membrane patch during the cell-attached patch clamp recording (Selyanko et al. 1992). Nevertheless local PIP2 depletion hypothesis may work for some type of cells (e.g. in cardiomyocytes; (Cho et al. 2005)) or in neurons with long processes. Another mechanism for specificity for the PIP2-mediated signalling may arise from the coincidence detection (as discussed above). Indeed, if PIP2 depletion requires a set of cofactors in order to mediate modulation of a given ion channel, and different ion channels require different sets of cofactors, than the functional outcome of the receptor-mediated PIP2 depletion will be defined by the availability and timing of the cofactor release (or withdrawal). All these interesting questions and theories require further research, which is well warranted given the fundamental nature of the phosphoinositide sensitivity of ion channels. A further focus on the interactions of the ion channels and phosphoinositides is brought about by the increasing evidence that mutations within the ion channel genes that disrupt channel interaction with phosphoinositides may underlie severe disorders in humans. Thus, three arrhytmogenic mutations within the Kv7.1 channel (Park et al. 2005) were suggested to impair cardiac IKs current by reducing apparent PIP2 affinity of Kv7.1. Likewise, mutations affecting channel-phosphoinositide interactions within several Kir channel genes were linked to diseases such as Andersen–Tawil syndrome (ATS), hyperprostaglandin E syndrome (HPS) and congenital hyperinsulinism (CHI), reviewed in Logothetis et al. (2010). Therefore comprehensive future studies of ion channel sensitivity to and regulation by phosphoinositides are necessary for elucidation of basic principles of membrane-associated cellular signalling in health and disease.
References
Akey JM, Swanson WJ, Madeoy J, Eberle M, Shriver MD (2006) TRPV6 exhibits unusual patterns of polymorphism and divergence in worldwide populations. Hum Mol Genet 15:2106–2113
Akopian AN, Ruparel NB, Jeske NA, Hargreaves KM (2007) Transient receptor potential TRPA1 channel desensitization in sensory neurons is agonist dependent and regulated by TRPV1-directed internalization. J Physiol 583:175–193
Albert AP, Saleh SN, Large WA (2008) Inhibition of native TRPC6 channel activity by phosphatidylinositol 4,5-bisphosphate in mesenteric artery myocytes. J Physiol 586:3087–3095
Bal M, Zhang J, Hernandez CC, Zaika O, Shapiro MS (2010) Ca2+/calmodulin disrupts AKAP79/150 interactions with KCNQ (M-Type) K+ channels. J Neurosci 30:2311–2323
Balla T (2001) Pharmacology of phosphoinositides, regulators of multiple cellular functions. Curr Pharm Des 7:475–507
Balla T (2009) Green light to illuminate signal transduction events. Trends Cell Biol 19:575–586
Balla T, Varnai P (2009) Visualization of cellular phosphoinositide pools with GFP-fused protein-domains. Curr Protoc Cell Biol Chapter 24:Unit 24.4
Bannister RA, Melliti K, Adams BA (2002) Reconstituted slow muscarinic inhibition of neuronal (Ca(v)1.2c) L-type Ca2+ channels. Biophys J 83:3256–3267
Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G (1996) K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384:78–80
Baukrowitz T, Schulte U, Oliver D, Herlitze S, Krauter T, Tucker SJ, Ruppersberg JP, Fakler B (1998) PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science 1141–1144
Bautista DM, Siemens J, Glazer JM, Tsuruda PR, Basbaum AI, Stucky CL, Jordt SE, Julius D (2007) The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 448:204–208
Bean BP (1989) Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature 340:153–156
Bernheim L, Beech DJ, Hille B (1991) A diffusible second messenger mediates one of the pathways coupling receptors to calcium channels in rat sympathetic neurons. Neuron 6:859–867
Bianco SD, Peng JB, Takanaga H, Suzuki Y, Crescenzi A, Kos CH, Zhuang L, Freeman MR, Gouveia CH, Wu J, Luo H, Mauro T, Brown EM, Hediger MA (2006) Marked disturbance of calcium homeostasis in mice with targeted disruption of the Trpv6 calcium channel gene. J Bone Miner Res 22:274–285
Brown DA, Adams PR (1980) Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 283:673–676
Brown DA, Passmore GM (2009) Neural KCNQ (Kv7) channels. Br J Pharmacol 156:1185–1195
Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389:816–824
Catterall WA (2000) Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol 16:521–555
Charpentier F, Merot J, Loussouarn G, Baro I (2010) Delayed rectifier K+ currents and cardiac repolarization. J Mol Cell Cardiol 48:37–44
Cho H, Kim YA, Yoon JY, Lee D, Kim JH, Lee SH, Ho WK (2005) Low mobility of phosphatidylinositol 4,5-bisphosphate underlies receptor specificity of Gq-mediated ion channel regulation in atrial myocytes. Proc Natl Acad Sci U S A 102:15241–15246
Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D (2001) Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2 mediated inhibition. Nature 411:957–962
Clapham DE, Runnels LW, Strubing C (2001) The TRP ion channel family. Nat Rev Neurosci 2:387–396
Colburn RW, Lubin ML, Stone DJ Jr, Wang Y, Lawrence D, D’Andrea MR, Brandt MR, Liu Y, Flores CM, Qin N (2007) Attenuated cold sensitivity in TRPM8 null mice. Neuron 54:379–386
Colquhoun D (1998) Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol 125:924–947
Cruzblanca H, Koh DS, Hille B (1998) Bradykinin inhibits M current via phospholipase C and Ca2+ release from PI3-sensitive Ca2+ stores in rat sympathetic neurons. Proc Natl Acad Sci U S A 95:7151–7156
D’Avanzo N, Cheng WW, Doyle DA, Nichols CG (2010) Direct and specific activation of human inward rectifier K+ channels by membrane phosphatidylinositol 4,5-bisphosphate. J Biol Chem
Dai Y, Wang S, Tominaga M, Yamamoto S, Fukuoka T, Higashi T, Kobayashi K, Obata K, Yamanaka H, Noguchi K (2007) Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. J Clin Invest 117:1979–1987
Daniels RL, Takashima Y, McKemy DD (2008) The activity of the neuronal cold sensor TRPM8 is regulated by phospholipase C via the phospholipid phosphoinositol-4,5-bisphosphate. J Biol Chem 284:1570–1582
Darian-Smith I, Johnson KO, Dykes R (1973) “Cold” fiber population innervating palmar and digital skin of the monkey: responses to cooling pulses. J Neurophysiol 36:325–346
Delmas P, Brown DA (2005) Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci 6:850–862
Delmas P, Wanaverbecq N, Abogadie FC, Mistry M, Brown DA (2002) Signaling microdomains define the specificity of receptor-mediated InsP3 pathways in neurons. Neuron 34:209–220
Delmas P, Coste B, Gamper N, Shapiro MS (2005) Phosphoinositide lipid second messengers: new paradigms for calcium channel modulation. Neuron 47:179–182
Derler I, Hofbauer M, Kahr H, Fritsch R, Muik M, Kepplinger K, Hack ME, Moritz S, Schindl R, Groschner K, Romanin C (2006) Dynamic but not constitutive association of calmodulin with rat TRPV6 channels enables fine tuning of Ca2+-dependent inactivation. J Physiol 577:31–44
Dhaka A, Murray AN, Mathur J, Earley TJ, Petrus MJ, Patapoutian A (2007) TRPM8 is required for cold sensation in mice. Neuron 54:371–378
Docherty RJ, Yeats JC, Bevan S, Boddeke HW (1996) Inhibition of calcineurin inhibits the desensitization of capsaicin-evoked currents in cultured dorsal root ganglion neurones from adult rats. Pflugers Arch 431:828–837
Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, Xu H (2010) PI(3,5)P2 controls membrane traffic by direct activation of mucolipin Ca release channels in the endolysosome. Nat Commun 1
Du X, Zhang H, Lopes CM, Mirshahi T, Rohacs T, Logothetis DE (2004) Characteristic interactions with phosphatidylinositol 4,5-bisphosphate determine regulation of Kir channels by diverse modulators. J Biol Chem 279:37271–37281
Eccles R (1994) Menthol and related cooling compounds. J Pharm Pharmacol 46:618–630
Estacion M, Sinkins WG, Schilling WP (2001) Regulation of Drosophila transient receptor potential-like (TrpL) channels by phospholipase C-dependent mechanisms. J Physiol 530:1–19
Falkenburger BH, Jensen JB, Dickson EJ, Suh BC, Hille B (2010a) Phosphoinositides: lipid regulators of membrane proteins. J Physiol 588:3179–3185
Falkenburger BH, Jensen JB, Hille B (2010b) Kinetics of M1 muscarinic receptor and G protein signaling to phospholipase C in living cells. J Gen Physiol 135:81–97
Falkenburger BH, Jensen JB, Hille B (2010c) Kinetics of PIP2 metabolism and KCNQ2/3 channel regulation studied with a voltage-sensitive phosphatase in living cells. J Gen Physiol 135:99–114
Fan Z, Makielski JC (1997) Anionic phospholipids activate ATP-sensitive potassium channels. J Biol Chem 272:5388–5395
Gamper N, Shapiro MS (2003) Calmodulin mediates Ca2+-dependent modulation of M-type K+ channels. J Gen Physiol 122:17–31
Gamper N, Shapiro MS (2007a) Regulation of ion transport proteins by membrane phosphoinositides. Nat Rev Neurosci 8:921–934
Gamper N, Shapiro MS (2007b) Target-specific PIP2 signalling: how might it work? J Physiol 582:967–975
Gamper N, Reznikov V, Yamada Y, Yang J, Shapiro MS (2004) Phosphotidylinositol 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J Neurosci 24:10980–10992
Gamper N, Li Y, Shapiro MS (2005) Structural requirements for differential sensitivity of KCNQ K+ channels to modulation by Ca2+/calmodulin. Mol Biol Cell 16:3538–3551
Greenwood IA, Ohya S (2009) New tricks for old dogs: KCNQ expression and role in smooth muscle. Br J Pharmacol 156:1196–1203
Gribble FM, Proks P, Corkey BE, Ashcroft FM (1998) Mechanism of cloned ATP-sensitive potassium channel activation by oleoyl-CoA. J Biol Chem 273:26383–26387
Gribkoff VK (2006) The role of voltage-gated calcium channels in pain and nociception. Semin Cell Dev Biol 17:555–564
Haley JE, Abogadie FC, Delmas P, Dayrell M, Vallis Y, Milligan G, Caulfield MP, Brown DA, Buckley NJ (1998) The alpha subunit of Gq contributes to muscarinic inhibition of the M-type potassium current in sympathetic neurons. J Neurosci 18:4521–4531
Haley JE, Delmas P, Offermanns S, Abogadie FC, Simon MI, Buckley NJ, Brown DA (2000) Muscarinic inhibition of calcium current and M current in Galpha q-deficient mice. J Neurosci 20:3973–3979
Hardie RC (2007) TRP channels and lipids: from Drosophila to mammalian physiology. J Physiol 578:9–24
Hardie RC, Raghu P (2001) Visual transduction in Drosophila. Nature 413:186–193
Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA (1996) Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature 380:258–262
Herlitze S, Hockerman GH, Scheuer T, Catterall WA (1997) Molecular determinants of inactivation and G protein modulation in the intracellular loop connecting domains I and II of the calcium channel alpha1A subunit. Proc Natl Acad Sci U S A 94:1512–1516
Hernandez CC, Zaika O, Shapiro MS (2008a) A carboxy-terminal inter-helix linker as the site of phosphatidylinositol 4,5-bisphosphate action on Kv7 (M-type) K+ channels. J Gen Physiol 132:361–381
Hernandez CC, Zaika O, Tolstykh GP, Shapiro MS (2008b) Regulation of neural KCNQ channels: signalling pathways, structural motifs and functional implications. J Physiol 586:1811–1821
Hernandez CC, Falkenburger B, Shapiro MS (2009) Affinity for phosphatidylinositol 4,5-bisphosphate determines muscarinic agonist sensitivity of Kv7K+ channels. J Gen Physiol 134:437–448
Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90:291–366
Hilgemann DW (1997) Cytoplasmic ATP-dependent regulation of ion transporters and channels: mechanisms and messengers. Annu Rev Physiol 59:193–220
Hilgemann DW, Ball R (1996) Regulation of cardiac Na+/Ca2+ exchange and KATP potassium channels by PIP2. Science 273:956–959
Hilgemann DW, Feng S, Nasuhoglu C (2001) The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE 2001:RE19
Hille B (1994) Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci 17:531–536
Hirose K, Kadowaki S, Tanabe M, Takeshima H, Iino M (1999) Spatiotemporal dynamics of inositol 1,4,5-trisphosphate that underlies complex Ca2+ mobilization patterns. Science 284:1527–1530
Hoenderop JG, Leeuwen JP van, Eerden BC van der, Kersten FF, Kemp AW van der, Merillat AM, Waarsing JH, Rossier BC, Vallon V, Hummler E, Bindels RJ (2003) Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J Clin Invest 112:1906–1914
Hoenderop JG, Nilius B, Bindels RJ (2005) Calcium absorption across epithelia. Physiol Rev 85:373–422
Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G (1999) Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397:259–263
Horowitz LF, Hirdes W, Suh BC, Hilgemann DW, Mackie K, Hille B (2005) Phospholipase C in living cells: activation, inhibition, Ca2+ requirement, and regulation of M current. J Gen Physiol 126:243–262
Hoshi N, Zhang JS, Omaki M, Takeuchi T, Yokoyama S, Wanaverbecq N, Langeberg LK, Yoneda Y, Scott JD, Brown DA, Higashida H (2003) AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat Neurosci 6:564–571
Hsuan JJ, Minogue S, dos Santos M (1998) Phosphoinositide 4- and 5-kinases and the cellular roles of phosphatidylinositol 4,5-bisphosphate. Adv Cancer Res 74:167–216
Huang CL (2007) Complex roles of PIP2 in the regulation of ion channels and transporters. Am J Physiol Renal Physiol 293:F1761–F1765
Huang CL, Feng S, Hilgemann DW (1998) Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature 391:803–806
Huang J, Liu CH, Hughes SA, Postma M, Schwiening CJ, Hardie RC (2010) Activation of TRP channels by protons and phosphoinositide depletion in Drosophila photoreceptors. Curr Biol 20:189–197
Iannotti FA, Panza E, Barrese V, Viggiano D, Soldovieri MV, Taglialatela M (2010) Expression, localization, and pharmacological role of Kv7 potassium channels in skeletal muscle proliferation, differentiation, and survival after myotoxic insults. J Pharmacol Exp Ther 332:811–820
Ikeda SR (1996) Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature 380:255–258
Inanobe A, Nakagawa A, Matsuura T, Kurachi Y (2010) A structural determinant for the control of PIP2-sensitivity in G protein-gated inward rectifier K+ channels. J Biol Chem
Iwasaki H, Murata Y, Kim Y, Hossain MI, Worby CA, Dixon JE, McCormack T, Sasaki T, Okamura Y (2008) A voltage-sensing phosphatase, Ci-VSP, which shares sequence identity with PTEN, dephosphorylates phosphatidylinositol 4,5-bisphosphate. Proc Natl Acad Sci U S A 105:7970–7975
Jardin I, Redondo PC, Salido GM, Rosado JA (2008) Phosphatidylinositol 4,5-bisphosphate enhances store-operated calcium entry through hTRPC6 channel in human platelets. Biochim Biophys Acta 1783:84–97
Jia Z, Bei J, Rodat-Despoix L, Liu B, Jia Q, Delmas P, Zhang H (2008) NGF inhibits M/KCNQ currents and selectively alters neuronal excitability in subsets of sympathetic neurons depending on their M/KCNQ current background. J Gen Physiol 131:575–587
Ju M, Shi J, Saleh SN, Albert AP, Large WA (2010) Ins(1,4,5)P3 interacts with PIP2 to regulate activation of TRPC6/C7 channels by diacylglycerol in native vascular myocytes. J Physiol 588:1419–1433
Karashima Y, Prenen J, Meseguer V, Owsianik G, Voets T, Nilius B (2008) Modulation of the transient receptor potential channel TRPA1 by phosphatidylinositol 4,5-bisphosphate manipulators. Pflugers Arch 457:77–89
Keselman I, Fribourg M, Felsenfeld DP, Logothetis DE (2007) Mechanism of PLC-mediated Kir3 current inhibition. Channels (Austin) 1:113–123
Kharkovets T, Hardelin JP, Safieddine S, Schweizer M, El Amraoui A, Petit C, Jentsch TJ (2000) KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc Natl Acad Sci U S A 97:4333–4338
Kharkovets T, Dedek K, Maier H, Schweizer M, Khimich D, Nouvian R, Vardanyan V, Leuwer R, Moser T, Jentsch TJ (2006) Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. EMBO J 25:642–652
Kim D, Cavanaugh EJ (2007) Requirement of a soluble intracellular factor for activation of transient receptor potential A1 by pungent chemicals: role of inorganic polyphosphates. J Neurosci 27:6500–6509
Kim AY, Tang Z, Liu Q, Patel KN, Maag D, Geng Y, Dong X (2008a) Pirt, a phosphoinositide-binding protein, functions as a regulatory subunit of TRPV1. Cell 133:475–485
Kim BJ, Kim MT, Jeon JH, Kim SJ, So I (2008b) Involvement of phosphatidylinositol 4,5-bisphosphate in the desensitization of canonical transient receptor potential 5. Biol Pharm Bull 31:1733–1738
Kim D, Cavanaugh EJ, Simkin D (2008c) Inhibition of transient receptor potential A1 channel by phosphatidylinositol-4,5-bisphosphate. Am J Physiol Cell Physiol 295:C92–C99
Klein RM, Ufret-Vincenty CA, Hua L, Gordon SE (2008) Determinants of molecular specificity in phosphoinositide regulation: PI(4,5)P2 is the endogenous lipid regulating TRPV1. J Biol Chem 283:26208–26216
Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El Amraoui A, Marlin S, Petit C, Jentsch TJ (1999) KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96:437–446
Kubista H, Kosenburger K, Mahlknecht P, Drobny H, Boehm S (2009) Inhibition of transmitter release from rat sympathetic neurons via presynaptic M(1) muscarinic acetylcholine receptors. Br J Pharmacol 156:1342–1352
Kwon Y, Hofmann T, Montell C (2007) Integration of phosphoinositide- and calmodulin-mediated regulation of TRPC6. Mol Cell 25:491–503
Langeslag M, Clark K, Moolenaar WH, Leeuwen FN van, Jalink K (2007) Activation of TRPM7 channels by PLC-coupled receptor agonists. J Biol Chem 282:232–239
Lechner SG, Hussl S, Schicker KW, Drobny H, Boehm S (2005) Presynaptic inhibition via a phospholipase C- and phosphatidylinositol bisphosphate-dependent regulation of neuronal Ca2+ channels. Mol Pharmacol 68:1387–1396
Lee J, Cha SK, Sun TJ, Huang C-L (2005) PIP2 activates TRPV5 and releases its inhibition by intracellular Mg2+. J Gen Physiol 126:439–451
Lemonnier L, Trebak M, Putney JW Jr (2007) Complex regulation of the TRPC3, 6 and 7 channel subfamily by diacylglycerol and phosphatidylinositol-4,5-bisphosphate. Cell Calcium 43:506–514
Leslie CC (2004) Regulation of the specific release of arachidonic acid by cytosolic phospholipase A2. Prostaglandins Leukot Essent Fatty Acids 70:373–376
Li Y, Gamper N, Shapiro MS (2004) Single-channel analysis of KCNQ K+ channels reveals the mechanism of augmentation by a cysteine-modifying reagent. J Neurosci 24:5079–5090
Li Y, Gamper N, Hilgemann DW, Shapiro MS (2005) Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5-bisphosphate. J Neurosci 25:9825–9835
Linley JE, Rose K, Patil M, Robertson B, Akopian AN, Gamper N (2008) Inhibition of M current in sensory neurons by exogenous proteases: a signaling pathway mediating inflammatory nociception. J Neurosci 28:11240–11249
Linley JE, Rose K, Ooi L, Gamper N (2010) Understanding inflammatory pain: ion channels contributing to acute and chronic nociception. Pflugers Arch 459:657–669
Lipscombe D, Kongsamut S, Tsien RW (1989) Alpha-adrenergic inhibition of sympathetic neurotransmitter release mediated by modulation of N-type calcium-channel gating. Nature 340:639–642
Lishko PV, Procko E, Jin X, Phelps CB, Gaudet R (2007) The ankyrin repeats of TRPV1 bind multiple ligands and modulate channel sensitivity. Neuron 54:905–918
Liu SJ (2007) Inhibition of L-type Ca2+ channel current and negative inotropy induced by arachidonic acid in adult rat ventricular myocytes. Am J Physiol Cell Physiol 293:C1594–C1604
Liu D, Liman ER (2003) Intracellular Ca2+ and the phospholipid PIP2 regulate the taste transduction ion channel TRPM5. Proc Natl Acad Sci U S A 100:15160–15165
Liu L, Rittenhouse AR (2003) Arachidonic acid mediates muscarinic inhibition and enhancement of N-type Ca2+ current in sympathetic neurons. Proc Natl Acad Sci U S A 100:295–300
Liu B, Qin F (2005) Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. J Neurosci 25:1674–1681
Liu L, Barrett CF, Rittenhouse AR (2001) Arachidonic acid both inhibits and enhances whole cell calcium currents in rat sympathetic neurons. Am J Physiol Cell Physiol 280:C1293–C1305
Liu B, Zhang C, Qin F (2005) Functional recovery from desensitization of vanilloid receptor TRPV1 requires resynthesis of phosphatidylinositol 4,5-bisphosphate. J Neurosci 25:4835–4843
Liu L, Zhao R, Bai Y, Stanish LF, Evans JE, Sanderson MJ, Bonventre JV, Rittenhouse AR (2006) M1 muscarinic receptors inhibit L-type Ca2+ current and M-current by divergent signal transduction cascades. J Neurosci 26:11588–11598
Liu B, Linley JE, Du X, Zhang X, Ooi L, Zhang H, Gamper N (2010) The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl− channels. J Clin Invest 120:1240–1252
Logothetis DE, Jin T, Lupyan D, Rosenhouse-Dantsker A (2007a) Phosphoinositide-mediated gating of inwardly rectifying K+ channels. Pflugers Arch 455:83–95
Logothetis DE, Lupyan D, Rosenhouse-Dantsker A (2007b) Diverse Kir modulators act in close proximity to residues implicated in phosphoinositide binding. J Physiol 582:953–965
Logothetis DE, Petrou VI, Adney SK, Mahajan R (2010) Channelopathies linked to plasma membrane phosphoinositides. Pflugers Arch 460:321–341
Lopes CMB, Zhang H, Rohacs T, Jin T, Logothetis DE (2002) Alterations in Conserved Kir Channel-PIP2 Interactions Underlie Channelopathies. Neuron 34:933–944
Loussouarn G, Park KH, Bellocq C, Baro I, Charpentier F, Escande D (2003) Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: a functional homology between voltage-gated and inward rectifier K+ channels. EMBO J 22:5412–5421
Lukacs V, Thyagarajan B, Balla A, Varnai P, Balla T, Rohacs T (2007) Dual regulation of TRPV1 by phosphoinositides. J Neurosci 27:7070–7080
Ma R, Li WP, Rundle D, Kong J, Akbarali HI, Tsiokas L (2005) PKD2 functions as an epidermal growth factor-activated plasma membrane channel. Mol Cell Biol 25:8285–8298
MacGregor GG, Dong K, Vanoye CG, Tang L, Giebisch G, Hebert SC (2002) Nucleotides and phospholipids compete for binding to the C terminus of KATP channels. Proc Natl Acad Sci U S A 99:2726–2731
Maljevic S, Wuttke TV, Seebohm G, Lerche H (2010) KV7 channelopathies. Pflugers Arch 460:277–288
Marrion NV (1997) Control of M-current. Annu Rev Physiol 59:483–504
Marrion NV, Smart TG, Marsh SJ, Brown DA (1989) Muscarinic suppression of the M-current in the rat sympathetic ganglion is mediated by receptors of the M1-subtype. Br J Pharmacol 98:557–573
Mathie A, Bernheim L, Hille B (1992) Inhibition of N- and L-type calcium channels by muscarinic receptor activation in rat sympathetic neurons. Neuron 8:907–914
McKemy DD, Neuhausser WM, Julius D (2002) Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416:52–58
Michailidis IE, Zhang Y, Yang J (2007) The lipid connection-regulation of voltage-gated Ca2+ channels by phosphoinositides. Pflugers Arch 455:147–155
Minke B, Cook B (2002) TRP channel proteins and signal transduction. Physiol Rev 82:429–472
Mitchell CA, Brown S, Campbell JK, Munday AD, Speed CJ (1996) Regulation of second messengers by the inositol polyphosphate 5- phosphatases. Biochem Soc Trans 24:994–1000
Mohapatra DP, Nau C (2005) Regulation of Ca2+-dependent desensitization in the vanilloid receptor TRPV1 by calcineurin and cAMP-dependent protein kinase. J Biol Chem 280:13424–13432
Montell C, Birnbaumer L, Flockerzi V (2002) The TRP channels, a remarkably functional family. Cell 108:595–598
Mucha M, Ooi L, Linley JE, Mordaka P, Dalle C, Robertson B, Gamper N, Wood IC (2010) Transcriptional control of KCNQ channel genes and the regulation of neuronal excitability. J Neurosci 30:13235–13245
Murata Y, Okamura Y (2007) Depolarization activates the phosphoinositide phosphatase Ci-VSP, as detected in Xenopus oocytes coexpressing sensors of PIP2. J Physiol 583:875–889
Niemeyer BA, Bergs C, Wissenbach U, Flockerzi V, Trost C (2001) Competitive regulation of CaT-like-mediated Ca2+ entry by protein kinase C and calmodulin. Proc Natl Acad Sci U S A 98:3600–3605
Nilius B, Owsianik G (2010) Channelopathies converge on TRPV4. Nat Genet 42:98–100
Nilius B, Voets T, Peters J (2005) TRP channels in disease. Sci STKE 2005:re8
Nilius B, Mahieu F, Prenen J, Janssens A, Owsianik G, Vennekens R, Voets T (2006) The Ca2+-activated cation channel TRPM4 is regulated by phosphatidylinositol 4,5-biphosphate. EMBO J 25:467–478
Nilius B, Owsianik G, Voets T (2008) Transient receptor potential channels meet phosphoinositides. EMBO J 27:2809–2816
Numazaki M, Tominaga T, Takeuchi K, Murayama N, Toyooka H, Tominaga M (2003) Structural determinant of TRPV1 desensitization interacts with calmodulin. Proc Natl Acad Sci U S A 100:8002–8006
Otsuguro KI, Tang J, Tang Y, Xiao R, Freichel M, Tsvilovskyy V, Ito S, Flockerzi V, Zhu MX, Zholos AV (2008) Isoform-specific inhibition of TRPC4 channel by phosphatidylinositol 4,5-bisphosphate. J Biol Chem 283:10026–10036
Park KH, Piron J, Dahimene S, Merot J, Baro I, Escande D, Loussouarn G (2005) Impaired KCNQ1-KCNE1 and phosphatidylinositol-4,5-bisphosphate interaction underlies the long QT syndrome. Circ Res 96:730–739
Pegan S, Arrabit C, Zhou W, Kwiatkowski W, Collins A, Slesinger PA, Choe S (2005) Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci 8:279–287
Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, Earley TJ, Dragoni I, McIntyre P, Bevan S, Patapoutian A (2002) A TRP channel that senses cold stimuli and menthol. Cell 108:705–715
Pingle SC, Matta JA, Ahern GP (2007) Capsaicin receptor: TRPV1 a promiscuous TRP channel. Handb Exp Pharmacol 155–171
Piron J, Choveau FS, Amarouch MY, Rodriguez N, Charpentier F, Merot J, Baro I, Loussouarn G (2010) KCNE1-KCNQ1 osmoregulation by interaction of phosphatidylinositol-4,5-bisphosphate with Mg2+ and polyamines. J Physiol 588:3471–3483
Prescott ED, Julius D (2003) A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science 300:1284–1288
Proudfoot CJ, Garry EM, Cottrell DF, Rosie R, Anderson H, Robertson DC, Fleetwood-Walker SM, Mitchell R (2006) Analgesia mediated by the TRPM8 cold receptor in chronic neuropathic pain. Curr Biol 16:1591–1605
Quinn KV, Behe P, Tinker A (2008) Monitoring changes in membrane phosphatidylinositol 4,5-bisphosphate in living cells using a domain from the transcription factor tubby. J Physiol 586:2855–2871
Ramsey IS, Delling M, Clapham DE (2006) An introduction to TRP channels. Annu Rev Physiol 68:619–647
Rapedius M, Soom M, Shumilina E, Schulze D, Schonherr R, Kirsch C, Lang F, Tucker SJ, Baukrowitz T (2005) Long chain CoA esters as competitive antagonists of phosphatidylinositol 4,5-bisphosphate activation in Kir channels. J Biol Chem 280:30760–30767
Raychowdhury MK, Gonzalez-Perrett S, Montalbetti N, Timpanaro GA, Chasan B, Goldmann WH, Stahl S, Cooney A, Goldin E, Cantiello HF (2004) Molecular pathophysiology of mucolipidosis type IV: pH dysregulation of the mucolipin-1 cation channel. Hum Mol Genet 13:617–627
Reid G, Babes A, Pluteanu F (2002) A cold- and menthol-activated current in rat dorsal root ganglion neurones: properties and role in cold transduction. J Physiol 545:595–614
Robbins J, Marsh SJ, Brown DA (1993) On the mechanism of M-current inhibition by muscarinic m1 receptors in DNA-transfected rodent neuroblastoma x glioma cells. J Physiol 469:153–178
Robbins J, Marsh SJ, Brown DA (2006) Probing the regulation of M (Kv7) potassium channels in intact neurons with membrane-targeted peptides. J Neurosci 26:7950–7961
Roberts-Crowley ML, Mitra-Ganguli T, Liu L, Rittenhouse AR (2009) Regulation of voltage-gated Ca2+ channels by lipids. Cell Calcium 45:589–601
Rohacs T (2007) Regulation of TRP channels by PIP2. Pflugers Arch 453:753–762
Rohacs T (2009) Phosphoinositide regulation of non-canonical transient receptor potential channels. Cell Calcium 45:554–565
Rohacs T, Chen J, Prestwich GD, Logothetis DE (1999) Distinct specificities of inwardly rectifying K+ channels for phosphoinositides. J Biol Chem 274:36065–36072
Rohacs T, Lopes C, Mirshahi T, Jin T, Zhang H, Logothetis DE (2002) Assaying phosphatidylinositol bisphosphate regulation of potassium channels. Methods Enzymol 345:71–92
Rohacs T, Lopes CM, Jin T, Ramdya PP, Molnar Z, Logothetis DE (2003) Specificity of activation by phosphoinositides determines lipid regulation of Kir channels. Proc Natl Acad Sci U S A 100:745–750
Rohacs T, Lopes CMB, Michailidis I, Logothetis DE (2005) PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat Neurosci 8:626–634
Rohacs T, Thyagarajan B, Lukacs V (2008) Phospholipase C mediated modulation of TRPV1 channels. Mol Neurobiol 37:153–163
Rosenbaum T, Gordon-Shaag A, Munari M, Gordon SE (2004) Ca2+/calmodulin modulates TRPV1 activation by capsaicin. J Gen Physiol 123:53–62
Rosenhouse-Dantsker A, Logothetis DE (2007) Molecular characteristics of phosphoinositide binding. Pflugers Arch 455:45–53
Rosenhouse-Dantsker A, Sui JL, Zhao Q, Rusinova R, Rodriguez-Menchaca AA, Zhang Z, Logothetis DE (2008) A sodium-mediated structural switch that controls the sensitivity of Kir channels to PtdIns(4,5)P(2). Nat Chem Biol 4:624–631
Rousset M, Cens T, Gouin-Charnet A, Scamps F, Charnet P (2004) Ca2+ and phosphatidylinositol 4,5-bisphosphate stabilize a Gβγ-sensitive state of Cav2 Ca 2+ channels. J Biol Chem 279:14619–14630
Runnels LW, Yue L, Clapham DE (2002) The TRPM7 channel is inactivated by PIP2 hydrolysis. Nat Cell Biol 4:329–336
Saleh SN, Albert AP, Large WA (2008) Obligatory role for phosphatidylinositol-4, 5-bisphosphate in activation of native TRPC1 store-operated channels in vascular myocytes. J Physiol 587:531–540
Saleh SN, Albert AP, Large WA (2009) Activation of native TRPC1/C5/C6 channels by endothelin-1 is mediated by both PIP3 and PIP2 in rabbit coronary artery myocytes. J Physiol 587:5361–5375
Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT (1996) Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature 384:80–83
Selyanko AA, Brown DA (1996) Intracellular calcium directly inhibits potassium M channels in excised membrane patches from rat sympathetic neurons. Neuron 16:151–162
Selyanko AA, Stansfeld CE, Brown DA (1992) Closure of potassium M-channels by muscarinic acetylcholine-receptor stimulants requires a diffusible messenger. Proc Biol Sci 250:119–125
Selyanko AA, Hadley JK, Brown DA (2001) Properties of single M-type KCNQ2/KCNQ3 potassium channels expressed in mammalian cells. J Physiol 534:15–24
Selyanko AA, Delmas P, Hadley JK, Tatulian L, Wood IC, Mistry M, London B, Brown DA (2002) Dominant-negative subunits reveal potassium channel families that contribute to M-like potassium currents. J Neurosci 22:RC212
Shapiro MS, Wollmuth LP, Hille B (1994) Angiotensin II inhibits calcium and M current channels in rat sympathetic neurons via G proteins. Neuron 12:1319–1329
Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B (2000) Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. J Neurosci 20:1710–1721
Shumilina E, Klocker N, Korniychuk G, Rapedius M, Lang F, Baukrowitz T (2006) Cytoplasmic accumulation of long-chain coenzyme A esters activates KATP and inhibits Kir2.1 channels. J Physiol 575:433–442
Shyng SL, Nichols CG (1998) Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 1138–1141
Soejima M, Noma A (1984) Mode of regulation of the ACh-sensitive K-channel by the muscarinic receptor in rabbit atrial cells. Pflugers Arch 400:424–431
Soom M, Schonherr R, Kubo Y, Kirsch C, Klinger R, Heinemann SH (2001) Multiple PIP2 binding sites in Kir2.1 inwardly rectifying potassium channels. FEBS Lett 490:49–53
Stein AT, Ufret-Vincenty CA, Hua L, Santana LF, Gordon SE (2006) Phosphoinositide 3-Kinase Binds to TRPV1 and Mediates NGF-stimulated TRPV1 Trafficking to the Plasma Membrane. J Gen Physiol 128:509–522
Stolz LE, Huynh CV, Thorner J, York JD (1998) Identification and characterization of an essential family of inositol polyphosphate 5-phosphatases (INP51, INP52 and INP53 gene products) in the yeast Saccharomyces cerevisiae. Genetics 148:1715–1729
Suh BC, Hille B (2002) Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35:507–520
Suh BC, Hille B (2007) Electrostatic interaction of internal Mg2+ with membrane PIP2 Seen with KCNQ K+ channels. J Gen Physiol 130:241–256
Suh BC, Hille B (2008) PIP2 is a necessary cofactor for ion channel function: how and why? Annu Rev Biophys 37:175–195
Suh BC, Inoue T, Meyer T, Hille B (2006) Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science 314:1454–1457
Suh BC, Leal K, Hille B (2010) Modulation of high-voltage activated Ca2+ channels by membrane phosphatidylinositol 4,5-bisphosphate. Neuron 67:224–238
Sui JL, Petit Jacques J, Logothetis DE (1998) Activation of the atrial KACh channel by the betagamma subunits of G proteins or intracellular Na+ ions depends on the presence of phosphatidylinositol phosphates. Proc Natl Acad Sci U S A 95:1307–1312
Suzuki Y, Pasch A, Bonny O, Mohaupt MG, Hediger MA, Frey FJ (2008) Gain-of-function haplotype in the epithelial calcium channel TRPV6 is a risk factor for renal calcium stone formation. Hum Mol Genet 17:1613–1618
Szentpetery Z, Balla A, Kim YJ, Lemmon MA, Balla T (2009) Live cell imaging with protein domains capable of recognizing phosphatidylinositol 4,5-bisphosphate; a comparative study. BMC Cell Biol 10:67
Takezawa R, Schmitz C, Demeuse P, Scharenberg AM, Penner R, Fleig A (2004) Receptor-mediated regulation of the TRPM7 channel through its endogenous protein kinase domain. Proc Natl Acad Sci U S A 101:6009–6014
Talavera K, Staes M, Janssens A, Droogmans G, Nilius B (2004) Mechanism of arachidonic acid modulation of the T-type Ca2+ channel alpha1G. J Gen Physiol 124:225–238
Thyagarajan B, Lukacs V, Rohacs T (2008) Hydrolysis of phosphatidylinositol 4,5-bisphosphate mediates calcium induced inactivation of TRPV6 channels. J Biol Chem 283:14980–14987
Thyagarajan B, Benn BS, Christakos S, Rohacs T (2009) Phospholipase C mediated regulation of TRPV6 channels: implications in active intestinal Ca2+ transport. Mol Pharmacol 75:608–616
Trebak M, Lemonnier L, Smyth JT, Vazquez G, Putney JW Jr (2007) Phospholipase C-coupled receptors and activation of TRPC channels. Handb Exp Pharmacol 593–614
Trebak M, Lemonnier L, Dehaven WI, Wedel BJ, Bird GS, Putney JW Jr (2008) Complex functions of phosphatidylinositol 4,5-bisphosphate in regulation of TRPC5 cation channels. Pflugers Arch
Tucker SJ, Baukrowitz T (2008) How highly charged anionic lipids bind and regulate ion channels. J Gen Physiol 131:431–438
Vallon V, Grahammer F, Volkl H, Sandu CD, Richter K, Rexhepaj R, Gerlach U, Rong Q, Pfeifer K, Lang F (2005) KCNQ1-dependent transport in renal and gastrointestinal epithelia. Proc Natl Acad Sci U S A 102:17864–17869
Wal J van der, Habets R, Varnai P, Balla T, Jalink K (2001) Monitoring agonist-induced phospholipase C activation in live cells by fluorescence resonance energy transfer. J Biol Chem 276:15337–15344
Varnai P, Balla T (1998) Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol 143:501–510
Varnai P, Balla T (2006) Live cell imaging of phosphoinositide dynamics with fluorescent protein domains. Biochim Biophys Acta 1761:957–967
Varnai P, Thyagarajan B, Rohacs T, Balla T (2006) Rapidly inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact cells. J Cell Biol 175:377–382
Vellani V, Reynolds AM, McNaughton PA (2000) Modulation of the synaptic Ca2+ current in salamander photoreceptors by polyunsaturated fatty acids and retinoids. J Physiol 529:333–344
Walder RY, Landau D, Meyer P, Shalev H, Tsolia M, Borochowitz Z, Boettger MB, Beck GE, Englehardt RK, Carmi R, Sheffield VC (2002) Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat Genet 31:171–174
Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, Jager T de, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT (1996) Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 12:17–23
Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D (1998) KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282:1890–1893
Wang YY, Chang RB, Waters HN, McKemy DD, Liman ER (2008) The nociceptor ion channel TRPA1 is potentiated and inactivated by permeating calcium ions. J Biol Chem 283:32691–32703
Watanabe H, Nagata E, Kosakai A, Nakamura M, Yokoyama M, Tanaka K, Sasai H (2000) Disruption of the epilepsy KCNQ2 gene results in neural hyperexcitability. J Neurochem 75:28–33
Wenk MR, Lucast L, Di Paolo G, Romanelli AJ, Suchy SF, Nussbaum RL, Cline GW, Shulman GI, McMurray W, De Camilli P (2003) Phosphoinositide profiling in complex lipid mixtures using electrospray ionization mass spectrometry. Nat Biotechnol 21:813–817
Wilson PD (2004) Polycystic kidney disease. N Engl J Med 350:151–164
Winks JS, Hughes S, Filippov AK, Tatulian L, Abogadie FC, Brown DA, Marsh SJ (2005) Relationship between membrane phosphatidylinositol-4,5-bisphosphate and receptor-mediated inhibition of native neuronal M channels. J Neurosci 25:3400–3413
Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB (2005) A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science
Wu L, Bauer CS, Zhen XG, Xie C, Yang J (2002) Dual regulation of voltage-gated calcium channels by PtdIns(4,5)P2. Nature 419:947–952
Xiao YF, Gomez AM, Morgan JP, Lederer WJ, Leaf A (1997) Suppression of voltage-gated L-type Ca2+ currents by polyunsaturated fatty acids in adult and neonatal rat ventricular myocytes. Proc Natl Acad Sci U S A 94:4182–4187
Yao J, Qin F (2009) Interaction with phosphoinositides confers adaptation onto the TRPV1 pain receptor. PLoS Biol 7:e46
Yaradanakul A, Hilgemann DW (2007) Unrestricted diffusion of exogenous and endogenous PIP2 in baby hamster kidney and Chinese hamster ovary cell plasmalemma. J Membr Biol 220:53–67
Yu FH, Catterall WA (2004) The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE 2004:re15
Yus-Najera E, Santana-Castro I, Villarroel A (2002) The identification and characterization of a noncontinuous calmodulin-binding site in noninactivating voltage-dependent KCNQ potassium channels. J Biol Chem 277:28545–28553
Zaika O, Lara LS, Gamper N, Hilgemann DW, Jaffe DB, Shapiro MS (2006) Angiotensin II regulates neuronal excitability via phosphatidylinositol 4,5-bisphosphate-dependent modulation of Kv7 (M-type) K+ channels. J Physiol 575:49–67
Zaika O, Tolstykh GP, Jaffe DB, Shapiro MS (2007) Inositol triphosphate-mediated Ca2+ signals direct purinergic P2Y receptor regulation of neuronal ion channels. J Neurosci 27:8914–8926
Zakharian E, Thyagarajan B, French RJ, Pavlov E, Rohacs T (2009) Inorganic polyphosphate modulates TRPM8 channels. PLoS One 4:e5404
Zakharian E, Cao C, Rohacs T (2010) Gating of transient receptor potential melastatin 8 (TRPM8) channels activated by cold and chemical agonists in planar lipid bilayers. J Neurosci 30:12526–12534
Zamponi GW, Snutch TP (1998) Decay of prepulse facilitation of N type calcium channels during G protein inhibition is consistent with binding of a single Gbeta subunit. Proc Natl Acad Sci U S A 95:4035–4039
Zhang H, He C, Yan X, Mirshahi T, Logothetis DE (1999) Activation of inwardly rectifying K+ channels by distinct PtdIns(4,5)P2 interactions. Nat Cell Biol 1:183–188
Zhang Y, Cribbs LL, Satin J (2000) Arachidonic acid modulation of alpha1H, a cloned human T-type calcium channel. Am J Physiol Heart Circ Physiol 278:H184–H193
Zhang H, Craciun LC, Mirshahi T, Rohacs T, Lopes CMB, Jin T, Logothetis DE (2003) PIP2 activates KCNQ channels and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37:963–975
Zhang Z, Okawa H, Wang Y, Liman ER (2005) Phosphatidylinositol 4,5-bisphosphate rescues TRPM4 channels from desensitization. J Biol Chem 280:39185–39192
Zhen XG, Xie C, Yamada Y, Zhang Y, Doyle C, Yang J (2006) A single amino acid mutation attenuates rundown of voltage-gated calcium channels. FEBS Lett 580:5733–5738
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Gamper, N., Rohacs, T. (2012). Phosphoinositide Sensitivity of Ion Channels, a Functional Perspective. In: Balla, T., Wymann, M., York, J. (eds) Phosphoinositides II: The Diverse Biological Functions. Subcellular Biochemistry, vol 59. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-3015-1_10
Download citation
DOI: https://doi.org/10.1007/978-94-007-3015-1_10
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-3014-4
Online ISBN: 978-94-007-3015-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)