
Abstract
The tight interplay between endoplasmic reticulum (ER) and mitochondria is a key determinant of cell function and survival through the control of intracellular calcium (Ca2+) signaling. The specific sites of physical association between ER and mitochondria are known as mitochondria-associated membranes (MAMs). It has recently become clear that MAMs are crucial for highly efficient transmission of Ca2+ from the ER to mitochondria, thus controlling fundamental processes involved in energy production and also determining cell fate by triggering or preventing apoptosis. In this contribution, we summarize the main features of the Ca2+-signaling toolkit, covering also the latest breakthroughs in the field, such as the identification of novel candidate proteins implicated in mitochondrial Ca2+ transport and the recent direct characterization of the high-Ca2+ microdomains between ER and mitochondria. We review the main functions of these two organelles, with special emphasis on Ca2+ handling and on the structural and molecular foundations of the signaling contacts between them. Additionally, we provide important examples of the physiopathological role of this cross-talk, briefly describing the key role played by MAMs proteins in many diseases, and shedding light on the essential role of mitochondria-ER interactions in the maintenance of cellular homeostasis and the determination of cell fate.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Akt
- Apoptosis
- Bap31
- Bip
- Ca2+ signaling
- Calcium ions
- Endoplasmic Reticulum
- Ero1α
- ERp44
- GM1-ganglioside
- grp75
- IP3Rs
- MCU
- Microdomains
- MICU1
- Mitochondria
- Mitochondria-Associated Membranes
- Mitofusin-1 and -2
- p66Shc
- PACS-2
- Plasma Membrane Associated Membranes
- PML
- PP2a
- Presenilin-1 and -2
- Sig-1R
- VDAC
The Ca2+-Signaling Toolkit
Calcium ions (Ca2+) are ubiquitous intracellular messengers that can set up and/or regulate many different cellular functions, including gene expression, cellular contraction, secretion, synaptic transmission, metabolism, differentiation and proliferation, as well as cell death. The universality of Ca2+-based signaling depends on its enormous versatility in terms of amplitude, duration, frequency and localization. The formation of the correct spatio-temporal Ca2+ signals is dependent on an extensive cellular machinery named the Ca2+ toolkit, which includes the various cellular Ca2+-binding and Ca2+-transporting proteins, present mainly in the cytosol, plasma membrane, endoplasmic reticulum (ER), and mitochondria [1].
The resting cytosolic Ca2+ concentration ([Ca2+]c) is maintained around the value of 100 nM, significantly lower than extracellular [Ca2+] (1 mM). This condition is achieved through active extrusion of Ca2+ by the plasma membrane Ca2+ ATPase (PMCA) and the Na+/Ca2+ exchanger (NCX) [2, 3]. The increase of intracellular [Ca2+] can be elicited by two fundamental mechanisms (or a combination of both). The first involves Ca2+ entry from the extracellular milieu, through the opening of plasma membrane Ca2+ channels (traditionally grouped into three classes: voltage operated Ca2+ channels (VOCs) [4], receptor operated Ca2+ channels (ROCs) [5] and second messenger operated Ca2+ channels (SMOCs) [6]); the second mechanism involves Ca2+ release from intracellular stores, mainly the ER and its specialized form in muscle, the sarcoplasmic reticulum (SR). In these intracellular stores, two main Ca2+-release channels exist that, upon stimulation, release Ca2+ into the cytosol, thus triggering Ca2+ signaling: the inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs) and the ryanodine receptors (RyRs) [7, 8]. IP3Rs are ligand-gated channels that function in releasing Ca2+ from ER Ca2+ stores in response to IP3 generation initiated by agonist binding to cell-surface G protein-coupled receptor [9, 10]. The subsequent rise in [Ca2+]c results in various Ca2+-dependent intracellular events. The exact cellular outcome depends on the spatiotemporal characteristics of the generated Ca2+ signal [11]. Once its downstream targets are activated, basal [Ca2+]c levels are regained by the combined activity of Ca2+ extrusion mechanisms, such as PMCA and NCX, and mechanisms that refill the intracellular stores, like sarco-endoplasmic reticulum Ca2+ ATPases (SERCAs) [2]. Due to SERCA activity and intraluminal Ca2+-binding proteins (CABPs), i.e., calnexin and calreticulin [12], the ER can accumulate Ca2+ more than a thousand-fold excess as compared to the cytosol.
While the role of the ER as a physiologically important Ca2+ store has long been recognized, a similar role for mitochondria have seen a reappraisal only in the past two decades [13]. The studies of Rizzuto, Pozzan and colleagues revealed that IP3-mediated Ca2+ release from the ER results in cytosolic Ca2+ increases that are accompanied by similar or even larger mitochondrial ones [14], driven by the large electrochemical gradient (mitochondrial membrane potential difference, ΔΨm = −180 mV, negative inside) generated by the respiratory chain [15]. The uptake of the Ca2+ ions into the mitochondrial matrix implies different transport systems responsible for the transfer of Ca2+ across the outer and the inner mitochondrial membrane (OMM and IMM respectively). Despite the surprisingly low affinity of the mitochondrial uptake systems (Kd around 10–20 μM) and the submicromolar global [Ca2+]c (which rarely exceed 2–3 μM) evoked by IP3-mediated Ca2+ release, mitochondrial Ca2+ concentration ([Ca2+]m) can undergo rapid changes upon cell stimulation, because their low affinity uptake systems are exposed to microdomains of high [Ca2+] in proximity to ER or plasma membrane Ca2+ channels [16–18]. The hypothesis, called “microdomain hypothesis” [19], was initially supported by a large body of indirect evidence, and its direct determination was carried out only very recently by two complementary studies that demonstrated the existence and amplitude of high Ca2+ microdomains on the surface of mitochondria. Giacomello et al. [20] targeted a new generation of FRET-based Ca2+ sensors [21] to the OMM and, through a sophisticated statistical analysis of the images, revealed the existence of small OMM regions whose [Ca2+] reaches values as high as 15–20 μM. The probe detected Ca2+ hotspots on about 10% of the OMM surface that were not observed in other parts of the cell. The Ca2+ hotspots were not uniform, and their frequency varied among mitochondria of the same cell. Moreover, classical epifluorescence and total internal reflection fluorescence (TIRF) microscopy experiments were combined in order to monitor the generation of high Ca2+ microdomains in mitochondria located near the plasma membrane. With this approach, it could be shown that Ca2+ hotspots on the surface of mitochondria occur upon opening of VOCs, but not upon capacitative Ca2+ entry (CCE). Csordás et al. [22] used a complementary approach in which they generated genetically encoded bifunctional linkers consisting of OMM and ER targeting sequences connected through a fluorescent protein, including a low-Ca2+-affinity pericam, and coupled with the two components of the FKBP-FRB heterodimerization system [23], respectively. Using rapamycin-assembled heterodimerization of the FKBP-FRB-based linker, they detected ER/OMM and plasma membrane/OMM junctions (the latter at a much lower frequency). In addition, the recruited low-Ca2+-affinity pericam reported Ca2+ concentrations as high as 25 μM at the ER/OMM junctions in response to IP3-mediated Ca2+ release, which is in excellent agreement with the values obtained by Giacomello et al..
The Ca2+-import system across the OMM occurs through the so-called voltage-dependent anion channels (VDAC) [24], traditionally considered as a large voltage-gated channel, fully opened with high-conductance and weak anion-selectivity at zero and low transmembrane potentials (<20–30 mV), but switching to cation selectivity and lower conductance at higher potentials (the so-called “closed” state) [25–27]. In contrast, the molecular identity of the IMM Ca2+-transport system, the mitochondrial Ca2+ uniporter (MCU), has been identified only very recently, preceded last year by the discovery of mitochondrial calcium uptake 1 (MICU1), an uniporter regulator which appears essential for mitochondrial Ca2+ uptake [28]. MICU1 has been identified in silico in the MitoCarta database [29]; it is a single-pass transmembrane protein which does not seem to participate in channel pore formation, so it is not known whether it actually forms (part of) a Ca2+ channel, or functions as Ca2+ buffer, or as a Ca2+-dependent regulatory protein acting as a Ca2+ sensor (it has a pair of Ca2+-binding EF-hand domains, the mutation of which eliminates the mitochondrial Ca2+ uptake). Then, this year, two independent papers identified the same protein, termed CCDC109A and renamed MCU, as the channel responsible for ruthenium-red-sensitive mitochondrial Ca2+ uptake. This protein shares the same tissue distribution with its regulator MICU1, and possesses two predicted transmembrane helices, which are separated by a highly conserved linker facing the intermembrane space. Just the protein’s orientation is the mainly discrepancy between the two papers, one affirming a C-terminus localization in the intermembrane space [30], the other in the matrix [31]. Further experiments have to be performed to solve this question. Interestingly, MCU can form multimers and blue native gel separation experiment shows how MCU migrates as a large complex, with an apparent molecular weight of 40 kDa [31].
In the IMM are also present the mitochondrial Na+/Ca2+ exchanger (mNCX) and the H+/Ca2+ exchanger (mHCX). Their main function is probably to export Ca2+ from the matrix, once mitochondrial Ca2+ has carried out its function, to reestablish resting conditions [32]. They have yet to be identified, although recently strong evidence has been provided that the Na+/Ca2+ exchanger isoform NCLX is the long-sought protein responsible for the mitochondrial Na+-dependent Ca2+ efflux [33]. Finally, the low conductance mode of the permeability transition pore (PTP), a channel of still debated nature localized in the IMM [34], can be also considered as a non-saturating mechanism for Ca2+ efflux from mitochondria. When open, PTP allows the passage of ions and molecules with a molecular weight up to 1.5 kDa, including Ca2+. Short-time openings may have a physiological function but its long-time activation leads to the demise of the cell, either by apoptosis or by necrosis, depending on whether PTP opening occurs in only a small fraction of the mitochondria or in all of them [35, 36].
The many efforts to better understand the Ca2+ toolkit and the role played by the relationship between ER and mitochondria in this elaborate signaling, are yielding a deeper understanding of how aberrant Ca2+ homeostasis is implicated in many diseases. A schematic view of the various processes described above is presented in Fig. 17.1.

Intracellular Ca 2+ signaling. Schematic model of intracellular Ca2+ homeostasis. Plasma membrane G-protein coupled receptors activate phospholipase C-β (PLC-β) to promote the generation of inositol 1,4,5-trisphosphate (IP3) and the release of Ca2+ from the endoplasmic reticulum (ER) into the cytosol. Mitochondrial surface directly interacts with the ER through contact sites defining hotspots Ca2+ signaling units. Ca2+ import across the outer mitochondrial membrane (OMM) occurs by the voltage-dependent anion channel (VDAC), and then enters the matrix through the mitochondrial Ca2+ uniporter (MCU), the main inner mitochondrial membrane (IMM) Ca2+-transport system (Ca2+ levels reached upon stimulation are indicated in square brackets). Mitochondrial Ca2+exchangers present in the IMM export Ca2+ from the matrix once mitochondrial Ca2+ has carried its function; another mechanism for Ca2+ efflux from mitochondria is the permeability transition pore (PTP). Ca2+ levels return to resting conditions (indicated in round brackets) through the concerted action of cytosolic Ca2+ buffers, plasma membrane Ca2+-ATPase (PMCA) and the Na+/Ca2+ exchanger (NCX) that permit the ion extrusion in the extracellular milieu. Sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) restablishes basal Ca2+ levels in intracellular stores. ANT adenine nucleotide translocase, Cyp D cyclophilin D, DAG diacylglycerol, PIP 2 phosphatidylinositol 4,5-bisphosphate
Mitochondrial Functions and Ca2+ Handling
Mitochondrial Ca2+ homeostasis has a key role in the regulation of aerobic metabolism and cell survival.
The first role assigned to the Ca2+ ions taken up into the mitochondrial matrix was the stimulation of the mitochondrial ATP production since important metabolic enzymes localized in the matrix, the pyruvate-, α-ketoglutarate- and isocitrate-dehydrogenases are activated by Ca2+, with different mechanisms: the first through a Ca2+-dependent dephosphorylation step, the others via direct binding to a regulatory site [37, 38]. Those three enzymes represent rate-limiting steps of the Krebs cycle thus controlling the feeding of electrons into the respiratory chain and the generation of the proton gradient across the inner membrane, in turn necessary for ATP production through oxidative phosphorylation (OXPHOS). As the ATP produced by mitochondria is subsequently transferred to the cytosol, mechanisms that control ATP production will not only affect overall cell life but, more specifically, will regulate the activity of ATP-sensitive proteins localized in the close vicinity of mitochondria, such as IP3Rs and SERCA which are stimulated by ATP [39, 40]. The bidirectional relation between Ca2+ release and ATP production allows for a positive feedback regulation between ER and mitochondria during increased energetic demand [41]. The uptake of Ca2+ in mitochondria will also affect Ca2+ signaling at both the local and the global level. Assuming the microdomain concept [16, 17], the local [Ca2+] will depend on both the amount of Ca2+ released by IP3Rs and that taken up by mitochondria. Since both SERCA pumps and IP3Rs are also regulated by Ca2+, the local [Ca2+] in the vicinity of mitochondria will determine the refilling of the ER and eventually the spatiotemporal characteristics of the subsequent Ca2+ signals [42]. This will in turn depend on the efficiency of the coupling between the ER and the mitochondrial network, as well as on the exact subcellular localization of mitochondria [43].
The connection between mitochondria and the ER can be highly dynamic as the local Ca2+ concentration can also affect mitochondrial motility and ER–mitochondria associations in various ways [44]. Mitochondrial movement may increase the chance of dynamic interactions between organelles and aid in the transportation of molecules between the cytoplasm and the organelle. Proteins involved in mitochondrial movement along microtubules, dynein and kinesin, are prone to high [Ca2+]c mediated by a Ca2+ sensor. Moreover, as the mitochondrial motility is inhibited by Ca2+ levels in the low micromolar range, it means that mitochondria will be trapped in the neighbourhood of active Ca2+-release sites, allowing for a more efficient uptake of Ca2+ by these mitochondria [45, 46].
Apart from organelles movement, mitochondria also continuously remodel their shape. Many of the gene products mediating the fission and fusion processes have been identified in yeast screens, and most are conserved in mammals, including the fission mediators dynamin-related protein 1 (Drp1, Dnm1 in yeast) and Fis1 (Fission 1 homologue), as well as the fusion mediators mitofusins (Mfn) 1 and 2 (Fzo1 in yeast) and optic atrophy 1 (OPA1, Mgm1 in yeast) [47]. Several previous studies have indicated that elevation of [Ca2+]c perturbs mitochondrial dynamics [48], and more recent works have clearly demonstrated that mitochondrial shape can be controlled by an ER-dependent signaling pathway [49, 50]. Mitochondria also undergo a more “macroscopic” remodelling of their shape during programmed cell death: after apoptosis induction, mitochondria become largely fragmented, resulting in small, rounded and numerous organelles. However, the relationship between mitochondrial fusion/fission and apoptosis is complex and mitochondrial fragmentation is not necessarily related to apoptosis [51]. Defects in fusion and fission processes are not to be underestimated, as they may have deleterious consequences on bioenergetic parameters and are likely to contribute to the pathogenesis of neurodegenerative diseases [52].
The mitochondrial Ca2+ signal also control the choice between cell survival and cell death, as it can participate in the induction and progression of apoptosis [53, 54]. Although the extrinsic pathway for apoptosis may or may not involve mitochondria, in the intrinsic pathway these organelles perform a pivotal role: they can release a number of proapototic factors - such as cytochrome c, apoptosis-inducing factor, Smac/Diablo, HtrA2/Omi and endonuclease G – from the intermembrane space (IMS) to the cytosol, which initiate the executor caspase cascade steering the cell towards the execution phase of apoptosis (for a review see [55]). Ca2+ uptake in mitochondria is crucial for multiple important cellular functions, but the risk of mitochondrial Ca2+ overload exists, and this may result in the induction of cell death. At a high concentration, mitochondrial Ca2+ favours the opening of the PTP, a mitochondrial megachannel likely to be located in the inner-outer contact sites of the mitochondrial membranes [35]. This event, also known as mitochondrial membrane permeabilization (MMP), leads to the subsequent release of various apoptogenic factors [56, 57]. MMP can also result from a distinct, yet partially overlapping process known as mitochondrial outer membrane permeabilization (MOMP) [55]. In MOMP, pro-apoptotic members of the Bcl-2 protein family may form protein-permeable pores in the OMM, and consequently release of IMS proteins into the cytosol. Bcl-2 family members function as regulators of Ca2+ signaling will be discussed later in this review (the interested reader should also refer to [58]).
Mitochondria are also an important source of ROS produced during OXPHOS. ROS can locally affect other systems, including Ca2+-signaling mechanisms, and increased levels of ROS within mitochondria are the principal trigger not only for mitochondrial dysfunctions but, more generally, for diseases associated with ageing. One of the key regulators of ROS production, mitochondrial dysfunction, and ageing is the 66-kDa isoform of the growth factor adapter shc (p66shc) [59]. The mechanisms by which p66shc increases intracellular ROS levels, inducing apoptosis and the deleterious effects of ageing have recently been clarified by Pinton et al.. Once imported into mitochondria, p66Shc causes alterations of organelle Ca2+ responses and three-dimensional structure, thus inducing apoptosis [60].
ER Functions and Ca2+ Handling
The ER is possibly the largest individual intracellular organelle comprising a three dimensional network of endomembranes arranged in a complex grid of microtubules and cisternae. It is made up of functionally and structurally distinct domains (reviewed extensively by a number of authors [61–64]), in relation to the variety of cellular functions played by the organelle, primarily concerning protein synthesis, maturation and delivery to their destination [65, 66]. Moreover, the ER is a dynamic reservoir of Ca2+ ions, which can be activated by both electrical and chemical cell stimulation [67, 68] making this organelle an indispensable component of Ca2+ signaling [69–71].
Modern analysis methods enabled the determination of the molecular profile of the ER. This profile reflects the ER’s role in signaling, as it comprises a number of components constituting the Ca2+ signaling pathway. It contains IP3Rs, RyRs, SERCAs, and in addition to these release channels and pumps, there are buffers (calnexin, calreticulin) and a number of ancillary proteins (FK 506-binding proteins, sorcin, triadin, phosholamban) that contribute to the ER Ca2+ signaling system [72].
The IP3Rs are activated after cell stimulation and play a crucial role in the initiation and propagation of the complex spatio-temporal Ca2+ signaling that control a myriad of cellular processes [73]. To achieve these various functions, often in a single cell, exquisite control of the Ca2+ release is needed. Ca2+ itself regulates channel activity in a biphasic manner: at low [Ca2+], the ion exerts an activating role while, at high [Ca2+], it has an opposite inhibitory effect, thus providing a fine dynamic feedback regulation during Ca2+ release [74]. In addition, also the ER Ca2+ content retains the capability to regulate the channel opening [75, 76]. Whereas IP3 and Ca2+ are essential for IP3R channel activation, other physiological ligands, such as ATP, are not necessary but can finely modulate the Ca2+-sensitivity of the channel [77]. As for Ca2+, the modulation of IP3R by ATP is biphasic: at micromolar concentrations, ATP exerts a stimulatory effect, while inhibiting channel opening in the millimolar range [78, 79]. Moreover, IP3R isoforms contain on their sequences multiple phosphorylation consensus sites and many docking sites for protein kinases and phosphatases. Currently, at least 12 different protein kinases are known to directly phosphorylate the IP3R [80], among them: Akt [81], protein kinase A (cAMP-dependent) [82], protein kinase G (cGMPdependent) [83], calmodulin-dependent protein kinase II (CaMKII) [84], protein kinase C (PKC) [85], and various protein tyrosine kinases [86].
Despite controlling many processes essential for life, Ca2+ arising from the ER can be a potent death-inducing signal [87, 88]. A clear impetus in the study of Ca2+ homeostasis in apoptosis came from the observation that important regulators of apoptosis, the proteins of the Bcl-2 family, are localized to ER and mitochondria, organelles deeply involved in Ca2+ handling. The role of the ER in supporting the mitochondrial apoptosis pathway is demonstrated by several findings, among which: (i) over-expression of anti-apoptotic proteins, such as Bcl-2, reduce the ER Ca2+ level, making the cells resistant to apoptosis [89–92]; (ii) genetic ablation of the pro-apoptotic proteins Bax and Bak (that drastically increases the resistance to death signals) also results in a dramatic reduction in ER Ca2+ content and consequently in a reduction of the Ca2+ that can be transferred to mitochondria [93, 94]; (iii) several different approaches resulting in decreases of ER Ca2+ content protect cells from apoptosis while, conversely, an increase in Ca2+ within the ER favours apoptosis triggered by a number of stimuli [95].
Hence, IP3R-mediated release of Ca2+ from ER appears to be a key sensitizing step in various apoptotic routes, but the precise molecular definition of this process still awaits a fine clarification of the macromolecular complex assembled at the interphase between the two organelles. As will be discussed shortly, significant research efforts have been made to shed some light on this signaling pathway.
ER and Mitochondria Physically and Functionally Interact at MAMs
Intracellular organelles coordinate complex pathways for signal transduction and metabolism in the cell through their functional or physical interactions with one another. The association between ER and mitochondria was first described by Copeland and Dalton over 50 years ago in pseudobranch gland cells [96]. By the beginning of the 1970s, the contacts between mitochondria and ER had been visualized by several groups [97, 98]. Electron micrograph images of quickly frozen samples [99] and experiments in living cells with the two organelles labelled by means of targeted spectral variants of GFP (mtBFP and erGFP) [17] demonstrated conclusively that such physical interactions between the two organelles indeed exist. These experiments revealed the presence of overlapping regions of the two organelles and allowed to estimate the area of the contact sites as 5–20% of the total mitochondrial surface. The distance between the ER and the OMM was originally estimated to be approximately 100 nm [100, 101]. More detailed morphological studies, carried out by Achleitner et al. in 1999, indicated that the distance between the ER and mitochondria in the areas of interaction varied between 10 and 60 nm [102]. Importantly, a direct fusion between membranes of the ER and mitochondria was not observed in any case, and the membranes invariably maintained their separate structures. The authors of this pioneering paper proposed that a distance of less than 30 nm between the two organelles could be considered as an association. More recently, electron tomography techniques allowed to estimate that the minimum distance is even shorter (e.g., 10–25 nm) [103]. This distance thus enables ER proteins to associate directly with proteins and lipids of the OMM. Further development of microscopic techniques enabled detailed analysis of such contacts with high resolution in three dimensions [104].
The interactions between these organelles at the contact sites are so tight and strong, that upon subcellular fractionation (at the step of mitochondria purification), a unique fraction, originally named “mitochondria-associated membranes” (MAMs) fraction, can be isolated [105, 106]. More recently, the isolation procedures was improved and adapted to isolate the MAMs fraction from yeast, different organs, tissues, and various cell lines [102, 107, 108]. Interestingly, the molecular analysis of both “crude” mitochondria and MAMs fractions demonstrated that, apart from specific ER and mitochondrial proteins, they also contain proteins which are abundant in the plasma membrane.
Research on the morphological organization of mitochondria and ER with respect to the plasma membrane is much less extensive. Modifications in the subcellular fractionation procedure enabled the isolation of the “plasma membrane associated membranes” (PAMs) fraction. In general, PAMs fractions have been described as the center of interactions between plasma membrane and the ER [109, 110], but the presence of mitochondrial proteins in these fractions indicates that mitochondria interact actively also with the plasma membrane [111, 112].
The MAMs have a pivotal role in several cellular functions related to bioenergetics and cell survival. MAMs have been originally shown to be enriched in enzymes involved in lipid synthesis and trafficking between ER and mitochondrial membranes, including long-chain fatty acid-CoA ligase type 4 (FACL4) and phosphatidylserine synthase-1 (PSS-1) [106, 113, 114]. The MAMs have since been shown to be enriched in functionally diverse enzymes involved not only in lipid metabolism but also in glucose metabolism (for recent reviews, see [115, 116]).
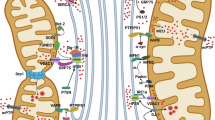
More recently, the same subcellular fraction has been shown to contain Ca2+-sensing ER chaperones and oxidoreductases, as well as key Ca2+ handling proteins of both organelles [117, 118] (a schematic representation of the interorganelle interactions and some of these proteins with the assigned functions is presented in Fig. 17.2). Together, these data have led to the conclusion that the MAMs are not only a site of lipid synthesis and transfer, but also function as a fundamental hub of cellular signaling that controls a growing number of processes associated with both organelles, ranging from ER chaperone-assisted folding of newly synthesized proteins to the fine-tuning of physiological and pathological Ca2+ signals from ER to mitochondria.

Schematic view of the interorganelle interactions and protein composition of the membranes contact sites. Possible contact sites between organelles are marked in dotted brown line. ER endoplasmic reticulum, ER lumen endoplasmic reticulum lumen, IMM inner mitochondrial membrane, MAMs mitochondria-associated membranes, OMM outer mitochondrial membrane, PAMs plasma membrane associated membranes, PM plasma membrane.The color indicates the function/role of the protein. Akt, the serine-threonine protein kinase Akt; ANT adenine nucleotide translocase, Bap31 B-cell receptor-associated protein 31 (or endoplasmic reticulum resident cargo receptor), Calr carleticulin, CRAC Ca2+ release-activated calcium channel, Cyp D cyclophilin D, cyt. c cytochrome c, ERp44 endoplasmic reticulum resident protein 44, grp75 glucose-regulated protein 75 (or mortalin), BiP Binding immunoglobulin Protein (or 78 kDa glucose-regulated protein (GRP78)), IP3R inositol 1,4,5-triphosphate receptor, MCU mitochondrial calcium uniporter, Mfn1/2 mitofusin-1/2, Ora i ORAI calcium release-activated calcium modulator, OSBP oxysterol binding protein, p66Shc 66-kDa isoform of the growth factor adapter shc, PACS-2 phosphofurin acidic cluster sorting protein 2, PEMT2 phosphatidylethanolamine N-methyltransferase 2, PP2a protein phosphatase 2a, PML promyelocytic leukemia protein, PS1/2 presenilin-1/2, PSS-1a phosphatidylserine synthase-1a, SERCA2b sarco-endoplasmic reticulum calcium ATPase 2b, Sig-1R Sigma-1 receptor, STIM1 stromal-interacting molecule 1, Stt4p phosphatidylinositol-4-kinase, t SERCA1 truncated sarco-endoplasmic reticulum Ca2+ ATPase, VDAC voltage-dependent anion channel, ?, unknown protein
MAMs Proteins and Ca2+ Homeostasis in Health and Disease
The aspect of functional interaction between the ER and mitochondria that has received most attention in recent decades is undoubtedly that involving Ca2+ ions. Ca2+-handling proteins such as IP3Rs (especially type 3 IP3Rs) are highly compartmentalized at MAMs [119], identifying these zones as “hotspots” of Ca2+ transfer from the ER to the closely adjacent mitochondrial network [17, 18]. Ca2+ signals arising from the ER are vital for regulating Ca2+ levels in mitochondria. Mitochondrial Ca2+ spikes and oscillations play a central role in energy production by regulating Ca2+-dependent enzymes involved in the ATP-producing Krebs cycle reactions [120, 121] and thus are important for cellular survival, although a mitochondrial Ca2+ overload will lead to the opening of the PTP, permeabilization of the OMM, eventually triggering cell death [122–124]. It remains unclear how the rise in mitochondrial Ca2+ (that has probably evolved to couple cell signaling to metabolic activation) can be transformed into a trigger of cell death. Both the amplitude and, most importantly, the duration of the Ca2+ rise in mitochondria, and perhaps even the concomitant insults that affect mitochondrial functions, play a major role in this transition. Therefore, ER Ca2+ handling on the MAMs acts as a double-edged sword, suggesting the existence of still not fully elucidated regulatory mechanisms, that are capable of discriminating between signals of life or death.
The connection between the ER and mitochondria is known to be highly dynamic as the local [Ca2+] itself can regulate ER-mitochondrial association in different ways [125], and increased [Ca2+]c blocks the motility of both organelles, enhancing their interaction [46]. Recently Hajnoczky et al. demonstrated that exposure to TGFβ affects Ca2+ transfer to the mitochondria through an impairment of the ER–mitochondrial coupling, further supporting the notion of a highly dynamic regulation of inter-organelle communication [126].
Several proteins may participate in the stabilization of those MAMs and, through this stabilization, affect Ca2+ transfer between ER and mitochondria, while other proteins may be directly involved in regulating the Ca2+-transport proteins described above. During the last years, research has focused on the identification of connecting structures between the ER and mitochondria at the MAMs, revealing that the interactions between the two organelles seem to be modulated both by a family of chaperone proteins and by a family of “mitochondria-shaping proteins”. One of the first advances was made in 2006, when Csordás et al. showed by electron tomography that ER and mitochondria are adjoined by tethers seemingly composed of proteins, since the in vitro incubation with proteinase not only detached the ER from mitochondria, but also disrupted Ca2+ transfer. Tightening of the connections sensitized mitochondria to Ca2+ overloading, ensuing permeability transition, and seemed relevant for several mechanisms of cell death. Thus, these results revealed an unexpected dependence of cell function and survival on the maintenance of a proper spacing between the ER and mitochondria [103].
At the same time, Szabadkai et al. found that the mitochondrial chaperone grp75 (glucose-regulated protein 75) mediates the molecular interaction of VDAC with the ER Ca2+-release channel IP3R. It was demonstrated that grp75 not only induces a chaperone-mediated conformational coupling of the proteins, but also allowed for a better transfer of the Ca2+ ions from the ER to the mitochondrial matrix [127]. In support of this view, we previously demonstrated that the overexpression of VDAC enhances Ca2+ signal propagation into the mitochondria, increasing the extent of mitochondrial Ca2+ uptake (also leading to a higher susceptibility for ceramide-induced cell death), acting at the ER–mitochondria contact sites [128]. Moreover, we have recently established that VDAC1, but not VDAC2 and VDAC3 isoforms, selectively interacts with IP3Rs; this interaction is further strengthened by apoptotic stimuli and thus VDAC1 is preferentially involved in the transmission of the low-amplitude apoptotic Ca2+ signals to mitochondria [129].
Subsequently, ER chaperones, particularly the Ca2+-binding chaperones calnexin, calreticulin, Sigma-1 receptor (Sig-1R) and Binding immunoglobulin Protein (BiP, also known as the glucose-regulated protein GRP78), were also found to be compartmentalized at the MAMs, yielding a new picture whereby chaperone machineries at both ER and mitochondria orchestrate the regulation of Ca2+ signaling between these two organelles. For instance, calnexin reversibly interacts with SERCA2b to block Ca2+ import [130]. Similarly, calreticulin inhibits uptake of Ca2+ by inhibiting the affinity for Ca2+ of the SERCA2b pump, but also regulates IP3-induced Ca2+ release [12, 131]. In vivo, these functions of calreticulin may very well be more crucial for survival than its chaperone activity, since calreticulin-deficient cells have impaired Ca2+ homoeostasis [132, 133].
Back in 2005, Simmen et al. reported the identification of a multifunctional cytosolic sorting protein, PACS-2 (phosphofurin acidic cluster sorting protein 2), that partially resides in the MAMs and maintains its integrity [134]. PACS-2 depletion induces mitochondria fragmentation and uncouples these organelles from the ER, raising the possibility that, in addition to mediating MAMs formation, PACS-2 might also influence Ca2+ homeostasis and apoptosis. Indeed, it has been shown that IP3Rs (and RyRs) possess potential PACS-2-binding sites [135]; hence, disruption of PACS-2 may cause mislocalization of IP3Rs, resulting in reduced Ca2+ transfer from the ER to mitochondria. Moreover, in response to apoptotic stimuli, PACS-2 has been demonstrated to be capable of inducing Bid recruitment to mitochondria, an event that leads to cytochrome c release and caspase 3 activation [134]. PACS-2 also interacts with and regulates the distribution and activity of calnexin. Under control conditions, >80% of calnexin localizes to the ER, mainly at the MAMs. However, through a protein–protein interaction, PACS-2 causes calnexin to distribute between the ER and the plasma membrane, affecting the homeostasis of ER Ca2+ [136]. PACS-2 and calnexin also interact with the MAMs-resident ER cargo receptor Bap31 (B-cell receptor-associated protein 31) and regulate its cleavage during the triggering of apoptosis [137]. Despite these observations, the exact role of PACS-2 in the regulation of Ca2+ transfer from the ER to the mitochondria remains to be further investigated.
Recently, Simmen’s group have also shown that the GTPase Rab32, a member of the Ras-related protein family of Rab, localizes to the ER and mitochondria and identified this protein as a regulator of MAMs properties. Its activity levels control MAMs composition, destroying the specific enrichment of calnexin at the MAMs, and consequently ER calcium handling. Furthermore, as a PKA-anchoring protein, Rab32 determines the targeting of PKA to mitochondrial and ER membranes, resulting in modulated PKA signaling. Together, these functions result in a delayed apoptosis onset with high Rab32 levels and, conversely, accelerated apoptosis with low Rab32 levels, explaining the possible mechanism by which it could act as an oncogene [138].
Also Sig-1R, an ER chaperone serendipitously identified in cellular distribution studies by Hayashi and Su, is enriched in the MAMs and seems to be involved in Ca2+-mediated stabilization of IP3Rs [139]. Under normal conditions in which the ER lumenal Ca2+ concentration is at 0.5–1.0 mM, it selectively resides at the MAMs and forms complexes with the ER Ca2+-binding chaperone BiP. Upon the activation of IP3Rs, which causes the decrease of the Ca2+ concentration at the MAMs, Sig-1R dissociates from BiP to chaperone IP3R, which would otherwise be degraded by proteasomes. Thus, Sig-1R appears to be involved in maintaining, on the ER luminal side, the integrity of the ER-mitochondrial Ca2+ cross-talk, as demonstrated by the fact that its silencing leads to impaired ER-mitochondrial Ca2+ transfer. Sig-1R has been implicated in several neuronal and non-neuronal pathological conditions [140], and is also upregulated in a wide variety of tumour cell lines [141]. Therefore, degenerative neurons or tissue might benefit by Sig-1R agonists which promote cell survival [142, 143]; conversely, its antagonists inhibit tumour-cell proliferation [144].
Another example of a folding enzyme regulating ER Ca2+ content is the oxidoreductase ERp44 (endoplasmic reticulum resident protein 44) that interacts with cysteines of the type 1 IP3R, thereby inhibiting Ca2+ transfer to mitochondria when ER conditions are reducing [145]. Recent results suggest that another oxidoreductase, Ero1α, might also perform such a function, since Ero1α interacts with the IP3R and potentiates the release of Ca2+ during ER stress [146]. This function of Ero1α could impact the induction of apoptosis that critically depends on ER-mitochondria Ca2+ communication [119, 147]. Gilady et al. showed that, despite Ero1α being an ER luminal protein, the targeting of Ero1α to the MAMs is quite stringent (>75%), consistent with its role in the regulation of Ca2+ homeostasis. Moreover, they found that localization of Ero1α on the MAMs is dependent on oxidizing conditions within the ER; indeed, hypoxia leads to a rapid and eventually complete depletion of Ero1α from the MAMs [148].
In the increasingly clear but complex picture that is emerging for MAMs, also the mitochondrial fusion protein Mfn2 has been shown to be enriched at contact sites between the ER and mitochondria. Mfn2 on the ER appeared to link the two organelles together: the connection depended on the interaction of the ER Mfn2 with either Mfn1 or Mfn2 on the OMM [104]. Moreover, its absence changes not only the morphology of the ER but also decreased by 40% the interactions between ER and mitochondria, thus affecting the transfer of Ca2+ signals to mitochondria. This may contribute to the Charcot-Marie-Tooth neuropathy type 2a in which missense mutations occur in Mfn2 [149]. A too strong ER–mitochondria interaction, and the concomitant improved Ca2+ transfer between the two organelles, may also be detrimental as overexpression of Mfn2 led to apoptosis in vascular smooth-muscle cells [150]. A recent report also propose the keratin-binding protein Trichoplein/mitostatin (TpMs), often downregulated in epithelial cancers [151], as a new regulator of mitochondria–ER juxtaposition in a Mfn2-dependent manner [152].
Also the mitochondrial fission protein Fis1 has been involved in ER-mitochondria coupling. Fis1 physically interacts with Bap31, an integral membrane protein expressed ubiquitously and highly enriched at the outer ER membrane), to bridge the mitochondria and the ER, setting up a platform for apoptosis induction. It appeared that the Fis1–Bap31 complex is required for the activation of procaspase-8. Importantly, as this signaling pathway can be initiated by Fis1, the Fis1-Bap31 complex establishes a feedback loop by releasing Ca2+ from the ER that is able to transmit an apoptosis signal from the mitochondria to the ER [153].
As described, it is now widely accepted that Ca2+ transfer between ER and mitochondria is a topic of major interest in physiology and pathology (Fig. 17.3). The release of Ca2+ from ER stores by IP3Rs has been implicated in multiple models of apoptosis as being directly responsible for mitochondrial Ca2+ overload. Apoptosis is a process of major biomedical interest, since its deregulation is involved in the pathogenesis of a broad variety of disorders (neoplasia, autoimmune disorders, viral and neurodegenerative diseases, to name a few).

Representation of MAMs proteins involved in ER-mitochondria Ca 2+ cross-talk and perturbations implicated in cell survival and cell death. Ca2+ release from the endoplasmic reticulum (ER) results in high-Ca2+ hot spots at the mitochondrial surface to allow efficient Ca2+ uptake through voltage-dependent anion channel - which is coupled to inositol 1,4,5-trisphosphate receptor by the chaperone glucose-regulated protein 75 (grp75) - and the mitochondrial Ca2+ uniporter. Mitochondrial Ca2+ activates organelle metabolism and ATP synthesis but also, when in excess, triggers apoptosis. Apoptosis deregulation is involved in the pathogenesis of neurodegenerative diseases as well as tumors development. Presenelin-1 (PS1) and Presenelin-2 (PS2), two proteins that when mutated cause familial Alzheimer’s disease (AD), have been recently found at MAMs, and familial AD (FAD) variants of PS2 (PS2FAD) seem to increase ER and mitochondria interaction; this could result in mitochondrial Ca2+ overload and subsequent excessive apoptosis. In addition, controlled apoptosis is likely to be important to eliminate cells, thereby avoiding tumor genesis. In this process the recently identified localization of the tumor suppressor promyelocytic leukemia protein (PML) at ER/MAMs plays a crucial role as it promotes IP3R-mediated Ca2+ transfer from ER into mitochondria. While Akt is known to suppress IP3R-channel activity by its phosphorylation, the recruitment of protein phosphatase PP2a via PML in a specific multi-protein complex (comprising PML, IP3R-3, PP2a, and Akt), dephosphorylates and inactivates Akt. This suppresses Akt-dependent phosphorylation of IP3R-3 and thus promotes Ca2+ release through this channel and Ca2+ transfer into the mitochondria. In cancer cells, where PML is often missing, IP3R-3 are hyper-phosphorylated due to an impaired PP2a activity, as a result the Ca2+ flux from ER to mitochondria is reduced and cells become resistant to apoptosis
Mitochondrial Ca2+ is therefore a central player in multiple neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease and Huntington’s disease [154]. It is noteworthy that alteration in Ca2+ homeostasis in sporadic AD patients started being reported in the middle of the 1980s, albeit in contrasting ways. Interestingly, very recent data have revealed that presenilin-1 (PS1) and presenilin-2 (PS2), two proteins that, when mutated, cause familial AD (FAD), have a strong effect on Ca2+ signaling (sometimes yielding contradictory experimental findings, as recently reviewed in [155]). Of particular interest on this topic, is the report that MAMs are the predominant subcellular location for PS1 and PS2, and for γ -secretase activity [156]. Moreover, it has recently been found that PS2 over-expression increases the interaction between ER and mitochondria and consequently Ca2+ transfer between these two organelles, an effect that is greater FAD variants [157]. It is possible to speculate that this favoured interaction could potentially result in a toxic mitochondrial Ca2+ overload (Fig. 17.3). A defect in Ca2+ signaling due to altered MAMs function could explain the well-known disturbances in Ca2+ homeostasis in AD [158, 159]. It also opens the door to new ways of thinking about complementary treatment; in addition, it may be possible to exploit aberrant MAMs function as a useful marker for the development of a diagnostic tool for AD [160].
Sano et al. also demonstrated that in GM1-gangliosidosis, a neurodegenerative disease, GM1-ganglioside (GM1) accumulates in brain within the MAMs, where it specifically interacts with phosphorylated IP3R-1, influencing its activity [161]. GM1 has been previously shown to modulate intracellular Ca2+ flux [162, 163]. As such, the recent discovery that MAMs are the sites where GM1 accumulates and influences ER-to-mitochondria Ca2+ flux, leading to Ca2+ overload and activation of the mitochondrial apoptotic pathway, explains the neuronal apoptosis and neurodegeneration that occurs in patients with GM1-gangliosidosis [161]. These findings may have important implications for targeting checkpoints of the GM1-mediated apoptotic cascade in the treatment of this catastrophic disease.
Modulation of the progression of cell death may therapeutically be also very important for the inhibition of tumour growth. Specific stimulation of the Ca2+ transfer between the IP3R and mitochondria could lead to increased cell death and so form a supplementary pathway to combat cancer. Our group has recently described that the tumor suppressor promyelocytic leukemia protein (PML) modulates the ER–mitochondria Ca2+-dependent cross-talk due to its unexpected and fundamental role at MAMs, highlighting a new extra-nuclear PML function critical for regulation of cell survival. This was demonstrated to be mediated by a specific multi-protein complex, localized at MAMs, including PML, IP3R-3, the protein phosphatase PP2a, and Akt. More than 50 different proteins can interact with and regulate the IP3Rs [80]; among these, a key role is played by the anti-apoptotic protein kinase Akt, which also phosphorylates IP3Rs, significantly reducing their Ca2+ release activity [81, 164]. In a previous work, we demonstrated that cells with the active form of Akt have a reduced cellular sensitivity to Ca2+-mediated apoptotic stimuli through a mechanism that involved diminished Ca2+ flux from the ER to mitochondria [165]. Our recent data show that PML mediates PP2a retention in the MAMs, which dephosphorylates and inactivates Akt. Thus, in the absence of PML, the unopposed action of Akt at ER, due to an impaired PP2a activity, leads to a hyperphosphorylation of IP3R-3 and in turn a reduced Ca2+ flux from ER to mitochondria, rendering cells resistant to apoptotic Ca2+-dependent stimuli [166] (Fig. 17.3). These findings may reveal a novel pharmacological target in apoptosis [167].
Interestingly, p66Shc, a cytosolic adaptor protein which is involved in the cellular response to oxidative stress (see above), has been found also in the MAMs fraction. In particular, we found that the level of p66Shc in MAMs fraction is age-dependent and corresponds well to the mitochondrial ROS production which is found to increase with age [168]. Finally, the functional significance of MAMs resident proteins in the regulation of ER-mitochondrial cross-talk is further supported by the finding that several viral proteins, such as the human cytomegalovirus vMIA [169], as well as the p7 and NS5B proteins of hepatitis C virus [170], are targeted to the MAMs and exert anti- or pro-apoptotic effects, respectively.
To conclude, whether or not mitochondria and MAMs contribute also to the Ca2+-dependent activation of autophagy is still unknown. If mitochondria actively contribute to the activation of autophagy through Ca2+ handling remains to be solved, but the close interaction between IP3Rs and mitochondria, on the one hand, and between IP3Rs and autophagy proteins, on the other hand, led to the hypothesis that IP3Rs could participate in the induction of this process [171, 172]. The study of the relation between IP3Rs, Ca2+ and the autophagic processes may become very important, since autophagy can protect the organism against various pathologies, including cancer and neurodegenerative diseases [118, 173].
The deeper understanding at the molecular level of the structural and functional links that are established at MAMs and the possibility to modulate them may in the future be of great importance in the treatment of many different human pathologies.
Abbreviations
- ΔΨm :
-
Mitochondrial membrane potential difference
- AD:
-
Alzheimer’s disease
- ANT:
-
Adenine nucleotide translocase
- Bap31:
-
(B-cell receptor-associated protein 31)
- BFP:
-
Blue fluorescent protein
- BiP:
-
Binding immunoglobulin Protein
- Ca2+ :
-
Calcium ions
- [Ca2+]:
-
Ca2+ concentration
- [Ca2+]c :
-
Cytosolic Ca2+ concentration
- [Ca2+]m :
-
Mitochondrial Ca2+ concentration
- CABPs:
-
Intraluminal Ca2+-binding proteins
- CaMKII:
-
Calmodulin-dependent protein kinase II
- CCE:
-
Capacitative Ca2+ entry
- Cyp D:
-
Cyclophilin D
- Drp1:
-
Dynamin-related protein 1
- ER:
-
Endoplasmic reticulum
- ERp44:
-
(Endoplasmic reticulum resident protein 44)
- FACL4:
-
Long-chain fatty acid-CoA ligase type 4
- FAD:
-
Familial Alzheimer’s disease
- Fhit:
-
Fragile histidine triad
- Fis1:
-
Fission 1 homologue
- FRET:
-
Fluorescence resonance energy transfer
- GFP:
-
Green fluorescent protein
- GM1:
-
GM1-ganglioside
- grp75:
-
Glucose-regulated protein 75
- HK:
-
Hexokinase
- IMM:
-
Inner mitochondrial membrane
- IMS:
-
Intermembrane space
- IP3:
-
Inositol 1,4,5-trisphosphate
- IP3R:
-
Inositol 1,4,5-trisphosphate receptor
- Letm1:
-
Leucine zipper-EF-hand containing transmembrane protein 1
- MAMs:
-
Mitochondria-associated membranes
- MCU:
-
Mitochondrial Ca2+ uniporter
- MICU1:
-
Mitochondrial calcium uptake 1
- Mfn:
-
Mitofusin
- mHCX:
-
Mitochondrial H+/Ca2+ exchanger
- MMP:
-
Mitochondrial membrane permeabilization
- mNCX:
-
Mitochondrial Na2+/Ca2+ exchanger
- MOMP:
-
Mitochondrial outer membrane permeabilization
- NADH:
-
Nicotinamide adenine dinucleotide
- NCX:
-
Na2+/Ca2+ exchanger
- NE:
-
nuclear envelope
- OMM:
-
Outer mitochondrial membrane
- OPA1:
-
Optic atrophy 1
- OXPHOS:
-
Oxidative phosphorylation
- p66shc:
-
66-kDa isoform of the growth factor adapter shc
- PACS-2:
-
Phosphofurin acidic cluster sorting protein 2
- PAMs:
-
Plasma membrane associated membranes
- PDH:
-
Pyruvate dehydrogenase
- PKA:
-
Protein kinase A
- PKC:
-
Protein kinase C
- PLC:
-
Phospholipase C
- PMCA:
-
Plasma membrane Ca2+ ATPase
- PML:
-
Promyelocytic leukemia protein
- PP2a:
-
Protein phosphatase 2a
- PS1:
-
Presenilin-1
- PS2:
-
Presenilin-2
- PSS-1:
-
Phosphatidylserine synthase-1
- PTP:
-
Permeability transition pore
- ROCs:
-
Receptor operated Ca2+ channels
- ROS:
-
Reactive oxygen species
- RyR:
-
Ryanodine receptor
- SERCA:
-
Sarco-endoplasmic reticulum Ca2+ ATPase
- Sig-1R:
-
Sigma-1 receptor
- SMOCs:
-
Second messenger operated Ca2+ channels
- SR:
-
Sarcoplasmic reticulum
- TIRF:
-
Total internal reflection fluorescence
- TpMs:
-
Trichoplein/Mitostatin
- UCP:
-
Uncoupling protein
- VDAC:
-
Voltage-dependent anion channel
- VOCs:
-
Voltage operated Ca2+ channels.
References
Berridge MJ, Lipp P, Bootman MD (2000) The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1:11–21
Brini M, Carafoli E (2009) Calcium pumps in health and disease. Physiol Rev 89:1341–1378
Blaustein MP, Lederer WJ (1999) Sodium/calcium exchange: its physiological implications. Physiol Rev 79:763–854
Bertolino M, Llinas RR (1992) The central role of voltage-activated and receptor-operated calcium channels in neuronal cells. Annu Rev Pharmacol Toxicol 32:399–421
McFadzean I, Gibson A (2002) The developing relationship between receptor-operated and store-operated calcium channels in smooth muscle. Br J Pharmacol 135:1–13
Meldolesi J, Pozzan T (1987) Pathways of Ca2+ influx at the plasma membrane: voltage-, receptor-, and second messenger-operated channels. Exp Cell Res 171:271–283
Foskett JK, White C, Cheung KH, Mak DO (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87:593–658
Sutko JL, Airey JA (1996) Ryanodine receptor Ca2+ release channels: does diversity in form equal diversity in function? Physiol Rev 76:1027–1071
Patel S, Joseph SK, Thomas AP (1999) Molecular properties of inositol 1,4,5-trisphosphate receptors. Cell Calcium 25:247–264
Patterson RL, Boehning D, Snyder SH (2004) Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu Rev Biochem 73:437–465
Berridge MJ (2006) Calcium microdomains: organization and function. Cell Calcium 40:405–412
John LM, Lechleiter JD, Camacho P (1998) Differential modulation of SERCA2 isoforms by calreticulin. J Cell Biol 142:963–973
Carafoli E (2003) Historical review: mitochondria and calcium: ups and downs of an unusual relationship. Trends Biochem Sci 28:175–181
Rizzuto R, Simpson AW, Brini M, Pozzan T (1992) Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358:325–327
Nicholls DG, Crompton M (1980) Mitochondrial calcium transport. FEBS Lett 111:261–268
Rizzuto R, Brini M, Murgia M, Pozzan T (1993) Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262:744–747
Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280:1763–1766
Csordas G, Thomas AP, Hajnoczky G (1999) Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J 18:96–108
Rizzuto R, Pozzan T (2006) Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev 86:369–408
Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, Pozzan T (2010) Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol Cell 38:280–290
Palmer AE, Giacomello M, Kortemme T, Hires SA, Lev-Ram V, Baker D, Tsien RY (2006) Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem Biol 13:521–530
Csordas G, Varnai P, Golenar T, Roy S, Purkins G, Schneider TG, Balla T, Hajnoczky G (2010) Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell 39:121–132
Inoue T, Heo WD, Grimley JS, Wandless TJ, Meyer T (2005) An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat Methods 2:415–418
Simamura E, Shimada H, Hatta T, Hirai K (2008) Mitochondrial voltage-dependent anion channels (VDACs) as novel pharmacological targets for anti-cancer agents. J Bioenerg Biomembr 40:213–217
De Pinto V, Reina S, Guarino F, Messina A (2008) Structure of the voltage dependent anion channel: state of the art. J Bioenerg Biomembr 40:139–147
Colombini M (2009) The published 3D structure of the VDAC channel: native or not? Trends Biochem Sci 34:382–389
Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N (2010) VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med 31:227–285
Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK (2010) MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467:291–296
Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, Hill DE, Vidal M, Evans JG, Thorburn DR, Carr SA, Mootha VK (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134:112–123
De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476:336–340
Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476:341–345
Bernardi P (1999) Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev 79:1127–1155
Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I (2010) NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci USA 107:436–441
Baines CP (2009) The molecular composition of the mitochondrial permeability transition pore. J Mol Cell Cardiol 46:850–857
Rasola A, Bernardi P (2007) The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 12:815–833
Rasola A, Sciacovelli M, Pantic B, Bernardi P (2010) Signal transduction to the permeability transition pore. FEBS Lett 584:1989–1996
McCormack JG, Halestrap AP, Denton RM (1990) Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev 70:391–425
Carafoli E (2010) The fateful encounter of mitochondria with calcium: how did it happen? Biochim Biophys Acta 1797:595–606
Maes K, Missiaen L, De Smet P, Vanlingen S, Callewaert G, Parys JB, De Smedt H (2000) Differential modulation of inositol 1,4,5-trisphosphate receptor type 1 and type 3 by ATP. Cell Calcium 27:257–267
Betzenhauser MJ, Wagner LE 2nd, Iwai M, Michikawa T, Mikoshiba K, Yule DI (2008) ATP modulation of Ca2+ release by type-2 and type-3 inositol (1, 4, 5)-triphosphate receptors. Differing ATP sensitivities and molecular determinants of action. J Biol Chem 283:21579–21587
Walsh C, Barrow S, Voronina S, Chvanov M, Petersen OH, Tepikin A (2009) Modulation of calcium signalling by mitochondria. Biochim Biophys Acta 1787:1374–1382
Arnaudeau S, Kelley WL, Walsh JV Jr (2001) Demaurex N: mitochondria recycle Ca(2+) to the endoplasmic reticulum and prevent the depletion of neighboring endoplasmic reticulum regions. J Biol Chem 276:29430–29439
Tinel H, Cancela JM, Mogami H, Gerasimenko JV, Gerasimenko OV, Tepikin AV, Petersen OH (1999) Active mitochondria surrounding the pancreatic acinar granule region prevent spreading of inositol trisphosphate-evoked local cytosolic Ca(2+) signals. EMBO J 18:4999–5008
Goetz JG, Genty H, St-Pierre P, Dang T, Joshi B, Sauve R, Vogl W, Nabi IR (2007) Reversible interactions between smooth domains of the endoplasmic reticulum and mitochondria are regulated by physiological cytosolic Ca2+ levels. J Cell Sci 120:3553–3564
Wang HJ, Guay G, Pogan L, Sauve R, Nabi IR (2000) Calcium regulates the association between mitochondria and a smooth subdomain of the endoplasmic reticulum. J Cell Biol 150:1489–1498
Yi M, Weaver D, Hajnoczky G (2004) Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol 167:661–672
Westermann B (2010) Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11:872–884
Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ (2003) Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci 23:7881–7888
Germain M, Mathai JP, McBride HM, Shore GC (2005) Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J 24:1546–1556
Alirol E, James D, Huber D, Marchetto A, Vergani L, Martinou JC, Scorrano L (2006) The mitochondrial fission protein hFis1 requires the endoplasmic reticulum gateway to induce apoptosis. Mol Biol Cell 17:4593–4605
Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R (2004) Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2 + -mediated apoptosis. Mol Cell 16:59–68
Lu B (2009) Mitochondrial dynamics and neurodegeneration. Curr Neurol Neurosci Rep 9:212–219
Giorgi C, Romagnoli A, Pinton P, Rizzuto R (2008) Ca2+ signaling, mitochondria and cell death. Curr Mol Med 8:119–130
Harr MW, Distelhorst CW (2010) Apoptosis and autophagy: decoding calcium signals that mediate life or death. Cold Spring Harb Perspect Biol 2:a005579
Kroemer G, Galluzzi L, Brenner C (2007) Mitochondrial membrane permeabilization in cell death. Physiol Rev 87:99–163
Tait SW, Green DR (2010) Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11:621–632
Armstrong JS (2006) The role of the mitochondrial permeability transition in cell death. Mitochondrion 6:225–234
Rong Y, Distelhorst CW (2008) Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol 70:73–91
Trinei M, Berniakovich I, Beltrami E, Migliaccio E, Fassina A, Pelicci P, Giorgio M (2009) P66Shc signals to age. Aging (Albany NY) 1:503–510
Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M, Contursi C, Minucci S, Mantovani F, Wieckowski MR, Del Sal G, Pelicci PG, Rizzuto R (2007) Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 315:659–663
Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA (2006) A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 124:573–586
Shibata Y, Voeltz GK, Rapoport TA (2006) Rough sheets and smooth tubules. Cell 126:435–439
English AR, Zurek N, Voeltz GK (2009) Peripheral ER structure and function. Curr Opin Cell Biol 21:596–602
Shibata Y, Shemesh T, Prinz WA, Palazzo AF, Kozlov MM, Rapoport TA (2010) Mechanisms determining the morphology of the peripheral ER. Cell 143:774–788
Chevet E, Cameron PH, Pelletier MF, Thomas DY, Bergeron JJ (2001) The endoplasmic reticulum: integration of protein folding, quality control, signaling and degradation. Curr Opin Struct Biol 11:120–124
Palade G (1975) Intracellular aspects of the process of protein synthesis. Science 189:347–358
Bootman MD, Petersen OH, Verkhratsky A (2002) The endoplasmic reticulum is a focal point for co-ordination of cellular activity. Cell Calcium 32:231–234
Verkhratsky A, Petersen OH (2002) The endoplasmic reticulum as an integrating signalling organelle: from neuronal signalling to neuronal death. Eur J Pharmacol 447:141–154
Jobsis FF, O’Connor MJ (1966) Calcium release and reabsorption in the sartorius muscle of the toad. Biochem Biophys Res Commun 25:246–252
Ridgway EB, Ashley CC (1967) Calcium transients in single muscle fibers. Biochem Biophys Res Commun 29:229–234
Ashley CC, Ridgway EB (1970) On the relationships between membrane potential, calcium transient and tension in single barnacle muscle fibres. J Physiol 209:105–130
MacKrill JJ (1999) Protein-protein interactions in intracellular Ca2 + -release channel function. Biochem J 337(Pt 3):345–361
Rizzuto R, Marchi S, Bonora M, Aguiari P, Bononi A, De Stefani D, Giorgi C, Leo S, Rimessi A, Siviero R, Zecchini E, Pinton P (2009) Ca(2+) transfer from the ER to mitochondria: when, how and why. Biochim Biophys Acta 1787:1342–1351
Iino M, Endo M (1992) Calcium-dependent immediate feedback control of inositol 1,4,5-triphosphate-induced Ca2+ release. Nature 360:76–78
Missiaen L, Taylor CW, Berridge MJ (1992) Luminal Ca2+ promoting spontaneous Ca2+ release from inositol trisphosphate-sensitive stores in rat hepatocytes. J Physiol 455:623–640
Nunn DL, Taylor CW (1992) Luminal Ca2+ increases the sensitivity of Ca2+ stores to inositol 1,4,5-trisphosphate. Mol Pharmacol 41:115–119
Smith JB, Smith L, Higgins BL (1985) Temperature and nucleotide dependence of calcium release by myo-inositol 1,4,5-trisphosphate in cultured vascular smooth muscle cells. J Biol Chem 260:14413–14416
Bezprozvanny I, Ehrlich BE (1993) ATP modulates the function of inositol 1,4,5-trisphosphate-gated channels at two sites. Neuron 10:1175–1184
Iino M (1991) Effects of adenine nucleotides on inositol 1,4,5-trisphosphate-induced calcium release in vascular smooth muscle cells. J Gen Physiol 98:681–698
Vanderheyden V, Devogelaere B, Missiaen L, De Smedt H, Bultynck G, Parys JB (2009) Regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by reversible phosphorylation and dephosphorylation. Biochim Biophys Acta 1793:959–970
Khan MT, Wagner L 2nd, Yule DI, Bhanumathy C, Joseph SK (2006) Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J Biol Chem 281:3731–3737
Bugrim AE (1999) Regulation of Ca2+ release by cAMP-dependent protein kinase. A mechanism for agonist-specific calcium signaling? Cell Calcium 25:219–226
Murthy KS, Zhou H (2003) Selective phosphorylation of the IP3R-I in vivo by cGMP-dependent protein kinase in smooth muscle. Am J Physiol Gastrointest Liver Physiol 284:G221–G230
Bagni C, Mannucci L, Dotti CG, Amaldi F (2000) Chemical stimulation of synaptosomes modulates alpha -Ca2+/calmodulin-dependent protein kinase II mRNA association to polysomes. J Neurosci 20:RC76
Vermassen E, Fissore RA, Nadif Kasri N, Vanderheyden V, Callewaert G, Missiaen L, Parys JB, De Smedt H (2004) Regulation of the phosphorylation of the inositol 1,4,5-trisphosphate receptor by protein kinase C. Biochem Biophys Res Commun 319:888–893
Jayaraman T, Ondrias K, Ondriasova E, Marks AR (1996) Regulation of the inositol 1,4,5-trisphosphate receptor by tyrosine phosphorylation. Science 272:1492–1494
Joseph SK, Hajnoczky G (2007) IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond. Apoptosis 12:951–968
Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R (2008) Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27:6407–6418
Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R (2000) Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J Cell Biol 148:857–862
Foyouzi-Youssefi R, Arnaudeau S, Borner C, Kelley WL, Tschopp J, Lew DP, Demaurex N, Krause KH (2000) Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc Natl Acad Sci USA 97:5723–5728
Palmer AE, Jin C, Reed JC, Tsien RY (2004) Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci USA 101:17404–17409
White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK (2005) The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol 7:1021–1028
Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300:135–139
Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, Korsmeyer SJ (2005) Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci USA 102:105–110
Pinton P, Rizzuto R (2006) Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ 13:1409–1418
Copeland DE, Dalton AJ (1959) An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J Biophys Biochem Cytol 5:393–396
Lewis JA, Tata JR (1973) A rapidly sedimenting fraction of rat liver endoplasmic reticulum. J Cell Sci 13:447–459
Morre DJ, Merritt WD, Lembi CA (1971) Connections between mitochondria and endoplasmic reticulum in rat liver and onion stem. Protoplasma 73:43–49
Mannella CA, Buttle K, Rath BK, Marko M (1998) Electron microscopic tomography of rat-liver mitochondria and their interaction with the endoplasmic reticulum. Biofactors 8:225–228
Soltys BJ, Gupta RS (1992) Interrelationships of endoplasmic reticulum, mitochondria, intermediate filaments, and microtubules–a quadruple fluorescence labeling study. Biochem Cell Biol 70:1174–1186
Mannella CA, Marko M, Penczek P, Barnard D, Frank J (1994) The internal compartmentation of rat-liver mitochondria: tomographic study using the high-voltage transmission electron microscope. Microsc Res Tech 27:278–283
Achleitner G, Gaigg B, Krasser A, Kainersdorfer E, Kohlwein SD, Perktold A, Zellnig G, Daum G (1999) Association between the endoplasmic reticulum and mitochondria of yeast facilitates interorganelle transport of phospholipids through membrane contact. Eur J Biochem 264:545–553
Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174:915–921
de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456:605–610
Dennis EA, Kennedy EP (1972) Intracellular sites of lipid synthesis and the biogenesis of mitochondria. J Lipid Res 13:263–267
Vance JE (1990) Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem 265:7248–7256
Vance JE, Stone SJ, Faust JR (1997) Abnormalities in mitochondria-associated membranes and phospholipid biosynthetic enzymes in the mnd/mnd mouse model of neuronal ceroid lipofuscinosis. Biochim Biophys Acta 1344:286–299
Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P (2009) Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat Protoc 4:1582–1590
Pichler H, Gaigg B, Hrastnik C, Achleitner G, Kohlwein SD, Zellnig G, Perktold A, Daum G (2001) A subfraction of the yeast endoplasmic reticulum associates with the plasma membrane and has a high capacity to synthesize lipids. Eur J Biochem 268:2351–2361
Wu MM, Buchanan J, Luik RM, Lewis RS (2006) Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol 174:803–813
Frieden M, Arnaudeau S, Castelbou C, Demaurex N (2005) Subplasmalemmal mitochondria modulate the activity of plasma membrane Ca2 + -ATPases. J Biol Chem 280:43198–43208
Koziel K, Lebiedzinska M, Szabadkai G, Onopiuk M, Brutkowski W, Wierzbicka K, Wilczynski G, Pinton P, Duszynski J, Zablocki K, Wieckowski MR (2009) Plasma membrane associated membranes (PAM) from Jurkat cells contain STIM1 protein is PAM involved in the capacitative calcium entry? Int J Biochem Cell Biol 41:2440–2449
Piccini M, Vitelli F, Bruttini M, Pober BR, Jonsson JJ, Villanova M, Zollo M, Borsani G, Ballabio A, Renieri A (1998) FACL4, a new gene encoding long-chain acyl-CoA synthetase 4, is deleted in a family with Alport syndrome, elliptocytosis, and mental retardation. Genomics 47:350–358
Stone SJ, Vance JE (2000) Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J Biol Chem 275:34534–34540
Lebiedzinska M, Szabadkai G, Jones AW, Duszynski J, Wieckowski MR (2009) Interactions between the endoplasmic reticulum, mitochondria, plasma membrane and other subcellular organelles. Int J Biochem Cell Biol 41:1805–1816
Hayashi T, Rizzuto R, Hajnoczky G, Su TP (2009) MAM: more than just a housekeeper. Trends Cell Biol 19:81–88
Giorgi C, De Stefani D, Bononi A, Rizzuto R, Pinton P (2009) Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int J Biochem Cell Biol 41:1817–1827
Decuypere JP, Monaco G, Bultynck G, Missiaen L, De Smedt H, Parys JB (2011) The IP(3) receptor-mitochondria connection in apoptosis and autophagy. Biochim Biophys Acta 1813:1003–1013
Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, Rodrigues MA, Gomez MV, Nathanson MH, Leite MF (2005) The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem 280:40892–40900
Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP (1995) Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82:415–424
Rizzuto R, Duchen MR, Pozzan T (2004) Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci STKE 2004:re1
Walter L, Hajnoczky G (2005) Mitochondria and endoplasmic reticulum: the lethal interorganelle cross-talk. J Bioenerg Biomembr 37:191–206
Green DR, Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science 305:626–629
Bernardi P, Rasola A (2007) Calcium and cell death: the mitochondrial connection. Subcell Biochem 45:481–506
Garcia-Perez C, Hajnoczky G, Csordas G (2008) Physical coupling supports the local Ca2+ transfer between sarcoplasmic reticulum subdomains and the mitochondria in heart muscle. J Biol Chem 283:32771–32780
Pacher P, Sharma K, Csordas G, Zhu Y, Hajnoczky G (2008) Uncoupling of ER-mitochondrial calcium communication by transforming growth factor-beta. Am J Physiol Renal Physiol 295:F1303–F1312
Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R (2006) Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 175:901–911
Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, Tuft RA, Fogarty KE, Rizzuto R (2002) Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol 159:613–624
De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, Rizzuto R (2011) VDAC1 selectively transfers apoptotic Ca(2+) signals to mitochondria. Cell Death Differ 2011 Jul 1. doi: 10.1038/cdd.2011.92. [Epub ahead of print]
Roderick HL, Lechleiter JD, Camacho P (2000) Cytosolic phosphorylation of calnexin controls intracellular Ca(2+) oscillations via an interaction with SERCA2b. J Cell Biol 149:1235–1248
Camacho P, Lechleiter JD (1995) Calreticulin inhibits repetitive intracellular Ca2+ waves. Cell 82:765–771
Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M (2009) Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem J 417:651–666
Molinari M, Eriksson KK, Calanca V, Galli C, Cresswell P, Michalak M, Helenius A (2004) Contrasting functions of calreticulin and calnexin in glycoprotein folding and ER quality control. Mol Cell 13:125–135
Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, Thomas G (2005) PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J 24:717–729
Kottgen M, Benzing T, Simmen T, Tauber R, Buchholz B, Feliciangeli S, Huber TB, Schermer B, Kramer-Zucker A, Hopker K, Simmen KC, Tschucke CC, Sandford R, Kim E, Thomas G, Walz G (2005) Trafficking of TRPP2 by PACS proteins represents a novel mechanism of ion channel regulation. EMBO J 24:705–716
Myhill N, Lynes EM, Nanji JA, Blagoveshchenskaya AD, Fei H, Carmine Simmen K, Cooper TJ, Thomas G, Simmen T (2008) The subcellular distribution of calnexin is mediated by PACS-2. Mol Biol Cell 19:2777–2788
Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC (2003) Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol 160:1115–1127
Bui M, Gilady SY, Fitzsimmons RE, Benson MD, Lynes EM, Gesson K, Alto NM, Strack S, Scott JD, Simmen T (2010) Rab32 modulates apoptosis onset and mitochondria-associated membrane (MAM) properties. J Biol Chem 285:31590–31602
Hayashi T, Su TP (2007) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131:596–610
Maurice T, Su TP (2009) The pharmacology of sigma-1 receptors. Pharmacol Ther 124:195–206
Aydar E, Palmer CP, Djamgoz MB (2004) Sigma receptors and cancer: possible involvement of ion channels. Cancer Res 64:5029–5035
Marrazzo A, Caraci F, Salinaro ET, Su TP, Copani A, Ronsisvalle G (2005) Neuroprotective effects of sigma-1 receptor agonists against beta-amyloid-induced toxicity. Neuroreport 16:1223–1226
Vagnerova K, Hurn PD, Bhardwaj A, Kirsch JR (2006) Sigma 1 receptor agonists act as neuroprotective drugs through inhibition of inducible nitric oxide synthase. Anesth Analg 103:430–434, table of contents
Spruce BA, Campbell LA, McTavish N, Cooper MA, Appleyard MV, O’Neill M, Howie J, Samson J, Watt S, Murray K, McLean D, Leslie NR, Safrany ST, Ferguson MJ, Peters JA, Prescott AR, Box G, Hayes A, Nutley B, Raynaud F, Downes CP, Lambert JJ, Thompson AM, Eccles S (2004) Small molecule antagonists of the sigma-1 receptor cause selective release of the death program in tumor and self-reliant cells and inhibit tumor growth in vitro and in vivo. Cancer Res 64:4875–4886
Higo T, Hattori M, Nakamura T, Natsume T, Michikawa T, Mikoshiba K (2005) Subtype-specific and ER lumenal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell 120:85–98
Li G, Mongillo M, Chin KT, Harding H, Ron D, Marks AR, Tabas I (2009) Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J Cell Biol 186:783–792
Szalai G, Krishnamurthy R, Hajnoczky G (1999) Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J 18:6349–6361
Gilady SY, Bui M, Lynes EM, Benson MD, Watts R, Vance JE, Simmen T (2010) Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM). Cell Stress Chaperones 15:619–629
Cartoni R, Martinou JC (2009) Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A. Exp Neurol 218:268–273
Guo X, Chen KH, Guo Y, Liao H, Tang J, Xiao RP (2007) Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circ Res 101:1113–1122
Vecchione A, Fassan M, Anesti V, Morrione A, Goldoni S, Baldassarre G, Byrne D, D’Arca D, Palazzo JP, Lloyd J, Scorrano L, Gomella LG, Iozzo RV, Baffa R (2009) MITOSTATIN, a putative tumor suppressor on chromosome 12q24.1, is downregulated in human bladder and breast cancer. Oncogene 28:257–269
Cerqua C, Anesti V, Pyakurel A, Liu D, Naon D, Wiche G, Baffa R, Dimmer KS, Scorrano L (2010) Trichoplein/mitostatin regulates endoplasmic reticulum-mitochondria juxtaposition. EMBO Rep 11:854–860
Iwasawa R, Mahul-Mellier AL, Datler C, Pazarentzos E, Grimm S (2011) Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J 30:556–568
Celsi F, Pizzo P, Brini M, Leo S, Fotino C, Pinton P, Rizzuto R (2009) Mitochondria, calcium and cell death: a deadly triad in neurodegeneration. Biochim Biophys Acta 1787:335–344
Contreras L, Drago I, Zampese E, Pozzan T (2010) Mitochondria: the calcium connection. Biochim Biophys Acta 1797:607–618
Area-Gomez E, de Groof AJ, Boldogh I, Bird TD, Gibson GE, Koehler CM, Yu WH, Duff KE, Yaffe MP, Pon LA, Schon EA (2009) Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol 175:1810–1816
Zampese E, Fasolato C, Kipanyula MJ, Bortolozzi M, Pozzan T, Pizzo P (2011) Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc Natl Acad Sci USA 108:2777–2782
Smith IF, Green KN, LaFerla FM (2005) Calcium dysregulation in Alzheimer’s disease: recent advances gained from genetically modified animals. Cell Calcium 38:427–437
Small DH (2009) Dysregulation of calcium homeostasis in Alzheimer’s disease. Neurochem Res 34:1824–1829
Yu JT, Chang RC, Tan L (2009) Calcium dysregulation in Alzheimer’s disease: from mechanisms to therapeutic opportunities. Prog Neurobiol 89:240–255
Sano R, Annunziata I, Patterson A, Moshiach S, Gomero E, Opferman J, Forte M, d’Azzo A (2009) GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol Cell 36:500–511
d’Azzo A, Tessitore A, Sano R (2006) Gangliosides as apoptotic signals in ER stress response. Cell Death Differ 13:404–414
Wu G, Lu ZH, Obukhov AG, Nowycky MC, Ledeen RW (2007) Induction of calcium influx through TRPC5 channels by cross-linking of GM1 ganglioside associated with alpha5beta1 integrin initiates neurite outgrowth. J Neurosci 27:7447–7458
Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L, Chastre E, Khan F, Landegren U, Soderberg O, Bootman MD, Roderick HL (2008) Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci USA 105:2427–2432
Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R, Pinton P (2008) Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2 + -dependent apoptotic stimuli. Biochem Biophys Res Commun 375:501–505
Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M, Bononi A, Bonora M, Duszynski J, Bernardi R, Rizzuto R, Tacchetti C, Pinton P, Pandolfi PP (2010) PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 330:1247–1251
Pinton P, Giorgi C, Pandolfi PP (2011) The role of PML in the control of apoptotic cell fate: a new key player at ER-mitochondria sites. Cell Death Differ 18:1450–1456
Lebiedzinska M, Duszynski J, Rizzuto R, Pinton P, Wieckowski MR (2009) Age-related changes in levels of p66Shc and serine 36-phosphorylated p66Shc in organs and mouse tissues. Arch Biochem Biophys 486:73–80
Bozidis P, Williamson CD, Colberg-Poley AM (2008) Mitochondrial and secretory human cytomegwalovirus UL37 proteins traffic into mitochondrion-associated membranes of human cells. J Virol 82:2715–2726
Sheikh MY, Choi J, Qadri I, Friedman JE, Sanyal AJ (2008) Hepatitis C virus infection: molecular pathways to metabolic syndrome. Hepatology 47:2127–2133
Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, Molgo J, Diaz J, Lavandero S, Harper F, Pierron G, di Stefano D, Rizzuto R, Szabadkai G, Kroemer G (2007) Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ 14:1029–1039
Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK (2010) Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142:270–283
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132:27–42
Acknowledgements
A.B. was supported by a research fellowship FISM – Fondazione Italiana Sclerosi Multipla – Cod. 2010/B/1. SP was supported by a training fellowship FISMJ.M.S. was supported by a PhD fellowship from The Foundation for Polish Science (FNP), EU, European Regional Development Fund and Operational Programme “Innovative economy”. This research was supported by: the Polish Ministry of Science and Higher Education under grant NN407 075 137 to M.R.W. and by Telethon (GGP09128), local funds from the University of Ferrara, the Italian Ministry of Education, University and Research (COFIN), the Italian Cystic Fibrosis Research Foundation and Italian Ministry of Health to P.P.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Bononi, A. et al. (2012). Mitochondria-Associated Membranes (MAMs) as Hotspot Ca2+ Signaling Units. In: Islam, M. (eds) Calcium Signaling. Advances in Experimental Medicine and Biology, vol 740. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-2888-2_17
Download citation
DOI: https://doi.org/10.1007/978-94-007-2888-2_17
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-2887-5
Online ISBN: 978-94-007-2888-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)