
Abstract
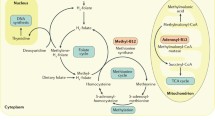
Vitamin B12, the “antipernicious anaemia factor”, is a crystallisable cobalt-complex, which belongs to a group of unique “complete” corrinoids, named cobalamins (Cbl). In humans, instead of the “vitamin”, two organometallic B12-forms are coenzymes in two metabolically important enzymes: Methyl-cobalamin, the cofactor of methionine synthase, and coenzyme B12 (adenosyl-cobalamin), the cofactor of methylmalonyl-CoA mutase. The cytoplasmatic methionine synthase catalyzes the transfer of a methyl group from N-methyl-tetrahydrofolate to homocysteine to yield methionine and to liberate tetrahydrofolate. In the mitochondrial methylmalonyl-CoA mutase a radical process transforms methylmalonyl-CoA (a remains e.g. from uneven numbered fatty acids) into succinyl-CoA, for further metabolic use. In addition, in the human mitochondria an adenosyl-transferase incorporates the organometallic group of coenzyme B12. In all these enzymes, the bound B12-derivatives engage (or are formed) in exceptional organometallic enzymatic reactions. This chapter recapitulates the physiological chemistry of vitamin B12, relevant in the context of the metabolic transformation of B12-derivatives into the relevant coenzyme forms and their use in B12-dependent enzymes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- 5′-adenosyl radical
- Coenzyme B12
- Methionine synthase
- Methylcobalamin
- Methylgroup transfer
- Methylmalonyl-CoA mutase
- Radical reaction
- Vitamin B12
17.1 Introduction
B12-coenzymes are conceivably Nature’s most broadly relevant and most complex organometallic cofactors (Eschenmoser 1988; Kräutler et al. 1998; Banerjee 1999). They are required (and are vitamins) in human metabolism, which depends on the binding and uptake of B12, on the controlled transport and metabolic transformation into the relevant B12-cofactors, and the catalysis by B12-dependent enzymes (Yamanishi et al. 2005). B12-derivatives (co)catalyze unique enzymatic reactions that directly depend upon the reactivity of the cobalt coordinated organic ligands (Kräutler and Ostermann 2003; Brown 2005).
About 60 years ago, the red cyanide-containing cobalt-complex vitamin B12 (cyano-cobalamin, CNCbl) was isolated as the (extrinsic) “anti-pernicious anaemia factor” (Fig. 17.1) (Rickes et al. 1948; Smith and Parker 1948). CNCbl is a relatively inert Co(III)-corrin, which crystallizes readily. It is the most important commercially available form of the naturally occurring B12-derivatives, although a direct physiological function of CNCbl appears not to exist (Ellenbogen and Cooper 1984). The physiologically relevant B12-derivatives are the light-sensitive and chemically more labile organometallic cofactors, coenzyme B12 (5′-deoxy-5′-adenosyl-cobalamin, AdoCbl) and methyl-cobalamin (MeCbl), as well as (in a formal sense) the “inorganic” and easily reducible B12-derivative aquo-cobalamin (H2OCbl+).

Left: structural formulae of selected cobalamins (Cbl); Right: symbol used. Vitamin B12 (CNCbl), R = CN; coenzyme B12 (AdoCbl), R = 5′-Ado; methyl-cobalamin (MeCbl), R = CH3; aquo-cobalamin (H2OCbl+), R = H2O+; cob(II)alamin (B12r), R = e–
Important historic contributions to the biologically relevant chemistry of B12 have been reviewed earlier (Dolphin 1982; Friedrich 1988). Remarkable scientific advances contributing to the solution of the major “B12-mysteries” were achieved during the last decades (Kräutler et al. 1998; Banerjee 1999; Brown 2005), including the elucidation of the microbial B12-biosynthesis (Battersby 1998; Scott et al. 2003), important structural and mechanistic studies of B12-binding proteins (Drennan et al. 1994; Ludwig and Evans 1999; Gruber and Kratky 2002), as well as genetic studies of inborn errors of Cbl metabolism (Rosenblatt and Fenton 1999).
17.2 B12: Structure and Reactivity
17.2.1 B12-Structural Studies
Vitamin B12 (CNCbl), and other B12-derivatives (where the cyanide ligand of CNCbl is replaced by a different “upper” or β-ligand) are all cobalamins (Cbl, 5′,6′-dimethylbenzimidazolyl-cobamides), the B12-derivatives relevant in human metabolism. Pioneering X-ray crystallographic studies of Hodgkin et al. (Hodgkin et al. 1955; Lenhert and Hodgkin 1961) established the structure of CNCbl (and of its unique corrin core), as well as the amazing nature of the organometallic cofactor coenzyme B12 (AdoCbl). Since the time of these landmark analyses, accurate structural studies of a variety of crystalline B12-derivatives have become available, as presented in recent reviews (Kratky and Kräutler 1999; Randaccio et al. 2006).
The replacement of the cobalt coordinated 5,6-dimethylimidazole (DMB) base by a protein-derived imidazole in B12-dependent enzymes, such as methionine synthase (Drennan et al. 1994) and methylmalonyl-CoA mutase (Mancia et al. 1996) (see below), has been a puzzling discovery. To study the effect of an imidazole in B12-derivatives, unnatural imidazolyl-cobamides (ImCba) were made available via “guided” biosynthesis, such as 5′-deoxy-5′-adenosyl-imidazolyl-cobamide (Ado-ImCba) (Brown et al. 2004), cyano-imidazolyl-cobamide (CN-ImCba) and methyl-imidazolyl-cobamide (Me-ImCba), both analyzed by x-ray crystallography (Kräutler et al. 1994; Fasching et al. 2000).

Structural formulae. Left: of cyano-imidazolylcobamide (CN-ImCba, R = CN), methyl-imidazolylcobamide (Me-ImCba, R = methyl) and of adenosyl-imidazolylcobamide (Ado-ImCba, R = 5′-Ado); Right: of cob(I)alamin (B12s)
Two structural characteristics of the corrin ligand are the saturated and direct trans-junction between two of its four five-membered rings and the non-planar nature of the corrin core in B12-derivatives. The bulky DMB-base was suggested to be a relevant enhancer to this latter “ligand-folding”, which is a main factor also to the variability in the conformation of the corrin ligand (Kratky and Kräutler 1999). Indeed, the folding of the corrin ligand in CN-ImCba (11.3°) is less than half of that of vitamin B12 (CNCbl). This finding could be rationalized, as the less bulky and more nucleophilic imidazole showed less steric interaction with corrin ligand in CN-ImCba, than the DMB base in CNCbl. However, the crystallographic studies could not provide a pertinent structure-based rationalization of the observed displacement of the DMB-base by histidine in the enzymes. Crystallographic information on the structure of cob(II)alamin (B12r) has been of particular interest (Kräutler et al. 1989), as it is the product of Co–C bond homolysis of coenzyme B12 (AdoCbl), relevant for the catalytic cycle of coenzyme B12-dependent enzymes (see below).
Nuclear Magnetic Resonance (NMR) spectroscopy has become an important methodology for the study of (diamagnetic) B12 derivatives, in (aqueous) solution. Frequently, it is complementary to crystal structure analysis and is particularly valuable in such cases, where crystals of the B12-derivative are not obtained (Konrat et al. 1998; Brown 1999). Natural corrinoids from bacteria were often characterized by NMR-spectroscopy (Hoffmann et al. 2000). By applying so called homo- and hetero-nuclear NMR-experiments and high field NMR-spectrometers, the structure of B12-derivatives in aqueous solution has been studied, and their behaviour as dynamic molecules could be analyzed (Summers et al. 1986; Konrat et al. 1998).
17.2.2 B12-Redoxchemistry
Under physiological conditions vitamin B12-derivatives have been observed in the oxidation states Co(III), Co(II), and Co(I), each possessing differing coordination properties and reactivities (Pratt 1972; Kräutler 1998).
Oxidation-reduction processes are of key importance in the metabolism of B12. Electrochemical methods have been crucial in analytical studies of B12-derivatives (Lexa and Savéant 1983) (Fig. 17.3). Axial coordination to the corrin-bound cobalt centre depends on the formal oxidation state of the cobalt ion (Kratky and Kräutler 1999): As a rule, the number of axial ligands decreases in parallel with the cobalt oxidation state. The diamagnetic Co(III) (coordination number 6) typically has two axial ligands bound, the paramagnetic (low spin) Co(II) (coordination number 5) has one axial ligand and the diamagnetic Co(I) (coordination number 4) has no axial ligands bound (Pratt 1972; Lexa and Savéant 1983). Redox reactions of B12-derivatives are, therefore, accompanied by a change in the number of axial ligands. In turn, the presence and the nature of axial ligands strongly influences the (thermodynamic and kinetic features of the) electron transfer of cobalt corrins (Lexa and Savéant 1983; Kräutler 1999).

Outline of the redox-transitions between the cob(III)alamin aquo-cobalamin (H2OCbl+, B12a), cob(II)alamin (B12r) and cob(I)alamin (B12s)
In Co(III)-corrins, such as vitamin B12 (CNCbl), coenzyme B12 (AdoCbl) and methyl-cobalamin (MeCbl), the corrin-bound cobalt centre is coordinatively saturated, when binding two axial ligands. In contrast, the metal centre in Co(I)-corrins, such as cob(I)alamin (B12s) is highly nucleophilic (with very low basicity) (Lexa and Savéant 1983). The intermediate oxidation level of the Co(II)-corrins, such as cob(II)alamin (B12r), provides a reactive metal-centred radicaloid species (Endicott and Netzel 1979; Kräutler et al. 1989). In neutral aqueous solution, cob(II)alamin (B12r) is present in its “base-on” form (i.e. the DMB-base is coordinating the Co(II)centre). B12r is converted into the “base-off” form, B12r-H+, by protonation of the DMB-base, with pKa (B12r-H+) = 2.9, and de-coordination from the Co(II)centre.
A standard potential vs. pH diagram has been established, which correlates the thermodynamics of the H2OCbl+-system (B12a /B12r /B12s) (Fig. 17.4) (Lexa and Savéant 1983). In the pH range 2.9–7.8, H2OCbl and (base-on) B12r represent the predominant Co(III)-/Co(II)-redox couple, with a standard potential of +0.20 V. For the Co(II)-/Co(I)-redox system there are two pH-independent standard potentials: at a pH less than 5.6 the Co(II)-/Co(I)-couple (base-off) B12r-H+/B12s-H predominates (standard potential of –0.50 V, at higher pH the redox couple (base-on) B12r / B12s exists with a more negative standard potential of –0.61 V.

Dependence of standard potentials of the redox system Co(III)-/Co(II)-/Co(I)-corrin (B12a/B12r/B12s) upon pH in aqueous solution (at 22°C), potentials are referenced to the normal hydrogen electrode (NHE) adapted from (Lexa and Savéant 1983)
The standard potential of the Co(III)-/Co(II)-redox pair of organometallic B12-derivatives, such as coenzyme B12 (AdoCbl) and methyl-cobalamin (MeCbl), is significantly more negative than that of B12r/B12s and out of the reach of biological reductants (Kräutler 1999). One-electron reduction of methyl-cobalamin to methyl-cob(II)alamin occurs at –1.36 V vs. NHE and demethylates MeCbl (at –30°C).
The thermodynamic trends of B12-redox systems can be summarized as:
-
(i)
Intra-molecular coordination of the nucleotide base and/or strongly coordinating or nucleophilic ligands (such as cyanide ions) stabilize the cobalt centre against reduction and shift the Co(III)-/Co(II)-redox couples to more negative potentials.
-
(ii)
The one-electron reduction of organometallic Co(III)-corrins typically occurs at more negative potentials than the Co(II)-/Co(I)-redox couple B12r/B12s and leads to loss of the organometallic ligand.
Electrochemistry is an excellent method for the selective production of reduced B12 forms. Since alkyl halides or alkyl tosylates react quickly and efficiently with Co(I)-corrins (Schrauzer and Deutsch 1969; Pratt 1972), which are cleanly generated at the electrodes, electrochemistry also is a method for the clean synthesis of most organometallic B12 derivatives (Kräutler 1999).
17.2.3 B12-Organometallic Reactivity
The reactivity of B12-derivatives in organometallic (redox-) reactions is the basis for their biological role as cofactors. Formation and cleavage of the Co–C bond in organometallic B12-cofactors are crucial steps in the reactions catalyzed by the B12-dependent enzymes (Kräutler et al. 1998; Banerjee 1999). In solution, cleavage and formation of the Co–C bond have been observed to occur in all of the basic oxidation levels for the cobalt centre of the corrin core (Kräutler and Ostermann 2003; Brown 2005).
Two mechanisms for these organometallic reactions have been found to be the basis for catalysis by the human cobalamin-dependent enzymes (Fig. 17.5):
-
(i)
the homolytic mode, which involves the cleavage or formation of a single axial bond, as is typical of the reactivity of coenzyme B12 (AdoCbl): 5′-Ado-Co(III)-corrin ⇄ Co(II)-corrin + 5′-deoxy-5′-adenosyl radical
-
(ii)
the nucleophile induced, heterolytic mode, which involves the cleavage or formation of two (trans-) axial bonds, as is typical of the reactivity of methyl-cobalamin (MeCbl): methyl-Co(III)-corrin + nucleophile ⇄ Co(I)-corrin + methylating agent

Patterns of reactivity and formal analysis of elementary reaction steps of “complete” corrinoids in organometallic (redox-)transformations, relevant for their cofactor functions in B12-dependent enzymes
The homolytic mode of the cleavage of the Co–C bond of coenzyme B12 (AdoCbl) is of particular importance in its role as a cofactor. Indeed, thermal decomposition of AdoCbl in aqueous solution occurs readily at higher temperatures and leads predominantly to an adenosyl radical and cob(II)alamin (B12r) (Fig. 17.6). AdoCbl is thus considered to be a “reversible carrier of an alkyl radical” (or a reversibly functioning “radical source”) (Halpern 1985). At room temperature (and in the dark), a neutral aqueous solution of AdoCbl is rather stable, with a calculated half life of 1010 s. The strength of the Co–C bond of AdoCbl (the homolytic Co–C bond dissociation energy = BDE) is about 30 kcal/mol (Halpern 1985; Hay and Finke 1986). The strength of the Co–C bond of AdoCbl is hardly influenced by the coordination of the nucleotide (Kräutler 1987, 1998).

Coenzyme B12 (AdoCbl), a reversibly functioning “radical source” (it provides a 5′-deoxy-5′-adenosly radical); in reverse, the “radical trap” B12r rapidly reacts with (5′-deoxy-5′-adenosyl) radicals to give organo-cob(III)alamins directly (such as AdoCbl)
The (crystal) structure of B12r is surprisingly similar to that of the cobalamin moiety of AdoCbl (Kräutler et al. 1989). The radicaloid B12r has a penta-coordinated Co(II)-centre, so that reactions with alkyl radicals (at its “upper” β-face) give hexa-coordinate organo-cob(III)alamins (such as AdoCbl) and occur with little restructuring of the corrin moiety: B12r thus fulfils the structural criteria of a very efficient “radical trap” and it reacts very fast (near diffusion control) with alkyl radicals (such as the 5′-deoxy-5′-adenosyl radical) (Endicott and Netzel 1979).
The second important type of organometallic reactivity of B12-derivatives represents a heterolytic mode of formation/cleavage of the Co–C bond and concerns the inter-conversion between Co(I)-corrins and methyl-Co(III)-corrins, important in enzyme-catalysed methyl-transfer reactions (Kräutler et al. 1998; Banerjee 1999; Matthews 2001): Co(I)-corrins react as strong nucleophiles with methylating agents (the Co–CH3 bond is formed) and methyl Co(III)-corrins may be demethylated by nucleophiles (the Co–CH3 bond is cleaved).
Alkylation at the corrin-bound Co(I) centre of B12s normally proceeds via the “classical” bimolecular nucleophilic substitution (SN2) mechanism (Fig. 17.7), where the Co(I)-corrin acts as a “supernucleophile” (Schrauzer and Deutsch 1969). However, in certain cases alkylation of Co(I)-corrins occurs via an alternative two-step one-electron transfer path, where Co(I)-corrins act as strong one-electron reducing agents and the process proceeds via Co(II)-corrin intermediates (Kräutler and Caderas 1984; Kräutler 1998).

The highly nucleophilic cob(I)alamin (B12s) reacts with methylating agents, to give methylcobalamin (MeCbl). For kinetic and thermodynamic reasons methylation of cob(I)alamin (B12s) occurs at the “upper” or β-face
In Co(I)-corrins, like B12s, the nucleophilicity in alkylation reactions is virtually independent of the presence of the DMB-nucleotide. Co(I)-corrins, both “complete” and “incomplete”, preferentially react at their β-face (Kräutler 1998). The immediate product of the β-alkylation is likely to be a penta-coordinate Coβ-alkyl-Co(III)-corrin (Fig. 17.7). Since most alkyl-cobalamins are hexa-coordinate in their more stable “base-on”-constitution, the methylation by the SN2-mode takes place in a two step mechanism. In aqueous solution and at room temperature methyl-cobalamin (MeCbl) is more stable in the “base-on”-form by about four kcal/mol than “base-off” Coα-aquo-Coβ-methyl-cobalamin (Kräutler 1987, 1998).
The reverse process, the nucleophile-induced dealkylations of methyl-Co(III)-corrins, is less studied. Intramolecular coordination of the nucleotide base stabilises MeCbl and slows the demethylation down: thiolates demethylate MeCbl to Co(I)corrinoids at approximately 1,000 times lower rate than methyl-corrinoids lacking a nucleotide base (Hogenkamp et al. 1985), reflecting the effect of the coordinated nucleotide (Kräutler 1987). Considerable axial base effects are thus expected for methyl-group transfer reactions involving enzyme-bound Co(I)- and methyl-Co(III)-corrins (Drennan et al. 1994; Kräutler 1998).
The two most relevant modes of formation and cleavage of the Co–C bond of the cobalt centre differ significantly in their structural requirements:
-
in the homolytic mode of cleavage, the complete cobalt-corrin part of organo-cobalamins (such as AdoCbl) hardly changes its structure (Fig. 17.6);
-
in the heterolytic mode of cleavage and formation of the Co–C bond significant reorganization occurs at both faces of the corrin-bound cobalt centre. Cleavage of the Co–CH3 bond is brought about by attack of a nucleophile at the readily accessible carbon of the cobalt-bound methyl group (Fig. 17.7).
Organocobalamins have long been know to be sensitive to visible light (Pailes and Hogenkamp 1968), which induces homolytic cleavage of the Co–C bond with a quantum yield of about 0.3 (Cole et al. 2002). Another mode of cleavage of the Co–C bond of organometallic B12-derivatives is the thermodynamically favourable radical-substitution at the cobalt-bound carbon centre, as observed in the demethylation of MeCbl by an organic radical (Kräutler et al. 1995; Mosimann and Kräutler 2000).
17.3 Occurrence of B12-Dependent Enzymes
B12-coenzymes are nowadays known to act as cofactors in four classes of distinct enzymes: B12-dependent methyl-transferases (Matthews 2001; Banerjee and Ragsdale 2003), B12-dependent isomerases and ribonucleotide reductases (both use AdoCbl) (Buckel and Golding 2006) and corrinoid dehalogenases (Siebert et al. 2002), some of which use a nor-vitamin B12-derivative (Kräutler et al. 2003). In addition, evidence for enzymatic catalysis of the biosynthetic incorporation of the organometallic groups of AdoCbl and of MeCbl (aside from such reactions in the methyl transferases and isomerases) is available (see below).
B12-dependent methyl transferases play an important role in amino acid metabolism in many organisms (including humans) as well as in one-carbon metabolism and CO2 fixation in anaerobic microbes (Banerjee and Ragsdale 2003). The reactivity of the “supernucleophilic” Co(I)-corrins and of methyl-Co(III)-corrins make B12-derivatives ideal as cofactors in enzymatic methyl-group transfer reactions (Kräutler et al. 1998; Banerjee 1999; Kräutler and Ostermann 2003; Brown 2005). B12-dependent methionine synthase has been particularly well studied (see below and refs (Matthews 1999, 2001; Banerjee and Ragsdale 2003)) as have methyl transferases in methanogenesis (Sauer and Thauer 1999) and in aerobic acetogenesis (Ragsdale et al. 1998). In human metabolism, methionine synthase is crucial for the endogenous formation of methionine (Banerjee 1997).
About ten coenzyme B12-dependent enzymes are now known, which are distributed rather disproportionately in the living world (see refs (Kräutler et al. 1998; Banerjee 1999; Brown 2005)). Only one of these, the carbon skeleton mutase methylmalonyl-CoA mutase (hMMCM), is indispensable in human metabolism (Banerjee 1997). Three more microbial carbon skeleton mutases are known (besides MMCM (Banerjee and Chowdhury 1999)): glutamate mutase (Buckel and Golding 1996; Gruber and Kratky 2002), methylene glutarate mutase (Buckel and Golding 1996) and isobutyryl-CoA mutase (Zerbe-Burkhardt et al. 1999). In addition, five bacterial isomerases, diol and glycerol dehydratase, ethanolamine ammonia lyase and two amino mutases are B12-dependent, as is an anaerobic form of ribonucleotide reductase (Frey and Chang 1999; Stubbe 2000; Toraya 2003).
The labile and light sensitive coenzyme B12 (AdoCbl) is not typically taken up with the nutrition, but is enzymatically produced in the human mitochondrion by adenosyltransferase. A feature of this enzyme is its ability to bind cob(II)alamin (B12r) in a remarkable four coordinate “base-off”-constitution and, thus, to render the bound B12r more accessible to reduction by biological reducing agents (Stich et al. 2005). This biosynthetic enzyme catalyzes the reduction of B12r to cob(I)alamin (B12s) and adenosylation by ATP to give AdoCbl, which appears to be bound in a “base-off”-form also (Fig. 17.8) (Yamanishi et al. 2005). Direct delivery of AdoCbl in the “base-off”-constitution has been suggested from adenosyl-transferase to its target (apo-)enzyme, methylmalonyl-CoA mutase.

Coenzyme B12 (AdoCbl, in its “base-off”-form) is biosynthesized (in humans) from “base-off” cob(II)alamin (B12r) by the mitochondrial enzyme adenosyltransferase
17.4 Methionine Synthase
Methionine synthase catalyzes the formation of methionine by methylation of homocysteine and demethylation of N5-methyl-tetrahydrofolate to tetrahydrofolate (Fig. 17.9). In B12-dependent methionine synthase, a methylcorrinoid, such as methyl-cobalamin (MeCbl) is the cofactor, whereas in cobalamin-independent methionine synthases, which have much slower turnover, homocysteine is merely activated by coordination to a zinc-centre (Matthews 2001).

Methionine synthase produces methionine and tetrahydrofolate from homocysteine and N5-methyl-tetrahydrofolate
17.4.1 B12-Dependent Methyl Group Transfer
In a catalytic cycle of B12-dependent methyl transferases (such as methionine synthase), the corrinoid cycles between enzyme-bound methyl-Co(III)- and Co(I)-forms (Matthews 2001). The transition between the hexa-coordinate methyl-Co(III)- form and the tetra-coordinate Co(I)-form must be accompanied by large structural changes (Figs. 17.5 and 17.7) and complementary changes in a protein environment.
This provides a means for controlling the organometallic reactivity of the bound cofactor at the same time (Kräutler 1998). The enzymatic methyl-group transfers (which formally involve nucleophile-induced heterolytic cleavage and formation of the organometallic Co–CH3 bond at the corrin bound cobalt centre) are thus expected to be subject to strict geometric control (mediated via the “regulatory” His-Asp-Ser-triad in methionine synthase (MetH) (Drennan et al. 1994; Matthews 2001), (Fig. 17.10 and below): The His-Asp-Ser-triad appears to have a dual role in MetH;
-
(i)
it may participate in maintaining conformational control of the mutual placement of the corrinoid cofactors and the bound substrates (in their respective enzyme-modules) by a H+-mediated conformational switch mechanism (Matthews 2001; Ludwig and Matthews 2002; Bandarian et al. 2003) and
-
(ii)
it may exert a relevant thermodynamic effect on the strength of the Coβ–CH3 bond by α-axial base-coordination (Drennan et al. 1994).

Illustration of methionine formation catalyzed by MetH (Enz signifies the MetH-apoenzyme), where the bound corrinoid shuttles between MeCbl, in a “base-off/His-on” form, and cob(I)alamin (B12s) (Matthews 2001). In addition, reductive methylation with SAM and a reduced flavodoxin reactivates oxidized enzyme (with catalytically inactive cob(II)alamin)
Solution studies had shown a significant thermodynamic trans-effect of the DMB-coordination in methyl-cobalamin (MeCbl) and stabilization of the methyl-Co(III)-corrin MeCbl by about 4 kcal/mol against nucleophilic abstraction of the methyl group (Kräutler 1987, 1998) (a similar effect on heterolytic methyl group transfer reactions was deduced for imidazole-coordination in methyl-imidazoyl-cobamide (Me-ImCba) (Fasching et al. 2000).
The methyl group may originate from N5-methyl-tetrahydropterins (such as N5-methyl-tetrahydromethanopterin or N5-methyl-tetrahydrofolate) and a variety of other methylated substrates (Stupperich et al. 1998; Matthews 2001). Typically, thiols are the methyl group acceptors in methionine synthesis (homocysteine) (Matthews 2001; Bandarian et al. 2003) and methanogenesis (coenzyme M) (Sauer and Thauer 1999). In the anaerobic biosynthesis of acetyl-coenzyme-A from one-carbon precursors the methyl group acceptor is suggested to be a nickel centre (Drennan et al. 2004).
17.4.2 Methionine Synthase from Escherichia coli
The methyl group transfer catalysed by methionine synthase from E. coli (MetH) (Matthews 2001; Ludwig and Matthews 2002) is indicated to proceed as a sequence of two nucleophilic displacement steps. This excludes free methyl cations or radicals as intermediates. The methyl group transfer relies on the inherent reactivities of enzyme-bound Co(I)corrins and methyl-Co(III)corrins (Matthews 2001). The great structural changes that must accompany the transitions from (tetra-coordinate) Co(I)corrins to (hexa-coordinate) methyl-Co(III)corrins (Kräutler 1998) also provide a means for control from the protein environment (on the methyl transfer, see above) (Ludwig and Matthews 2002).
MetH represents the most thoroughly studied B12-dependent methyl transferase (Matthews 1999, 2001; Bandarian et al. 2003). It is a modular enzyme containing four separate (and independently functioning) domains for binding (beginning at the N-terminus) homocysteine, N5-methyl-tetrahydrofolate, the B12-cofactor or S-adenosyl-methionine (SAM). The B12-binding domain interacts specifically with each of the other three domains (depending upon its three oxidation states): The Co(I) form with the N5-methyl-tetrahydrofolate domain, the Co(II) form with the SAM domain, and the CH3–Co(III) form with the homocysteine domain.
MetH catalyses the methylation of the bound and reduced cob(I)alamin cofactor by (N5-protonated) N5-methyltetrahydrofolate to give enzyme-bound MeCbl in a “base-off/His-on” form (see later) (Matthews 2001; Ludwig and Matthews 2002; Bandarian et al. 2003). The bound MeCbl is demethylated by homocysteine (Fig. 17.10), whose sulfur is activated and deprotonated due to the coordination to a zinc ion (held by three cysteine residues) of the homocysteine domain. The two methyl-transfer reactions occur in a rapid sequential mechanism (kcat (MetH) = 27 s–1) (Jarrett et al. 1996; Ludwig and Matthews 2002). The bound cob(I)alamin (B12s) may adventitiously be oxidized to inactive cob(II)alamin (B12r) and requires reactivation by reductive methylation with SAM and a reduced flavodoxin (Ludwig and Matthews 2002).
X-ray crystal analysis of the 28 kDa B12-binding domain of MetH provided the first insight into the three-dimensional structure of a B12-binding protein (Drennan et al. 1994, 1998; Ludwig and Matthews 2002). The cobalt-coordinating DMB-nucleotide tail of the protein-bound cofactor MeCbl was displaced by a histidine and extended into the core of the “Rossmann fold”, two most astounding revelations of this work (Drennan et al. 1994). Consequently, in MetH the corrinoid cofactor is bound “base-off” and in a “base-off/His-on”-mode, due to histidine ligation to the metal centre. The crucial cobalt-ligating histidine residue is part of a Gly–X–X–His–X–Asp sequence, which was noticed to be a common sequence in some B12-binding proteins (Marsh and Holloway 1992). The B12-binding domain of MetH, therefore, provides both an anchoring site for the nucleotide tail and cobalt-ligation via the residues of the (“regulatory”) His-Asp-Ser triad, holding the corrinoid cofactor with its “catalytic” β-side exposed at an inter-domain interface.
Further crystallographic studies provided more evidence for how the selection between the two ways of methylating the bound corrinoid (the “productive” methylation by N5-methyl-tetrahydrofolate or the “regenerative” methylation by enzyme bound SAM (Ludwig and Matthews 2002)) are achieved. This is controlled by a dynamic domain alternation (Bandarian et al. 2002). As shown by the crystal structure of the N-terminal (two domain) substrate-binding modules of MetH, the two substrates, homocysteine and N5-methyl-tetrahydrofolate, are bound and activated in orientations that position them for reaction with the bound corrinoid. However, the two active sites are separated by ≈ 50 Å and the B12-binding domain must shuttle back and forth between these distant active sites to complete the catalytic cycle (Evans et al. 2004).
The crystal structure and the finding of the “base-off/His-on” binding of the B12-cofactor in MetH explained ESR-spectroscopic evidence for histidine binding to the cobalt centre of p-cresolyl-cobamide (a “base-off” cobamide, with a non-coordinating cresolyl-“nucleotide”) in the acetogen Sporomusa ovata (Stupperich et al. 1990).
17.4.3 Human Methionine Synthase
Human methionine synthase (hMS, E.C. 2.1.1.13) is a cytoplasmic enzyme with 4 domains, which shares 55% identity in deduced amino acid sequence with MetH (from E. coli). hMS shows extensive structural similarities with MetH and has all residues implicated in relevant B12-binding to MetH. hMS also catalyzes the methylation of homocysteine (with demethylation of N5-methyl-tetrahydrofolate) in an apparently related mechanism to that of MetH (Banerjee 1997; Banerjee 1998). In both methionine synthases occasional oxidation (to an inactive cob(II)alamin carrying form) occurs (in the micro-aerophilic conditions of mammalian cells, hMS is oxidized about once every 2,000 turnovers). In contrast to MetH, which is reactivated via reduced ferredoxin (Matthews 2001), hMS is reactivated by a specific reductase, called hMS-reductase (hMSR) (Yamada et al. 2006). hMSR is a dual flavoprotein homologous to flavodoxin and appears to function as a chaperone in the assembly of holo-hMS from apo-hMS and B12a in the presence of NADPH (it also achieves the reduction of aquo-cobalamin (B12a) to B12r) (Yamada et al. 2006) and to reactivate oxidized hMS.
17.5 Methylmalonyl-CoA Mutase
The reversible isomerisation of methylmalonyl-CoA to succinyl-CoA is an enzyme catalyzed radical process (Fig. 17.11). Related isomerisations (malonates to succinates) have been achieved in model radical processes in solution (Halpern 1982; Buckel and Golding 2006).

(2R)-methylmalonyl-CoA is reversibly converted into succinyl-CoA (see below)
17.5.1 Coenzyme B12-Dependent Enzymes
Coenzyme B12-dependent enzymes, such as methylmalonyl-CoA mutase (Fig. 17.11), thus performs “difficult” transformations. With the exception of the enzymatic ribonucleotide reduction (Stubbe et al. 1998), the results of coenzyme B12-catalysed enzymatic reactions correspond to isomerisations with vicinal exchange of a hydrogen atom and of a larger moiety (Buckel and Golding 1996). Homolytic cleavage of the Co–C bond of the protein-bound AdoCbl to a 5′-deoxy-5′-adenosyl radical and cob(II)alamin (B12r) (Fig. 17.6) was shown to be the entry to H-abstraction reactions induced by the 5′-deoxy-5′-adenosyl radical (Rétey 1999). Therefore, homolysis of the Co–C bond of AdoCbl is its biologically most significant reactivity (Halpern 1985; Finke 1998). The coenzyme B12-catalysed enzyme reactions occur with maximal rates of approximately 100 s–1 (Licht et al. 1999; Marsh and Drennan 2001). With MMCM, rapid formation of Co(II)corrins was observed when substrate was added to a solution of holoenzyme (or of apoenzymes and AdoCbl) (Rétey 1998).
Homolysis of the Co–C bond of protein-bound AdoCbl needs to be accelerated by a factor of >1012 to relate the observed rates of catalysis by the coenzyme B12-dependent enzymes to the rate of homolysis of AdoCbl in aqueous solution (Halpern 1985; Finke 1998). This dramatic labilisation of the bound cofactor towards homolysis of the Co–C bond is an intriguing feature of coenzyme B12-dependent enzymes (Halpern 1985; Finke 1998). The mechanism of the (enzyme- and substrate-) induced labilisation of AdoCbl towards cleavage of its Co–C bond is a much discussed problem. A “conformational distortion” of the cobalt-corrin part of AdoCbl was suggested as its basis (Halpern 1985). However, the crystal structures of several coenzyme B12-dependent enzymes (Mancia et al. 1996; Reitzer et al. 1999; Shibata et al. 1999) and of cob(II)alamin (B12r) (Kräutler et al. 1989) have not supported this mode of action. Labilisation of the Co–C bond appears to mostly result from protein- and substrate-induced strain on the organometallic group, separation of the largely non-strained homolysis fragments and strong binding of the separated pair, 5′-deoxy-5′-adenosyl radical and B12r (in “base-off/His-on” or “base-on” form) (Kräutler et al. 1989; Toraya 2003; Gschösser et al. 2005). Fixed placement of the corrin moiety at the interface of the B12-binding and substrate-binding/activating domains is given a high significance and movements of the corrin moiety are not required. The nature of the axial trans ligand (histidine or DMB) has a lesser effect on the homolytic Co–C bond dissociation (Fasching et al. 2000). The “regulatory triad” appears not to be involved in proton-transfer steps and may conserve its structure largely during enzymatic turnover (Ludwig and Evans 1999).
The 5′-deoxy-5′-adenosyl radical is the established reactive partner in the actual coenzyme B12-dependent enzymatic reactions, so that AdoCbl functions as a “pre-catalyst” (or catalyst precursor) (Kräutler 1998). The further enzyme reactions all involve bound organic radicals, which are formed (directly or indirectly) by a H-atom abstraction by the 5′-deoxy-5′-adenosyl radical (that originates from AdoCbl). The rearrangement steps of B12-dependent enzymatic rearrangements are assumed to be accomplished by tightly protein-bound radicals that are controlled in their reaction space by the protein (Toraya 2003; Buckel and Golding 2006). The role of the Co(II)-corrin fragment of the coenzyme (as a “spectator” or a “conductor”) is a matter of ongoing discussions (Buckel et al. 2006). The major functions of the enzyme concern not only the catalysis of its proper reactions but also the reversible generation of the radical intermediates and the effective protection from non-specific radical chemistry, called “negative catalysis” (Rétey 1990).
Four of the known coenzyme B12-dependent enzymatic reactions concern carbon skeleton rearrangements, in which two vicinal groups (a hydrogen atom and an organic substituent) exchange their positions in a (pseudo-) intra-molecular fashion (Marsh and Drennan 2001; Buckel and Golding 2006). The B12-cofactor is bound “base-off/His-on” at an interface between two modules, the B12-binding and substrate activating domains (or subunits) as revealed by the analysis of the crystal structures of methylmalonyl-CoA mutase (MMCM) from Propionibacterium shermanii (Hodgkin et al. 1956) and of glutamate mutase from Clostridium cochlearium (Reitzer et al. 1999; Gruber and Kratky 2002). The B12-binding motif (Gly–X–X–His–X–Asp) (Marsh and Holloway 1992) is common to the carbon skeleton mutases (Marsh and Drennan 2001). The B12-binding domain of MMCM exhibits considerable sequence homology, even with the B12-binding domain of MetH (Ludwig and Evans 1999). Such homology can be rationalized by the related way of “base-off/His-on” binding the cobalamin cofactor and does not extend to the substrate binding domains (subunits) of the carbon skeleton mutases (Marsh 2000).
17.5.2 Methylmalonyl-CoA Mutase from Propionibacterium shermanii
Methylmalonyl-CoA mutase has been most thoroughly studied as the P. shermanii mutase (MMCM), which interconverts (2R)-methylmalonyl-CoA and succinyl-CoA (Banerjee and Chowdhury 1999) (Fig. 17.12). The main function of MMCM in P. shermanii concerns the formation of (2R)-methylmalonyl-CoA from succinyl-CoA. Binding of the substrate to holo-MMCM triggers a very fast homolysis of the Co–C bond of the bound AdoCbl. The overall turnover for the radical rearrangement occurs with a rate of about 60 s–1 at 25°C. The 5′-deoxy-5′-adenosyl (5′-Ado) radical induces the rearrangement reaction by abstracting an H-atom from the methyl group of enzyme-bound methylmalonyl-CoA. A large deuterium isotope effect of the abstracted H-atoms on the rate of homolysis of the Co–C bond was observed (Chowdhury and Banerjee 2000a). Labilisation of the Co–C bond towards homolysis by about 16 kcal/mol is due largely to a decrease of the activation enthalpy (Chowdhury and Banerjee 2000b). Recent studies (Brooks et al. 2004) indicated the stabilisation of the separated homolysis fragments, 5′-Ado radical and cob(II)alamin (B12r) to be a dominant contribution to the activation (for homolysis) of the Co–C bond of AdoCbl, consistent with an earlier suggestion, based on the crystal structure of B12r (Kräutler et al. 1989).

Methylmalonyl-CoA mutase (MMCM) interconverts (2R)-methylmalonyl-CoA and succinyl-CoA. Proposed reaction mechanism, involving H-atom abstraction (step a), radical rearrangement (step b) and back transfer of H-atom (step c). The 5′-deoxy-5′-adenosyl radical (and cob(II)alamin (B12r)) originate(s) from homolysis of protein bound AdoCbl (Fig. 17.6) (Banerjee and Chowdhury 1999; Buckel and Golding 2006)
H-atom abstraction gives the 2-methylmalon-2′-yl-CoA radical which rearranges rapidly to the succin-3-yl-CoA radical (Golding et al. 1998; Banerjee and Chowdhury 1999). Both, fragmentation/ recombination and intramolecular addition/elimination, via a cyclopropyloxyl radical, pathways have been considered for this rearrangement (Buckel and Golding 2006). However, computational studies indicate that the energetic barrier for the addition/elimination pathway is lower than the dissociative pathway (Smith et al. 1999). Experimental evidence has also been used to support this theory, in which (i) the succin-3-yl-CoA radical arises from an intramolecular radical rearrangement and (ii) occurs without noticeable participation of the bound cob(II)alamin (B12r) (Golding et al. 1998). The succin-3-yl-CoA radical (resulting from the rearrangement) then re-abstracts an H-atom from 5′-deoxyadenosine to give succinyl-CoA and the 5′-deoxy-5′-adenosyl radical (which recombines with B12r to give enzyme-bound AdoCbl).
X-ray analysis of the 150 kDa heterodimeric MMCM from P. shermanii was the first accurate structure of a coenzyme B12-dependent enzyme. It showed the B12-cofactor to be bound “base-off/His-on”. The cobalt centre was coordinated to the histidine of the “regulatory triad” His-Asp-Lys. As in MetH the nucleotide tail of the corrinoid was tightly inserted into the protein and the corrinoid was bound at an interface between two domains (Evans and Mancia 1998; Ludwig and Evans 1999).
The question of how enzymes recognize and bind their corrinoid cofactor in a “base-off”-form is intriguing (in aqueous solution, AdoCbl and MeCbl prefer to be “base-on”). The solution structure of the cofactor-free B12-binding subunit of glutamate mutase from Cl. tetanomorphum (Tollinger et al. 1998, 2001), derived by NMR-spectroscopy, showed the B12-binding subunit to be largely pre-organized for B12-binding. However, the apo-protein was seen to include a flexible loop and a “nascent” helix, which were both suggested to structure upon binding of the B12-cofactor. A model for the events in binding of the “base-off/His-on” corrinoid by the B12-binding subunit was derived, in which the “base-off”-form of the B12-cofactor was trapped by its nucleotide tail and the bound nucleotide moiety, in turn, stabilized the protein (Tollinger et al. 2001).
In crystallographic work, substrate free MMCM was investigated, as well as with bound pseudo-substrate: both structures indicated the organometallic group to be strained or detached from the cobalt centre (Mancia and Evans 1998; Mancia et al. 1999). Substrate binding appears to squeeze the adenosyl group off from the cobalt-corrin and to assist Co–C bond homolysis (Ludwig and Evans 1999).
17.5.3 Human Methylmalonyl-CoA Mutase
Human methylmalonyl-CoA mutase (hMMCM, EC 5.4.99.2) is a mitochondrial enzyme and catalyzes the isomerisation of (2R)-methylmalonyl-CoA to succinyl-CoA (as final part of the catabolic pathway of uneven numbered and branched chain fatty acids) (Banerjee and Chowdhury 1999). Impairment of hMMCM leads to methyl-malonic aciduria, a metabolic error that leads to developmental retardation and mortality in infants. hMMCM binds two AdoCbl per (homodimeric) enzyme (predicted mass: about 159 kDa) and is specific for the 2R-epimer of methylmalonyl-CoA. The (monomer-)chain of hMMCM and the B12-binding α-chain of MMCM are homologous; evidence for a related mechanism in both mutases has been provided (Buckel and Golding 2006). Apo-hMMCM has been suggested to be loaded with its cofactor, AdoCbl, in a direct and “targeted” interaction with adenosyl-transferase. In the presence of the latter enzyme, hMMCM exhibits a striking resistance towards deactivation (Yamanishi et al. 2005).
Abbreviations
- Ado:
-
5′-(deoxy)-adenosyl
- AdoCbl:
-
5′-deoxy-5′-adenosyl-cobalamin (coenzyme B12, also named adenosyl-cobalamin)
- Ado-ImCba:
-
5′-deoxy-5′-adenosyl-imidazolyl-cobamide
- B12r:
-
cob(II)alamin
- B12s:
-
cob(I)alamin
- BDE:
-
(homolytic) bond dissociation energy
- Cba:
-
cobamide
- Cbl:
-
cobalamin (a DMB-cobamide)
- CNCbl:
-
cyano-cobalamin (vitamin B12)
- CN-ImCba:
-
cyano-imidazolyl-cobamide
- DMB:
-
5,6-dimethylbenzimidazole
- H2OCbl+ :
-
aquo-cobalamin (B12a)
- HOCbl:
-
hydroxo-cobalamin
- ImCba:
-
imidazolyl-cobamide
- MeCbl:
-
methyl-cobalamin
- Me-ImCba:
-
methyl-imidazolyl-cobamide
- MetH:
-
methionine synthase (from E. coli)
- MMCM:
-
methylmalonyl-CoA mutase (from Propionibacterium shermanii)
- NHE:
-
normal hydrogen electrode
- NMR:
-
nuclear magnetic resonance
- SAM:
-
S-adenosyl-methionine (AdoMet)
- UV/Vis:
-
ultraviolet/visible absorbance spectrum
References
Bandarian V, Ludwig ML, Matthews RG (2003) Factors modulating conformational equilibria in large modular proteins: a case study with cobalamin-dependent methionine synthase. Proc Natl Acad Sci USA 100:8156–8163
Bandarian V, Pattridge KA, Lennon BW, Huddler DP, Matthews RG, Ludwig ML (2002) Domain alternation switches B12-dependent methionine synthase to the activation conformation. Nat Struct Biol 9:53–56
Banerjee R (1997) The Yin-Yang of cobalamin biochemistry. Chem Biol 4:175–186
Banerjee R (1998) Spectroscopic and molecular genetic characterization of the two mammalian B12-dependent enzymes. In: Kräutler B, Arigoni D, Golding BT (eds) Vitamin B12 and B12-proteins, Wiley-VCH, Weinheim, pp. 189–197
Banerjee R (ed) (1999) Chemistry and biochemistry of B12. Wiley, New York
Banerjee R, Chowdhury S (1999) Methylmalonyl-CoA mutase. In: Banerjee R (ed) Chemistry and biochemistry of B12. Wiley, New York, pp. 707–730
Banerjee R, Ragsdale SW (2003) The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu Rev Biochem 72:209–247
Battersby AR (1998) B12-biosynthesis in an aerobic organism: how the pathway was elicudated. In: Kräutler B, Arigoni D, Golding BT (eds) Vitamin B12 and B12-proteins. Wiley-VCH, Weinheim, pp. 47–61
Brooks AJ, Vlasie M, Banerjee R, Brunold TC (2004) Spectroscopic and computational studies on the adenosylcobalamin-dependent methylmalonyl-CoA mutase: evaluation of enzymatic contributions to Co–C bond activation in the Co3 + ground state. J Am Chem Soc 126:8167–8180
Brown KL (1999) NMR Spectroscopy of B12. In: Banerjee R (ed) Chemistry and biochemistry of B12. Wiley, New York, pp. 197–237
Brown KL (2005) Chemistry and enzymology of vitamin B12. Chem Rev 105:2075–2149
Brown KL, Zou X, Banka RR, Perry CB, Marques HM (2004) Solution structure and thermolysis of Coβ-5′-deoxyadenosylimidazolylcobamide, a coenzyme B12 analogue with an imidazole axial nucleoside. Inorg Chem 43:8130–8142
Buckel W, Golding BT (1996) Glutamate and 2-methyleneglutarate mutase: from microbial curiosities to paradigms for coenzyme B12-dependent enzymes. Chem Soc Rev 25:329
Buckel W, Golding BT (2006) Radical enzymes in anaerobes. Ann Rev Microbiol 60:27–49
Buckel W, Kratky C, Golding BT (2006) Stabilization of methylene radicals by Cob(II)alamin in coenzyme B12 dependent mutases. Chem Eur J 12:352–362
Chowdhury S, Banerjee R (2000a) Evidence for quantum mechanical tunneling in the coupled cobalt-carbon bond homolysis-substrate radical generation reaction catalyzed by methylmalonyl-CoA mutase. J Am Chem Soc 122:5417–5418
Chowdhury S, Banerjee R (2000b) Thermodynamic and kinetic characterization of Co–C bond homolysis catalyzed by coenzyme B12-dependent methylmalonyl-CoA mutase. Biochemistry 39:7998–8006
Cole AG, Yoder LM, Shiang JJ, Anderson NA, Walker LA, Holl MMB, Sension RJ (2002) Time-resolved spectroscopic studies of B12 coenzymes: a comparison of the primary photolysis mechanism in methyl-, ethyl-, n-propyl-, and 5′-deoxyadenosylcobalamin. J Am Chem Soc 124:434–41
Dolphin D (ed) (1982) B12, vols I and II. Wiley, New York
Drennan CL, Dixon MM, Hoover DM, Jarrett JT, Goulding CW, Matthews RG, Ludwig ML (1998) Cobalamin-dependent methionine synthase from Escherichia coli: structure and reactivity. In: Kräutler B, Arigoni D, Golding BT (eds) Vitamin B12 and B12-proteins. Weinheim, Wiley-VCH, pp. 133–155
Drennan CL, Doukov TI, Ragsdale SW (2004) The metalloclusters of carbon monoxide dehydrogenase/acetyl-CoA synthase: a story in pictures. J Biol Inorg Chem 9:511–515
Drennan CL, Huang S, Drummond JT, Matthews RG, Ludwig ML (1994) How a protein binds B12 – a 3.0 Å X-ray structure of B12-binding domains of methionine synthase. Science 266:1669–1674
Drennan CL, Matthews RG, Ludwig ML (1994) Cobalamin-dependent methionine synthase – the structure of a methylcobalamin-binding fragment and implications for other B12-dependent enzymes. Curr Opin Struct Biol 4:919–929
Ellenbogen L, Cooper BA (1984) Vitamin B12. In: Machlin LJ (ed) Handbook of vitamins, nutritional and clinical aspects. Food Science and Technology. Marcel Dekker, New York, pp. 491–536
Endicott JF, Netzel TL (1979) Early events and transient chemistry in the photohomolysis of alkylcobalamins. J Am Chem Soc 101:4000–4002
Eschenmoser A (1988) Vitamin-B12 – Experiments concerning the origin of its molecular structure. Angew Chem Int Ed 27:5–39
Evans JC, Huddler DP, Hilgers MT, Romanchuk G, Matthews RG, Ludwig ML (2004) Structures of the N-terminal modules imply large domain motions during catalysis by methionine synthase. Proc Natl Acad Sci USA 101:3729–3736
Evans PR, Mancia F (1998) Insights on the reaction mechanism of methylmalonyl-CoA mutase from the crystal structure. In: Kräutler B, Golding BT, Arigoni D (eds) Vitamin B12 and B12 proteins. Wiley-VCH, Weinheim, pp. 217–226
Fasching M, Schmidt W, Kräutler B, Stupperich E, Schmidt A, Kratky C (2000) Coα-(1H-imidazolyl)-Coβ-methylcob(III)amide: model for protein-bound corrinoid cofactors. Helv Chim Acta 83:2295–2316
Finke RG (1998) Coenzyme B12-based chemical precedent for Co–C bond homolysis and other key elementary step. In: Kräutler B, Arigoni D, Golding BT (eds) Vitamin B12 and B12-proteins. Wiley-VCH, Weinheim, pp. 383–402
Frey PA, Chang CH (1999) Aminomutases. In: Banerjee R (ed) Chemistry and biochemistry of B12. Wiley, New York, pp. 835–858
Friedrich, W. (1988) Vitamins. Walter de Gruyter, Berlin
Golding BT, Anderson RJ, Ashwell S, Edwards CH, Garnett I, Kroll F, Buckel W (1998) A mechanistic overview of B12 dependent processes. In: Kräutler B, Arigoni D, Golding BT (eds) Vitamin B12 and B12 proteins. Wiley-VCH, Weinheim, pp. 201–216
Gruber K, Kratky C (2002) Coenzyme B12 dependent glutamate mutase. Curr Opin Chem Biol 6:598–603
Gschösser S, Hannak RB, Konrat R, Gruber K, Mikl C, Kratky C, Kräutler B (2005) Homocoenzyme B12 and bishomocoenzyme B12, covalent structural mimics for homolyzed, enzyme-bound coenzyme B12. Chem Eur J 11:81–93
Halpern J (1982) Chemistry and significance of vitamin B12 model systems. In: Dolphin D (ed) B12. Wiley, New York, pp. 501–541
Halpern J (1985) Mechanisms of coenzyme B12-dependent rearrangements. Science 227:869–875
Hay BP, Finke RG (1986) Thermolysis of the Co–C bond of adenosylcobalamin. 2. products, kinetics, and Co–C bond-dissociation energy in aqueous-solution. J Am Chem Soc 108:4820–4829
Hodgkin DC, Kamper J, Mackay M, Pickworth J, Trueblood KN, White JG (1956) Structure of Vitamin-B12. Nature 178:64–66
Hodgkin DC, Pickworth J, Robertson JH, Trueblood KN, Prosen RJ, White JG (1955) Crystal structure of the hexacarboxylic acid derived from B12 and the molecular structure of the vitamin. Nature 176:325–328
Hoffmann B, Oberhuber. M., Stupperich E, Bothe H, Buckel W, Konrat R, Kräutler B (2000) Native corrinoids from Clostridium cochlearium are adeninylcobamides: spectroscopic analysis and identification of pseudovitamin B12 and factor A. J Bacteriol 182:4773–4782
Hogenkamp HPC, Bratt GT, Sun S (1985) Methyl transfer from methylcobalamin to thiols – a reinvestigation. Biochemistry 24:6428–6432
Jarrett JT, Amaratunga M, Drennan CL, Scholten JD, Sands RH, Ludwig ML, Matthews RG (1996) Mutations in the B12-binding region of methionine synthase: how the protein controls methylcobalamin reactivity. Biochemistry 35:2464–2475
Konrat R, Tollinger M, Kräutler B (1998) New NMR structural and dynamical probes of organometallic B12 derivatives. In: Kräutler B, Arigoni D, Golding BT (eds) Vitamin B12 and B12-proteins. Wiley-VCH, Weinheim, pp. 349–368
Kratky C, Kräutler B (1999) Molecular structure of B12 cofactors and other B12 derivatives. In: Banerjee R (ed) Chemistry and biochemistry of B12. Wiley, New York, pp. 6–41
Kräutler B (1987) Thermodynamic trans-effects of the nucleotide base in the B12 coenzymes. Helv Chim Acta 70:1268–1278
Kräutler B (1998) B12 coenzymes, the central theme. In: Kräutler B, Arigoni D, Golding BT (eds) Vitamin B12 and B12 proteins. Wiley-VCH, Weinheim, pp. 3–43
Kräutler B (1999) Electrochemistry and organometallic electrochemical synthesis. In: Banerjee R (ed) Chemistry and biochemistry of B12. Wiley, New York, pp. 315–339
Kräutler B, Arigoni D, Golding BT (eds) (1998) Vitamin B12 and B12-proteins. Wiley-VCH, Weinheim
Kräutler B, Caderas C (1984) Complementary diastereoselective cobalt methylations of the Vitamin-B12 derivative cobester. Helv Chim Acta 67:1891–1896
Kräutler B, Dérer T, Liu PL, Mühlecker W, Puchberger M, Kratky C, Gruber K (1995) Oligomethylene-bridged vitamin-B12 dimers. Angew Chem Int Ed 34:84–86
Kräutler B, Fieber W, Ostermann S, Fasching M, Ongania KH, Gruber K, Kratky C, Mikl C, Siebert A, Diekert G (2003) The cofactor of tetrachloroethene reductive dehalogenase of Dehalospirillum multivorans is norpseudo-B12, a new type of a natural corrinoid. Helv Chim Acta 86:3698–36716
Kräutler B, Keller W, Kratky C (1989) Coenzyme B12-chemistry: the crystal and molecular structure of Cob(II)alamin. J Am Chem Soc 111:8936–8938
Kräutler B, Konrat R, Stupperich E, Färber G, Gruber K, Kratky C (1994) Direct evidence for the conformational deformation of the corrin ring by the nucleotide base in vitamin-B12 – synthesis and solution spectroscopic and crystal-structure analysis of Coβ-cyanoimidazolylcobamide. Inorg Chem 33:4128–4139
Kräutler B, Ostermann S (2003) Structure, reactions and functions of B12 and B12-proteins. In: Kadish KM, Smith KM, Guilard R (eds) The porphyrin handbook. Elsevier Science, Oxford, pp. 229–276
Lenhert PG, Hodgkin DC (1961) Structure of 5,6-dimethylbenzimidazolylcobamide coenzyme. Nature 192:937
Lexa D, Savéant JM (1983) The electrochemistry of vitamin-B12. Acc Chem Res 16:235–243
Licht SS, Booker S, Stubbe J (1999) Studies on the catalysis of carbon-cobalt bond homolysis by ribonucleoside triphosphate reductase: evidence for concerted carbon-cobalt bond homolysis and thiyl radical formation. Biochemistry 38:1221–1233
Ludwig ML, Evans PR (1999) X-ray crystallography of B12 enzymes: methylmalonyl-CoA mutase and methionine synthase. In: Banerjee R (ed) Chemistry and biochemistry of B12. Wiley, New York, pp. 595–632
Ludwig ML, Matthews RG (2002) B12-dependent methionine synthase: a structure that adapts to catalyze multiple methyl transfer reactions. Struct Mech: from Ashes to Enzymes 827:186–201
Mancia F, Evans PR (1998) Conformational changes on substrate binding to methylmalonyl CoA mutase and new insights into the free radical mechanism. Structure 6:711–720
Mancia F, Keep NH, Nakagawa A, Leadlay PF, McSweeney S, Rasmussen B, Bösecke B, Diat O, Evans PR (1996) How coenzyme B12 radicals are generated: the crystal structure of methylmalonyl-coenzyme A mutase at 2 Å resolution. Structure 4:339–350
Mancia F, Smith GA, Evans PR (1999) Crystal structure of substrate complexes of methylmalonyl-CoA mutase. Biochemistry 38:7999–8005
Marsh ENG (2000) Coenzyme-B12-dependent glutamate mutase. Bioorg Chem 28:176–189
Marsh ENG, Drennan CL (2001) Adenosylcobalamin-dependent isomerases: new insights into structure and mechanism. Curr Opin Chem Biol 5:499–505
Marsh ENG, Holloway DE (1992) Cloning and sequencing of glutamate mutase component-S from Clostridium tetanomorphum – homologies with other cobalamin-dependent enzymes. Febs Lett 310:167–170
Matthews RG (1999) Cobalamin-dependent methionine synthase. In: Banerjee R (ed) Chemistry and biochemistry of B12. Wiley, New York, pp. 681–706
Matthews RG (2001) Cobalamin-dependent methyltransferases. Acc Chem Res 34:681–689
Mosimann H, Kräutler B (2000) Methylcorrinoids methylate radicals – Their second biological mode of action? Angew Chem Int Ed 39:393–200
Pailes WH, Hogenkamp HPC (1968) Photolability of Co-alkylcobinamides. Biochemistry 7:4160–4166
Pratt JM (1972) Inorganic chemistry of vitamin B12. Academic, New York
Ragsdale SW, Kumar M, Zhao S, Menon S, Seravalli J, Doukov T (1998) Discovery of a biological organometallic reaction sequence involving vitamin B12. In: Kräutler B, Arigoni D, Golding BT (eds) Vitamin B12 and B12-proteins. Wiley-VCH, Weinheim, pp. 167–178
Randaccio L, Geremia S, Nardin G, Würges J (2006) X-ray structural chemistry of cobalamins. Coord Chem Rev 250:1332–1250
Reitzer R, Gruber K, Jogl G, Wagner, UG, Bothe H, Buckel W, Kratky C (1999) Glutamate mutase from Clostridium cochlearium: the structure of a coenzyme B12-dependent enzyme provides new mechanistic insights. Structure 7:891–902
Rétey J (1990) Enzymatic-reaction selectivity by negative catalysis or how do enzymes deal with highly reactive intermediates. Angew Chem Int Ed 29:355–361
Rétey J (1998) Coenzyme B12-dependent enzymes and their models. In: Kräutler B, Arigoni D, Golding BT (eds) Vitamin B12 and B12-proteins. Wiley-VCH, Weinheim, pp. 273–288
Rétey J (1999) Stereospecifity of the coenzyme B12-catalyzed rearrangements and the role of negative catalysis. In: Banerjee R (ed) Chemistry and biochemistry of B12. Wiley, New York, pp. 271–288
Rickes EL, Brink NG, Koniuszy FR, Wood TR, Folkers K (1948) Crystalline vitamin B12. Science 107:396–397
Rosenblatt DS, Fenton WA (1999) Inborn errors of cobalamin metabolism. In: Banerjee R (ed) Chemistry and biochemistry of B12. Wiley, New York, pp. 367–384
Sauer K, Thauer RK (1999) The role of corrinoids in methanogenesis. In: Banerjee R (ed) Chemistry and biochemistry of B12. Wiley, New York, pp. 655–679
Schrauzer GN, Deutsch E (1969) Reactions of cobalt(I) supernucleophiles. The alkylation of vitamin B12s cobaloximes(I), and related compounds. J Am Chem Soc 91:3341–3350
Scott AI, Roessner CA, Santander PJ (2003) Genetic and mechanistic exploration of the two pathways of vitamin B12 biosynthesis. In: Kadish KM, Smith KM Guilard R (eds) Porphyrin handbook. Elsevier Science, Amsterdam, pp. 211–228
Shibata N, Masuda J, Tobimatsu T, Toraya T, Suto K, Morimoto Y, Yasuoka N (1999) A new mode of B12 binding and the direct participation of a potassium ion in enzyme catalysis: x-ray structure of diol dehydratase. Structure 7:997–1008
Siebert A, Neumann A, Schubert T, Diekert G (2002) A non-dechlorinating strain of Dehalospirillum multivorans: evidence for a key role of the corrinoid cofactor in the synthesis of an active tetrachloroethene dehalogenase. Arch Microbiol 178:443–449
Smith DM, Golding BT, Radom L (1999) Facilitation of enzyme-catalyzed reactions by partial proton transfer: application to coenzyme-B12-dependent methylmalonyl-CoA mutase. J Am Chem Soc 121:1383–1384
Smith EL, Parker LFJ (1948) Purification of anti-pernicious anaemia factor. Biochem J 43:R8–R9
Stich TA, Yamanishi M, Banerjee R, Brunold TC (2005) Spectroscopic evidence for the formation of a four-coordinate Co2 + cobalamin species upon binding to the human ATP: Cobalamin adenosyltransferase. J Am Chem Soc 127:7660–7661
Stubbe J (2000) Ribonucleotide reductases: the link between an RNA and a DNA world? Curr Opin Struct Biol 10:731–736
Stubbe J, Licht S, Gerfen G, Silva D, Booker S (1998) Adenosylcobalamin-dependent ribonucleotide reductases: still amazing but no longer confusing. In: Kräutler B, Arigoni D, Golding BT (eds) Vitamin B12 and B12-proteins. Wiley-VCH, Weinheim, pp. 321–331
Stupperich E, Eisinger HJ, Albracht SPJ (1990) Evidence for a super-reduced cobamide as the major corrinoid fraction in vivo and a histidine residue as a cobalt ligand of the p-cresolyl cobamide in the acetogenic bacterium Sporomusa ovata. Eur J Biochem 193:105–109
Stupperich E, Konle R, Lehle M (1998) Corrinoid-dependent methyl transfer reactions in Sporomusa ovata. In: Kräutler B, Arigoni, D, Golding BT (eds) Vitamin B12 and B12 proteins. Wiley VCH, Weinheim, pp. 179–187
Summers MF, Marzilli LG, Bax A (1986) Complete 1H and 13C assignments of coenzyme-B12 through the use of new two-dimensional NMR experiments. J Am Chem Soc 108:4285–4294
Tollinger M, Eichmüller C, Konrat R, Huhta MS, Marsh ENG, Kräutler B (2001) The B12-binding subunit of glutamate mutase from Clostridium tetanomorphum traps the nucleotide moiety of coenzyme B12. J Mol Biol 309:777–791
Tollinger M, Konrat R, Hilbert BH, Marsh ENG, Kräutler B (1998) How a protein prepares for B12 binding: structure and dynamics of the B12-binding subunit of glutamate mutase from Clostridium tetanomorphum. Structure 6:1021–1033
Toraya T (2003) Radical catalysis in coenzyme B12-dependent isomerization (eliminating) reactions. Chem Rev 103:2095–2127
Yamada K, Gravel, RA, Toraya T, Matthews RG (2006) Human methionine synthase reductase is a molecular chaperone for human methionine synthase. Proc Natl Acad Sci USA 103:9476–9481
Yamanishi M, Vlasie M, Banerjee R (2005) Adenosyltransferase: an enzyme and an escort for coenzyme B12? Trends Biochem Sci 30:304–308
Zerbe-Burkhardt K, Ratnatilleke A, Vrijbloed JW, Robinson JA (1999) Isobutyryl-CoA mutase. In: Banerjee R (ed) Chemistry and biochemistry of B12. Wiley, New York, pp. 859–870
Acknowledgements
This work was supported by the Austrian National Science Foundation (project No P13595) and by the European Commission (project No HPRN-CT-2002-00195).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Kräutler, B. (2012). Biochemistry of B12-Cofactors in Human Metabolism. In: Stanger, O. (eds) Water Soluble Vitamins. Subcellular Biochemistry, vol 56. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-2199-9_17
Download citation
DOI: https://doi.org/10.1007/978-94-007-2199-9_17
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-2198-2
Online ISBN: 978-94-007-2199-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)