
Abstract
Cholesterol is a major constituent of the plasma membrane in eukaryotic cells. It regulates the physical state of the phospholipid bilayer and is crucially involved in the formation of membrane microdomains. Cholesterol also affects the activity of several membrane proteins, and is the precursor for steroid hormones and bile acids. Here, methods are described that are used to explore the binding and/or interaction of proteins to cholesterol. For this purpose, a variety of cholesterol probes bearing radio-, spin-, photoaffinity- or fluorescent labels are currently available. Examples of proven cholesterol binding molecules are polyene compounds, cholesterol-dependent cytolysins, enzymes accepting cholesterol as substrate, and proteins with cholesterol binding motifs. Main topics of this report are the localization of candidate membrane proteins in cholesterol-rich microdomains, the issue of specificity of cholesterol– protein interactions, and applications of the various cholesterol probes for these studies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1.1 Introduction
Cholesterol is a major constituent of the plasma membrane in most eukaryotic cells where it fulfils several functions. It regulates membrane fluidity, increases membrane thickness, establishes the permeability barrier of the membrane, modulates the activity of various membrane proteins, and is the precursor for steroid hormones and bile acids (Burger et al., 2000; Pucadyil and Chattopadhyay, 2006). Cholesterol is non-randomly distributed in cells and membranes (Yeagle, 1985) and plays an essential role in the formation of lateral membrane domains, often designated as ‘lipid rafts’ (Simons and Ikonen, 1997; Simons and Toomre, 2000).
Here, methods are described that are used to explore the binding/interaction of proteins to cholesterol. In case of membrane proteins such as receptors, transporters or ion channels, researchers often like to know whether their candidate protein is associated with cholesterol-rich microdomains or not. To address this issue, different subcellular fractionation protocols are usually performed. These approaches will be briefly compared herein. Even if a given membrane protein is shown to be excluded from cholesterol-rich microdomains, it is unlikely for any membrane protein to avoid molecular contacts with cholesterol. This is due to the fact that cholesterol is in large excess over any membrane protein within the plasma membrane. Some membrane proteins show functional changes dependent on the amounts of cholesterol. In this case, one has to discriminate between direct cholesterol–protein interactions and indirect effects caused by the influence of cholesterol on the biophysical state of the membrane. Approaches are described that help to prove the specificity of putative cholesterol–protein interactions. Various cholesterol-binding molecules are currently known. Among these are polyene compounds, cholesterol-dependent cytolysins, enzymes accepting cholesterol as substrate, and proteins with cholesterol binding motifs, many of which are covered in detail within the chapters of this book. In Table 1.1, proteins with defined or putative cholesterol binding motifs are listed. For cholesterol modifying enzymes cholesterol binding is obvious as they use cholesterol as substrate. For other proteins, cholesterol binding/interaction has been shown by various techniques such as binding studies, affinity labelling with photoreactive cholesterol analogues, or crystallography. Convincing proof for cholesterol binding has only been demonstrated for a handful of proteins, e.g. for NPC2 using various approaches such as binding studies with [3H]cholesterol (Okamura et al., 1999; Ko et al., 2003; Infante et al., 2008c), spectroscopical measurements (Friedland et al., 2003; Liou et al., 2006; Cheruku et al., 2006), and crystallography (Friedland et al., 2003). The following criteria and techniques could support evidence for cholesterol–protein interaction: (i) presence in cholesterol-rich microdomains; (ii) alterations in protein function induced by changes of the cholesterol content in membranes/cells; (iii) alterations in protein function induced by substitution of cholesterol by sterol analogues; (iv) influence of cholesterol binding molecules (e.g. polyenes) as functional cholesterol ‘competitors’; (v) binding studies with [3H]cholesterol; (vi) spectroscopic binding assays using fluorescent sterol analogues (e.g. dehydroergosterol); (vii) affinity labelling of the protein with photoreactive cholesterol analogues; (viii) identification of cholesterol binding domains. The above mentioned criteria and topics will be critically discussed below. Finally, I will focus on currently available cholesterol probes bearing radio-, spin-, photoaffinity- or fluorescent labels and describe their utility for cholesterol research.
1.2 Cholesterol-Rich Microdomains
According to our current understanding, biomembranes are much more ordered than postulated in the fluid mosaic model proposed by Singer and Nicolson in 1972. Flippases are involved to generate and maintain an asymmetric distribution of lipids across the bilayer of the plasma membrane. Moreover, within the plane of the membrane, lipids and proteins are unevenly distributed. The type of lateral membrane organization that exists in vivo is, however, still controversial. The concept of lipid ‘rafts’ is a widespread microdomain model that was originally developed by Simons and van Meer to explain the sorting of proteins to the apical membrane in polarized epithelial cells (Simons and van Meer, 1988; Simons and Ikonen, 1997; Simons and Toomre, 2000). This concept has been modified over the years, a process that is still going on. In a 2006 Keystone Symposium, membrane rafts were defined as ‘small (10–200 nm), heterogeneous, highly dynamic, sterol- and sphingolipid-enriched domains that compartmentalize cellular processes. Small rafts can sometimes coalesce to form larger platforms through protein–protein and protein–lipid interactions.’
The flask-shaped caveolae that are seen in electron micrographs are also enriched in glycolipids and cholesterol and are regarded as subdomains of lipid rafts. The scaffolding protein caveolin-1 is necessary for the formation of the typical caveolae structures (Kurzchalia and Parton, 1999). Raft and caveolar microdomains may participate in signal transduction, cholesterol trafficking, and vesicular sorting. A variety of hormone-receptor complexes, toxins, viruses, and bacteria are internalized into cells by a caveolar/raft-dependent endocytosis pathway that is clathrin-independent, but requires dynamin and cholesterol (Nabi and Le, 2003). Biochemically, rafts are primarily based on the criterion of cholesterol enrichment. They are assumed to be composed of lipids that exist in a cholesterol-enriched liquid-ordered (L o) state, separated from and coexisting with cholesterol-poor liquid-disordered (L d) domains. Detergent resistance is often used as an experimental hallmark of L o structures. However, there is no definitive evidence to identify detergent-resistant biomembranes with raft and L o domains (Lichtenberg et al., 2005). Recently, the entire raft model has raised some criticism, and alternatives to this model have been developed (Munro, 2003; Shaw, 2006; Kenworthy, 2008). In these alternative models, lipid ordering plays a minor role. Instead, formation of microdomains (submicrometer-sized clusters) is mainly driven by protein–protein interactions, either through diffusional trapping or self-organization (Douglass and Vale, 2005; Sieber et al., 2007).
The following detergent-based criteria are used to verify the localization of a candidate membrane protein in lipid rafts: (i) Insolubility of the protein in non-ionic detergents (Triton X-100) at cold (and vice versa, solubility in Triton X-100 at 37°C). (ii) Flotation of the detergent-insoluble proteins to the upper low-density fractions following sucrose (or OptiPrep) gradient centrifugation. (iii) Decrease or disappearance of detergent-insolubility after removal of cholesterol (e.g. by cyclodextrins). Different subcellular fractionation protocols are employed to isolate membrane microdomains and to verify or exclude the raft association of a certain membrane protein. Basically, we can distinguish detergent-based and detergent-free fractionation methods.
1.2.1 Detergent-Based Methods
Detergent-based methods utilize the property of insolubility of raft proteins in cold non-ionic detergents, typically Triton X-100. In contrast, at 37°C, solubilization of raft proteins occurs in Triton X-100 (Brown and Rose, 1992). After incubation of the cells with detergent, the extract is fractionated in a density gradient (e.g. sucrose, OptiPrep) and the cholesterol-enriched low-density gradient fractions are harvested. Marker proteins such as caveolin or flotillin are used to identify these low-density fractions. Rafts prepared accordingly are also designated as ‘detergent-insoluble (or detergent-resistant) glycosphingolipid-enriched’ membrane domains (DIGs or DRMs). Different types of raft domains may be isolated when using detergents other than Triton X-100 (Roper et al., 2000; Chamberlain, 2004). Detergents that have been employed for this purpose were Triton X-114, Lubrol PX, Lubrol WX, Brij58, Brij96, Brij98, CHAPS, Nonident P40, and octylglucoside (Brown and Rose, 1992; Madore et al., 1999; Roper et al., 2000; Drevot et al., 2002; Slimane et al., 2003; Schuck et al., 2003). Recently, we introduced a novel cholesterol-based detergent (termed Chapsterol) to improve the isolation of cholesterol dependent raft proteins (Gehrig-Burger et al., 2005). Unfortunately, this detergent is not yet available commercially. The pentaspan protein prominin, for example, was soluble in Triton X-100, but insoluble in Lubrol WX. Nevertheless, several other properties of prominin classified it as a bona fide raft protein (Roper et al., 2000). Since rafts become solubilized at high detergent:lipid ratios (Chamberlain and Gould, 2002), it is necessary to use the lowest amount of detergent that maintains insolubility for raft proteins but completely solubilizes non-raft proteins (e.g. the transferrin receptor) (Chamberlain, 2004). Moreover, the level of detergent insolubility can change for some raft proteins (e.g. for H-ras at GTP-loading) (Prior et al., 2001). Some proteins shown to be raft-associated by other criteria (e.g. the insulin receptor residing in caveolae) could be solubilized by detergent (Gustavsson et al., 1999). Thus, proteins excluded from DRM fractions can still be associated with raft domains. In addition, detergents such as Triton X-100, can themselves promote domain formation in lipid mixtures (Heerklotz, 2002). In each case, the name of the employed detergent should be included to specify the type of microdomains that has been isolated, i.e. designate them as Triton X-100 rafts, Lubrol WX-rafts, etc. To exclude potential artifacts associated with the use of detergents, various detergent-free fractionation protocols have been developed.
1.2.2 Detergent-Free Methods
It is clear that the composition of proteins and lipids from detergent and non-detergent-based preparations significantly differ from each other. To avoid confusion, we have designated the low-buoyant density gradient fractions obtained by detergent-free preparations as low-density microdomains (LDM) (Gimpl and Fahrenholz, 2000). Two widely applied protocols use sonication steps to disrupt the cellular membranes (Smart et al., 1995; Song et al., 1996). In one approach, the cells were sonicated in sodium carbonate buffer (pH 11) prior to centrifugation in a discontinuous 5–45% sucrose gradient (Song et al., 1996). Smart et al. (1995) prepared LDM rafts according to the following subsequent steps: lysis of the cells in an isotonic buffer, purification of plasma membranes therefrom on a Percoll gradient, sonication of these membranes, and isolation of LDM rafts by flotation through a 10–20% OptiPrep gradient. Each of these protocols has subsequently been modified. The time-consuming OptiPrep protocol has been simplified (Macdonald and Pike, 2005). For the isolation of rafts from brain tissues, the usage of sucrose was preferred before OptiPrep in density gradients (Persaud-Sawin et al., 2009). Antibody-based immunoisolation approaches allow isolation of subpopulations of rafts enriched for different markers, as caveolin-1 or flotillin (Shah and Sehgal, 2007).
1.2.3 Receptors in Cholesterol-Rich Microdomains (Lipid Rafts)
Membrane proteins of different families such as G protein coupled receptors (GPCRs), transporters, or ion channels have been shown to be localized or enriched in lipid rafts. For GPCRs, Chini and Parenti (2004) have recently summarized their signaling, coupling efficacy, and trafficking in dependence on their distribution in lipid rafts and caveolae. We have studied in more detail the oxytocin receptor, a typical GPCR, that requires cholesterol to maintain its high-affinity state for oxytocin (Klein et al., 1995; Gimpl et al., 1995, 1997). Thus, a partial localization of this receptor in lipid rafts was expected. For this purpose, HEK293 cells expressing the human oxytocin receptor (HEKOTR) were fractionated by detergent-free and detergent-based methods (Gimpl and Fahrenholz, 2000). Only a minor fraction (~1%) of the receptor was found in Triton X-100 rafts, whereas substantially more oxytocin receptors (10–15%) were found in LDM rafts produced according to a detergent-free protocol. To analyze the amounts of functional receptors, we modified the fractionation method based on sodium carbonate buffer (pH 11) (Song et al., 1996), because ligand-receptor binding is normally inhibited at such an alkaline pH. For this purpose, a subcellular fractionation protocol was developed in which the sodium carbonate buffer was substituted by sodium chloride (1 M) in 20 mM Hepes (pH 7.4)/EDTA (Gimpl and Fahrenholz, 2000). The distribution profile of total proteins and of the raft marker caveolin was shown to be similar for sodium carbonate and sodium chloride based fractionation (Fig. 1.1). In addition, the majority of cholesterol was found in the low-density fractions for both methods. The cholesterol enrichment of low-density fractions is one of the most important properties for lipid rafts. Vice versa, rafts and caveolae are disrupted when cholesterol is extracted from these microdomains, e.g. via cyclodextrins.


Detergent-free subcellular fractionation of HEK293 cells expressing the human oxytocin receptor (HEKOTR). The cells were fractionated in a sucrose flotation gradient using either a sodium carbonate (Song et al., 1996) or sodium chloride (Gimpl and Fahrenholz, 2000) protocol. Aliquots of each of the 13 fractions were analyzed with respect to caveolin immunodetection (A) and determination of total protein contents (B). For methodical details see Gimpl and Fahrenholz (2000)
1.3 Manipulation of the Membrane Cholesterol Content by Cyclodextrins
Cyclodextrins (CDs) are torus-shaped cyclic oligosaccharides linked by α-1,4 glycosidic bonds (Fig. 1.2A). They are produced from the enzymatic degradation of starch. Cyclodextrins comprised of 6, 7 and 8 D-glucopyranosyl residues units (termed α-, β- and γ-forms, respectively) were used to alter the lipid composition of cells (Ohtani et al., 1989). They possess a hydrophilic outer surface and a hydrophobic inner cavity. In aqueous solution, this hydrophobic cavity contains low entropy and easily displaceable water molecules. Cyclodextrins enhance the solubility of non-polar substances (e.g. cholesterol) by incorporating them (at least partly) into their hydrophobic cavity and forming non-covalent water-soluble inclusion complexes. Particularly, β-cyclodextrins (βCDs) and its derivatives, such as methyl-β-cyclodextrin (MβCD) or 2-hydroxypropyl-β-cyclodextrin (HPβCD), were found to selectively extract cholesterol from the plasma membrane, in preference to other membrane lipids (Irie et al., 1992; Klein et al., 1995; Gimpl et al., 1995, 1997; Kilsdonk et al., 1995). However, the extraction of cholesterol does not seem to be selective for lipid rafts (Mahammad and Parmryd, 2008). Figure 1.2 shows that the size of the cholesterol molecule is too large to be fully incorporated into the cavity of βCDs. The exact structure of the soluble cholesterol–βCD inclusion complex is still unknown. An excellent review to the use of cyclodextrins for manipulation of the cholesterol content in membranes has been published (Zidovetzki and Levitan, 2007).


Structure and dimensions of cyclodextrins (A) and cholesterol (B). Cyclodextrins (CDs) typically possess 6, 7 or 8 D-glucopyranosyl residues (α-, β-, and γ-cyclodextrin respectively). Their overall shape is like a truncated cone that accommodates guest molecules into a hydrophobic cavity. The hydrophilic OH groups are on the outside of the cavity with the C2- and C3-hydroxyls located around the wider ring and the C6-OH groups aligned around the smaller opening. The hydroxyl groups may be derivatized to modify the physical and chemical properties of the cyclodextrins. The 6-OH groups (black arrows) are most easily derivatized. Methyl-β-cyclodextrin (MβCD) and (2-Hydroxypropyl)-β-cyclodextrin (HPβCD) are often used for cholesterol depletion experiments. They normally contain about 0.5-2 mol methyl- or hydroxymethyl groups per unit anhydroglucose. Cholesterol is too large to be completely encapsulated within the cavity of a single βCD. So, cholesterol may be either partly incorporated into the cavity, or two stacked βCDs may be required for the complete complexation of cholesterol
The kinetics of cyclodextrin-mediated cholesterol efflux provided information about cholesterol pools in cells (Yancey et al., 1996). While ‘empty’ β-cyclodextrins function as rather selective cholesterol acceptors, cholesterol–cyclodextrin complexes serve as very efficient sterol donors in vitro and in vivo (Gimpl et al., 1995, 1997; Kilsdonk et al., 1995). For example, up to 80% of the cholesterol can be extracted from living cells via MβCD within 10–30 min (Fig. 1.3). Vice versa, using cholesterol-MβCD as donor, cholesterol-depleted cells can be reloaded with cholesterol within the same time scale. Thereby, substantial cholesterol overloading of cells easily occurs, as shown in Fig. 1.3. The efficiency of cholesterol extraction and reloading varies with incubation time, temperature, cell type, and concentration of the cholesterol acceptor. It is also possible to stabilize membranes or cells at a certain cholesterol concentration by varying the molar ratio between βCD and cholesterol in the complex. The experimental conditions to achieve this ‘cholesterol equilibrium’ have to be determined for each cell system (Zidovetzki and Levitan, 2007). However, one should be aware that even when the total cholesterol levels are held constant, the distribution of intracellular cholesterol pools (e.g. between plasma membrane and endoplasmic reticulum) may change.


Depletion and reloading of cholesterol in living HEKOTR cells. To extract cholesterol, the cells were incubated with 10 mM MβCD (stock 200 mM) for 0–40 min at 37°C in serum-free culture medium. The cells were then washed twice with medium. Cholesterol enrichment of the cholesterol-depleted cells was started using 0.3 mM Chol-MβCD (stock 10 mM) for 0–60 min in serum-free culture medium at 37°C. Cholesterol levels were determined using a diagnostic kit (data are means ± SD, n=3). Methods are described in detail (see Gimpl et al., 1997)
Membrane cholesterol can also be rapidly substituted with sterol analogues by adding the corresponding sterol–MβCD complexes to the cholesterol-depleted membranes or cells. Thus, cyclodextrin-based exchanges enable the researcher to explore the cholesterol specificity of a candidate membrane protein in a precise structure-activity analysis. We have performed such studies for two GPCRs: the oxytocin receptor (OTR) and cholecystokinin B receptor (CCKBR) (Gimpl et al., 1997). A similar study has been performed for SCAP, a protein with a sterol sensing domain (Table 1.1) (Brown et al., 2002). As shown in Table 1.2, the sterol requirements reveal some similarities between the GPCRs and SCAP, although these are unrelated proteins. Sterols supporting low membrane fluidity (equal to a high level of anisotropy of diphenylhexatriene, Table 1.2) (e.g. desmosterol, dihydrocholesterol, β-sitosterol, 5α-cholest-7-en-3βol) were most effective to maintain the binding function of the GPCRs and a certain conformation of SCAP. The structure-activity analysis also permits discrimination between cholesterol effects that are due to specific sterol–protein interactions or due to changes in the physical state of the membrane bilayer. In case of the CCKBR, the effects of sterols correlated with changes in membrane fluidity. For the oxytocin receptor and for SCAP, a unique requirement for cholesterol was observed suggesting that these proteins are regulated by specific cholesterol–protein interactions. For example, epicholesterol that maintains membrane fluidity moderately-good was completely inactive for both the OTR and SCAP. The data for lanosterol and 19-hydroxycholesterol indicate a distinct cholesterol specificity of SCAP and the OTR, respectively (Table 1.2). The cholesterol analogue 4-cholesten-3-one supports the membrane fluidity moderately and maintains the ligand binding of the CCKBR. However, 4-cholesten-3-one was found to be inactive for all cholesterol-dependent membrane proteins reported so far. Examples include the oxytocin receptor, SCAP (Table 1.2), the hippocampal 5HT1A receptor (Pucadyil et al., 2005), the galanin receptor (Pang et al., 1999), or ecto-nucleotidase CD39 (Papanikolaou et al., 2005).
Overall, the administration of β-cyclodextrins and sterol-β-cyclodextrins as cholesterol acceptor and sterol donor complexes, respectively, allows one to alter and exchange the cholesterol content in membranes and cells. This is now a standard methodology in the research of ‘lipid rafts’ (Simons and Toomre, 2000). Moreover, cyclodextrins have found a wide range of applications in food, pharmaceutical and textile industry, cosmetics, environmental engineering, and agrochemistry. For example, cyclodextrins are employed for the preparation of cholesterol-free products or for the delivery of drugs (Challa et al., 2005). Finally, cyclodextrins offer great therapeutic potential as recently been demonstrated for HPβCD. The cholesterol acceptor was able to reverse the defective lysosomal transport in a mouse model of Niemann Pick C disease (Liu et al., 2009a). However, one should be aware that application of β-cyclodextrins induce several ill-defined side effects that are associated with their non-specific action. They can extract a wide range of hydrophobic compounds including phospholipids, sphingomyelin, even GPI-anchored proteins and some protein kinases (Ilangumaran and Hoessli, 1998; Ottico et al., 2003; Monnaert et al., 2004). Some of these undesirable effects of β-cyclodextrins may be caused by inhibition of tyrosine kinases and/or vesicle shedding (Sheets et al., 1999). Thus, careful control experiments and the avoidance of overly high concentrations of cyclodextrins are recommended (Zidovetzki and Levitan, 2007).
1.4 Cholesterol-Binding Molecules
1.4.1 Polyenes
Among the family of polyenes, filipin is certainly the most important tool to visualize the localization of free cholesterol in cells. Filipin is an antibiotic with antifungal properties and a mixture of four macrolides with minor differences in their structure, the fraction known as filipin III being the major component (Bolard, 1986) (Fig. 1.4A). Filipin performs its antibiotic action by inducing a structural disorder in sterol containing membranes. The disintegration of the membranes then leads to the leakage of cellular components. Filipin requires a sterol partner with a free 3′-OH group. So it does not recognize esterified cholesterol. Further details concerning the filipin–sterol interaction are unclear. Different models have been generated to explain the organization of the filipin-sterol complexes within the membrane bilayer (Elias et al., 1979; Lopes et al., 2004). Filipin has been used for decades to localize the distribution of free cholesterol in cells and tissues (Kinsky et al., 1967; Elias et al., 1979; Orci et al., 1983; Butler et al., 1992). Filipin staining has been and still is a prominent diagnostic tool for the identification of cholesterol mislocalization in lysosomes of the Niemann-Pick C (NPC) phenotype (Butler et al., 1987, 1992) (Fig. 1.4B). However, filipin is a cytotoxic compound disrupting the integrity of sterol-containing membranes (Behnke et al., 1984). So, staining with filipin can only be used in prefixed cells or tissues. Moreover, it possesses unfavorable spectroscopic properties, e.g. high photobleaching and excitation within the UV range. Other polyene antibiotics such as nystatin and amphotericin B share the cholesterol binding property of filipin. Both substances form pores, unlike filipin. Electrophysiologists use nystatin for the so-called ‘perforated patch clamping’ technique. Nystatin forms complexes with cholesterol that lead to ion-selective ‘perforations’ in the bilayer inside the patch pipette. Polyenes, particularly filipin, can also be employed as cholesterol competitors in binding assays or functional interaction studies in order to verify or falsify a putative cholesterol interaction of candidate proteins. The oxytocin and galanin receptors, for example, showed a dose-dependent decrease in ligand binding in the presence of increasing concentrations of filipin (Gimpl et al., 1997; Pang et al., 1999).


Filipin (structure in panel A) staining is the standard method to visualize free cholesterol in cells and tissues. Human fibroblasts from a control person (B, ‘fibroblasts’) and from a patient with Niemann Pick C disease (B, ‘NPC fibroblasts’) were fixed by paraformaldehyde (3.7%), incubated with the filipin III fluorophore (0.05% in PBS/10% FCS) for 60 min, washed twice with PBS, and were mounted in Moviol. NPC fibroblasts show intense filipin accumulation in lysosomes/late endosomes whereas in control fibroblasts the filipin (and thus free cholesterol) is concentrated on the cell surface. [bar, 5 μm]
Overall, although filipin is routinely used as to visualize free cholesterol, it is not entirely clear whether it’s staining reflects the correct distribution of cholesterol, particularly at intracellular sites that are not easily accessible and/or prone to artefacts from fixation techniques. It has also been reported that some sterol-containing membranes are not labelled by filipin (Pelletier and Vitale, 1994; Steer et al., 1984; Severs and Simons, 1983).
1.4.2 Cholesterol-Dependent Cytolysins
Cholesterol-dependent cytolysins (CDCs) (see also Chap. 20) are a family of protein toxins produced by a variety of pathogenic Gram-positive bacteria including Streptococcus pyogenes (streptolysin O), Streptococcus pneumoniae (pneumolysin), Listeria monocytogenes (listeriolysin O), Clostridium perfringens (perfringolysin O), and Bacillus anthracis (anthrolysin) (Rossjohn et al., 1997; Palmer, 2001; Tweten et al., 2001). When the water-soluble cytolysin monomers bind to cholesterol-containing membranes, they self-associate to form large oligomeric complexes and aqueous pores in the bilayer. Structural features of the cholesterol molecule required for interaction with the toxins include the 3β-OH group, the stereochemistry of the sterol ring system, and the isooctyl side chain (Watson and Kerr, 1974; Prigent and Alouf, 1976; Nelson et al., 2008). This suggests that CDCs possess a specific cholesterol binding site.
Among the CDCs, perfringolysin O is one of the best studied cholesterol binding molecules. It is comprised of four domains. The C-terminal portion of perfringolysin O (D4 domain) folds into a separate β-sandwich domain composed of two four-stranded β-sheets at one end of the elongated molecule (Rossjohn et al., 1997). This D4 domain is involved in cholesterol recognition and binding (see also Chap. 21). Specifically, only the short hydrophobic loops at the tip of the D4 β-sandwich are responsible for mediating the interaction of the CDCs with cholesterol-rich membranes, whereas the remainder of the structure remains close to the membrane surface (Ramachandran et al., 2002; Soltani et al., 2007). Interestingly, the mammalian immune defense system, the complement membrane attack complex perforin, was found to be structurally homologous to the bacterial CDCs (Hadders et al., 2007; Anderluh and Lakey, 2008). A protease-nicked and biotinylated derivative of perfringolysin O (termed BCθ-toxin) was shown to retain specific binding to cholesterol without cytolytic activity. BCθ-toxin combined with fluorophore-labelled avidin has been introduced as a cholesterol reporter system (Fujimoto et al., 1997). This probe was used for the localization of membrane cholesterol in various cells by fluorescence microscopy and by electron microscopy in cryosections (Iwamoto et al., 1997; Waheed et al., 2001; Mobius et al., 2002; Sugii et al., 2003; Reid et al., 2004; Tashiro et al., 2004). Visualization of the D4 probe has been achieved both by conjugation of a fluorophore to D4 and by N-terminal fusion of green fluorescent protein (EGFP variant) to the protein (Shimada et al., 2002). Perfringolysin O derivatives detected cholesterol primarily in cholesterol-rich membrane microdomains (e.g. caveolae or ‘lipid rafts’) or liposomes with high (>20–25 mol%) cholesterol (Ohno-Iwashita et al., 1992; Waheed et al., 2001; Shimada et al., 2002; Sugii et al., 2003). Studying the pathophysiological cholesterol accumulation in the Niemann-Pick C mouse brain, staining with BCθ-toxin was found to be superior to that achieved by filipin. In brain regions known to be affected by the neurodegenerative NPC disease, cholesterol accumulations were observed both at a better signal-to-noise ratio and at earlier time points with BCθ-toxin as compared with filipin (Reid et al., 2004). In contrast, in a hippocampal culture system, cholesterol was detectable by BCθ only at the cell surface of fully matured neurons, whereas filipin stained intracellular and cell surface cholesterol in neurons at all developmental stages. Additionally, the two cholesterol reporters showed different labelling patterns in cultured hippocampal neurons. While BCθ staining was observed mainly on axons, filipin labeled axons, dendrites and somata (Tashiro et al., 2004). These authors reported also that neurons that were induced to the NPC phenotype by administration of certain reagents, lose cell surface BCθ staining on axons. Obviously, the distribution of cholesterol at the axonal surface is critical for recognition by BCθ. In addition, when membranes or cells were depleted of cholesterol by β-cyclodextrins, the binding of θ-toxin was completely abolished whereas significant filipin staining was retained (Waheed et al., 2001; Shimada et al., 2002). Thus, the toxin might recognize a certain arrangement of cholesterol at the outer leaflet of the membrane bilayer (Mobius et al., 2002).
Taken together, perfringolysin O derivatives might be good and selective probes for cholesterol-rich domains such as caveolae or rafts, but are neither suitable to label cholesterol-poor organelles nor for quantitative in situ determination of membrane cholesterol. In addition to their potency as cholesterol probes, cytolysins are useful tools in cell biology due to their pore-forming capacity. For example, application of a low concentration of streptolysin O facilitates the entry of macromolecules into cells of interest (Lafont et al., 1995).
1.4.3 Enzymes with Cholesterol as Substrate
Of course, all enzymes that accept cholesterol as substrate possess a specific cholesterol binding site and are potential candidate proteins to measure cholesterol amounts or to detect cholesterol in membranes and cells. These comprise cholesterol oxidases, the cholesterol esterifying ACAT enzymes, cholesterol sulfotransferase SULT2B1b (Lee et al., 2003), the cytochrome P450 family proteins (e.g. cholesterol hydroxylases like CYP46A1) (Mast et al., 2008), and other less well characterized enzymes (e.g. cholesterol transferase linking cholesterol to sonic hedgehog). However, most of these enzymes are very hydrophobic, require detergents for solubilization or are inconvenient for the development of highly sensitive enzymatic assays. So far, primarily the cholesterol oxidases (see also Chap. 5) have been used to measure cholesterol concentrations or to gain information about cellular and membrane cholesterol distribution.
The flavoenzyme cholesterol oxidase converts cholesterol and oxygen into the products 4-cholesten-3-one and hydrogen peroxide that can be quantitated by spectrophotometry (or fluorometry) via an oxidative coupling reaction in the presence of peroxidase to form a chromogen (or fluorophore). Currently, cholesterol oxidase is immobilized onto different surfaces for the fabrication of cholesterol biosensors. The properties of different cholesterol oxidases have been reviewed (MacLachlan et al., 2000). Cholesterol oxidase is produced by several microorganisms, e.g. Nocardia erythropolis, Brevibacterium sterolicum, Streptomyces hygroscopicus. Structures of cholesterol oxidase from Brevibacterium sterolicum and Streptomyces hygroscopicus in its free and substrate-bound states have been determined at atomic resolution (Vrielink et al., 1991; Li et al., 1993; Yue et al., 1999). The water-soluble enzyme associates peripherally with the surface of the membrane. Probably, it forms a complex with the lipid bilayer that allows cholesterol to move directly from the membrane into the active site (Bar et al., 1989; Ahn and Sampson, 2004). In fact, it is known that the properties of the membrane strongly influence the accessibility of the enzyme to its substrate. Thus, cholesterol oxidase is a valuable probe for studying membrane organization and a sensor of the bilayer lipid phase, with a preferential binding to the solid phase (Patzer and Wagner, 1978; Ahn and Sampson, 2004). Cellular cholesterol can be tracked by using its susceptibility to cholesterol oxidase (Lange, 1992). In living intact cells, cholesterol is only a poor substrate for cholesterol oxidase. This changes markedly, when certain substances or enzymes were added to the cells. Among the agents stimulating the enzymatic turnover are cholesterol, glutaraldehyde, low ionic strength buffer, phospholipase C, sphingomyelinase, detergents, and membrane intercalators such as decane or octanol (Lange et al., 1984; Slotte et al., 1989). Lysophosphatides are shown to inhibit the activity of cholesterol oxidase (Lange et al., 1984; Lange et al., 2005). It has been proposed that in the unperturbed plasma membrane, cholesterol is kept at a low chemical potential by its association with bilayer phospholipids (Radhakrishnan and McConnell, 2000). Thus, stimulators of enzyme activity might act by increasing the chemical activity of cholesterol leading to a better accessibility of cholesterol to the enzyme, whereas inhibitors such as lysophosphatidylcholine might associate with excess cholesterol and thereby lower its chemical activity (Lange et al., 2004). Variations of cholesterol oxidase accessibility have been explained by sterol superlattices in membranes (Wang et al., 2004). According to this model, cholesterol within sterol superlattices is tightly packed and more accessible to the aqueous phase (i.e. to cholesterol oxidase) as compared with cholesterol localized in irregularly distributed lipid areas (Wang et al., 2004). The cholesterol oxidase-accessible plasma membrane pool may be the same pool of cholesterol removed by high density lipoproteins (Vaughan and Oram, 2005). The susceptibility to cholesterol oxidase has been exploited to gain information about the localization, transfer kinetics, and transbilayer distribution of cholesterol (Lange, 1992). In human fibroblasts, 90% of the cholesterol in fixed (e.g. glutaraldehyde treated) cells was oxidized by the enzyme within ~1 min. The residual 10% of cholesterol resistant to cholesterol oxidase coincided with markers of endocytic membranes that are also in large part derived from the plasma membrane (Lange et al., 1989; Lange, 1991). This would indicate that in fibroblasts almost all of the cellular cholesterol is localized in the plasma membrane pool, whereas only minor cholesterol amounts (1% or less) are distributed to other organelles (e.g. the endoplasmic reticulum). Application of cholesterol oxidase in human erythrocytes suggested that cholesterol flips very rapidly (half time < 3 s at 37°C) across the plasma membrane (Lange et al., 1981). In contrast, another group reported a half-time of 1–2 h for the transmembrane movement of cholesterol using susceptibility to cholesterol oxidase as reporter (Brasaemle et al., 1988). The transfer of newly synthesized cholesterol to the cell surface occurred with a half-time of 10–60 min as measured by the cholesterol oxidase approach in fibroblasts (Lange and Matthies, 1984; Lange, 1991).
Overall, the application of cholesterol oxidase as a cholesterol probe requires careful selection of reaction conditions and rigorous control experiments. When the enzyme is used on living cells, alterations in protein localization or receptor signaling are possible (Smart et al., 1994; Gimpl et al., 1997; Okamoto et al., 2000). Inherent difficulties in this approach are related with the fact that the enzyme converts cholesterol to a steroid with substantially altered properties. For example, 4-cholesten-3-one does not condense a phospholipid monolayer to the same extent as cholesterol (Gronberg and Slotte, 1990). In addition, 4-cholestene-3-one is a raft-dissolving steroid that, unlike cholesterol, favours the liquid disordered phase (Xu and London, 2000). Its action promotes a certain rate of leakage of the plasma membrane (Ghoshroy et al., 1997). Mutant enzymes with unimpaired membrane binding and no catalytic activity may overcome this limitation (Yin et al., 2002). Also, other enzymes with cholesterol as substrate, e.g. cholesterol dehydrogenases or cholesterol sulfatases, should be evaluated for their applicability as cellular cholesterol reporters.
1.4.4 Other Cholesterol-binding Proteins
Enzymes using cholesterol as substrate usually possess binding cavities to accommodate large parts of the cholesterol molecule (Mast et al., 2008; Chen et al., 2008). In these and other cholesterol binding proteins, a couple of binding motifs have been described and may be classified as follows (Table 1.1):
(i) Cholesterol binding tunnels/cavities: typically these structures are hydrophobic pockets, sometimes closable by a lid. They accommodate a single cholesterol molecule with medium- to high-affinity and are found in enzymes and other unrelated proteins (e.g. NPC2, the retinoic acid-related orphan receptor (RORα)) (Kallen et al., 2002). (ii) Sterol-sensing domain (SSD): this domain comprises a pentahelical region that is weakly conserved across different polytopic proteins, such as sterol regulatory element-binding protein cleavage-activating protein (SCAP), hydroxymethylglutaryl-CoA reductase (HMG-CoA reductase), the Niemann-Pick C (NPC) disease protein NPC1, and the hedgehog receptor patched (Kuwabara and Labouesse, 2002). Although all these proteins are implicated in some aspects of cholesterol metabolism, proof of cholesterol binding at this domain are rather weak. In the case of NPC1, photoaffinity approaches suggested that the cholesterol binding site is present within the SSD domain (Ohgami et al., 2004), whereas cholesterol binding was localized to the large Cys-rich luminal loop-1 according to radioligand binding assays (Infante et al., 2008b). (iii) START domain: START proteins represent a superfamily of hydrophobic ligand binding proteins. The name-giving member of proteins bearing this ~200 amino acids domain is ‘Steroid Acute Regulatory’ Protein (StAR=STARD1). STARD1 is essentially involved in the rate-limiting step of steroidogenesis, the transport of cholesterol to the mitochondria. The structure of the cholesterol binding cavity of the related StARD3 (=MLN64) has been resolved (Tsujishita and Hurley, 2000). The START domain is however not a specific cholesterol binding domain as in other START proteins other lipids are bound in the cavity (e.g. phosphatidylcholine in StARD2). (iv) ‘Cholesterol recognition amino acids consensus’ (CRAC) domain: the peptide ‘ATVLNYYVWRDNS’ (underlined amino acids are purported to interact with cholesterol) has first been identified as a high-affinity cholesterol binding motif in the C-terminus of the peripheral benzodiazepine receptor (PBR) (= TSPO, ‘Translocator protein, 18 kDa’) (Li et al., 2001). Together with STARD1, this receptor enables the translocation of cholesterol into mitochondria (see above). In its generalized form (– L/V – (X)1−5 − Y − (X)1−5 – R/K), this motif is found in many proteins such as cholesterol-dependent GPCRs (receptors for oxytocin and 5HT1A possess this motif in their fifth transmembrane helix), the sigma-1 receptor (with two consecutive CRAC motifs), HIV-1 env gp41 protein, and others (Table 1.1). (v) ‘Cholesterol Consensus Motif’ (CCM): this motif has been described by the crystal structure of the β2-adrenergic receptor (Hanson et al., 2008). Two cholesterol molecules were found in a receptor cleft formed by the segments of transmembrane helices 1-4. Based on homology, the following CCM has been defined: [4.39-4.43(R,K)]—[4.50(W,Y)]—[4.46(I,V,L)]—[2.41(F,Y)] (according to Ballesteros-Weinstein nomenclature). This motif is found in more than 40 class A GPCRs, including the cholesterol dependent oxytocin and 5HT1A receptor (Table 1.1).
Presumably, many proteins exist that interact with cholesterol but do not possess the above-mentioned motifs. The knowledge about these cholesterol binding motifs can be exploited to develop improved cholesterol binding assays. One of the main obstacles in cholesterol binding assays results from the low water solubility of cholesterol. Solubility of cholesterol can for example be achieved through inclusion in cyclodextrin complexes or stabilization by low amounts of detergents such as Triton X-100. However, these additional compounds can disturb binding reactions. For this purpose, it would be beneficial to have water-soluble cholesterol binding modules available that can act both as ‘solubilizers’ and competitors for cholesterol binding. To function as cholesterol donor, the affinity of these molecules to cholesterol must be lower than that of the candidate cholesterol binding protein under investigation. Further applications of these cholesterol binding proteins concern their potency to act as specific cholesterol donors or acceptors, that once may substitute the non-specific cholesterol donor and acceptor complexes MβCD/Chol-MβCD.
1.5 Binding Studies with Radiolabelled Cholesterol
In classical binding studies, radioligand and receptor protein are incubated to equilibrium, bound is separated from free radioligand by means of centrifugation or rapid filtration through presoaked glass fiber filters, and the pelleted membranes or the filters are washed and counted. However, when water-insoluble lipids such as cholesterol are the radioligands, direct binding assays are difficult and often not reproducible. Saturation and kinetic data have successfully been obtained when recombinant His-tagged proteins (e.g. NPC1 and SCAP) were used for cholesterol binding assays (Table 1.3). This allows the separation of bound and free radioligand via affinity chromatography on nickel agarose columns: adsorption of the assay solution to the affinity matrix, washing off the free and elution of the bound radioligand by imidazol (>0.2 M). If the candidate protein is not His-tagged, separation in gel filtration columns may be possible. However, these assays are tedious because each data point requires a separate column. To characterize the cholesterol binding of CRAC domains in HIV-1 env gp 41 and sigma-1 receptor, cholesteryl-hemisuccinate coupled to agarose was used as the affinity matrix for these proteins (Vincent et al., 2002; Palmer et al., 2007). However, this adsorption method will not be generally applicable because most cholesterol binding proteins require the free hydroxyl group of cholesterol, which is esterified in the cholesteryl affinity matrix.
The K D values obtained for cholesterol binding can vary markedly when different binding assays are used. This is well documented for NPC2 for which dissociation constants between 30 nM and 2.3 μM have been obtained (Table 1.3). The lowest affinity (2.3 μM) has been reported using a cholesterol binding assay where free [3H]cholesterol was bound onto dextran-coated charcoal and was removed by centrifugation, a separation method often used in radioimmunoassays. Spectroscopic assays with NPC2 using dehydroergosterol resulted in K D values of 120–660 nM (Friedland et al., 2003). When detergents are required, their usage can lead to dramatic alterations in cholesterol binding as documented for SCAP and NPC1 (Radhakrishnan et al., 2004; Infante et al., 2008a, 2008b). Detergent micelles may themselves sequester radiolabeled cholesterol and can thus disturb binding kinetics. The best detergents for SCAP and NPC1 were Fos-Choline 13 and Nonidet P-40. Recombinant NPC1 bound cholesterol, but binding was inhibited when the concentration of Nonidet P-40 exceeded the micellar threshold. In case of the more water soluble oxysterols (e.g. [3H]25-hydroxycholesterol), traditional filter assays have been successfully performed in radioligand binding studies with NPC1 (Infante et al., 2008a). In these binding assays, NPC1 was actually identified as an oxysterol binding protein. The K D for [3H]25-hydroxycholesterol binding was ~10 nM in 0.004% Nonidet P-40, whereas at higher detergent concentrations (1%), the K D increased several-fold to 80 nM (Infante et al., 2008a). Again, the binding results markedly depended on the concentrations of the employed detergent. Further studies showed that one (the putative second site within the SSD?) cholesterol binding site of NPC1 is localized in the N-terminal luminal loop-1 domain of the protein. NPC1 luminal loop-1 bound [3H]cholesterol with a K D of 130 nM, whereas binding of [3H]25-hydroxycholesterol occurred with >10 fold higher affinity compared to [3H]cholesterol (Infante et al., 2008b). More qualitative than quantitative cholesterol binding assays can be achieved by lipid protein overlay assays (Dowler et al., 2002; Rodriguez-Agudo et al., 2008). In this case, serial dilutions of cholesterol or other sterols are spotted onto a nitrocellulose membrane and are incubated with the candidate cholesterol binding protein possessing an epitope tag. After washing steps, the protein is detected with an anti-epitope antibody and a secondary fluorescence- or radiolabelled antibody. The method allows sensitive readouts and can be performed even if no radiolabelled sterol is available.
1.6 Fluorescent Cholesterol Analogues
The cholesterol molecule has achieved evolutionary perfection to fulfil its different functions in membrane organization (Yeagle, 1985). Features that have been found to be necessary for a biologically active cholesterol analogue are a free 3β-OH, a planar tetracyclic ring system with a Δ5(6) double bond, angular methyl groups, and an isooctyl side chain at the 17β-position (Schroeder, 1984) (Fig. 1.2B). The aliphatic side chain may be necessary to allow flip-flops and/or tail-to-tail transbilayer interaction of cholesterol molecules, whereas the 3′-OH group could interact with the head group of phospholipids. The structural features of a biologically active cholesterol substitute supporting ordered lipid domains are very stringent (Yeagle, 1985; Schroeder et al., 1995; Vainio et al., 2006; Megha et al., 2006). None of the cholesterol probes designed so far can claim to mimic all properties of the multifunctional cholesterol molecule. Among the fluorescent sterols two classes of probes can be distinguished: (i) intrinsically fluorescent sterols (e.g. dehydroergosterol and cholestatrienol); (ii) cholesterol probes with chemically linked fluorophores (Fig. 1.5). Both classes of probes have their specific advantages and disadvantages. Sterols belonging to the first class are more cholesterol-like but possess unfavourable spectroscopic properties. To compensate for the low quantum yield and severe photobleaching of these fluorophores, cells must be loaded with a relatively high sterol concentration. It cannot be excluded that the high amounts required for these probes preferentially force them into pathways which are untypical for cholesterol. Sterol analogues of the second class bear bulky reporter groups. However, their fluorescence properties are much better so that these probes can be applied at lower concentrations. Several fluorescent cholesterol analogues have been employed to address fundamental issues of distribution and trafficking of cholesterol. In particular, they enable the researcher to design pulse-chase experiments and/or to image the sterol in living cells. Moreover, binding assays can be developed by exploiting specific fluorescence properties such as sensitized emission, polarization, lifetime, quenching behaviour, or resonance energy transfer (Table 1.4).


Fluorescent cholesterol reporters used for cholesterol imaging and/or cholesterol binding assays: dehydroergosterol, cholestatrienol (=cholesta-5,7,9(11)-triene-3β-ol), 22-NBD-cholesterol (=22-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-23,24-bisnor-5-cholen-3-ol), 25-NBD-cholesterol (=25-[N-[(7-nitro-2-1,3-benzoxadiazol-4-yl)methyl]amino]-27-norcholesterol); 6-dansyl-cholestanol; Bodipy-cholesterol, 23-(dipyrrometheneboron difluoride)-24-norcholesterol
1.6.1 Dehydroergosterol
Dehydroergosterol (=ergosta-5,7,9(11),22-tetraene-3β-ol) is a cholesterol analogue with intrinsic fluorescence that naturally occurs in yeasts and certain sponges. Its structure differs from cholesterol only in possessing three additional double bonds and a methyl group at C-24 (Fig. 1.5). Dehydroergosterol is one of the best studied cholesterol probes concerning its physico-chemical properties (Schroeder, 1984; Schroeder et al., 1995). In many respects, it faithfully mimics cholesterol. For example, it co-distributes with cholesterol in both model and biological membranes, exhibits the same exchange kinetics as cholesterol in membranes, is nontoxic to cultured cells or animals, and is accepted by ACAT as a substrate for esterification (Schroeder, 1984; Smutzer et al., 1986; Schroeder et al., 1996; Frolov et al., 1996). It can also replace up to 85% of L-cell fibroblast cholesterol without causing significant detrimental effects (Schroeder, 1984). So, its employment as a cholesterol reporter should be a good choice. However, dehydroergosterol has unfavorable spectroscopic properties including a low quantum yield, excitation and emission in the UV region, and rapid photobleaching. Several imaging studies with dehydroergosterol have now been performed (Mukherjee et al., 1998; Frolov et al., 2000; Hao et al., 2002; Wustner et al., 2002, 2004; McIntosh et al., 2003; Zhang et al., 2005; Pipalia et al., 2007; Wustner, 2007; Mondal et al., 2009). Even short pulse-chase experiments were possible using dehydroergosterol complexed with methyl-β-cyclodextrin. In mouse L-fibroblasts, dehydroergosterol applied in the form of large unilamellar vesicles as donor was rapidly targeted from the plasma membrane to lipid droplets (Frolov et al., 2000; Zhang et al., 2005). In most studies, UV-microscopy was performed using dehydroergosterol complexed with methyl-β-cyclodextrin as donor (Hao et al., 2002; Wustner et al., 2002, 2004). Hao et al. (2002) found that in a CHO cell line, dehydroergosterol was preferentially incorporated into the endocytic recycling compartment within minutes and remained there for hours. Likewise, in polarized HepG2 hepatocytes and in J774 macrophages, the influx of dehydroergosterol was reported to occur via a vesicular pathway with enrichment in recycling endosomes. In macrophages but not in hepatocytes, dehydroergosterol was also translocated to lipid droplets (Wustner et al., 2002, 2004). Since dehydroergosterol possesses a significantly higher esterification rate (>7-fold) as compared with [3H]cholesterol, one may expect that after hours the sterol will be stored as ester into lipid droplets (Frolov et al., 2000). Because the enzymes responsible for esterification are localized in the endoplasmic reticulum, its substrate should also be localized there for some time. From unknown reasons dehydroergosterol has not been observed to be translocated into the endoplasmic reticulum. Dehydroergosterol has also been employed to address the question whether cholesterol-rich microdomains are present in vivo. The results of these studies provided some evidence in favour of the ‘lipid rafts’ concept (McIntosh et al., 2003; Zhang et al., 2005). Additionally, cholesterol binding assays for various cholesterol-binding proteins such as fatty acid binding protein (Nemecz and Schroeder, 1991), sterol carrier protein-2 (Schroeder et al., 1990), NPC1 (Liu et al., 2009b), and NPC2 (Friedland et al., 2003; Liou et al., 2006) have been established using the fluorescence properties of dehydroergosterol.
1.6.2 Cholestatrienol
Cholestatrienol (=cholesta-5,7,9(11)-triene-3β-ol) is a fluorescent cholesterol probe similar to dehydroergosterol. It differs from dehydroergosterol in the absence of both the double bond Δ22 and the methyl group at C-24 (Fig. 1.5). Thus, cholestatrienol possesses an isooctyl side chain like cholesterol and should therefore mimic cholesterol better than dehydroergosterol. This was indeed the case when their effects on phospholipid condensation by NMR spectroscopy were compared (Scheidt et al., 2003) Both fluorescent sterol analogues have been introduced at the same time as membrane and lipoprotein probes and can be used to measure the sterol exchange between membranes (Bergeron and Scott, 1982; Nemecz et al., 1988). In each case, cholestatrienol is regarded as a cholesterol analogue that mimics the membrane behaviour of cholesterol quite well (Fischer et al., 1984; Smutzer et al., 1986; Schroeder et al., 1988; Hyslop et al., 1990; Yeagle et al., 1990; Scheidt et al., 2003; Bjorkqvist et al., 2005).
Cholestatrienol associates with liquid ordered domains and its quenching by nitroxide-labelled lipids can report on the formation or separation of lipid domains (Bjorkqvist et al., 2005; Heczkova and Slotte, 2006). Cholestatrienol has also been evaluated as an appropriate reporter for sterol–protein interactions (Schroeder et al., 1985). A close interaction between cholesterol and rhodopsin has been demonstrated by fluorescence energy transfer from protein tryptophans to cholestatrienol in retinal rod outer segment disk membranes (Albert et al., 1996). Recently, even the imaging of cholestatrienol-specific fluorescence by confocal microscopy has been reported (Tserentsoodol et al., 2006). Low-density lipoproteins labelled with cholestatrienol crossed the blood-retina barrier and were taken up by the retina within 2 h of intravenous injection. The fluorescent sterol was observed to remain in photoreceptor outer segments for at least 24 h. Presumably, cholestatrienol became highly concentrated in retinal tissues, because imaging could not otherwise be expected under the described conditions (Tserentsoodol et al., 2006). In CHO cells, recent quenching studies performed with cholestatrienol and dehydroergosterol as cholesterol reporters and 2,4,6-trinitrobenzene sulfonic acid as membrane impermeant quencher, provided evidence that cholesterol is preferentially (~60–70%) localized in the cytoplasmic leaflet of the plasma membrane (Mondal et al., 2009).
1.6.3 NBD-Cholesterol
The 7-nitrobenz-2-oxa-1,3-diazol-4-yl (=NBD) fluorophore has been widely used as a reporter group for lipids (Chattopadhyay, 1990). The term NBD-cholesterol causes some confusion in the literature, because a couple of fluorescent cholesterol analogues are available with this name. The two predominantly applied analogues will be designated herein as 22-NBD-cholesterol and 25-NBD-cholesterol, respectively (Fig. 1.5). In further less appropriate NBD-cholesterol variants, the fluorophore including a spacer has been attached at the C-3 OH via an ester linkage. Both 22- and 25-NBD-cholesterol have been employed to study the distribution and dynamics of cholesterol in different systems. The behaviour of lateral phases in cholesterol and phosphatidylcholine monolayers has been visualized by fluorescence microscopy using 22-NBD-cholesterol (Slotte and Mattjus, 1995). Results with model membranes and 25-NBD-cholesterol as reporter indicated that cholesterol may form trans-bilayer, tail-to-tail dimers even at low sterol concentrations (Mukherjee and Chattopadhyay, 1996; Rukmini et al., 2001). McIntyre and Sleight (1991) introduced the fluorescence quenching of NBD by dithionite as an approach to measure the membrane lipid asymmetry. If the NBD group is localized at the outer leaflet of the membrane and is accessible from the aqueous phase, it can be chemically reduced to a non-fluorescent state by water-soluble dithionite. In lipid vesicles, which were selectively labelled with 22-NBD-cholesterol at the outer leaflet, dithionite reduced 95% of the NBD fluorescence (McIntyre and Sleight, 1991). Schroeder et al. (1991) explored the trans-bilayer cholesterol distribution of human erythrocytes using two approaches: photobleaching of 22-NBD-cholesterol and quenching of dehydroergosterol fluorescence. These results suggested an enrichment of cholesterol in the inner leaflet of the erythrocytes (Schroeder et al., 1991) similar to that observed in quenching studies with CHO cells (Mondal et al., 2009). In aggregates and micelles, taurocholic acid quenches the fluorescence of 22-NBD-cholesterol (Cai et al., 2002). Based on the dequenching of the NBD fluorescence, an in vitro assay has been developed to measure the exit of cholesterol from bile acid micelles (Cai et al., 2002). Concerning the quenching behavior of the NBD group, it was observed that the photostability of a NBD-labelled ceramide was strongly dependent on the cholesterol status of the Golgi apparatus where the ceramide accumulates. Cholesterol deprivation of the cells accelerated the photobleaching of the NBD-labelled ceramide several-fold, suggesting that this lipid may be used to monitor cholesterol at the Golgi compartment (Martin et al., 1993).
In recent years, 22-NBD-cholesterol has been mainly used, although the 25-NBD variant has some advantages as compared with 22-NBD-cholesterol. For example, 22-NBD-cholesterol revealed an anomalous distribution behaviour in phosphatidylcholine/cholesterol bilayers (Loura et al., 2001). 25-NBD-cholesterol contains the full isooctyl side chain like cholesterol. A chain length of at least 5 carbons at the 17β-position was necessary for sterols to form visible sterol/phospholipid domains in lipid monolayers (Mattjus et al., 1995). The spectroscopic properties of 25-NBD-cholesterol have been characterized in detail (Chattopadhyay and London, 1987, 1988). A critical evaluation of both NBD-cholesterols has been published by Scheidt et al. (2003), who observed that they adopt a reverse (up-side-down) orientation within a phospholipid bilayer. In contrast, Chattopadhyay and London (1987) found that the fluorophore of 25-NBD-cholesterol was deeply buried within the bilayer. Possibly, the high mobility of the sterol may explain these discrepant results. 22-NBD-cholesterol has successfully been employed to prove and characterize the cholesterol binding of the cholesterol-binding proteins ‘steroidogenic acute regulatory protein’ and ‘sterol carrier protein-2’ by spectroscopic techniques (Avdulov et al., 1999; Petrescu et al., 2001). When applied to CHO cells, a mistargeting to mitochondria has been observed for both 22- and 25-NBD-cholesterol (Mukherjee et al., 1998). However, in L-cell fibroblasts, 22-NBD-cholesterol distributed similarly as dehydroergosterol from the plasma membrane into lipid droplets (Frolov et al., 2000; Atshaves et al., 2000). In hamsters fed with a diet containing 22-NBD-cholesterol, the sterol was found to be absorbed (less efficiently than cholesterol) by intestinal epithelial cells and packaged into lipoproteins (Sparrow et al., 1999). Within the enterocytes most of the sterol was translocated into large apical droplets and was presumably stored there in esterified form. 22-NBD-cholesterol was verified as a good substrate for esterification in different cells (Sparrow et al., 1999; Frolov et al., 2000; Lada et al., 2004). HDL-associated 22-NBD-cholesterol was imaged in 3T3-L1 fibroblasts differentiating to adipocytes (Dagher et al., 2003). At early stages of differentiation, 22-NBD-cholesterol colocalized with scattered Golgi structures, while in developing adipocytes, the fluorescent sterol gradually concentrated in lipid droplets (Dagher et al., 2003).
1.6.4 Bodipy-Cholesterol
Several cholesterol analogs have been synthesized in which the Bodipy group was inserted into the aliphatic chain of cholesterol. The most promising reporter of this series is cholesterol linked to a Bodipy moiety at position C-24 (Fig. 1.5). This compound preferentially partitioned into liquid-ordered domains in model membranes and giant unilamellar vesicles (Li et al., 2006; Shaw et al., 2006; Li and Bittman, 2007). Recently, this compound was used as promising tool to visualize sterol trafficking in living cells and organisms. When compared with [3H]cholesterol, Bodipy-cholesterol has a higher tendency to be released from cells and its esterification rate was markedly lower (Holtta-Vuori et al., 2008).
1.6.5 Fluorescent PEG-Cholesterol
The fluorescein ester of poly(ethyleneglycol)cholesteryl ether (fPEG-Cholesterol) (Fig. 1.5) has been introduced as a special cholesterol probe (Ishiwata et al., 1997; Sato et al., 2004; Takahashi et al., 2007). Due to its water solubility and the absence of a hydroxyl group at C3, fPEG-Cholesterol is certainly not a good cholesterol mimic. However, this probe exclusively incorporates into the outer leaflet of the plasma membrane, co-localizes to some degree with rafts markers, and is thus useful to monitor the dynamics of cholesterol-rich membrane microdomains. The trafficking of fPEG-cholesterol was found to be different from that of dehydroergosterol. FPEG-cholesterol was not observed in the endocytic recycling compartment. Instead, it internalizes slowly via clathrin-independent pathways into endosomes and the Golgi region together with some raft markers. In fixed and permeabilized fibroblasts, the fluorescence pattern of fPEG-cholesterol was similar to that of filipin. The probe was also able to detect the mislocalization of cholesterol in NPC fibroblasts (Sato et al., 2004). However, one should be aware that the ester bond in fPEG-cholesterol could be easily cleaved by intracellular esterases.
1.6.6 Dansyl-Cholestanol
With the synthesis of 6-dansyl-cholestanol we have recently introduced a novel fluorescent cholesterol probe (Fig. 1.5) (Wiegand et al., 2003). The introduction of a photoreactive azo-group at the same position (6-azi-5α-cholestanol, see below) has proven to be a useful tool for cholesterol–protein interaction studies, as described below. Derivatization at position 6 did not change the biophysical parameters of the cholesterol analogue in model membranes (Mintzer et al., 2002). The ‘dansyl’-group was chosen because it is one of the smallest fluorescent groups available. Using CHO cells we compared the behaviour of dansyl-cholestanol versus [3H]cholesterol with respect to esterification rate, efflux kinetics, and distribution in detergent-insoluble lipid domains (‘rafts’). Dansyl-cholestanol showed the same kinetics of esterification by ACAT as compared with [3H]cholesterol. Also, the efflux kinetics and subcellular distribution profile were found to be same for both sterols (Wiegand et al., 2003). Further observations indicated the quality of dansyl-cholestanol as a probe for cholesterol: (i) The cellular influx of dansyl-cholestanol occurred rapidly by an energy-independent pathway via the endoplasmic reticulum. In previous biochemical studies with [3H]cholesterol, it had been proposed that plasma membrane-derived cholesterol passed through the endoplasmic reticulum prior to its transfer to other intracellular sites (Lange et al., 1993; Liscum and Munn, 1999). (ii) Following inhibition of ACAT the unesterified dansyl-cholestanol accumulated in the endoplasmic reticulum in accordance with earlier predictions for cholesterol (Butler et al., 1992; Blanchette-Mackie, 2000). (iii) Dansyl-cholestanol was finally translocated to lipid droplets. This agrees well with the trafficking behavior of 22-NBD-cholesterol and dehydroergosterol as described above. (iv) In a recent imaging study, dansyl-cholestanol was also observed in cholesterol-rich microdomains and showed overall distribution patterns similar as dehydroergosterol (Petrescu et al., 2009). (v) Analysis of the membrane penetration depth revealed that the dansyl group of the probe is localized at the interfacial region of the membrane in agreement with the location of cholesterol in fluid-phase membranes (Shrivastava et al., 2009). Thus, 6-dansyl-cholestanol is certainly a promising cholesterol probe. One disadvantage concerns its substantial photobleaching that shortens the imaging time.
1.7 Spin-Labelled Cholesterol
Spin-labeled lipids provide information about the structure of biological membranes by using nuclear magnetic and electron spin resonance spectroscopy. Cholesterol analogues with a nitroxide spin-label (doxyl moiety) attached at the C-3 or C-25 position have been synthesized to analyze the orientation, distribution and transbilayer movements of the cholesterol probe in liposomes and biological membranes. Spin–spin interaction of 3β-doxyl-5α-cholestane in liposomes provided evidence for the formation of cholesterol-enriched domains (Tampe et al., 1991). Using this probe, it was also observed that cholesterol undergoes a rapid transbilayer movement (<1 min) in liposomes and human erythrocytes (Muller and Herrmann, 2002). An even faster flip-flop (<1 s) of cholesterol has been reported (Steck et al., 2002). Concerning its condensing effect on phospholipids the spin-labeled compound 25-doxyl-cholesterol (Fig. 1.6) was found to be an excellent cholesterol analogue. This sterol probe revealed a cholesterol-like orientation, with the doxyl group at C-25 facing the chain termini of the phospholipids (Scheidt et al., 2003). The localization of the doxyl group in the membrane interior was confirmed by the finding that the nitroxide label was inaccessible from the aqueous phase as it could not be reduced by ascorbate (Scheidt et al., 2003). Probes with the spin-label group accessible from the aqueous phase (e.g. at C-3 as in 3β-doxyl-5α-cholestane) allow to measure cholesterol flip-flop by chemical reduction of the nitroxide radical with ascorbate (Morrot et al., 1987). However, cholesterol analogues with modifications at C-3 are not regarded as faithful mimics of cholesterol. For example, 3β-doxyl-5α-cholestane was not able to exert a comparable condensing effect on phospholipids as cholesterol (Scheidt et al., 2003). Investigations on the transmembrane diffusion of lipids obtained with spin-labeled and fluorescent lipid probes have recently been summarized (Devaux et al., 2002).


Spin-labelled (A) and photoreactive (B) cholesterol analogues: (A) 25-doxyl-cholestanol; (B) [3H]6-azi-5α-cholestanol, [3H]7-azi-5α-cholestanol, 22-(p-benzoylphenoxy)-23,24-bisnorcholan-5-en-3β-ol (D, R=R1), the fluorenone moiety (D, R=R2)
1.8 Affinity Labelling with Photoreactive Cholesterol
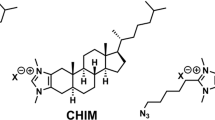
Specific cholesterol binding proteins can be directly identified by the usage of photoreactive cholesterol analogues. In the first photoreactive cholesterol analogues that were synthesized, the photoreactive groups were incorporated either at the C-3 position (cholesteryl diazoacetate, 3α-azido-5-cholestene, or 3α-(4-azido-3-iodosalicylic)-cholest-5-ene) (Middlemas and Raftery, 1987; Corbin et al., 1998) or at the aliphatic side chain of cholesterol (25-azidonorcholesterol or sterols with diazoacetate, aryldiazirines or fluorodiazirine attached at C-22 or C-24) (Stoffel and Klotzbucher, 1978; Terasawa et al., 1986). Unfortunately, not many applications have been described for most of these compounds. The nicotinic acetylcholine receptor binds cholesterol but reveals very low structure–activity requirements for cholesterol. Even analogues derivatized at the C-3 positions with a broad range of substituents or bile acid derivatives support receptor activity (Fernandez et al., 1993; Corbin et al., 1998). Using 3α-(4-azido-3-iodosalicylic)-cholest-5-ene or the bile acid p-azidophenacyl 3α-hydroxy-5β-cholan-24-ate as photoreactive probes, all subunits of the nicotinic acetylcholine receptor could be labeled in membranes or proteoliposomes (Corbin et al., 1998). Photoreactive cholesteryl diazoacetate also labeled the nicotinic acetylcholine receptor (Middlemas and Raftery, 1987). Although this probe is modified at C-3, it immobilized in lipid bilayers like cholesterol and upon irradiation incorporated into the choline head group of phosphatidylcholine (Keilbaugh and Thornton, 1983). However, cholesteryl diazoacetate behaved differently to cholesterol concerning its exchange kinetics from unilamellar vesicles (Kan et al., 1992). The acetylcholine receptor may be an exception concerning its broad tolerance for cholesterol substitutes. To develop a probe with more general applicability, we synthesized [3H]6-azi-5α-cholestanol in which both the C-3 and the isooctyl side chain left unattached (Fig. 1.6B) (Gimpl and Gehrig-Burger, 2007). The azi-group was introduced at position C-6 because this modification was functionally tolerated by the oxytocin receptor that we studied in detail with respect to its specific requirement for cholesterol (Klein et al., 1995; Gimpl et al., 1995, 1997; Burger, 2000). The first application of this compound (designated as photocholesterol) was published by Thiele et al. (2000). Up to now, several putative cholesterol binding proteins have been labelled with 6-azi-5α-cholestanol, among these are synaptophysin, caveolin (Thiele et al., 2000), vitellogenins (Matyash et al., 2001), proteolipid protein (Simons et al., 2000; Kramer-Albers et al., 2006), tetraspanins (Charrin et al., 2003), cholesterol absorption proteins in enterocytes (Kramer et al., 2003), STARD3 (Alpy et al., 2005; Reitz et al., 2008), and the E1 fusion protein from Semliki Forest virus (Umashankar et al., 2008). In all these studies, 6-azi-5α-cholestanol was primarily used to identify or confirm the cholesterol binding of a candidate protein. Recently, we demonstrated for STARD1 that this photoreactive probe could also be used to identify cholesterol binding sites within a protein (Reitz et al., 2008). Another related tritiated photoreactive cholesterol analogue, [3H]7-azi-5α-cholestanol (Fig. 1.6B), has been synthesized by Cruz et al. (2002). A direct binding of this analogue with caveolin-1 and Niemann-Pick C1 (NPC1) protein has been demonstrated (Cruz et al., 2002; Ohgami et al., 2004; Liu et al., 2009b).
Spencer et al. (2004) synthesized a series of eight benzophenone-containing photoreactive cholesterol analogues. Due to the larger size of these photophores compared with the diazirines, these sterol analogues have the disadvantage of being less cholesterol-like. On the other hand, benzophenone derivatives show a high crosslinking yield and a preferential reaction with C-H bonds, which may be beneficial for the sterol labelling of some proteins. In one group of benzophenone-containing cholesterol probes, the photophore moiety extended, or replaced, most of the cholesterol isooctyl side chain. In another group of analogues, the photophore was attached at C-3 via an amide linkage. Surprisingly, for all of these analogues even those with modifications at C-3 were similarly effective as cholesterol when tested in an apolipoprotein A-I dependent sterol efflux assay. This indicates that at least in relation to certain transport pathways of cholesterol, biological membranes show an unexpected tolerance for cholesterol substitutes (Spencer et al., 2004). One of these analogues, tritiated 22-(p-benzoylphenoxy)-23,24-bisnorcholan-5-en-3β-ol (Fig. 1.6B, R=R1), photolabelled caveolin effectively (Fielding et al., 2002). Fluorenone-containing cholesterol probes that are structurally similar to the corresponding benzophenone derivatives (Fig. 1.6B, R=R2) represent a further interesting group of compounds since they are both photoreactive and fluorescent (Spencer et al., 2006). Two such analogues behaved similar to cholesterol in the above mentioned sterol efflux assay (Spencer et al., 2006).
In experiments with photoreactive cholesterol analogues, one has to consider that at least in the plasma membrane cholesterol is always present in large amounts, so that each integral membrane protein faces cholesterol in its direct environment. One expects that only membrane proteins are photolabelled that possess one ore more specific cholesterol docking site(s). Membrane proteins residing in cholesterol-enriched microdomains are good candidates. However, even if they are functionally dependent on cholesterol, their affinity for cholesterol could be low (~ millimolar range), since embedded in a cholesterol-rich environment they will always be in contact with cholesterol. In contrast, cholesterol-dependent membrane proteins residing in cholesterol-poor organelles such as the mitochondrion or the endoplasmic reticulum, should be evolutionary selected towards higher affinity for cholesterol. Therefore, it should be considered that the employment of high concentrations of photoreactive cholesterol could artefactually label proteins that do not possess a specific cholesterol binding site.
1.9 Concluding Remarks
A couple of membrane proteins such as GPCRs, receptor tyrosine kinases and ion channels, have been shown to reside in cholesterol-rich microdomains. Some of them may directly interact with cholesterol. However, due to the abundance of cholesterol in the plasma membrane, particularly in lipid rafts, the affinity of these proteins to cholesterol may be very low and therefore difficult to determine with traditional radioligand binding assays. Therefore, alternative binding protocols are required. To explore the binding or interaction of proteins to cholesterol, a variety of cholesterol probes bearing radio-, spin-, photoaffinity- or fluorescent labels are currently available. Examples of proven cholesterol binding molecules are polyene compounds, cholesterol-dependent cytolysins, enzymes accepting cholesterol as substrate, and proteins with cholesterol binding motifs. As far as we know to date, cholesterol binding domains are heterogenous structures existing either as hydrophobic cavities, an assembly of several transmembrane helices, or small stretches of amino acids. Possibly, water-soluble cholesterol binding modules could be applied in the future as ‘solubilizers’ and competitors for cholesterol binding. Of course, to be useful as potential cholesterol donors in binding assays, the affinity of these binding modules to cholesterol has to be lower than that of the candidate cholesterol binding protein. In contrast, high-affinity cholesterol binding domains might be useful as efficient cholesterol acceptors and could then substitute for the widely used β-cyclodextrins that currently function as non-specific cholesterol carrier molecules. Among the cholesterol modifying enzymes, the cholesterol oxidases have achieved a wide range of applications. They are used to determine the cholesterol concentration in all kinds of biological samples such as membranes, cells, serum, or food. Additionally, susceptibility to cholesterol oxidase provides information about the localization of cholesterol and the structure of cholesterol-containing membranes. Filipin, a member of the polyene compounds, is the standard reporter for the distribution of free cholesterol in fixed cells. Among the family of cholesterol-dependent cytolysin, perfringolysin O and fragments therefrom have been introduced to selectively label the localization of cholesterol-rich microdomains. Hopefully, other members of this family will be added as cholesterol probes in the future. Cholesterol research has been markedly stimulated by the development of different cholesterol derivatives such as photoreactive, spin-labelled, and fluorescent cholesterol analogues. The administration of photoreactive cholesterol probes offers a direct approach to identify and define cholesterol binding sites, whereas fluorescent sterols allow us to explore the trafficking and distribution of cholesterol in vivo. In addition, fluorescent cholesterol analogues have been established as important tools to analyze cholesterol-protein binding. Due to tremendous progress in microscopy/spectroscopy (e.g. FRET analysis, lifetime microscopy) in recent years, cholesterol research will focus more and more on living cells, thereby expanding our knowledge on all facets of cholesterol–protein interaction in the future.
Abbreviations
- ACAT:
-
acyl-coenzyme A:cholesterol acyltransferase
- BCθ-toxin:
-
a biotinylated and carlsberg protease-nicked derivative of perfringolysin O
- Benzophenone-cholesterol:
-
22-(p-benzoylphenoxy)-23,24-bisnorcholan-5-en-3β-ol
- Bodipy:
-
boron dipyrromethene(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diazara-s-indacene)
- CCKBR:
-
cholecystokinin receptor type B
- CCM:
-
cholesterol consensus motif
- CDCs:
-
cholesterol-dependent cytolysins
- CRAC:
-
cholesterol recognition/interaction amino acid consensus
- DIG, (= DRM):
-
detergent-insoluble (= detergent resistant) glycosphingolipid-enriched membrane domains
- GPCR:
-
G protein coupled receptor
- HDL:
-
high-density lipoprotein
- HPβCD:
-
2-Hydroxypropyl)-β-cyclodextrin
- 5HT1A:
-
5-hydroxytryptamine 1A
- LDL:
-
low-density lipoprotein
- LDM:
-
low-density microdomains
- MβCD:
-
methyl-β-cyclodextrin
- 22-NBD Cholesterol:
-
22-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-23,24-bisnor-5-cholen-3β-ol
- 25-NBD Cholesterol:
-
25-[N-[(7-nitro-2-1,3-benzoxadiazol-4-yl)methyl]amino]-27-norcholesterol
- NMR:
-
nuclear magnetic resonance
- NPC:
-
Niemann-Pick C
- OTR:
-
oxytocin receptor
- PBR:
-
peripheral benzodiazepine receptor (= TSPO)
- SCAP:
-
SREBP cleavage activating protein
- SREBP:
-
sterol regulatory element binding protein
- SSD:
-
sterol sensing domain
- StAR:
-
steroid acute regulatory protein
- START:
-
StAR related lipid transfer
- TSPO:
-
mitochondrial translocator protein (18 kDa) (= PBR)
References
Adams, C.M., Reitz, J., De Brabander, J.K., Feramisco, J.D., Li, L., Brown, M.S., Goldstein, J.L., 2004, Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J. Biol. Chem. 279: 52772–52780.
Ahn, K.W., Sampson, N.S., 2004, Cholesterol oxidase senses subtle changes in lipid bilayer structure. Biochemistry 43: 827–836.
Albert, A.D., Young, J.E., Yeagle, P.L., 1996, Rhodopsin-cholesterol interactions in bovine rod outer segment disk membranes. Biochim. Biophys. Acta 1285: 47–55.
Alpy, F., Latchumanan, V.K., Kedinger, V., Janoshazi, A., Thiele, C., Wendling, C., Rio, M.C., Tomasetto, C., 2005, Functional characterization of the MENTAL domain. J. Biol. Chem. 280: 17945–17952.
Anderluh, G., Lakey, J.H., 2008, Disparate proteins use similar architectures to damage membranes. Trends Biochem. Sci. 33: 482–490.
Atshaves, B.P., Starodub, O., McIntosh, A., Petrescu, A., Roths, J.B., Kier, A.B., Schroeder, F., 2000, Sterol carrier protein-2 alters high density lipoprotein-mediated cholesterol efflux. J. Biol. Chem. 275: 36852–36861.
Avdulov, N.A., Chochina, S.V., Igbavboa, U., Warden, C.S., Schroeder, F., Wood, W.G., 1999, Lipid binding to sterol carrier protein-2 is inhibited by ethanol. Biochim. Biophys. Acta 1437: 37–45.
Baker, B.Y., Epand, R.F., Epand, R.M., Miller, W.L., 2007, Cholesterol binding does not predict activity of the steroidogenic acute regulatory protein, StAR. J. Biol. Chem. 282: 10223–10232.
Bar, L.K., Chong, P.L., Barenholz, Y., Thompson, T.E., 1989, Spontaneous transfer between phospholipid bilayers of dehydroergosterol, a fluorescent cholesterol analog. Biochim. Biophys. Acta 983: 109–112.
Bari, M., Battista, N., Fezza, F., Finazzi-Agro, A., Maccarrone, M., 2005, Lipid rafts control signaling of type-1 cannabinoid receptors in neuronal cells. Implications for anandamide-induced apoptosis. J. Biol. Chem. 280: 12212–12220.
Behnke, O., Tranum-Jensen, J., van, D.B., 1984, Filipin as a cholesterol probe. II. Filipin-cholesterol interaction in red blood cell membranes. Eur. J. Cell Biol. 35: 200–215.
Bergeron, R.J., Scott, J., 1982, Cholestatriene and ergostatetraene as in vivo and in vitro membrane and lipoprotein probes. J. Lipid Res. 23: 391–404.
Bjorkqvist, Y.J., Nyholm, T.K., Slotte, J.P., Ramstedt, B., 2005, Domain formation and stability in complex lipid bilayers as reported by cholestatrienol. Biophys. J. 88: 4054–4063.
Blanchette-Mackie, E.J., 2000, Intracellular cholesterol trafficking: role of the NPC1 protein. Biochim. Biophys. Acta 1486: 171–183.
Bolard, J., 1986, How do the polyene macrolide antibiotics affect the cellular membrane properties? Biochim. Biophys. Acta 864: 257–304.
Brannigan, G., Henin, J., Law, R., Eckenhoff, R., Klein, M.L., 2008, Embedded cholesterol in the nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. U. S. A 105: 14418–14423.
Brasaemle, D.L., Robertson, A.D., Attie, A.D., 1988, Transbilayer movement of cholesterol in the human erythrocyte membrane. J. Lipid Res. 29: 481–489.
Brown, A.J., Sun, L., Feramisco, J., Brown, M.S., Goldstein, J.L., 2002, Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol. Cell 10: 237–245.
Brown, D.A., Rose, J.K., 1992, Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 68: 533–544.
Burger, K., 2000, Cholesterin und Progesteron – Modulatoren G-Protein gekoppelter Signaltransduktionswege. Dissertation, Mainz.
Burger, K., Gimpl, G., Fahrenholz, F., 2000, Regulation of receptor function by cholesterol. Cell Mol. Life Sci. 57: 1577–1592.
Butler, J.D., Blanchette-Mackie, J., Goldin, E., O’Neill, R.R., Carstea, G., Roff, C.F., Patterson, M.C., Patel, S., Comly, M.E., Cooney, A., 1992, Progesterone blocks cholesterol translocation from lysosomes. J. Biol. Chem. 267: 23797–23805.
Butler, J.D., Comly, M.E., Kruth, H.S., Vanier, M., Filling-Katz, M., Fink, J., Barton, N., Weintroub, H., Quirk, J.M., Tokoro, T., 1987, Niemann-Pick variant disorders: comparison of errors of cellular cholesterol homeostasis in group D and group C fibroblasts. Proc. Natl. Acad. Sci. U. S. A 84: 556–560.
Cai, T.Q., Guo, Q., Wong, B., Milot, D., Zhang, L., Wright, S.D., 2002, Protein-disulfide isomerase is a component of an NBD-cholesterol monomerizing protein complex from hamster small intestine. Biochim. Biophys. Acta 1581: 100–108.
Challa, R., Ahuja, A., Ali, J., Khar, R.K., 2005, Cyclodextrins in drug delivery: an updated review. AAPS PharmSciTech. 6: E329–E357.
Chamberlain, L.H., 2004, Detergents as tools for the purification and classification of lipid rafts. FEBS Lett. 559: 1–5.
Chamberlain, L.H., Gould, G.W., 2002, The vesicle- and target-SNARE proteins that mediate Glut4 vesicle fusion are localized in detergent-insoluble lipid rafts present on distinct intracellular membranes. J. Biol. Chem. 277: 49750–49754.
Chang, C.C., Lee, C.Y., Chang, E.T., Cruz, J.C., Levesque, M.C., Chang, T.Y., 1998, Recombinant acyl-CoA:cholesterol acyltransferase-1 (ACAT-1) purified to essential homogeneity utilizes cholesterol in mixed micelles or in vesicles in a highly cooperative manner. J. Biol. Chem. 273: 35132–35141.
Charrin, S., Manie, S., Thiele, C., Billard, M., Gerlier, D., Boucheix, C., Rubinstein, E., 2003, A physical and functional link between cholesterol and tetraspanins. Eur. J. Immunol. 33: 2479–2489.
Chattopadhyay, A., 1990, Chemistry and biology of N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-labeled lipids: fluorescent probes of biological and model membranes. Chem. Phys. Lipids 53: 1–15.
Chattopadhyay, A., London, E., 1987, Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry 26: 39–45.
Chattopadhyay, A., London, E., 1988, Spectroscopic and ionization properties of N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-labeled lipids in model membranes. Biochim. Biophys. Acta 938: 24–34.
Chen, L., Lyubimov, A.Y., Brammer, L., Vrielink, A., Sampson, N.S., 2008, The binding and release of oxygen and hydrogen peroxide are directed by a hydrophobic tunnel in cholesterol oxidase. Biochemistry 47: 5368–5377.
Cheruku, S.R., Xu, Z., Dutia, R., Lobel, P., Storch, J., 2006, Mechanism of cholesterol transfer from the Niemann-Pick type C2 protein to model membranes supports a role in lysosomal cholesterol transport. J. Biol. Chem. 281: 31594–31604.
Chiang, Y.R., Ismail, W., Heintz, D., Schaeffer, C., Van, D.A., Fuchs, G., 2008, Study of anoxic and oxic cholesterol metabolism by Sterolibacterium denitrificans. J. Bacteriol. 190: 905–914.
Chini, B., Parenti, M., 2004, G-protein coupled receptors in lipid rafts and caveolae: how, when and why do they go there? J. Mol. Endocrinol. 32: 325–338.
Corbin, J., Wang, H.H., Blanton, M.P., 1998, Identifying the cholesterol binding domain in the nicotinic acetylcholine receptor with [125I]azido-cholesterol. Biochim. Biophys. Acta 1414: 65–74.
Cruz, J.C., Thomas, M., Wong, E., Ohgami, N., Sugii, S., Curphey, T., Chang, C.C., Chang, T.Y., 2002, Synthesis and biochemical properties of a new photoactivatable cholesterol analog 7,7-azocholestanol and its linoleate ester in Chinese hamster ovary cell lines. J. Lipid Res. 43: 1341–1347.
Dagher, G., Donne, N., Klein, C., Ferre, P., Dugail, I., 2003, HDL-mediated cholesterol uptake and targeting to lipid droplets in adipocytes. J. Lipid Res. 44: 1811–1820.
Das, A., Davis, M.A., Rudel, L.L., 2008, Identification of putative active site residues of ACAT enzymes. J. Lipid Res. 49: 1770–1781.
Devaux, P.F., Fellmann, P., Herve, P., 2002, Investigation on lipid asymmetry using lipid probes: Comparison between spin-labeled lipids and fluorescent lipids. Chem. Phys. Lipids 116: 115–134.
Douglass, A.D., Vale, R.D., 2005, Single-molecule microscopy reveals plasma membrane microdomains created by protein-protein networks that exclude or trap signaling molecules in T cells. Cell 121: 937–950.
Dowler, S., Kular, G., Alessi, D.R., 2002, Protein lipid overlay assay. Sci. STKE 2002: L6.
Drevot, P., Langlet, C., Guo, X.J., Bernard, A.M., Colard, O., Chauvin, J.P., Lasserre, R., He, H.T., 2002, TCR signal initiation machinery is pre-assembled and activated in a subset of membrane rafts. EMBO J. 21: 1899–1908.
Elias, P.M., Friend, D.S., Goerke, J., 1979, Membrane sterol heterogeneity. Freeze-fracture detection with saponins and filipin. J. Histochem. Cytochem. 27: 1247–1260.
Epshtein, Y., Chopra, A.P., Rosenhouse-Dantsker, A., Kowalsky, G.B., Logothetis, D.E., Levitan, I., 2009, Identification of a C-terminus domain critical for the sensitivity of Kir2.1 to cholesterol. Proc. Natl. Acad. Sci. U. S. A 106: 8055–8060.
Eroglu, C., Brugger, B., Wieland, F., Sinning, I., 2003, Glutamate-binding affinity of Drosophila metabotropic glutamate receptor is modulated by association with lipid rafts. Proc. Natl. Acad. Sci. U. S. A 100: 10219–10224.
Fernandez, A.M., Fernandez-Ballester, G., Ferragut, J.A., Gonzalez-Ros, J.M., 1993, Labeling of the nicotinic acetylcholine receptor by a photoactivatable steroid probe: effects of cholesterol and cholinergic ligands. Biochim. Biophys. Acta 1149: 135–144.
Fielding, P.E., Russel, J.S., Spencer, T.A., Hakamata, H., Nagao, K., Fielding, C.J., 2002, Sterol efflux to apolipoprotein A-I originates from caveolin-rich microdomains and potentiates PDGF-dependent protein kinase activity. Biochemistry 41: 4929–4937.
Fischer, R.T., Stephenson, F.A., Shafiee, A., Schroeder, F., 1984, delta 5,7,9(11)-Cholestatrien-3 beta-ol: a fluorescent cholesterol analogue. Chem. Phys. Lipids 36: 1–14.
Florek, M., Bauer, N., Janich, P., Wilsch-Braeuninger, M., Fargeas, C.A., Marzesco, A.M., Ehninger, G., Thiele, C., Huttner, W.B., Corbeil, D., 2007, Prominin-2 is a cholesterol-binding protein associated with apical and basolateral plasmalemmal protrusions in polarized epithelial cells and released into urine. Cell Tissue Res. 328: 31–47.
Friedland, N., Liou, H.L., Lobel, P., Stock, A.M., 2003, Structure of a cholesterol-binding protein deficient in Niemann-Pick type C2 disease. Proc. Natl. Acad. Sci. U. S. A 100: 2512–2517.
Frolov, A., Petrescu, A., Atshaves, B.P., So, P.T., Gratton, E., Serrero, G., Schroeder, F., 2000, High density lipoprotein-mediated cholesterol uptake and targeting to lipid droplets in intact L-cell fibroblasts. A single- and multiphoton fluorescence approach. J. Biol. Chem. 275: 12769–12780.
Frolov, A., Woodford, J.K., Murphy, E.J., Billheimer, J.T., Schroeder, F., 1996, Spontaneous and protein-mediated sterol transfer between intracellular membranes. J. Biol. Chem. 271: 16075–16083.
Fujimoto, T., Hayashi, M., Iwamoto, M., Ohno-Iwashita, Y., 1997, Crosslinked plasmalemmal cholesterol is sequestered to caveolae: analysis with a new cytochemical probe. J. Histochem. Cytochem. 45: 1197–1205.
Gehrig-Burger, K., Kohout, L., Gimpl, G., 2005, CHAPSTEROL. A novel cholesterol-based detergent. FEBS J. 272: 800–812.
Ghoshroy, K.B., Zhu, W., Sampson, N.S., 1997, Investigation of membrane disruption in the reaction catalyzed by cholesterol oxidase. Biochemistry 36: 6133–6140.
Gimpl, G., Burger, K., Fahrenholz, F., 1997, Cholesterol as modulator of receptor function. Biochemistry 36: 10959–10974.
Gimpl, G., Fahrenholz, F., 2000, Human oxytocin receptors in cholesterol-rich vs. cholesterol-poor microdomains of the plasma membrane. Eur. J. Biochem. 267: 2483–2497.
Gimpl, G., Gehrig-Burger, K., 2007, Cholesterol reporter molecules. Biosci. Rep. 27: 335–358.
Gimpl, G., Klein, U., Reilander, H., Fahrenholz, F., 1995, Expression of the human oxytocin receptor in baculovirus-infected insect cells: high-affinity binding is induced by a cholesterol- cyclodextrin complex. Biochemistry 34: 13794–13801.
Gronberg, L., Slotte, J.P., 1990, Cholesterol oxidase catalyzed oxidation of cholesterol in mixed lipid monolayers: effects of surface pressure and phospholipid composition on catalytic activity. Biochemistry 29: 3173–3178.
Gustavsson, J., Parpal, S., Karlsson, M., Ramsing, C., Thorn, H., Borg, M., Lindroth, M., Peterson, K.H., Magnusson, K.E., Stralfors, P., 1999, Localization of the insulin receptor in caveolae of adipocyte plasma membrane. FASEB J. 13: 1961–1971.
Hadders, M.A., Beringer, D.X., Gros, P., 2007, Structure of C8alpha-MACPF reveals mechanism of membrane attack in complement immune defense. Science 317: 1552–1554.
Hanson, M.A., Cherezov, V., Griffith, M.T., Roth, C.B., Jaakola, V.P., Chien, E.Y., Velasquez, J., Kuhn, P., Stevens, R.C., 2008, A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 16: 897–905.
Hao, M., Lin, S.X., Karylowski, O.J., Wustner, D., McGraw, T.E., Maxfield, F.R., 2002, Vesicular and non-vesicular sterol transport in living cells. The endocytic recycling compartment is a major sterol storage organelle. J. Biol. Chem. 277: 609–617.
Heczkova, B., Slotte, J.P., 2006, Effect of anti-tumor ether lipids on ordered domains in model membranes. FEBS Lett. 580: 2471–2476.
Heerklotz, H., 2002, Triton promotes domain formation in lipid raft mixtures. Biophys. J. 83: 2693–2701.
Holtta-Vuori, M., Uronen, R.L., Repakova, J., Salonen, E., Vattulainen, I., Panula, P., Li, Z., Bittman, R., Ikonen, E., 2008, BODIPY-cholesterol: a new tool to visualize sterol trafficking in living cells and organisms. Traffic 9: 1839–1849.
Huang, P., Xu, W., Yoon, S.I., Chen, C., Chong, P.L., Liu-Chen, L.Y., 2007, Cholesterol reduction by methyl-beta-cyclodextrin attenuates the delta opioid receptor-mediated signaling in neuronal cells but enhances it in non-neuronal cells. Biochem. Pharmacol. 73: 534–549.
Hyslop, P.A., Morel, B., Sauerheber, R.D., 1990, Organization and interaction of cholesterol and phosphatidylcholine in model bilayer membranes. Biochemistry 29: 1025–1038.
Ilangumaran, S., Hoessli, D.C., 1998, Effects of cholesterol depletion by cyclodextrin on the sphingolipid microdomains of the plasma membrane. Biochem. J. 335 (Pt 2): 433–440.
Infante, R.E., bi-Mosleh, L., Radhakrishnan, A., Dale, J.D., Brown, M.S., Goldstein, J.L., 2008a, Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J. Biol. Chem. 283: 1052–1063.
Infante, R.E., Radhakrishnan, A., bi-Mosleh, L., Kinch, L.N., Wang, M.L., Grishin, N.V., Goldstein, J.L., Brown, M.S., 2008b, Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J. Biol. Chem. 283: 1064–1075.
Infante, R.E., Wang, M.L., Radhakrishnan, A., Kwon, H.J., Brown, M.S., Goldstein, J.L., 2008c, NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. U. S. A 105: 15287–15292.
Irie, T., Fukunaga, K., Pitha, J., 1992, Hydroxypropylcyclodextrins in parenteral use. I: Lipid dissolution and effects on lipid transfers in vitro. J. Pharm. Sci. 81: 521–523.
Ishiwata, H., Sato, S.B., Vertut-Doi, A., Hamashima, Y., Miyajima, K., 1997, Cholesterol derivative of poly(ethylene glycol) inhibits clathrin-independent, but not clathrin-dependent endocytosis. Biochim. Biophys. Acta 1359: 123–135.
Iwamoto, M., Morita, I., Fukuda, M., Murota, S., Ando, S., Ohno-Iwashita, Y., 1997, A biotinylated perfringolysin O derivative: a new probe for detection of cell surface cholesterol. Biochim. Biophys. Acta 1327: 222–230.
Jamin, N., Neumann, J.M., Ostuni, M.A., Vu, T.K., Yao, Z.X., Murail, S., Robert, J.C., Giatzakis, C., Papadopoulos, V., Lacapere, J.J., 2005, Characterization of the cholesterol recognition amino acid consensus sequence of the peripheral-type benzodiazepine receptor. Mol. Endocrinol. 19: 588–594.
Kallen, J.A., Schlaeppi, J.M., Bitsch, F., Geisse, S., Geiser, M., Delhon, I., Fournier, B., 2002, X-ray structure of the hRORalpha LBD at 1.63 A: structural and functional data that cholesterol or a cholesterol derivative is the natural ligand of RORalpha. Structure 10: 1697–1707.
Kan, C.C., Yan, J., Bittman, R., 1992, Rates of spontaneous exchange of synthetic radiolabeled sterols between lipid vesicles. Biochemistry 31: 1866–1874.
Keilbaugh, S.A., Thornton, E.R., 1983, Synthesis and photoreactivity of cholesteryl diazoacetate: a novel photolabeling reagent. J. Am. Chem. Soc. 105: 3283–3286.
Kenworthy, A.K., 2008, Have we become overly reliant on lipid rafts? Talking point on the involvement of lipid rafts in T-cell activation. EMBO Rep. 9: 531–535.
Kilsdonk, E.P., Yancey, P.G., Stoudt, G.W., Bangerter, F.W., Johnson, W.J., Phillips, M.C., Rothblat, G.H., 1995, Cellular cholesterol efflux mediated by cyclodextrins. J. Biol. Chem. 270: 17250–17256.
Kinsky, S.C., Luse, S.A., Zopf, D., van Deenen, L.L., Haxby, J., 1967, Interaction of filipin and derivatives with erythrocyte membranes and lipid dispersions: electron microscopic observations. Biochim. Biophys. Acta 135: 844–861.
Kishi, K., Watazu, Y., Katayama, Y., Okabe, H., 2000, The characteristics and applications of recombinant cholesterol dehydrogenase. Biosci. Biotechnol. Biochem. 64: 1352–1358.
Klein, U., Gimpl, G., Fahrenholz, F., 1995, Alteration of the myometrial plasma membrane cholesterol content with beta-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry 34: 13784–13793.
Ko, D.C., Binkley, J., Sidow, A., Scott, M.P., 2003, The integrity of a cholesterol-binding pocket in Niemann-Pick C2 protein is necessary to control lysosome cholesterol levels. Proc. Natl. Acad. Sci. U. S. A. 100: 2518–2525.
Kramer, W., Girbig, F., Corsiero, D., Burger, K., Fahrenholz, F., Jung, C., Muller, G., 2003, Intestinal cholesterol absorption: identification of different binding proteins for cholesterol and cholesterol absorption inhibitors in the enterocyte brush border membrane. Biochim. Biophys. Acta 1633: 13–26.
Kramer-Albers, E.M., Gehrig-Burger, K., Thiele, C., Trotter, J., Nave, K.A., 2006, Perturbed interactions of mutant proteolipid protein/DM20 with cholesterol and lipid rafts in oligodendroglia: implications for dysmyelination in spastic paraplegia. J. Neurosci. 26: 11743–11752.
Kurzchalia, T.V., Parton, R.G., 1999, Membrane microdomains and caveolae. Curr. Opin. Cell Biol. 11: 424–431.
Kuwabara, P.E., Labouesse, M., 2002, The sterol-sensing domain: multiple families, a unique role? Trends Genet. 18: 193–201.
Lada, A.T., Davis, M., Kent, C., Chapman, J., Tomoda, H., Omura, S., Rudel, L.L., 2004, Identification of ACAT1- and ACAT2-specific inhibitors using a novel, cell based fluorescence assay: individual ACAT uniqueness. J. Lipid Res. 45: 378–386.
Lafont, F., Simons, K., Ikonen, E., 1995, Dissecting the molecular mechanisms of polarized membrane traffic: reconstitution of three transport steps in epithelial cells using streptolysin-O permeabilization. Cold Spring Harb. Symp. Quant. Biol. 60: 753–762.
Lange, Y., 1991, Disposition of intracellular cholesterol in human fibroblasts. J. Lipid Res. 32: 329–339.
Lange, Y., 1992, Tracking cell cholesterol with cholesterol oxidase. J. Lipid Res. 33: 315–321.
Lange, Y., Dolde, J., Steck, T.L., 1981, The rate of transmembrane movement of cholesterol in the human erythrocyte. J. Biol. Chem. 256: 5321–5323.
Lange, Y., Matthies, H., Steck, T.L., 1984, Cholesterol oxidase susceptibility of the red cell membrane. Biochim. Biophys. Acta 769: 551–562.
Lange, Y., Matthies, H.J., 1984, Transfer of cholesterol from its site of synthesis to the plasma membrane. J. Biol. Chem. 259: 14624–14630.
Lange, Y., Strebel, F., Steck, T.L., 1993, Role of the plasma membrane in cholesterol esterification in rat hepatoma cells. J. Biol. Chem. 268: 13838–13843.
Lange, Y., Swaisgood, M.H., Ramos, B.V., Steck, T.L., 1989, Plasma membranes contain half the phospholipid and 90% of the cholesterol and sphingomyelin in cultured human fibroblasts. J. Biol. Chem. 264: 3786–3793.
Lange, Y., Ye, J., Steck, T.L., 2004, How cholesterol homeostasis is regulated by plasma membrane cholesterol in excess of phospholipids. Proc. Natl. Acad. Sci. U. S. A 101: 11664–11667.
Lange, Y., Ye, J., Steck, T.L., 2005, Activation of membrane cholesterol by displacement from phospholipids. J. Biol. Chem. 280: 36126–36131.
Lascombe, M.B., Ponchet, M., Venard, P., Milat, M.L., Blein, J.P., Prange, T., 2002, The 1.45 A resolution structure of the cryptogein-cholesterol complex: a close-up view of a sterol carrier protein (SCP) active site. Acta Crystallogr. D. Biol. Crystallogr. 58: 1442–1447.
Lee, K.A., Fuda, H., Lee, Y.C., Negishi, M., Strott, C.A., Pedersen, L.C., 2003, Crystal structure of human cholesterol sulfotransferase (SULT2B1b) in the presence of pregnenolone and 3'-phosphoadenosine 5'-phosphate. Rationale for specificity differences between prototypical SULT2A1 and the SULT2BG1 isoforms. J. Biol. Chem. 278: 44593–44599.
Li, H., Yao, Z., Degenhardt, B., Teper, G., Papadopoulos, V., 2001, Cholesterol binding at the cholesterol recognition/ interaction amino acid consensus (CRAC) of the peripheral-type benzodiazepine receptor and inhibition of steroidogenesis by an HIV TAT-CRAC peptide. Proc. Natl. Acad. Sci. U. S. A 98: 1267–1272.
Li, J., Vrielink, A., Brick, P., Blow, D.M., 1993, Crystal structure of cholesterol oxidase complexed with a steroid substrate: implications for flavin adenine dinucleotide dependent alcohol oxidases. Biochemistry 32: 11507–11515.
Li, Z., Bittman, R., 2007, Synthesis and spectral properties of cholesterol- and FTY720-containing boron dipyrromethene dyes. J. Org. Chem. 72: 8376–8382.
Li, Z., Mintzer, E., Bittman, R., 2006, First synthesis of free cholesterol-BODIPY conjugates. J. Org. Chem. 71: 1718–1721.
Lichtenberg, D., Goni, F.M., Heerklotz, H., 2005, Detergent-resistant membranes should not be identified with membrane rafts. Trends Biochem. Sci. 30: 430–436.
Liou, H.L., Dixit, S.S., Xu, S., Tint, G.S., Stock, A.M., Lobel, P., 2006, NPC2, the protein deficient in Niemann-Pick C2 disease, consists of multiple glycoforms that bind a variety of sterols. J. Biol. Chem. 281: 36710–36723.
Liscum, L., Munn, N.J., 1999, Intracellular cholesterol transport. Biochim. Biophys. Acta 1438: 19–37.
Liu, B., Turley, S.D., Burns, D.K., Miller, A.M., Repa, J.J., Dietschy, J.M., 2009a, Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1-/- mouse. Proc. Natl. Acad. Sci. U. S. A 106: 2377–2382.
Liu, R., Lu, P., Chu, J.W., Sharom, F.J., 2009b, Characterization of fluorescent sterol binding to purified human NPC1. J. Biol. Chem. 284: 1840–1852.
Lopes, S.C., Goormaghtigh, E., Cabral, B.J., Castanho, M.A., 2004, Filipin orientation revealed by linear dichroism. Implication for a model of action. J. Am. Chem. Soc. 126: 5396–5402.
Loura, L.M., Fedorov, A., Prieto, M., 2001, Exclusion of a cholesterol analog from the cholesterol-rich phase in model membranes. Biochim. Biophys. Acta 1511: 236–243.
Macdonald, J.L., Pike, L.J., 2005, A simplified method for the preparation of detergent-free lipid rafts. J. Lipid Res. 46: 1061–1067.
MacLachlan, J., Wotherspoon, A.T., Ansell, R.O., Brooks, C.J., 2000, Cholesterol oxidase: sources, physical properties and analytical applications. J. Steroid Biochem. Mol. Biol. 72: 169–195.
Madore, N., Smith, K.L., Graham, C.H., Jen, A., Brady, K., Hall, S., Morris, R., 1999, Functionally different GPI proteins are organized in different domains on the neuronal surface. EMBO J. 18: 6917–6926.
Mahammad, S., Parmryd, I., 2008, Cholesterol homeostasis in T cells. Methyl-beta-cyclodextrin treatment results in equal loss of cholesterol from Triton X-100 soluble and insoluble fractions. Biochim. Biophys. Acta 1778: 1251–1258.
Martin, O.C., Comly, M.E., Blanchette-Mackie, E.J., Pentchev, P.G., Pagano, R.E., 1993, Cholesterol deprivation affects the fluorescence properties of a ceramide analog at the Golgi apparatus of living cells. Proc. Natl. Acad. Sci. U. S. A 90: 2661–2665.
Marzesco, A.M., Wilsch-Brauninger, M., Dubreuil, V., Janich, P., Langenfeld, K., Thiele, C., Huttner, W.B., Corbeil, D., 2009, Release of extracellular membrane vesicles from microvilli of epithelial cells is enhanced by depleting membrane cholesterol. FEBS Lett. 583: 897–902.
Mast, N., White, M.A., Bjorkhem, I., Johnson, E.F., Stout, C.D., Pikuleva, I.A., 2008, Crystal structures of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc. Natl. Acad. Sci. U. S. A 105: 9546–9551.
Mattjus, P., Bittman, R., Vilcheze, C., Slotte, J.P., 1995, Lateral domain formation in cholesterol/phospholipid monolayers as affected by the sterol side chain conformation. Biochim. Biophys. Acta 1240: 237–247.
Matyash, V., Geier, C., Henske, A., Mukherjee, S., Hirsh, D., Thiele, C., Grant, B., Maxfield, F.R., Kurzchalia, T.V., 2001, Distribution and transport of cholesterol in Caenorhabditis elegans. Mol. Biol. Cell 12: 1725–1736.
McIntosh, A.L., Gallegos, A.M., Atshaves, B.P., Storey, S.M., Kannoju, D., Schroeder, F., 2003, Fluorescence and multiphoton imaging resolve unique structural forms of sterol in membranes of living cells. J. Biol. Chem. 278: 6384–6403.
McIntyre, J.C., Sleight, R.G., 1991, Fluorescence assay for phospholipid membrane asymmetry. Biochemistry 30: 11819–11827.
Megha, B.O., London, E., 2006, Cholesterol precursors stabilize ordinary and ceramide-rich ordered lipid domains (lipid rafts) to different degrees. Implications for the Bloch hypothesis and sterol biosynthesis disorders. J. Biol. Chem. 281: 21903–21913.
Middlemas, D.S., Raftery, M.A., 1987, Identification of subunits of acetylcholine receptor that interact with a cholesterol photoaffinity probe. Biochemistry 26: 1219–1223.
Mikes, V., Milat, M.L., Ponchet, M., Ricci, P., Blein, J.P., 1997, The fungal elicitor cryptogein is a sterol carrier protein. FEBS Lett. 416: 190–192.
Mintzer, E.A., Waarts, B.L., Wilschut, J., Bittman, R., 2002, Behavior of a photoactivatable analog of cholesterol, 6- photocholesterol, in model membranes. FEBS Lett. 510: 181–184.
Mobius, W., Ohno-Iwashita, Y., van Donselaar, E.G., Oorschot, V.M., Shimada, Y., Fujimoto, T., Heijnen, H.F., Geuze, H.J., Slot, J.W., 2002, Immunoelectron microscopic localization of cholesterol using biotinylated and non-cytolytic perfringolysin O. J. Histochem. Cytochem. 50: 43–55.
Mondal, M., Mesmin, B., Mukherjee, S., Maxfield, F.R., 2009, Sterols are mainly in the cytoplasmic leaflet of the plasma membrane and the endocytic recycling compartment in CHO cells. Mol. Biol. Cell 20: 581–588.
Monnaert, V., Tilloy, S., Bricout, H., Fenart, L., Cecchelli, R., Monflier, E., 2004, Behavior of alpha-, beta-, and gamma-cyclodextrins and their derivatives on an in vitro model of blood-brain barrier. J. Pharmacol. Exp. Ther. 310: 745–751.
Morrot, G., Bureau, J.F., Roux, M., Maurin, L., Favre, E., Devaux, P.F., 1987, Orientation and vertical fluctuations of spin-labeled analogues of cholesterol and androstanol in phospholipid bilayers. Biochim. Biophys. Acta 897: 341–345.
Mukherjee, S., Chattopadhyay, A., 1996, Membrane organization at low cholesterol concentrations: a study using 7-nitrobenz-2-oxa-1,3-diazol-4-yl-labeled cholesterol. Biochemistry 35: 1311–1322.
Mukherjee, S., Zha, X., Tabas, I., Maxfield, F.R., 1998, Cholesterol distribution in living cells: fluorescence imaging using dehydroergosterol as a fluorescent cholesterol analog. Biophys. J. 75: 1915–1925.
Muller, P., Herrmann, A., 2002, Rapid transbilayer movement of spin-labeled steroids in human erythrocytes and in liposomes. Biophys. J. 82: 1418–1428.
Munro, S., 2003, Lipid rafts: elusive or illusive? Cell 115: 377–388.
Murata, M., Peranen, J., Schreiner, R., Wieland, F., Kurzchalia, T.V., Simons, K., 1995, VIP21/caveolin is a cholesterol-binding protein. Proc. Natl. Acad. Sci. U. S. A 92: 10339–10343.
Murcia, M., Faraldo-Gomez, J.D., Maxfield, F.R., Roux, B., 2006, Modeling the structure of the StART domains of MLN64 and StAR proteins in complex with cholesterol. J. Lipid Res. 47: 2614–2630.
Nabi, I.R., Le, P.U., 2003, Caveolae/raft-dependent endocytosis. J. Cell Biol. 161: 673–677.
Nelson, L.D., Johnson, A.E., London, E., 2008, How interaction of perfringolysin O with membranes is controlled by sterol structure, lipid structure, and physiological low pH: insights into the origin of perfringolysin O-lipid raft interaction. J. Biol. Chem. 283: 4632–4642.
Nemecz, G., Fontaine, R.N., Schroeder, F., 1988, A fluorescence and radiolabel study of sterol exchange between membranes. Biochim. Biophys. Acta 943: 511–521.
Nemecz, G., Schroeder, F., 1991, Selective binding of cholesterol by recombinant fatty acid binding proteins. J. Biol. Chem. 266: 17180–17186.
Ohgami, N., Ko, D.C., Thomas, M., Scott, M.P., Chang, C.C., Chang, T.Y., 2004, Binding between the Niemann-Pick C1 protein and a photoactivatable cholesterol analog requires a functional sterol-sensing domain. Proc. Natl. Acad. Sci. U. S. A 101: 12473–12478.
Ohno-Iwashita, Y., Iwamoto, M., Ando, S., Iwashita, S., 1992, Effect of lipidic factors on membrane cholesterol topology–mode of binding of theta-toxin to cholesterol in liposomes. Biochim. Biophys. Acta 1109: 81–90.
Ohtani, Y., Irie, T., Uekama, K., Fukunaga, K., Pitha, J., 1989, Differential effects of alpha-, beta- and gamma-cyclodextrins on human erythrocytes. Eur. J. Biochem. 186: 17–22.
Okamoto, Y., Ninomiya, H., Miwa, S., Masaki, T., 2000, Cholesterol oxidation switches the internalization pathway of endothelin receptor type A from caveolae to clathrin-coated pits in Chinese hamster ovary cells. J. Biol. Chem. 275: 6439–6446.
Okamura, N., Kiuchi, S., Tamba, M., Kashima, T., Hiramoto, S., Baba, T., Dacheux, F., Dacheux, J.L., Sugita, Y., Jin, Y.Z., 1999, A porcine homolog of the major secretory protein of human epididymis, HE1, specifically binds cholesterol. Biochim. Biophys. Acta 1438: 377–387.
Orci, L., Perrelet, A., Montesano, R., 1983, Differential filipin labeling of the luminal membranes lining the pancreatic acinus. J. Histochem. Cytochem. 31: 952–955.
Ottico, E., Prinetti, A., Prioni, S., Giannotta, C., Basso, L., Chigorno, V., Sonnino, S., 2003, Dynamics of membrane lipid domains in neuronal cells differentiated in culture. J. Lipid Res. 44: 2142–2151.
Palmer, C.P., Mahen, R., Schnell, E., Djamgoz, M.B., Aydar, E., 2007, Sigma-1 receptors bind cholesterol and remodel lipid rafts in breast cancer cell lines. Cancer Res. 67: 11166–11175.
Palmer, M., 2001, The family of thiol-activated, cholesterol-binding cytolysins. Toxicon 39: 1681–1689.
Pang, L., Graziano, M., Wang, S., 1999, Membrane cholesterol modulates galanin-GalR2 interaction. Biochemistry 38: 12003–12011.
Papanikolaou, A., Papafotika, A., Murphy, C., Papamarcaki, T., Tsolas, O., Drab, M., Kurzchalia, T.V., Kasper, M., Christoforidis, S., 2005, Cholesterol-dependent lipid assemblies regulate the activity of the ecto-nucleotidase CD39. J. Biol. Chem. 280: 26406–26414.
Patzer, E.J., Wagner, R.R., 1978, Cholesterol oxidase as a probe for studying membrane organisation. Nature 274: 394–395.
Pelletier, R.M., Vitale, M.L., 1994, Filipin vs enzymatic localization of cholesterol in guinea pig, mink, and mallard duck testicular cells. J. Histochem. Cytochem. 42: 1539–1554.
Persaud-Sawin, D.A., Lightcap, S., Harry, G.J., 2009, Isolation of rafts from mouse brain tissue by a detergent-free method. J. Lipid Res. 50: 759–767.
Petrescu, A.D., Gallegos, A.M., Okamura, Y., Strauss, J.F., III, Schroeder, F., 2001, Steroidogenic acute regulatory protein binds cholesterol and modulates mitochondrial membrane sterol domain dynamics. J. Biol. Chem. 276: 36970–36982.
Petrescu, A.D., Vespa, A., Huang, H., McIntosh, A.L., Schroeder, F., Kier, A.B., 2009, Fluorescent sterols monitor cell penetrating peptide Pep-1 mediated uptake and intracellular targeting of cargo protein in living cells. Biochim. Biophys. Acta 1788: 425–441.
Pipalia, N.H., Hao, M., Mukherjee, S., Maxfield, F.R., 2007, Sterol, protein and lipid trafficking in Chinese hamster ovary cells with Niemann-Pick type C1 defect. Traffic. 8: 130–141.
Prigent, D., Alouf, J.E., 1976, Interaction of steptolysin O with sterols. Biochim. Biophys. Acta 443: 288–300.
Prior, I.A., Harding, A., Yan, J., Sluimer, J., Parton, R.G., Hancock, J.F., 2001, GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat. Cell Biol. 3: 368–375.
Pucadyil, T.J., Chattopadhyay, A., 2004, Cholesterol modulates ligand binding and G-protein coupling to serotonin(1A) receptors from bovine hippocampus. Biochim. Biophys. Acta 1663: 188–200.
Pucadyil, T.J., Chattopadhyay, A., 2006, Role of cholesterol in the function and organization of G-protein coupled receptors. Prog. Lipid Res. 45: 295–333.
Pucadyil, T.J., Shrivastava, S., Chattopadhyay, A., 2005, Membrane cholesterol oxidation inhibits ligand binding function of hippocampal serotonin(1A) receptors. Biochem. Biophys. Res. Commun. 331: 422–427.
Qiu, L., Lewis, A., Como, J., Vaughn, M.W., Huang, J., Somerharju, P., Virtanen, J., Cheng, K.H., 2009, Cholesterol modulates the interaction of beta-amyloid peptide with lipid bilayers. Biophys. J. 96: 4299–4307.
Radhakrishnan, A., McConnell, H.M., 2000, Chemical activity of cholesterol in membranes. Biochemistry 39: 8119–8124.
Radhakrishnan, A., Sun, L.P., Kwon, H.J., Brown, M.S., Goldstein, J.L., 2004, Direct binding of cholesterol to the purified membrane region of SCAP: mechanism for a sterol-sensing domain. Mol. Cell 15: 259–268.
Raghuraman, H., Chattopadhyay, A., 2004, Interaction of melittin with membrane cholesterol: a fluorescence approach. Biophys. J. 87: 2419–2432.
Ramachandran, R., Heuck, A.P., Tweten, R.K., Johnson, A.E., 2002, Structural insights into the membrane-anchoring mechanism of a cholesterol-dependent cytolysin. Nat. Struct. Biol. 9: 823–827.
Reid, P.C., Sakashita, N., Sugii, S., Ohno-Iwashita, Y., Shimada, Y., Hickey, W.F., Chang, T.Y., 2004, A novel cholesterol stain reveals early neuronal cholesterol accumulation in the Niemann-Pick type C1 mouse brain. J. Lipid Res. 45: 582–591.
Reitz, J., Gehrig-Burger, K., Strauss, J.F., III, Gimpl, G., 2008, Cholesterol interaction with the related steroidogenic acute regulatory lipid-transfer (START) domains of StAR (STARD1) and MLN64 (STARD3). FEBS J. 275: 1790–1802.
Rodriguez-Agudo, D., Ren, S., Hylemon, P.B., Redford, K., Natarajan, R., Del, C.A., Gil, G., Pandak, W.M., 2005, Human StarD5, a cytosolic StAR-related lipid binding protein. J. Lipid Res. 46: 1615–1623.
Rodriguez-Agudo, D., Ren, S., Wong, E., Marques, D., Redford, K., Gil, G., Hylemon, P., Pandak, W.M., 2008, Intracellular cholesterol transporter StarD4 binds free cholesterol and increases cholesteryl ester formation. J. Lipid Res. 49: 1409–1419.
Romanowski, M.J., Soccio, R.E., Breslow, J.L., Burley, S.K., 2002, Crystal structure of the Mus musculus cholesterol-regulated START protein 4 (StarD4) containing a StAR-related lipid transfer domain. Proc. Natl. Acad. Sci. U. S. A 99: 6949–6954.
Roper, K., Corbeil, D., Huttner, W.B., 2000, Retention of prominin in microvilli reveals distinct cholesterol-based lipid micro-domains in the apical plasma membrane. Nat. Cell Biol. 2: 582–592.
Rossjohn, J., Feil, S.C., McKinstry, W.J., Tweten, R.K., Parker, M.W., 1997, Structure of a cholesterol-binding, thiol-activated cytolysin and a model of its membrane form. Cell 89: 685–692.
Rossjohn, J., Polekhina, G., Feil, S.C., Morton, C.J., Tweten, R.K., Parker, M.W., 2007, Structures of perfringolysin O suggest a pathway for activation of cholesterol-dependent cytolysins. J. Mol. Biol. 367: 1227–1236.
Rukmini, R., Rawat, S.S., Biswas, S.C., Chattopadhyay, A., 2001, Cholesterol organization in membranes at low concentrations: effects of curvature stress and membrane thickness. Biophys. J. 81: 2122–2134.
Sato, S.B., Ishii, K., Makino, A., Iwabuchi, K., Yamaji-Hasegawa, A., Senoh, Y., Nagaoka, I., Sakuraba, H., Kobayashi, T., 2004, Distribution and transport of cholesterol-rich membrane domains monitored by a membrane-impermeant fluorescent polyethylene glycol-derivatized cholesterol. J. Biol. Chem. 279: 23790–23796.
Scheidt, H.A., Muller, P., Herrmann, A., Huster, D., 2003, The potential of fluorescent and spin-labeled steroid analogs to mimic natural cholesterol. J. Biol. Chem. 278: 45563–45569.
Schroeder, F., 1984, Fluorescent sterols: probe molecules of membrane structure and function. Prog. Lipid Res. 23: 97–113.
Schroeder, F., Butko, P., Nemecz, G., Scallen, T.J., 1990, Interaction of fluorescent delta 5,7,9(11),22-ergostatetraen-3 beta-ol with sterol carrier protein-2. J. Biol. Chem. 265: 151–157.
Schroeder, F., Dempsey, M.E., Fischer, R.T., 1985, Sterol and squalene carrier protein interactions with fluorescent delta 5,7,9(11)-cholestatrien-3 beta-ol. J. Biol. Chem. 260: 2904–2911.
Schroeder, F., Frolov, A.A., Murphy, E.J., Atshaves, B.P., Jefferson, J.R., Pu, L., Wood, W.G., Foxworth, W.B., Kier, A.B., 1996, Recent advances in membrane cholesterol domain dynamics and intracellular cholesterol trafficking. Proc. Soc. Exp. Biol. Med. 213: 150–177.
Schroeder, F., Nemecz, G., Gratton, E., Barenholz, Y., Thompson, T.E., 1988, Fluorescence properties of cholestatrienol in phosphatidylcholine bilayer vesicles. Biophys. Chem. 32: 57–72.
Schroeder, F., Nemecz, G., Wood, W.G., Joiner, C., Morrot, G., yraut-Jarrier, M., Devaux, P.F., 1991, Transmembrane distribution of sterol in the human erythrocyte. Biochim. Biophys. Acta 1066: 183–192.
Schroeder, F., Woodford, J.K., Kavecansky, J., Wood, W.G., Joiner, C., 1995, Cholesterol domains in biological membranes. Mol. Membr. Biol. 12: 113–119.
Schuck, S., Honsho, M., Ekroos, K., Shevchenko, A., Simons, K., 2003, Resistance of cell membranes to different detergents. Proc. Natl. Acad. Sci. U. S. A 100: 5795–5800.
Severs, N.J., Simons, H.L., 1983, Failure of filipin to detect cholesterol-rich domains in smooth muscle plasma membrane. Nature 303: 637–638.
Shah, M.B., Sehgal, P.B., 2007, Nondetergent isolation of rafts. Methods Mol. Biol. 398: 21–28.
Shaw, A.S., 2006, Lipid rafts: now you see them, now you don’t. Nat. Immunol. 7: 1139–1142.
Shaw, J.E., Epand, R.F., Epand, R.M., Li, Z., Bittman, R., Yip, C.M., 2006, Correlated fluorescence-atomic force microscopy of membrane domains: structure of fluorescence probes determines lipid localization. Biophys. J. 90: 2170–2178.
Sheets, E.D., Holowka, D., Baird, B., 1999, Critical role for cholesterol in Lyn-mediated tyrosine phosphorylation of FcepsilonRI and their association with detergent-resistant membranes. J. Cell Biol. 145: 877–887.
Shimada, Y., Maruya, M., Iwashita, S., Ohno-Iwashita, Y., 2002, The C-terminal domain of perfringolysin O is an essential cholesterol-binding unit targeting to cholesterol-rich microdomains. Eur. J. Biochem. 269: 6195–6203.
Shrivastava, S., Haldar, S., Gimpl, G., Chattopadhyay, A., 2009, Orientation and dynamics of a novel fluorescent cholesterol analogue in membranes of varying phase. J. Phys. Chem. B 113: 4475–4481.
Sieber, J.J., Willig, K.I., Kutzner, C., Gerding-Reimers, C., Harke, B., Donnert, G., Rammner, B., Eggeling, C., Hell, S.W., Grubmuller, H., Lang, T., 2007, Anatomy and dynamics of a supramolecular membrane protein cluster. Science 317: 1072–1076.
Simons, K., Ikonen, E., 1997, Functional rafts in cell membranes. Nature 387: 569–572.
Simons, K., Toomre, D., 2000, Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1: 31–39.
Simons, K., van, M.G., 1988, Lipid sorting in epithelial cells. Biochemistry 27: 6197–6202.
Simons, M., Kramer, E.M., Thiele, C., Stoffel, W., Trotter, J., 2000, Assembly of myelin by association of proteolipid protein with cholesterol- and galactosylceramide-rich membrane domains. J. Cell Biol. 151: 143–154.
Sjogren, B., Hamblin, M.W., Svenningsson, P., 2006, Cholesterol depletion reduces serotonin binding and signaling via human 5-HT(7(a)) receptors. Eur. J. Pharmacol. 552: 1–10.
Slimane, T.A., Trugnan, G., Van IJzendoorn, S.C., Hoekstra, D., 2003, Raft-mediated trafficking of apical resident proteins occurs in both direct and transcytotic pathways in polarized hepatic cells: role of distinct lipid microdomains. Mol. Biol. Cell 14: 611–624.
Slotte, J.P., Hedstrom, G., Rannstrom, S., Ekman, S., 1989, Effects of sphingomyelin degradation on cell cholesterol oxidizability and steady-state distribution between the cell surface and the cell interior. Biochim. Biophys. Acta 985: 90–96.
Slotte, J.P., Mattjus, P., 1995, Visualization of lateral phases in cholesterol and phosphatidylcholine monolayers at the air/water interface–a comparative study with two different reporter molecules. Biochim. Biophys. Acta 1254: 22–29.
Smart, E.J., Ying, Y.S., Conrad, P.A., Anderson, R.G., 1994, Caveolin moves from caveolae to the Golgi apparatus in response to cholesterol oxidation. J. Cell Biol. 127: 1185–1197.
Smart, E.J., Ying, Y.S., Mineo, C., Anderson, R.G., 1995, A detergent-free method for purifying caveolae membrane from tissue culture cells. Proc. Natl. Acad. Sci. U. S. A 92: 10104–10108.
Smutzer, G., Crawford, B.F., Yeagle, P.L., 1986, Physical properties of the fluorescent sterol probe dehydroergosterol. Biochim. Biophys. Acta 862: 361–371.
Soltani, C.E., Hotze, E.M., Johnson, A.E., Tweten, R.K., 2007, Structural elements of the cholesterol-dependent cytolysins that are responsible for their cholesterol-sensitive membrane interactions. Proc. Natl. Acad. Sci. U. S. A 104: 20226–20231.
Song, K.S., Li, S., Okamoto, T., Quilliam, L.A., Sargiacomo, M., Lisanti, M.P., 1996, Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent-free purification of caveolae microdomains. J. Biol. Chem. 271: 9690–9697.
Sparrow, C.P., Patel, S., Baffic, J., Chao, Y.S., Hernandez, M., Lam, M.H., Montenegro, J., Wright, S.D., Detmers, P.A., 1999, A fluorescent cholesterol analog traces cholesterol absorption in hamsters and is esterified in vivo and in vitro. J. Lipid Res. 40: 1747–1757.
Spencer, T.A., Wang, P., Li, D., Russel, J.S., Blank, D.H., Huuskonen, J., Fielding, P.E., Fielding, C.J., 2004, Benzophenone-containing cholesterol surrogates: synthesis and biological evaluation. J. Lipid Res. 45: 1510–1518.
Spencer, T.A., Wang, P., Popovici-Muller, J.V., Peltan, I.D., Fielding, P.E., Fielding, C.J., 2006, Preparation and biochemical evaluation of fluorenone-containing lipid analogs. Bioorg. Med. Chem. Lett. 16: 3000–3004.
Steck, T.L., Ye, J., Lange, Y., 2002, Probing red cell membrane cholesterol movement with cyclodextrin. Biophys. J. 83: 2118–2125.
Steer, C.J., Bisher, M., Blumenthal, R., Steven, A.C., 1984, Detection of membrane cholesterol by filipin in isolated rat liver coated vesicles is dependent upon removal of the clathrin coat. J. Cell Biol. 99: 315–319.
Stoffel, W., Klotzbucher, R., 1978, Inhibition of cholesterol synthesis in cultured cells by 25-azidonorcholesterol. Hoppe Seylers. Z. Physiol Chem. 359: 199–209.
Sugii, S., Reid, P.C., Ohgami, N., Shimada, Y., Maue, R.A., Ninomiya, H., Ohno-Iwashita, Y., Chang, T.Y., 2003, Biotinylated theta toxin derivative as a probe to examine intracellular cholesterol-rich domains in normal and Niemann-pick type C1 cells. J. Lipid Res. 44: 1033–1041.
Takahashi, M., Murate, M., Fukuda, M., Sato, S.B., Ohta, A., Kobayashi, T., 2007, Cholesterol controls lipid endocytosis through Rab11. Mol. Biol. Cell 18: 2667–2677.
Tampe, R., von, L.A., Galla, H.J., 1991, Glycophorin-induced cholesterol-phospholipid domains in dimyristoylphosphatidylcholine bilayer vesicles. Biochemistry 30: 4909–4916.
Tashiro, Y., Yamazaki, T., Shimada, Y., Ohno-Iwashita, Y., Okamoto, K., 2004, Axon-dominant localization of cell-surface cholesterol in cultured hippocampal neurons and its disappearance in Niemann-Pick type C model cells. Eur. J. Neurosci. 20: 2015–2021.
Terasawa, T., Ikekawa, N., Morisaki, M., 1986, Syntheses of cholesterol analogs with a carbene-generating substituent on the side chain. Chem. Pharm. Bull. 34: 931–934.
Thiele, C., Hannah, M.J., Fahrenholz, F., Huttner, W.B., 2000, Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicles. Nat. Cell Biol. 2: 42–49.
Tserentsoodol, N., Sztein, J., Campos, M., Gordiyenko, N.V., Fariss, R.N., Lee, J.W., Fliesler, S.J., Rodriguez, I.R., 2006, Uptake of cholesterol by the retina occurs primarily via a low density lipoprotein receptor-mediated process. Mol. Vis. 12: 1306–1318.
Tsujishita, Y., Hurley, J.H., 2000, Structure and lipid transport mechanism of a StAR-related domain. Nat. Struct. Biol. 7: 408–414.
Tweten, R.K., Parker, M.W., Johnson, A.E., 2001, The cholesterol-dependent cytolysins. Curr. Top. Microbiol. Immunol. 257: 15–33.
Umashankar, M., Sanchez-San, M.C., Liao, M., Reilly, B., Guo, A., Taylor, G., Kielian, M., 2008, Differential cholesterol binding by class II fusion proteins determines membrane fusion properties. J. Virol. 82: 9245–9253.
Vainio, S., Jansen, M., Koivusalo, M., Rog, T., Karttunen, M., Vattulainen, I., Ikonen, E., 2006, Significance of sterol structural specificity. Desmosterol cannot replace cholesterol in lipid rafts. J. Biol. Chem. 281: 348–355.
Vaughan, A.M., Oram, J.F., 2005, ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid-depleted apolipoproteins. J. Biol. Chem. 280: 30150–30157.
Vincent, N., Genin, C., Malvoisin, E., 2002, Identification of a conserved domain of the HIV-1 transmembrane protein gp41 which interacts with cholesteryl groups. Biochim. Biophys. Acta 1567: 157–164.
Vrielink, A., Lloyd, L.F., Blow, D.M., 1991, Crystal structure of cholesterol oxidase from Brevibacterium sterolicum refined at 1.8 A resolution. J. Mol. Biol. 219: 533–554.
Waheed, A.A., Shimada, Y., Heijnen, H.F., Nakamura, M., Inomata, M., Hayashi, M., Iwashita, S., Slot, J.W., Ohno-Iwashita, Y., 2001, Selective binding of perfringolysin O derivative to cholesterol-rich membrane microdomains (rafts). Proc. Natl. Acad. Sci. U. S. A 98: 4926–4931.
Wang, M.M., Olsher, M., Sugar, I.P., Chong, P.L., 2004, Cholesterol superlattice modulates the activity of cholesterol oxidase in lipid membranes. Biochemistry 43: 2159–2166.
Watson, K.C., Kerr, E.J., 1974, Sterol structural requirements for inhibition of streptolysin O activity. Biochem. J. 140: 95–98.
Wiegand, V., Chang, T.Y., Strauss, J.F., III, Fahrenholz, F., Gimpl, G., 2003, Transport of plasma membrane-derived cholesterol and the function of Niemann-Pick C1 Protein. FASEB J. 17: 782–784.
Wustner, D., 2007, Plasma membrane sterol distribution resembles the surface topography of living cells. Mol. Biol. Cell 18: 211–228.
Wustner, D., Herrmann, A., Hao, M., Maxfield, F.R., 2002, Rapid nonvesicular transport of sterol between the plasma membrane domains of polarized hepatic cells. J. Biol. Chem. 277: 30325–30336.
Wustner, D., Mondal, M., Huang, A., Maxfield, F.R., 2004, Different transport routes for high density lipoprotein and its associated free sterol in polarized hepatic cells. J. Lipid Res. 45: 427–437.
Xu, S., Benoff, B., Liou, H.L., Lobel, P., Stock, A.M., 2007, Structural basis of sterol binding by NPC2, a lysosomal protein deficient in Niemann-Pick type C2 disease. J. Biol. Chem. 282: 23525–23531.
Xu, X., London, E., 2000, The effect of sterol structure on membrane lipid domains reveals how cholesterol can induce lipid domain formation. Biochemistry 39: 843–849.
Yancey, P.G., Rodrigueza, W.V., Kilsdonk, E.P., Stoudt, G.W., Johnson, W.J., Phillips, M.C., Rothblat, G.H., 1996, Cellular cholesterol efflux mediated by cyclodextrins. Demonstration of kinetic pools and mechanism of efflux. J. Biol. Chem. 271: 16026–16034.
Yeagle, P.L., 1985, Cholesterol and the cell membrane. Biochim. Biophys. Acta 822: 267–287.
Yeagle, P.L., Albert, A.D., Boesze-Battaglia, K., Young, J., Frye, J., 1990, Cholesterol dynamics in membranes. Biophys. J. 57: 413–424.
Yin, Y., Liu, P., Anderson, R.G., Sampson, N.S., 2002, Construction of a catalytically inactive cholesterol oxidase mutant: investigation of the interplay between active site-residues glutamate 361 and histidine 447. Arch. Biochem. Biophys. 402: 235–242.
Yue, Q.K., Kass, I.J., Sampson, N.S., Vrielink, A., 1999, Crystal structure determination of cholesterol oxidase from Streptomyces and structural characterization of key active site mutants. Biochemistry 38: 4277–4286.
Zhang, W., McIntosh, A.L., Xu, H., Wu, D., Gruninger, T., Atshaves, B., Liu, J.C., Schroeder, F., 2005, Structural analysis of sterol distributions in the plasma membrane of living cells. Biochemistry 44: 2864–2884.
Zidovetzki, R., Levitan, I., 2007, Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim. Biophys. Acta 1768: 1311–1324.
Acknowledgements
I thank Christa Wolpert for technical assistance and Falk Fahrenholz, Katja Gehrig-Burger, Volker Wiegand, Conny Trossen, and Julian Reitz for discussions and cooperations in cholesterol research over recent years.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Gimpl, G. (2010). Cholesterol–Protein Interaction: Methods and Cholesterol Reporter Molecules. In: Harris, J. (eds) Cholesterol Binding and Cholesterol Transport Proteins:. Subcellular Biochemistry, vol 51. Springer, Dordrecht. https://doi.org/10.1007/978-90-481-8622-8_1
Download citation
DOI: https://doi.org/10.1007/978-90-481-8622-8_1
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-90-481-8621-1
Online ISBN: 978-90-481-8622-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)