
Abstract
Cobalamin (Cbl, vitamin B12) consists of a corrinoid structure with cobalt in the centre of the molecule. Neither humans nor animals are able to synthesize this vitamin. Foods of animal source are the only natural source of cobalamin in human diet. There are only two enzymatic reactions in mammalian cells that require cobalamin as cofactor. Methylcobolamin is a cofactor for methionine synthase. The enzyme methylmalonyl-CoA-mutase requires adenosylcobalamin as a cofactor. Therefore, serum concentrations of homocysteine (tHcy) and methylmalonic acid (MMA) will increase in cobalamin deficiency. The cobalamin absorption from diet is a complex process that involves different proteins: haptocorrin, intrinsic factor and transcobalamin (TC). Cobalamin that is bound to TC is called holotranscobalamin (holoTC) which is the metabolically active vitamin B12 fraction. HoloTC consists 6 and 20% of total cobalamin whereas 80% of total serum cobalamin is bound to another binding protein, haptocorrin. Cobalamin deficiency is common worldwide. Cobalamin malabsorption is common in elderly subjects which might explain low vitamin status. Subjects who ingest low amount of cobalamin like vegetarians develop vitamin deficiency. No single parameter can be used to diagnose cobalamin deficiency. Total serum cobalamin is neither sensitive nor it is specific for cobalamin deficiency. This might explain why many deficient subjects would be overlooked by utilizing total cobalamin as status marker. Concentration of holotranscobalamin (holoTC) in serum is an earlier marker that becomes decreased before total serum cobalamin. Concentrations of MMA and tHcy increase in blood of cobalamin deficient subjects. Despite limitations of these markers in patients with renal dysfunction, concentrations of MMA and tHcy are useful functional markers of cobalamin status. The combined use of holoTC and MMA assays may better indicate cobalamin status than either of them. Because Cbl deficiency is a risk factor for neurodegenerative diseases an early diagnosis of a low B12 status is required which should be followed by an effective treatment in order to prevent irreversible damages.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
16.1 Cobalamin and Its Metabolism
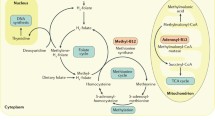
The discovery of cobalamin (Cbl; vitamin B12) was after several years of intensive studies on pernicious anemia, a previously fatal disease. In the early 1920s Minot and Murphy demonstrated that they were able to cure pernicious anaemia by whole liver extract. Later, it was shown that liver is an important source of cobalamin. Cobalamin was then isolated and the crystallized molecule was identified. Cobalamin belongs to a group of compounds of similar chemical structure but completely different biological functions. Cbl consists of a corrinoid molecule with cobalt in the centre of this molecule. The synthetic forms of cobalamin are cyanocobalamin and hydroxycobalamin. There are only two forms of Cbl that have biological activity as cofactors in enzyme reactions (Fig. 16.1). These are adenosylcobalamin (AdoCbl) and methylcobalamin (MeCbl) (Herzlich and Herbert 1988). In mammalian metabolism, Cbl is required for only two key enzymatic reactions. The first reaction occurs in the cytosol and involves synthesis of methionine from homocysteine. Methionine synthase and its cofactor MeCbl catalyze this reaction. The second pathway takes place in the mitochondria and involves isomerisation of methylmalonyl-CoA to succinyl-CoA. This reaction is catalyzed by methylmalonyl-CoA mutase with AdoCbl as a cofactor (Stroinsky and Schneider 1987). The latter reaction is part of the catabolism of odd-chained fatty acids, cholesterol and several amino acids. The excess of methymalonyl-CoA is converted into methylmalonic acid (MMA). Therefore, cobalamin deficiency leads to MMA elevation that is considered a sensitive marker for this deficiency. A reduced flux through the methylmalonyl-CoA mutase reaction is discussed to contribute to subsequent neurological tissue damage.

The metabolic pathways enhanced by cobalamin and inherited disorders of cobalamin (see text for details). Hcy; homocysteine, Met; methionine, SAH; S-adenosylhomocysteine, SAM; S-adenosylmethionin, methyl-Cbl; methyl cobalamin, Ado-Cbl; adenosyl cobalamin
16.1.1 Sources of Cobalamin
Cobalamin synthesis is very complex and restricted to a certain stain of bacteria. Neither humans, nor animals are able to synthesize this complex molecule. Plants and fungi are thought to neither synthesize, nor to use the vitamin as well. Animals can get cobalamin by consuming foods contaminated with synthesizing bacteria and then incorporate the vitamin into their body organs. Foods of animal source are the only natural source of cobalamin in human diet. The main sources of cobalamin in humans diet are meet (2–5 μg/100 g), fish (2–8 μg/100 g), milk (1.5 μg/100 ml), cheese (1–2 μg/100 g) and eggs (2 μg/100 g) (Scott 1997). Cobalamin is essential for normal maturation and development of all DNA synthesizing cells including blood cells and cells of the central nervous system. The ultimate source of cobalamin in human diet is from foods contaminated with B12-synthesizing bacteria. Not all forms of cobalamin formed by microbes are metabolically active for mammalian cells. Some naturally occurring forms of corrinoids have similar structure but no biological roles in humans. These are produced by some algae (spirulina) and are termed analogous. Cobalamin-analogous may even block the normal metabolism of the vitamin (Herbert and Drivas 1982).
16.1.2 Absorption, Excretion and Haemostasis of Cobalamin
Cobalamin is bound to food proteins and must be released before that the vitamin can be absorbed. This is achieved by the action of gastric acid and proteolytic enzymes in the stomach. In the stomach cobalamin is captured by haptocorrin, an R-binder protein made in the saliva and stomach. In the upper small intestine, pancreatic enzymes and an alkaline pH degrade the haptocorrin-cobalamin complex. The free vitamin is then captured by intrinsic factor, another B12-binding protein. The intrinsic factor-cobalamin complex is transported to the terminal ileum where the complex is recognised and internalised by specific membrane receptors of the enterocytes (intrinsic factor receptor). The receptor-mediated absorption of cobalamin is a saturable process and a maximal amount of 3 μg of the vitamin per meal can be internalized via this pathway. After absorption of cobalamin-intrinsic factor complex into the enterocytes, the complex is degraded and cobalamin is transferred to a third binding protein, transcobalamin (TC).
Transcobalamin is synthesized within the enterocytes and is the only binder that can deliver cobalamin into cells via TC-receptor. The TC-cobalamin complex is released into the portal circulation and is subsequently recognised by TC-receptors that are expressed by all cell types. The part of cobalamin which is bound to TC is named holotranscobalamin (holoTC) (Carmel 1985). Only 6–20% of total plasma cobalamin is present as holoTC (Hall 1977). The remaining part of cobalamin is bound to haptocorrin and is called holohaptocorrin (holoHC) (England et al. 1976). Despite that haptocorrin binds almost 80% of total plasma cobalamin, the functions of this protein are not well investigated.
The bioavailability of cobalamin from vitamin preparations is greater than that from foods. In contrast to the receptor mediated absorption, about 1% of free cobalamin is absorbed by passive diffusion even in the absence of intrinsic factor. Therefore, even patients with pernicious anemia, or those with disturbed gastrointestinal pH or atrophic gastritis may benefit from relatively high doses of oral cobalamin treatment. Furthermore, a considerable amount of cobalamin is secreted into the bile. Two thirds of the secreted cobalamin in the bile is reabsorbed in the ileum. The liver contains most of the body’s cobalamin. It is calculated that 2–3 mg of cobalamin are stored in the liver (Markle 1996). The kidney and the brain are also two important organs that accumulate cobalamin. The kidney can release cobalamin in case of short term depletion of the vitamin. Cobalamin is excreted into urine and this can be reabsorbed in the proximal tubules via a specific receptor (megalin) (Moestrup et al. 1996). The major route by which cobalamin is lost from the body is through the feces.
16.2 Cobalamin Deficiency
Frank cobalamin deficiency is common worldwide (Morris et al. 2002; Obeid et al. 2002). In developed countries, cobalamin deficiency is restricted to patients with malabsorption, intestinal resection, or those who do not ingest a sufficient amount of the vitamin (Herrmann et al. 2003; Carmel 1997). Cobalamin deficiency is common in elderly people and in those who ingest a strict vegetarian diet. In contrast, cobalamin deficiency has been reported in children, middle age, and elderly people from developing countries.
Pre-clinical cobalamin deficiency is the state in which metabolic evidence of insufficiency exists without symptoms of anemia or neurological complications. Measurement of serum concentrations of metabolic markers allow detection of a large number of asymptomatic subjects who are depleted of the vitamin.
16.2.1 Cobalamin Deficiency in the Elderly
16.2.1.1 Factors Contributing to Low Vitamin Status with Aging
Cobalamin deficiency in geriatric population has gained a particular importance in recent years (Baik and Russell 1999; Obeid et al. 2004). Serum concentration of cobalamin decreases with age and that of MMA and tHcy increases. On the one hand, the decline in cobalamin status with age could not be explained by a low dietary intake of the vitamin. On the other hand, it is well recognized that low cobalamin status in the elderly is associated with an increased incidence of pernicious anemia (type A atrophic gastritis), and type B atrophic gastritis. In western countries, approximately 2% to 3% of free living elderly people (>60 years) had undiagnosed pernicious anemia (Carmel 1996; Krasinski et al. 1986). Ethnic differences in the incidence of pernicious anemia and the age of onset have been reported (Carmel and Johnson 1978).
16.2.2 Causes of Cobalamin Deficiency
16.2.2.1 Inherited Defects
The most important inherited disorders of cobalamin absorption, transport, metabolism, or utilization are presented in Table 16.1.
Congenital transcobalamin deficiency: This congenital recessive defect causes a lack of the binding protein, TC. Several cases have been described in the literature. Although babies are born asymptomatic, severe haematological and neurological manifestations are expressed few days to weeks after birth. This case is characterized by irritation, failure to thrive, and severe elevation of methylmalonic acid in blood and urine of affected children. Skin fibroblasts or lymphocytes isolated from affected children can not take up cobalamin, in contrast to cells isolated from healthy subjects. In most cases, high doses of cobalamin can improve clinical symptoms (Bibi et al. 1999; Teplitsky et al. 2003; Cooper and Rosenblatt 1987; Hakami et al. 1971). Abnormal transcobalamin has also been reported and is associated with similar symptoms.
Imerslund-Grasbeck-syndrom: This disorder is caused by a defective transport of cobalamin by enterocytes. This disease is characterized by proteinuria and lose of cobalamin in urine (Ben-Ami et al. 1990). Cobalamin malabsorption in this case is not related to intrinsic factor deficiency or to an abnormal cobalain-intrinsic factor complex. The cause behind cobalamin malabsorption and lose is related to a defective cubulin, the receptor for IF-cobalamin that is expressed in the enterocytes and the renal tubulus.
Disorders of cobalamin utilization: There are several inherited defects in cobalamin utilization. These are summarized in Fig. 16.1. Methylmalonic aciduria is related to a functional defect or a deficiency of the mithochondrial enzyme, methylmalonyl CoA mutase. This defect is characterized by severe elevation of blood and urine concentrations of MMA. Cultures of patients fibroblasts have shown that some patients do not response to cobalamin treatment, and others have a residual enzyme activity and response to cobalamin treatment. Cobalamin mutant A and B are related to adenosylcobalamin deficiency and cause methylmalonic aciduria. Cobalamin A defect is related to a failure in a reduction step and the second disorder is caused by a failure to transfer adenosyl to cobalamin.
Cobalamin mutant C, D, and F diseases are associated with combined adenosylcobalamin and methylcobalain deficiencies. Therefore, patients have usually, homocysteinuria, hypomethioninemia, and methylmalonic aciduria. The C and D defects involve a defective reductase step; whereas the F mutant involves inability of releasing cobalamin from the lysosome. The last two disorders in cobalamin utilization are, cobalamin mutant E and G that are related to methylcobalamin deficiency.
Haptocorrin deficiency (deficiency of R binder): This case has been recently reported and is associated with a very low total serum cobalamin. Nevertheless, subjects who express this phenotype remain asymptomatic and concentrations of holotranscobalain remain within the normal range (Lin et al. 2001).
16.2.2.2 Acquired Causes of Cobalamin Deficiency
Food cobalamin malabsorption: Food cobalamin malabsorption is the most common cause of cobalamin deficiency in elderly people. There are several diseases that may cause cobalamin malabsorption. Pernicious anemia (anti IF antibodies) is the most famous disorder of cobalamin absorption. In this case, cobalamin can not be absorbed because of the lack of IF. Antiparietal cell antibodies can cause lack of intrinsic factor and thus cobalamin deficiency.
Type B chronic atrophic gastritis is related to Helicobacter pylori infection and is described to cause cobalamin malabsorption. This disorder results in a low acid-pepsin production and food cobalamin malabsorption. The release of cobalamin from food protein is decreased in case of lowered gastric acidity (Doscherholmen et al. 1977). Importantly, because the production of IF is not affected in Type B atrophic gastritis, those subjects may benefit from crystalline cobalamin. Oral cobalamin therapy (3–5 mg/week) was effective in treatment of food-cobalamin malabsorption (Andres et al. 2001, 2003).
Celiac disease and tropical spree are also associated with cobalamin deficiency. In both cases, recurrent diarrhea causes severe damage to the gastrointestinal tract and interferes with cobalamin absorption by the enterocytes. Importantly, several drugs are known to alter gastrointestinal pH thus causing cobalamin malabsorption. Table 16.2 summarizes the most important acquired causes of cobalamin deficiency.
Cobalamin deficiency related to a low intake of the vitamin: The Recommended Dietary Intake (RDI) of cobalamin for adults is 2.4 μg/day (Institute of Medicine 2000). Recent studies indicated that a total daily intake of 6 μg/day can be necessary for preventing metabolic signs of deficiency in older women (Bor et al. 2006). Chronic low intake of cobalamin can cause severe deficiency. Cobalamin deficiency takes years to develop, even when one stops to ingest the vitamin. This is related to the relatively large body stores of cobalamin, in addition to the effective enterohepatic circulation that ensures reabsorption of the vitamin. Subjects who adhere to a life long strict vegetarian diet develop cobalamin deficiency (Herrmann et al. 2003; Herbert 1994). Studies have shown that the severity of cobalamin deficiency is related to the degree of animal food restriction and to the duration of the vegetarian lifestyle (Herrmann et al. 2003; Herbert 1994). Because milk and eggs contain a low amount of cobalamin, the intake of cobalamin in a lacto- and lacto-ovo-vegetarian diet might be sufficient. Nevertheless, studies have shown that cobalamin status (indicated by MMA, tHcy, holoTC) was lower in lacto- and lacto-ovo-vegetarians compared with omnivorous subjects (Herrmann et al. 2003). In addition, metabolic signs of cobalamin deficiency were more common in vegans compared with lacto- and lacto-ovo-vegetarians (Herrmann et al. 2003). Chronic low intake of cobalamin and its deficiency may be endemic in developing countries and is related to poverty and malnutrition in this case (Refsum et al. 2001).
The RDI for cobalamin is higher for pregnant and lactating women. Cobalamin deficiency may occur in women who do not ingest a sufficient amount of the vitamin. Metabolic and clinical signs of cobalamin deficiency have been reported in newborn babies from strict vegetarian mothers or in breast fed infants from deficient mothers (Bjorke et al. 2001; Schneede et al. 1994; Graham et al. 1992; Dagnelie et al. 1989). The content of cobalamin in mother milk was related to markers of cobalamin deficiency in the infants. These conditions were partly reversible after cobalamin supplementation. In a study that included adolescents who were fed a macrobiotic diet until the age of 6 years, metabolic signs of cobalamin deficiency were observed even several years after ingesting an omnivorous diet (Dhonukshe-Rutten et al. 2005). These data show that metabolic signs of cobalamin deficiency can not be resolved after mild modifications of the diet. Moreover, clinical signs might be irreversible.
16.2.3 Diagnosis of Cobalamin Malabsorption
Schilling test was the first test described for diagnosis of cobalamin malabsorption. This test depends on using an oral dose of radioactively-labeled cobalamin and following the radioactivity in urine or blood of patients. Schilling test can be applied with or without IF. Furthermore, the test has been improved to address the issue of malabsorption of protein-bound cobalamin (Nickoloff 1988; Doscherholmen et al. 1983). The use of radio-labeled cobalamin has limited the use of Schilling test in recent years.
Other approaches have been suggested recently to assess cobalamin absorption. One approach is to measure the response of holoTC (cobalamin saturated part) after a small oral dose of cobalamin (Bor et al. 2005). This test might be also applied with recombinant human IF. Preliminary results indicated that recombinant human IF promotes B12 absorption among patients with evident cobalamin deficiency (Hvas et al. 2001, 2006). Although early results are encouraging, studies that include patients with malabsorption disorders should be conducted before setting this test to the routine use.
16.2.4 Diagnosis of Cobalamin Deficiency
16.2.4.1 Determination of Total Serum Cobalamin
Despite its serious limitations, total serum cobalamin is the first parameter that is measured when cobalamin deficiency is suspected. As mentioned above, 80% of total cobalamin is bound to haptocorrin. This part is metabolically inert and can not be delivered into the cells. Therefore, minor changes in cobalamin status in the blood can not be reflected by concentrations of haptocorrin. Total serum cobalamin has several major limitations. On the one hand, low serum concentrations of cobalamin were not observed in all subjects with clinically evident cobalamin deficiency (false negative). On the other hand, there are many subjects with low serum cobalamin who are not deficient (false positive). Therefore, the low sensitivity and specificity of total cobalamin should be considered, particularly when serum cobalamin concentrations range between 150 and 350 pmol/L (Green 1995; Herrmann et al. 2000, 2001a). Approximately 45% of cobalamin-deficient subjects (MMA > 300 nmol/L and holoTC < 40 pmol/L) would have been overlooked, using cobalamin as a screening parameter. Obviously, the insensitivity of the total cobalamin test can not be resolved by simply shifting the cut-off value towards a higher level, because more subjects with normal B12 status will be falsely diagnosed as deficient (Fig. 16.2). Because of its low costs compared to other available markers, cobalamin assay can be used as a first line parameter. Nevertheless, the results should be interpreted individually.

Distribution of total serum cobalamin in deficient (upper plot) and non deficient (lower plot) subjects. cobalamin deficiency was defined as holoTC < 40 pmol/L and MMA ≥ 300 nmol/L
16.2.4.2 Determination of Holotranscobalamin
HoloTC represents the biologically active cobalamin fraction. Recent studies indicated that holoTC might be the earliest marker that indicate cobalamin status in blood (Bor et al. 2004).
Currently, the estimation of holoTC has been made possible by several methods (Ulleland et al. 2002; Refsum et al. 2006). Studies on cobalamin-deficient and non-deficient subjects have confirmed that holoTC has a better specificity and sensitivity compared with total B12 (Herrmann et al. 2005).
Limitations of holoTC should be also considered when interpreting the results of the test. Oral contraceptives have been reported to be associated with a 25% lower concentrations of holoTC without affecting serum concentrations of tHcy or MMA (Riedel et al. 2005). Moreover, renal insufficiency is associated with increased concentrations of holoTC in blood (Herrmann et al. 2005; Carmel et al. 2001). This metabolic condition is also associated with higher concentrations of MMA. Therefore, normal or elevated concentrations of holoTC in renal patients or in elderly people with mild renal insufficiency might not exclude cobalamin deficiency.
16.2.4.3 Metabolic Markers of Cobalamin Deficiency
The most important markers for cobalamin status are summarized in Table 16.3.
Plasma concentrations of methylmalonic acid (MMA): Because cobalamin is a cofactor for methylmalonyl-CoA mutase, cobalamin deficiency causes accumulation of methylmalonyl-CoA. The last compound is converted into methylmalonic acid (MMA). Serum concentration of MMA increase in case of cobalamin deficiency and thus MMA is considered a functional marker for cobalamin status. Concentrations of MMA (> 300 nmol/L) are more sensitive and specific for cobalamin deficiency than total serum cobalamin.
Limitations of this metabolic marker should be also kept in mind. Concentrations of MMA increase in renal dysfunction. Even sub-clinical decline in renal function (i.e., common in elderly people) may cause artificial increase in concentrations of MMA (Herrmann et al. 2001a). Intestinal bacterial overgrowth may also cause artificial elevation of MMA because of increased production of propionic acid, a precursor of MMA. Nevertheless, in these cases cobalamin deficiency can not be confirmed or excluded. A significant decrease in concentrations of MMA by cobalamin treatment indicates a pre-clinical deficiency.
Plasma concentrations of tHcy: Concentrations of tHcy increase in cobalamin deficiency because of the role of methylcobalamin as a cofactor for methionine synthase that converts tHcy into methionine. Nevertheless, concentrations of tHcy increase also in folate and vitamin B6 deficiency or renal insufficiency (Herrmann et al. 2001b). Folate deficiency has more impact on tHcy compared with cobalamin deficiency. Unlike MMA elevation, tHcy elevation is not common in B12-deficient vegetarians, because of relatively high folate concentrations found in those subjects. High folate status partly compensates for the effect of cobalamin deficiency on tHcy elevation.
Figure 16.3 shows a diagnostic algorithm suggested for diagnosis of cobalamin deficiency.

Suggested algorithm for diagnosing cobalamin deficiency
16.2.4.4 Staging of Cobalamin Deficiency
Cobalamin deficiency is thought to be developed in four stages (Herbert 1994). In the early stages (I and II), the plasma and cell stores become depleted of the vitamin. This is reflected by a low plasma concentration of holoTC (Herbert et al. 1990; Lindgren et al. 1999). If the negative balance continues (stage III), metabolic markers of cobalamin deficiency become elevated (tHcy and MMA). This is explained by impairment of the B12-dependent enzymes (methionine synthase and methylmalonyl-CoA mutase). Clinical signs of cobalamin deficiency become obvious in stage IV (macrocytic anemia, neurological symptoms).
16.2.4.5 Diagnosis of Cobalamin Deficiency Utilizing Available Markers
There is no single parameter that can be reliably used to diagnose cobalamin deficiency. Megaloblastic anemia and neurological symptoms are neither sensitive nor they are specific for cobalamin deficiency. Metabolic signs (elevated MMA and tHcy) of deficiency were observed in the absence of haematological or clinical manifestations. Concentrations of total cobalamin in serum are also too insensitive (Herrmann et al. 2000, 2001b). Measurement of serum concentration of MMA alone or in conjunction with tHcy has partly resolved the demand for a sensitive and a specific test. In general, results of the metabolites should be interpreted with caution, because it is difficult to determine to which extent the impaired kidney function may participate in MMA and tHcy elevation. Subjects with serum cobalamin concentrations up to 350 pmol/L may have increased MMA and low holoTC indicating functional cobalamin deficiency. Studies on vegetarian subjects have shown that the majority of them had low holoTC in addition to metabolic signs indicating cobalamin deficiency (stage III) (elevated tHcy and MMA) (Herrmann et al. 2003).
16.2.4.6 Cobalamin Deficiency Causes Folate Trap
Because of the role of cobalamin in folate metabolism, cobalamin deficiency can cause a secondary folate deficiency. Cobalamin deficiency inhibits the activity of methionine synthase, and causes the retention of 5-MTHF. 5-MTHF becomes trapped because the transfer of the methyl group is inhibited (Fig. 16.1). The level of folate in serum or plasma of cobalamin deficient subjects may be normal to high normal, however, this is mostly as 5-MTHF. This phenomenon is called “folate trap”. Folate and cobalamin deficiency have similar clinical (megaloblastic red cells) and metabolic (elevation of tHcy) signs. This explains why cobalamin deficient subjects were frequently treated with folate. This treatment may even relief the hematological symptoms. Nevertheless, neurological signs of cobalamin deficiency may be worsen. Figure 16.4 shows this phenomenon in strict vegetarians compared with omnivorous subjects.

Scatter plot showing concentrations of tHcy in relation to serum folate in omnivorous (left plot) and vegetarians (right plot). Numbers on the axis are anti-log
16.3 Cobalamin Deficiency in Renal Patients
Cobalamin deficiency is common in renal patients. Nevertheless, diagnosing cobalamin deficiency in renal patients remains a challenge. Renal insufficiency and hypovolemia cause elevation of MMA and tHcy (Herrmann et al. 2001b; Lindgren et al. 1999; Savage et al. 1994). Unexpectedly low serum concentrations of cobalamin or holoTC are uncommon in patients with renal insufficiency (Herrmann et al. 2005). A very important finding is that renal patients show significant metabolic improvements (reduction of MMA) after treatment with cobalamin (Obeid et al. 2005). The reasons for elevated serum cobalamins (total B12 and holoTC) in renal patients are not known. An abnormal distribution of holoTC (Carmel et al. 2001), a disturbed receptor activity for renal TC uptake and the possibility that TC is functionally altered by renal failure are possible explanations for accumulation of cobalamin in serum of renal patients.
16.3.1 Cobalamin Status and Vegetarian Diet
Cobalamin status is directly correlated with dietary intake and length of time following a vegetarian diet (Chanarin et al. 1985; Miller et al. 1991). According to the strictness of the diet, approximately 30–60% of vegetarian subjects might have metabolic evidence indicating cobalamin deficiency (Donaldson 2000; Rauma et al. 1995). Serum concentrations of MMA, holoTC, tHcy and total cobalamin are related to the type of the diet (Table 16.4). Vegan subjects had the lowest cobalamin status. This is expected, because a vegan diet includes no kind animal foods. Although lacto- and lactoovo-vegetarians consume some animal foods (egg, milk, and milk products), metabolic signs of cobalamin deficiency were also common in this group (Herrmann et al. 2003) (Table 16.4). This group of vegetarians show intermediate cobalamin status compared with vegans and omnivorous (Herrmann et al. 2003). Approximately, 45% of subjects with low holoTC and elevated MMA had normal serum B12 (Fig. 16.2). It should be noticed that low doses of cobalamin such as the one usually found in the multivitamin preparations are not likely to prevent the depletion of the vitamin in individuals who ingest little or no animal products.
16.4 Cobalamin Status and Markers in Elderly People
Cobalamin deficiency is common in elderly people. A low dietary intake of cobalamin was not common in cobalamin deficient-elderly people. Nevertheless, several age-related physiological factors may influence negatively the absorption of the vitamin from the intestine. Cobalamin deficiency is associated with a poor cognitive performance and neurological damage. Approximately, 20% of elderly people were found to have a low concentration of holoTC and an elevated MMA indicating a relatively late stage of B12 deficiency. Importantly, a relatively large proportion of elderly subjects were found to have an elevated concentration of MMA and a normal holoTC, in addition to a higher concentration of serum creatinine. This typical profile associated with impaired renal function may delay the diagnosis of cobalamin deficiency. The relation between holoTC and MMA has been examined in relation to age and renal function (Obeid et al. 2004). In general, elderly people display higher MMA concentrations in ranges of holoTC comparable to that in younger subjects. This phenomenon was more pronounced in elderly with renal insufficiency. The last findings suggest that cellular-B12-delivery may be challenged in elderly with renal insufficiency, leading to MMA increment and holoTC retention.
Available evidence suggests that elderly people seem to be resistant to low oral doses of cobalamin (Rajan et al. 2002). Serum concentrations of MMA were not normalized in most B12-deficienct elderly people who received crystalline B12 in doses between 25 and 100 μg. The RDI of B12 for older adults (2.4 μg/day) is far below the dose likely to normalize serum concentrations of the metabolites in elderly people. Taken together, a low cobalamin status in elderly people with renal insufficiency may be overlooked by using holoTC as a sole marker. Moreover, the occurrence of cobalamin deficiency may be overestimated using MMA test. The presence of a low concentration of holoTC and an elevated MMA in elderly people may confirm B12 deficiency and the absence of this combined results may not exclude a deficiency of this vitamin. A significant reduction of MMA after cobalamin treatment might indicate a pre-treatment low status of the vitamin.
Cobalamin deficiency in elderly people was also associated with increased plasma SAH. This marker of methylation status was lowered by high oral dose of cobalamin (1 mg/day for 3 months) (Stabler et al. 1996, 2006).
16.5 Clinical Manifestations of Cobalamin Deficiency
16.5.1 Hematological Symptoms
Megaloblastic anemia is the classical finding of cobalamin deficiency. The underling mechanism of anemia in cobalamin deficiency is a slowed DNA synthesis in rapidly dividing blood cell. This will lead to large red cells (increased mean corpuscular volume, MCV). Nevertheless, megaloblastic anemia is neither sensitive, nor it is specific for cobalamin deficiency. Recent studies have shown that many cobalamin deficient subjects may develop neurological disorders without macrocytosis of red blood cells. Furthermore, because cobalamin and iron deficiencies might coincide in many cases, depending on MCV to diagnose cobalamin deficiency is not reliable. Microcytic anemia of iron deficiency can mask macrocytic anemia of cobalamin deficiency. The result is hypochromic red blood cells of normal size. In addition, the hematological manifestations of cobalamin deficiency can be mistaken by that of folate deficiency. Hypersegmented neutrophils are also common in cobalamin deficient subjects. Nevertheless, in general haematological indices are not reliable in diagnosing early stages of cobalamin deficiency.
16.5.2 Neurological Complications
Concentrations of tHcy > 12.0 μmol/L and that of MMA > 271 nmol/L are common in neuro-psychiatric population even in the absence of haematological manifestations (Lindenbaum et al. 1988). Cobalamin deficiency can cause lesions in spinal cord, peripheral nerves, and cerebrum and improvements have been reported after initiation of vitamin treatment (Masalha et al. 2001; Lorenzl et al. 2003). The most common symptoms are sensory disturbances in the extremities, memory loss, dementia, psychosis.
The association between cobalamin deficiency and depression has been documented in elderly women (Penninx et al. 2000). Metabolically significant cobalamin deficiency was present in approximately 15% of non-depressed women, 17% of mildly depressed women and in 27% of the severely depressed women (Penninx et al. 2000). Women with cobalamin deficiency were 2.05 times as likely to be severely depressed as were non-deficient women. Moreover, peripheral neuropathy occurred in 40% of cobalamin-deficient subjects (Shorvon et al. 1980).
Cobalamin deficiency can cause serious neurological symptoms in infants (Graham et al. 1992; Higginbottom et al. 1978; Kuhne et al. 1991). The most common cause of vitamin deficiency at early age is being born or lactated by a vitamin deficient mother. A serious neurological syndrome and developmental disorders have been described in few exclusively breast-fed infants of strict vegetarian mothers who were cobalamin deficient (Graham et al. 1992; Higginbottom et al. 1978; Kuhne et al. 1991). Vitamin B12 deficiency in infants is associated with a marked developmental regression, a poor brain growth or a poor intellectual outcome. Other signs include impaired communicative reactions and fine and gross motor functions. Low vitamin B12 status had also negative influence on school achievement in schoolchildren.
Prolonged insufficient intake of the vitamin is also a common case of cobalamin deficiency in children. Adolescences previously consumed a macrobiotic diet had lower scores in some measures of cognitive performance as compared to adolescences consumed omnivorous diet from birth onward (Schneede et al. 1994; Dagnelie et al. 1989).
Available studies emphasize the need for early recognition and prevention or treatment of B-vitamins deficiency in children. Although, large scale screening programs are currently not recommended (Refsum et al. 2004), cases suspected for cobalamin deficiency should be individually judged. Cobalamin deficiency should be suspected in children with unexplained neurological symptoms, failure to thrive, or poor intellectual performance. A great attention should be paid to familial factors (maternal vitamin status), and predisposing environmental factors (poverty, vegetarian diet).
16.6 Treatment of Cobalamin Deficiency
A typical western diet provides an average intake of 2–6 μg/day of B12. An additional source of the vitamin is highly recommended for subjects with malabsorption or a low intake of the vitamin. Most elderly people who have already developed metabolic signs indicating cobalamin deficiency (i.e., elevated MMA) require more than 100 μg of oral B12 to normalize serum MMA, which is a larger dose than is available in most standard multivitamins and cobalamin supplements.
Folate treatment for cobalamin deficient subject may delay the diagnosis of B12 deficiency and cause irreversible neurological symptoms. Therefore, addition of cobalamin to folate treatment is strongly recommended. Doses and duration of vitamin supplementation should be individually determined, because malabsorption is a major limiting factor for the bioavailability of the vitamins in elderly people. Table 16.5 shows a few examples on cobalamin doses and responses to treatment in different clinical conditions.
References
Andres E, Kaltenbach G, Noel E, Noblet-Dick M, Perrin AE, Vogel T et al. (2003) Efficacy of short-term oral cobalamin therapy for the treatment of cobalamin deficiencies related to food-cobalamin malabsorption: a study of 30 patients. Clin Lab Haematol 25:161–166
Andres E, Kurtz JE, Perrin AE, Maloisel F, Demangeat C, Goichot B, Schlienger JL (2001) Oral cobalamin therapy for the treatment of patients with food-cobalamin malabsorption. Am J Med 111:126–129
Baik HW, Russell RM (1999) Vitamin B12 deficiency in the elderly. Annu Rev Nutr 19:357–377
Ben-Ami M, Katzuni E, Koren A (1990) Imerslund syndrome with dolichocephaly. Pediatr Hematol Oncol 7:177–181
Bibi H, Gelman-Kohan Z, Baumgartner ER, Rosenblatt DS (1999) Transcobalamin II deficiency with methylmalonic aciduria in three sisters. J Inherit Metab Dis 22:765–772
Bjorke Monsen AL, Ueland PM, Vollset SE, Guttormsen AB, Markestad T, Solheim E, Refsum H (2001) Determinants of cobalamin status in newborns. Pediatrics 108:624–630
Bolann BJ, Solli JD, Schneede J, Grottum KA, Loraas A, Stokkeland M et al. (2000) Evaluation of indicators of cobalamin deficiency defined as cobalamin-induced reduction in increased serum methylmalonic acid. Clin Chem 46:1744–1750
Bor MV, Cetin M, Aytac S, Altay C, Nexo E (2005) Nonradioactive vitamin B12 absorption test evaluated in controls and in patients with inherited malabsorption of vitamin B12. Clin Chem 51:2151–2155
Bor MV, Lydeking-Olsen E, Moller J, Nexo E (2006) A daily intake of approximately 6 microg vitamin B-12 appears to saturate all the vitamin B-12-related variables in Danish postmenopausal women. Am J Clin Nutr 83:52–58
Bor MV, Nexo E, Hvas AM (2004) Holo-transcobalamin concentration and transcobalamin saturation reflect recent vitamin B12 absorption better than does serum vitamin B12. Clin Chem 50:1043–1049
Carmel R (1985) The distribution of endogenous cobalamin among cobalamin-binding proteins in the blood in normal and abnormal states. Am J Clin Nutr 41:713–719
Carmel R (1996) Prevalence of undiagnosed pernicious anemia in the elderly. Arch Intern Med 156:1097–1100
Carmel R (1997) Cobalamin, the stomach, and aging. Am J Clin Nutr 66:750–759
Carmel R, Johnson CS (1978) Racial patterns in pernicious anemia. Early age at onset and increased frequency of intrinsic-factor antibody in black women. N Engl J Med 298:647–650
Carmel R, Vasireddy H, Aurangzeb I, George K (2001) High serum cobalamin levels in the clinical setting – clinical associations and holo-transcobalamin changes. Clin Lab Haematol 23:365–371
Chanarin I, Malkowska V, O’Hea AM, Rinsler MG, Price AB (1985) Megaloblastic anaemia in a vegetarian Hindu community. Lancet 2:1168–1172
Cooper BA, Rosenblatt DS (1987) Inherited defects of vitamin B12 metabolism. Annu Rev Nutr 7:291–320
Dagnelie PC, van Staveren WA, Vergote FJ, Dingjan PG, van den BH, Hautvast JG (1989) Increased risk of vitamin B-12 and iron deficiency in infants on macrobiotic diets. Am J Clin Nutr 50:818–824
Dhonukshe-Rutten RA, Van DM, Schneede J, de Groot LC, van Staveren WA (2005) Low bone mineral density and bone mineral content are associated with low cobalamin status in adolescents. Eur J Nutr 44:341–347
Donaldson MS (2000) Metabolic vitamin B12 status on a mostly raw vegan diet with follow-up using tablets, nutritional yeast, or probiotic supplements. Ann Nutr Metab 44:229–234
Doscherholmen A, Ripley D, Chang S, Silvis SE (1977) Influence of age and stomach function on serum vitamin B12 concentration. Scand J Gastroenterol 12:313–319
Doscherholmen A, Silvis S, McMahon J (1983) Dual isotope Schilling test for measuring absorption of food-bound and free vitamin B12 simultaneously. Am J Clin Pathol 80:490–495
England JM, Down MC, Wise IJ, Linnell JC (1976) The transport of endogenous vitamin B12 in normal human serum. Clin Sci Mol Med 51:47–52
Graham SM, Arvela OM, Wise GA (1992) Long-term neurologic consequences of nutritional vitamin B12 deficiency in infants. J Pediatr 121:710–714
Green R (1995) Metabolite assays in cobalamin and folate deficiency. Baillieres Clin Haematol 8:533–566
Hakami N, Neiman PE, Canellos GP, Lazerson J (1971) Neonatal megaloblastic anemia due to inherited transcobalamin II deficiency in two siblings. N Engl J Med 285:1163–1170
Hall CA (1977) The carriers of native vitamin B12 in normal human serum. Clin Sci Mol Med 53:453–457
Herbert V (1994) Staging vitamin B-12 (cobalamin) status in vegetarians. Am J Clin Nutr 59:1213S–1222S
Herbert V, Drivas G (1982) Spirulina and vitamin B 12. JAMA 248:3096–3097
Herbert V, Fong W, Gulle V, Stopler T (1990) Low holotranscobalamin II is the earliest serum marker for subnormal vitamin B12 (cobalamin) absorption in patients with AIDS. Am J Hematol 34:132–139
Herrmann W, Obeid R, Schorr H, Geisel J (2005) The usefulness of holotranscobalamin in predicting vitamin B12 status in different clinical settings. Curr Drug Metab 6:47–53
Herrmann W, Schorr H, Bodis M, Knapp JP, Muller A, Stein G, Geisel J (2000) Role of homocysteine, cystathionine and methylmalonic acid measurement for diagnosis of vitamin deficiency in high-aged subjects. Eur J Clin Invest 30:1083–1089
Herrmann W, Schorr H, Geisel J, Riegel W (2001a) Homocysteine, cystathionine, methylmalonic acid and B-vitamins in patients with renal disease. Clin Chem Lab Med 39:739–746
Herrmann W, Schorr H, Obeid R, Geisel J (2003) Vitamin B-12 status, particularly holotranscobalamin II and methylmalonic acid concentrations, and hyperhomocysteinemia in vegetarians. Am J Clin Nutr 78:131–136
Herrmann W, Schorr H, Purschwitz K, Rassoul F, Richter V (2001b) Total homocysteine, vitamin B-12, and total antioxidant status in vegetarians. Clin Chem 47:1094–1101
Herzlich B, Herbert V (1988) Depletion of serum holotranscobalamin II. An early sign of negative vitamin B12 balance. Lab Invest 58:332–337
Higginbottom MC, Sweetman L, Nyhan WL (1978) A syndrome of methylmalonic aciduria, homocystinuria, megaloblastic anemia and neurologic abnormalities in a vitamin B12-deficient breast-fed infant of a strict vegetarian. N Engl J Med 299:317–323
Hvas AM, Buhl H, Laursen NB, Hesse B, Berglund L, Nexo E (2006) The effect of recombinant human intrinsic factor on the uptake of vitamin B12 in patients with evident vitamin B12 deficiency. Haematologica 91:805–808
Hvas AM, Ellegaard J, Nexo E (2001) Vitamin B12 treatment normalizes metabolic markers but has limited clinical effect: a randomized placebo-controlled study. Clin Chem 47:1396–1404
Institute of Medicine (2000) Dietary reference intakes for thiamin, riboflavin, niacin, vitamin B6, folate, vitamin B12, pantothenic acid, biotin, and choline. National Academy Press, Washington, DC, pp. 150–195
Krasinski SD, Russell RM, Samloff IM, Jacob RA, Dallal GE, McGandy RB, Hartz SC (1986) Fundic atrophic gastritis in an elderly population. Effect on hemoglobin and several serum nutritional indicators. J Am Geriatr Soc 34:800–806
Kuhne T, Bubl R, Baumgartner R (1991) Maternal vegan diet causing a serious infantile neurological disorder due to vitamin B12 deficiency. Eur J Pediatr 150:205–208
Kuzminski AM, Del Giacco EJ, Allen RH, Stabler SP, Lindenbaum J (1998) Effective treatment of cobalamin deficiency with oral cobalamin. Blood 92:1191–1198
Lewerin C, Nilsson-Ehle H, Matousek M, Lindstedt G, Steen B (2003) Reduction of plasma homocysteine and serum methylmalonate concentrations in apparently healthy elderly subjects after treatment with folic acid, vitamin B12 and vitamin B6: a randomised trial. Eur J Clin Nutr 57:1426–1436
Lin JC, Borregaard N, Liebman HA, Carmel R (2001) Deficiency of the specific granule proteins, R-binder/transcobalamin I and lactoferrin, in plasma and saliva: a new disorder. Am J Med Genet 100:145–151
Lindenbaum J, Healton EB, Savage DG, Brust JC, Garrett TJ, Podell ER et al. (1988) Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med 318:1720–1728
Lindgren A, Kilander A, Bagge E, Nexo E (1999) Holotranscobalamin – a sensitive marker of cobalamin malabsorption. Eur J Clin Invest 29:321–329
Lorenzl S, Vogeser M, Muller-Schunk S, Pfister HW (2003) Clinically and MRI documented funicular myelosis in a patient with metabolical vitamin B12 deficiency but normal vitamin B12 serum level. J Neurol 250:1010–1011
Markle HV (1996) Cobalamin. Crit Rev Clin Lab Sci 33:247–356
Masalha R, Chudakov B, Muhamad M, Rudoy I, Volkov I, Wirguin I (2001) Cobalamin-responsive psychosis as the sole manifestation of vitamin B12 deficiency. Isr Med Assoc J 3:701–703
Moestrup SK, Birn H, Fischer PB, Petersen CM, Verroust PJ, Sim RB et al. (1996) Megalin-mediated endocytosis of transcobalamin-vitamin-B12 complexes suggests a role of the receptor in vitamin-B12 homeostasis. Proc Natl Acad Sci USA 93:8612–8617
Miller DR, Specker BL, Ho ML, Norman EJ (1991) Vitamin B-12 status in a macrobiotic community. Am J Clin Nutr 53:524–529
Morris MS, Jacques PF, Rosenberg IH, Selhub J (2002) Elevated serum methylmalonic acid concentrations are common among elderly Americans. J Nutr 132:2799–2803
Nickoloff E (1988) Schilling test: physiologic basis for and use as a diagnostic test. Crit Rev Clin Lab Sci 26:263–276
Obeid R, Jouma M, Herrmann W (2002) Cobalamin status (holo-transcobalamin, methylmalonic acid) and folate as determinants of homocysteine concentration. Clin Chem 48:2064–2065
Obeid R, Kuhlmann MK, Kohler H, Herrmann W (2005) Response of homocysteine, cystathionine, and methylmalonic acid to vitamin treatment in dialysis patients. Clin Chem 51:196–201
Obeid R, Schorr H, Eckert R, Herrmann W (2004) Vitamin B12 status in the elderly as judged by available biochemical markers. Clin Chem 50:238–241
Penninx BW, Guralnik JM, Ferrucci L, Fried LP, Allen RH, Stabler SP (2000) Vitamin B(12) deficiency and depression in physically disabled older women: epidemiologic evidence from the Women’s Health and Aging Study. Am J Psychiatry 157:715–721
Rajan S, Wallace JI, Brodkin KI, Beresford SA, Allen RH, Stabler SP (2002) Response of elevated methylmalonic acid to three dose levels of oral cobalamin in older adults. J Am Geriatr Soc 50:1789–1795
Rauma AL, Torronen R, Hanninen O, Mykkanen H (1995) Vitamin B-12 status of long-term adherents of a strict uncooked vegan diet (“living food diet”) is compromised. J Nutr 125:2511–2515
Refsum H, Grindflek AW, Ueland PM, Fredriksen A, Meyer K, Ulvik A et al. (2004) Screening for serum total homocysteine in newborn children. Clin Chem 50:1769–1784
Refsum H, Johnston C, Guttormsen AB, Nexo E (2006) Holotranscobalamin and total transcobalamin in human plasma: determination, determinants, and reference values in healthy adults. Clin Chem 52:129–137
Refsum H, Yajnik CS, Gadkari M, Schneede J, Vollset SE, Orning L et al. (2001) Hyperhomocysteinemia and elevated methylmalonic acid indicate a high prevalence of cobalamin deficiency in Asian Indians. Am J Clin Nutr 74:233–241
Riedel B, Bjorke Monsen AL, Ueland PM, Schneede J (2005) Effects of oral contraceptives and hormone replacement therapy on markers of cobalamin status. Clin Chem 51:778–781
Savage DG, Lindenbaum J, Stabler SP, Allen RH (1994) Sensitivity of serum methylmalonic acid and total homocysteine determinations for diagnosing cobalamin and folate deficiencies [see comments]. Am J Med 96:239–246
Schneede J, Dagnelie PC, van Staveren WA, Vollset SE, Refsum H, Ueland PM (1994) Methylmalonic acid and homocysteine in plasma as indicators of functional cobalamin deficiency in infants on macrobiotic diets. Pediatr Res 36:194–201
Scott JM (1997) Bioavailability of vitamin B12. Eur J Clin Nutr 51(Suppl 1):S49–S53
Shorvon SD, Carney MW, Chanarin I, Reynolds EH (1980) The neuropsychiatry of megaloblastic anaemia. Br Med J 281:1036–1038
Stabler SP, Allen RH, Dolce ET, Johnson MA (2006) Elevated serum S-adenosylhomocysteine in cobalamin-deficient elderly and response to treatment. Am J Clin Nutr 84:1422–1429
Stabler SP, Lindenbaum J, Allen RH (1996) The use of homocysteine and other metabolites in the specific diagnosis of vitamin B-12 deficiency. J Nutr 126:1266S–1272S
Stroinsky A, Schneider Z (1987) Cobamide dependant enzymes. In: Schneider Z, Stroinsky A (eds) Comprehensive B-12. Printing house de Gruyter, Berlin, pp. 225–266
Teplitsky V, Huminer D, Zoldan J, Pitlik S, Shohat M, Mittelman M (2003) Hereditary partial transcobalamin II deficiency with neurologic, mental and hematologic abnormalities in children and adults. Isr Med Assoc J 5:868–872
Ulleland M, Eilertsen I, Quadros EV, Rothenberg SP, Fedosov SN, Sundrehagen E, Orning L (2002) Direct assay for cobalamin bound to transcobalamin (holo- transcobalamin) in serum. Clin Chem 48:526–532
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Herrmann, W., Obeid, R. (2012). Cobalamin Deficiency. In: Stanger, O. (eds) Water Soluble Vitamins. Subcellular Biochemistry, vol 56. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-2199-9_16
Download citation
DOI: https://doi.org/10.1007/978-94-007-2199-9_16
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-2198-2
Online ISBN: 978-94-007-2199-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)